

Summary
The mammalian brain has over 10,000 types of neurons. Therefore, studying gene regulation in the brain requires effective strategies for targeting specific cell types, especially those in low abundance. Cell isolation may alter gene expression and is disruptive to mature neurons with extensive processes. This protocol describes cell-type-specific expression of tagged ribosome and the use of ribosome tagging followed by RNA-seq to identify translatome of low number and sparse cells in mouse brains without disruptive cell isolation.
For complete details on the use and execution of this protocol, please refer to Gao et al. (2020)
Subject Areas: RNA-seq, Gene expression, Neuroscience
Graphical Abstract
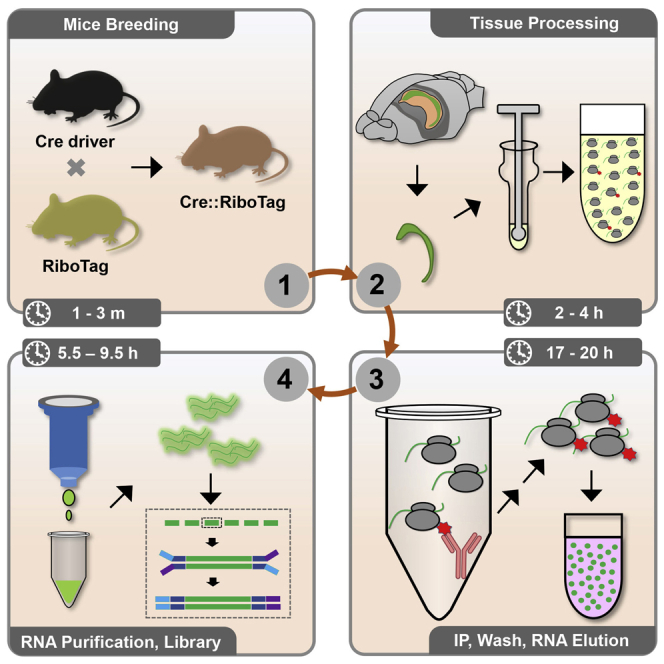
Highlights
-
•
The protocol is optimized to identify translating mRNAs in low abundance cell types
-
•
Useful in cell types that normally require pooling tissue from multiple animals
-
•
Tissue processing and immunoprecipitation are optimized for low-abundance cell types
-
•
RNA purification and library synthesis are modified for small amount of RNA
The mammalian brain has over 10,000 types of neurons. Therefore, studying gene regulation in the brain requires effective strategies for targeting specific cell types, especially those in low abundance. Cell isolation may alter gene expression and is disruptive to mature neurons with extensive processes. This protocol describes cell-type-specific expression of tagged ribosome and the use of ribosome tagging followed by RNA-seq to identify translatome of low number and sparse cells in mouse brains without disruptive cell isolation.
Before you begin
Inducing RiboTag expression in specific cell types in the brain
Timing: 1–3 months
-
1.Strategy 1: Cell-type-specific RiboTag expression using RiboTag mice (Figure 1, left)
-
a.Crossing RiboTag mice (RPL22HA/WT) with specific Cre-driver mice: Cre-lox recombination approach is commonly used as a tool for cell-type-specific gene expression. The RiboTag mice express HA-tagged ribosome protein RPL22 in Cre-expressing cells. Although this strategy is straightforward and efficient, there are several considerations in choosing a correct Cre-driver line:
-
i.Specificity of Cre expression in a given cell type: The specificity or expression pattern of Cre in the given mouse Cre-driver line is critical for the cell-type-specific gene expression profiling. Researchers must evaluate the specificity of the Cre-driver before using it. Immunostaining of Cre protein or in situ hybridization of Cre mRNA can be used to check Cre expression patterns. The researcher can also cross the Cre line with a Cre reporter mouse line (e.g., Ai14, ZsGreen1, etc.) to check the specificity of the Cre line. However, the most precise method is evaluating the RiboTag expression in double transgenic mice that the researcher plans to use for RiboTag-Seq.
-
ii.Efficiency of RiboTag expression in Cre-expressing cells: Some Cre-driver lines, especially inducible Cre (e.g., CreERT2) lines suffer low efficiency for recombination. Therefore, it is important to assess the efficiency of RiboTag expression using histology before RiboTag-Seq. In addition, since immunostaining signals do not fully reflect protein expression levels, a more precise method to evaluate the levels of RiboTag expression is quantifying immunoprecipitated RNA.
-
i.
-
b.RiboTag mice injected with virus expressing Cre: Using virus to deliver Cre to induce RiboTag expression is faster than mouse crossing. A unique advantage is that virus can be injected in a specific brain region. There are multiple options for the viruses that can be used for Cre delivery, such as retrovirus, lentivirus, and AAV.
-
a.
-
2.Strategy 2: Cell-type-specific RiboTag expression using Recombinant viruses (Figure 1, right)
-
a.Virus expressing RiboTag driven by a cell-type-specific promoter: If cell-type-specific promoter is available, this method can be used in wild type mice, rats, or other species.
-
b.Virus expressing Cre-dependent RiboTag.
-
i.If cell-type-specific promoter is unavailable but a Cre-driver mouse line is available, injecting Cre-dependent RiboTag virus in a specific Cre-driver can effectively induce ribosome tagging in a given cell type
-
ii.Brain pathway or connectivity-specific RiboTag expression: The potential presynaptic neurons can be infected by AAV expressing Cre-dependent RiboTag (e.g., DIO-RiboTag). Cre can be expressed using engineered monosynaptic retrograde rabies virus to target presynaptic neurons.
-
i.
-
a.
Figure 1.

Strategies to achieve cell-type-specific RiboTag expression
Cell-type-specific expression of RiboTag (HA-RPL22) can be achieved by either crossing transgenic mouse lines or delivering virus carrying either RiboTag or Cre into the brain.
Fast genotyping methods for Cre and/or RiboTag mice
Figure 2.

Schematic representation of the fast genotyping protocol for Cre and/or RiboTag mice
-
3.
Cut a small piece of mouse tail and put it in a DNase-free PCR tube.
CRITICAL: Minimize the amount of tissue removed. A small piece of mouse tail (around 2 mm long) would be sufficient for genotyping. Removing longer tail would stress the mouse too much.
-
4.
Add 20 μL of the QuickExtract DNA Extraction Solution and quickly spin the tube to ensure the entire tail is submerged in the extraction solution.
-
5.
Place the tube in a preheated hot-lid thermal cycler with the following program:
| Digestion program | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial digestion | 65°C | 15 min | 1 |
| Digestion | 68°C | 15 min | 1 |
| Inactivation of enzyme | 95°C | 10 min | 1 |
| Hold | 4°C | Forever | |
-
6.
Dilute the tail digestion mix by 10 times
-
7.
Prepare genotyping PCR by mixing following reagents in a DNase-free PCR tube:
| Reagent | Volume (μL) |
|---|---|
| UltraPure water (RNase and DNase free) UltraPure DNase/RNase-free distilled water | 7.2 |
| Forward primer (10 μM) | 0.4 |
| Reverse primer (10 μM) | 0.4 |
| EmeraldAmp Master Mix (2×) | 10 |
| Diluted Tail Digestion Mix | 2 |
-
8.
Run the genotyping PCR in a preheated hot-lid thermal cycler with the following program:
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 98°C | 2 min | 1 |
| Denaturation | 98°C | 10 s | 35 cycles |
| Annealing | 60°C | 30 s | |
| Extension | 72°C | 30 s | |
| Final extension | 72°C | 5 min | 1 |
| Hold | 4°C | Forever | |
-
9.
Run gel and identify the genotypes: immediately after PCR, the reaction mixture can be directly used for agarose gel electrophoresis.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| UltraPure 1 M Tris-HCI buffer, pH 7.5 | Thermo Fisher Scientific | Cat# 15567027 |
| KCl (2 M), RNase-free | Thermo Fisher Scientific | Cat# AM9640G |
| MgCl2 (1 M) | Thermo Fisher Scientific | Cat# AM9530G |
| IGEPAL CA-630 | Sigma-Aldrich | Cat# I8896 |
| DL-Dithiothreitol solution (DTT) | Sigma-Aldrich | Cat# 43816 |
| Protector RNase inhibitor | Roche | Cat# 3335402001 |
| Cycloheximide | Sigma-Aldrich | Cat# C7698 |
| Heparin | Sigma-Aldrich | Cat# H3393 |
| UltraPure DNase/RNase-free distilled water | Thermo Fisher Scientific | Cat# 10977015 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11873580001 |
| TRIzol reagent | Thermo Fisher Scientific | Cat# 15596018 |
| HBSS | Gibco | Cat# 14175103 |
| AMPure XP beads | Beckman Coulter | Cat# A63881 |
| Dynabeads Protein G for immunoprecipitation | Thermo Fisher Scientific | Cat# 10004D |
| 1 kb Plus DNA ladder | Thermo Fisher Scientific | Cat# 10787018 |
| SYBR Safe DNA gel stain | Thermo Fisher Scientific | Cat# S33102 |
| Absolute ethanol | Fisher Scientific | Cat# BP2818100 |
| Antibodies | ||
| Anti-HA.11 Epitope Tag Antibody | BioLegend | Cat# MMS-101R |
| Oligonucleotides | ||
| RiboTag forward primer (see Table 1) | Integrated DNA Technologies | N/A |
| RiboTag reverse primer (see Table 1) | Integrated DNA Technologies | N/A |
| Cre forward primer (see Table 1) | Integrated DNA Technologies | N/A |
| Cre reverse primer (see Table 1) | Integrated DNA Technologies | N/A |
| Experimental models: organisms/strains | ||
| Mouse: Tg(Nestin-cre/ERT2) | JAX | RRID:IMSR_JAX:016261 |
| Mouse: RPL22HA/WT | JAX (Sanz et al., 2009) | RRID:IMSR_JAX:029977 |
| Critical commercial assays | ||
| QuickExtract DNA extraction solution | Epicentre | Cat# QE09050 |
| EmeraldAmp GT PCR master mix | Takara Bio | Cat# RR310A |
| Direct-zol RNA Microprep Kit | Zymo Research | Cat# R2061 |
| Agilent RNA 6000 Pico Kit | Agilent Technologies | Cat# 5067-1513 |
| Agilent High Sensitivity DNA Kit | Agilent Technologies | Cat# 5067-4626 |
| Qubit dsDNA HS kit | Thermo Fisher Scientific | Cat# Q32851 |
| Pico Input Mammalian SMARTer Stranded Total RNA-Seq Kit v2 | Takara Bio | Cat# 634413 |
| Other | ||
| KONTES Dounce tissue grinders | Kimble Chase | Cat# KT885300-0002 |
| DynaMag-2 magnet | Thermo Fisher Scientific | Cat# 12321D |
| DynaMag-PCR magnet | Thermo Fisher Scientific | Cat# 492025 |
| TipOne filter tip 10 μL | USA Scientific | Cat# 1120-3810 |
| TipOne filter tip 20 μL | USA Scientific | Cat# 1120-1810 |
| TipOne filter tip 200 μL | USA Scientific | Cat# 1120-8810 |
| TipOne filter tip 1,000 μL | USA Scientific | Cat# 1120-1830 |
| PCR tube | USA Scientific | Cat# 14024700 |
| DNA LoBind tubes (1.5 mL) | Eppendorf | Cat# Z666548 |
| Refrigerated centrifuge | Eppendorf | Cat# 5430R |
| Agilent 2100 bioanalyzer | Agilent Technologies | Cat# G2939BA |
| Centrifuge | Eppendorf | Cat# 5424 |
| Qubit 2.0 fluorometer | Thermo Fisher Scientific | Cat# Q32866 |
| Veriti 96-well thermal cycler | Applied Biosystems | Cat# 4375786 |
| AirClean Systems 600 PCR Workstation | AirClean Systems | Model# AC632DB |
| Leica Microsystems 10447197 EZ4 stereo microscope | Leica | SKU# 10447197 |
Table 1.
Primers used in this protocol
| Name | Sequence (5′–3′) | Purpose |
|---|---|---|
| RiboTag Forward primer | GGGAGGCTTGCTGGATATG | RiboTag mouse genotyping (Sanz et al., 2009) |
| RiboTag reverse primer | TTTCCAGACACAGGCTAAGTACAC | RiboTag mouse genotyping (Sanz et al., 2009) |
| Cre forward primer | TTCGCAAGAACCTGATGGACATG | Cre mouse genotyping |
| Cre reverse primer | AGCATTGCTGTCACTTGGTCGTG | Cre mouse genotyping |
Materials and equipment
Preparation of stock solutions heparin (50 mg/mL)
| Reagent | Weight (g) or volume (mL) |
|---|---|
| Heparin | 1 g |
| UltraPure water (RNase and DNase free) | 20 mL |
Note: Dissolve and the solution can be stored at 4°C for at least 2 months.
Cycloheximide (5 mg/mL)
| Reagent | Weight (g) or volume (mL) |
|---|---|
| Cycloheximide | 10 mg |
| UltraPure water (RNase and DNase free) | 2 mL |
Protease inhibitors (25×)
| Reagent | Quantity or volume (mL) |
|---|---|
| cOmplete, EDTA-free Protease Inhibitor Cocktail | 1 tablet |
| UltraPure water (RNase and DNase free) | 2 mL |
Note: Dissolve, can be stored at −20°C for at least 2 weeks.
Preparation of buffers rinse buffer
| Reagent | Final concentration | Add to 50 mL |
|---|---|---|
| Tris-HCl, pH 7.5 or 7.4 (1 M) | 50 mM | 2,500 μL |
| KCl (2 M) | 100 mM | 2,500 μL |
| MgCl2 (1 M) | 12 mM | 600 μL |
| IGEPAL | 1% | 5,000 μL |
| DTT (1 M) | 1 mM | 50 μL |
| Protease inhibitors (25×) | 1× | 2,000 μL |
| Cycloheximide (5 mg/mL) | 100 μg/mL | 1,000 μL |
| Heparin (50 mg/mL) | 1 mg/mL | 1,000 μL |
| UltraPure water (RNase and DNase free) | N/A | 35,350 μL |
Sterile, on ice, before use
Homogenization buffer
| Reagent | Final concentration | Add to 50 mL |
|---|---|---|
| Tris-HCl, pH 7.5 or 7.4 (1 M) | 50 mM | 2,500 μL |
| KCl (2 M) | 100 mM | 2,500 μL |
| MgCl2 (1 M) | 12 mM | 600 μL |
| IGEPAL | 1% | 5,000 μL |
| DTT (1 M) | 1 mM | 50 μL |
| Protease inhibitors (25×) | 1× | 2,000 μL |
| Protector RNase inhibitor (40 U/μL) | 0.2 U/μL | 250 μL |
| Cycloheximide (5 mg/mL) | 100 μg/mL | 1,000 μL |
| Heparin (50 mg/mL) | 1 mg/mL | 1,000 μL |
| UltraPure water (RNase and DNase free) | N/A | 35,100 μL |
Sterile, on ice, before use
Wash buffer
| Reagent | Final concentration | Add to 50 mL |
|---|---|---|
| Tris-HCl, pH 7.5 or 7.4 (1 M) | 50 mM | 2,000 μL |
| KCl (2 M) | 300 mM | 7,500 μL |
| MgCl2 (1 M) | 12 mM | 600 μL |
| IGEPAL | 1% | 5,000 μL |
| DTT (1 M) | 1 mM | 50 μL |
| Cycloheximide (5 mg/mL) | 100 μg/mL | 1,000 μL |
| Protease inhibitors (25×) | 1× | 2,000 μL |
| UltraPure water (RNase and DNase free) | N/A | 31,350 μL |
Sterile, on ice, before use
Step-by-step method details
Tissue processing and homogenization
Figure 3.

Experimental scheme for tissue processing and homogenization
In this procedure, a specific brain region of an adult mouse (8 weeks old or older) is dissected and homogenized immediately.
-
1.
Turn on refrigerated centrifuge at least 15 min before using to allow sufficient time for cooling down.
-
2.
Make working homogenization buffer and working rinse buffer from the stock solutions (please see in the “Preparation of stock solutions” and “Preparation of Buffers”) and keep them on ice.
-
3.Rigorous wash of Dounce homogenizers (2 mL or 7 mL):
-
a)Fill the empty mortar with milliQ water. Put two pestles in two separate conical tubes, fill the tubes with milliQ water, and let them sit for 10 min at room temperature (21°C–26°C).
-
b)Wash with milliQ water for 3 times by homogenization with both loose pestles and tight pestles.
-
c)Wash once with 70% Ethanol by homogenization with both loose pestles and tight pestles.
-
d)Wash with UltraPure water for 3 times by homogenization with both loose pestles and tight pestles.
-
e)Place on ice.
-
f)Rinse with ice-cold rinse buffer right before use.
-
a)
-
4.
Discard the filled rinse buffer from the mortar, remove the rinse buffer as much as possible, fill it with 500 μL cold homogenization buffer and keep the mortar in ice-water all the time.
Note: It is important to consider the ratio of tissue to buffer. For example, 2%–3% weight per volume of tissue or 25 mg of tissue in 1 mL of buffer would be sufficient for tissue homogenization and lysis. Too much tissue would reduce homogenization and immunoprecipitation efficiency. Making the homogenate too diluted does not affect homogenization but would waste valuable reagents. Balancing cost and dilution depend on the tissue size and RiboTag expression levels.
-
5.
Bring one animal at a time to the surgical room and decapitate the animal immediately. Write down the ear tag number. Cut the tail for re-genotyping to confirm the genotype of the mouse. If live decapitation without CO2 euthanasia is not approved in your animal protocol, do a CO2 euthanasia before decapitation.
Note: We suggest adding one control animal without RiboTag expression to serve as a negative control.
Note: We highly recommend that you cut tails from the mice you dissect the brains and confirm the genotypes of the mice. The previous genotyping results might have been wrong due to various reasons.
-
6.
Quickly remove the brain out of the skull and put it into a petri dish containing ice-cold HBSS.
-
7.
Dissect out the brain region as quickly as possible under a dissecting microscope.
-
8.
Rinse the dissected tissue with ice-cold rinse buffer quickly.
-
9.
Transfer the dissected tissue into the mortar filled with cold homogenization buffer (with Protector RNase Inhibitor).
-
10.
Use a pre-cooled Dounce and a loose pestle to homogenize the tissue immediately for 20 times.
-
11.
Homogenize the tissue with a tight pestle for 20 times.
-
12.
Let the foam (generated by homogenization) and homogenization lysate go back to the bottom of the mortar by waiting 10 min.
-
13.
Transfer the lysate into a pre-cooled RNase-free microcentrifuge tube.
-
14.
Centrifuge in the pre-cooled refrigerated centrifuge, 10,000 × g, 4°C, for 10 min.
-
15.
Transfer the supernatant into another pre-cooled microcentrifuge tube and put the tube on ice.
-
16.
Repeat from step 4 to step 15 for next animal until all animals are done.
Optional: Alternatively, you can finish all animals to step 12 and then centrifuge all of them (steps 13–15) at the same time.
Immunoprecipitation, washing, and RNA elution
Figure 4.

Experimental scheme for immunoprecipitation, washing, and RNA elution
In this procedure, the HA-tagged ribosomes together with their bound translating mRNA is immunoprecipitated by using an anti-HA antibody. After washing off unbound protein and RNA, the RiboTag bound mRNA is isolated.
-
17.
Mix the lysate by inverting up and down a few times.
-
18.
Transfer 5–10 μL of the lysate to a pre-cooled microcentrifuge tube with ice-cold 100 μL TRIzol and vortex for 30 s. Save it in a −80°C freezer. This lysate can be used for isolating Input sample RNA.
-
19.
Add 3 μL of the HA antibody into the rest of the lysate and put on a rotator and rotate for 6 h.
-
20.Washing magnetic G-protein beads:
-
a)Resuspend the magnetic G-protein beads and transfer 300 μL of the beads to an RNase-free tube.
-
b)Put the tube on a magnetic stand and wait for 3–5 min until the liquid becomes clear. Remove and discard the supernatant and fill the tube with 500 μL of the rinse buffer.
-
c)Place the tube on a rotator and let it rotate for 5 min.
-
d)Repeat steps b and c one time.
-
a)
Note: To further reduce potential impact of non-specific bindings on magnetic beads, you may consider adding one negative control that uses IgG instead of HA antibody for IP.
-
21.
Place the tube containing rotated G-protein beads (from step 20) on a magnetic stand and wait for 3–5 min until it becomes clear, remove the supernatant.
-
22.
Transfer the HA antibody incubated lysate into the tube.
-
23.
Place the tube on a rotator and let it rotate at 4°C overnight (12–16 h).
-
24.
(Next day) Make fresh Wash Buffer using stock solutions.
-
25.
Cool down the wash buffer on ice. Ensure the buffer is ice-cold before use.
-
26.
Place the tube with incubated lysate on a magnetic stand, wait until the lysate become clear.
-
27.
Keep the tube on the magnetic stand and remove the supernatant.
Optional: In step 27, the supernatant can be saved in – 80°C if needed. This can be used for evaluating the IP efficiency if you are carrying out optimization experiments.
-
28.
Add 1 mL ice-cold wash buffer and rotate the tube on a rotator for 5 min.
-
29.
Repeat steps 26–28.
-
30.
Repeat steps 26 and 27.
-
31.
Add 0.5 mL ice-cold wash buffer and rotate the tube on a rotator for 5 min.
-
32.
Transfer the fully resuspended RiboTag ribosome-Ab-Beads particles into a pre-cooled tube.
-
33.
Add 0.5 mL ice-cold wash buffer to the “old” tube and invert the tube up and down to collect the residual resuspended RiboTag ribosome-Ab-Beads particles.
-
34.
Transfer the 0.5 mL wash buffer with the residual resuspended RiboTag ribosome-Ab-Beads particles and combine with the previous 0.5 mL wash buffer in the “new” tube.
-
35.
With the “new” tube, repeat steps 26–28.
-
36.
Place the tube with incubated lysate on a magnetic stand, wait until the lysate become clear.
-
37.
Remove and discard the supernatant.
-
38.
Quick mild spin (200 × g, 5 s) the tube to bring down all the residual wash buffer to the bottom of the tube.
-
39.
Place the tube on a magnetic stand, carefully remove the residual wash buffer at the bottom of the tube.
-
40.
Carefully remove the tube from the magnetic stand.
-
41.
Add 100 μL of TRIzol right onto the beads.
-
42.
Vortex for 30 s at room temperature (21°C–26°C).
-
43.
Quick spin the tube (500 × g, 5 s) to pellet the beads.
-
44.
Place the tube on a magnetic stand, wait until the supernatant becomes clear.
-
45.
Transfer the supernatant into a new pre-cooled tube and put it on ice.
-
46.
Add 50 μL of TRIzol right onto the same beads.
-
47.
Repeat steps 42–44.
-
48.
Transfer the supernatant and combine it with the previous eluted 100 μL in the “new” tube.
-
49.
Continue to the RNA purification procedure or store the eluted RNA in TRIzol at −80°C for subsequent RNA purification.
Pause Point: The eluted RNA in TRIzol can be stored at −80°C for at least 6 months (we tested).
RNA purification
Figure 5.

Experimental scheme for RNA purification
In this procedure, the immunoprecipitated RNA is purified from the RNA TRIzol elute. To purify RNA (small amount of RNA) from TRIzol elute, use column-based Direct-zol RNA Microprep Kits. We compared several kits for small-amount-RNA purification, the TRIzol based RNA purification yielded the highest quality of RNA. This is also the best way to protect the small amount of RNA from degradation. Besides, the smaller diameter of membrane in the elution column allows to elute RNA in a small volume of elutes therefore there in no need to add an extra step to concentrate the purified RNA.
-
50.
Prepare DNase I digestion mix by mixing 5 μL DNase I (provided in the Direct-zol RNA Microprep Kit, 6 U/μL) and 35 μL DNA digestion buffer in an RNase-free tube.
-
51.
Thaw the eluted RNA in TRIzol at room temperature (21°C–26°C), process to next step immediately after the sample thawed.
-
52.
Combine TRIzol elutes into one sample if needed.
-
53.
Add an equal volume of ethanol (95%–100%) to the TRIzol lysate sample and mix thoroughly.
-
54.
Transfer the mixture into a Zymo-Spin IC Column in a Collection Tube and centrifuge at 13,000 × g for 30 s, at room temperature (21°C–26°C).
-
55.
Transfer the flow-through in the collection tube back into the Zymo-Spin IC Column and centrifuge at 13,000 × g for 30 s, at room temperature (21°C–26°C).
-
56.
Add 40 μL of the DNase I digestion mix onto the center of the membrane in the column and incubate at room temperature (20°C–30°C) for 10 min.
-
57.
Wash with 400 μL Direct-zol RNA PreWash two times, then wash with 700 μL RNA Wash Buffer according to the instruction of the Direct-zol RNA Microprep Kit (https://www.zymoresearch.com/pages/direct-zol).
-
58.
To elute RNA, add 11 μL of DNase/RNase-Free Water directly onto the center of the membrane in the column and incubate for 3 min at room temperature (21°C–26°C).
-
59.
Centrifuge at 13,000 × g for 2 min.
-
60.
Transfer 1 μL of the purified RNA in a tube for determining the quality of the purified RNA (RIN score or DV200) and immediately store the rest of the purified RNA at −80°C for subsequent RNA fragmentation.
-
61.
Determine the quality of the purified RNA with an Agilent bioanalyzer with the Agilent RNA 6000 Pico Kit (https://www.agilent.com/en/product/automated-electrophoresis/bioanalyzer-systems/bioanalyzer-rna-kits-reagents/bioanalyzer-high-sensitivity-rna-analysis-228255).
RNA-seq library synthesis
In this procedure, the RNA sequencing library is synthesized. For synthesizing sncRiboTag-Seq sequencing library, the Pico Input Mammalian SMARTer Stranded Total RNA-Seq Kit v2 requires less input RNA and therefore works well for sncRiboTag-Seq. There are several other advantages of using this kit for low input RNA, which will be emphasized in following procedures. We have customized the generic manufactural instruction with several modifications.
-
62.
Preheat a hot-lid thermal cycler at 94°C, ready for use.
-
63.
Prepare the fragmentation reaction mix in a pre-cooled RNase- and DNase-free PCR tube on ice and mix well:
| Reagent | Volume (μL) |
|---|---|
| Purified RNA (250 pg–10 ng, RIN > 7) and RNase-free water | 8 |
| SMART Pico Oligos Mix v2 | 1 |
| 5× First-Strand Buffer | 4 |
Note: The sncRiboTag-Seq library synthesis allows for the use of small amount of input RNA (as less as 250 pg) and RNA with wider range of qualities: The quality of RNA purified using our procedure is normally very high (RIN > 8). Since RNA needs to be fragmented, the starting RNA can be partially degraded (RIN > 4) or even highly degraded (RIN < 4).
-
64.
Fragmentation of RNA based on the quality of the RNA (see CRITICAL). Incubate the fragmentation reaction mix at 94°C for 4 min, then place the tube on ice for 2–3 min to cool down the fragmented RNA immediately.
Note: This kit incorporates SMART (Switching Mechanism At 5′ end of RNA Template) cDNA synthesis technology and generates Illumina-compatible libraries via PCR amplification, avoiding the need for adapter ligation. The directionality of the template-switching reaction preserves the strand orientation of the original RNA, making it possible to obtain strand-specific sequencing data from the synthesized cDNA.
-
65.
Prepare the First-Strand cDNA Synthesis mix before the RNA fragmentation. Combine the following reagents in following order in a pre-cooled RNase- and DNase-free PCR tube on ice:
| Reagent | Volume (μL) |
|---|---|
| SMART TSO Mix v2 | 4.5 |
| RNase Inhibitor | 0.5 |
| SMARTScribe Reverse Transcriptase | 2 |
-
66.
Right after cooling down the fragmented RNA, add the First-Strand cDNA Synthesis mix (7 μL) and mix well, quick spin and incubate the final reaction mix in a preheated hot-lid thermal cycler with the following program:
| Reaction conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Reverse transcription | 42°C | 90 min | 1 |
| Inactivation Enzymes | 70°C | 10 min | 1 |
| Hold | 4°C | Forever | |
-
67.
Continue to the next procedure or store the cDNA (20 μL) at −20°C.
-
68.
Prepare PCR master mix to add Illumina Adapters and Indexes by mixing following reagents in a DNase-free PCR tube:
| Reagent | Volume (μL) |
|---|---|
| Nuclease-Free Water | 2 |
| SeqAmp CB PCR Buffer (2×) | 25 |
| SeqAmp DNA Polymerase | 1 |
| 5′ index oligos | 1 |
| 3′ index oligos | 1 |
-
69.
Mix the PCR master mix with the synthesized cDNA (20 μL) and place the PCR reaction mix tube in a preheated hot-lid thermal cycler to run the following program:
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 94°C | 1 min | 1 |
| Denaturation | 98°C | 15 s | 5 cycles |
| Annealing | 55°C | 15 s | |
| Extension | 68°C | 30 s | |
| Final extension | 68°C | 2 min | 1 |
| Hold | 4°C | Forever | |
-
70.
Continue to the next procedure or store the PCR product at −20°C.
-
71.
Take out AMPure beads from fridge to warm up to room temperature (it normally needs at least 30 min to warm up).
-
72.
Prepare freshly made 80% ethanol.
-
73.
Resuspend the room temperature AMPure beads completely, add 40 μL of the AMPure beads to the 50 μL synthesized library (50 μL PCR product), mix well, and incubate at room temperature (21°C–26°C) for 8 min.
-
74.
Quick spin the tube and place the tube on a magnetic stand for 8 min to separate the beads and the solution.
-
75.
Remove the supernatant while keeping the tube on the magnetic stand.
-
76.
Keep the tube on the magnetic stand, add 200 μL of freshly made 80% ethanol, wait 30 s, and then discard the 80% ethanol.
-
77.
Repeat step 76 once.
-
78.
Remove the tube from the magnetic stand and quick spin the tube.
-
79.
Put the tube back on the magnetic stand for 30 s and remove the residual ethanol.
-
80.
Keep the tube open on the magnetic stand for 5 min.
-
81.
Add 52 μL of Nuclease-Free Water onto the beads pellet and remove the tube from the magnetic stand.
-
82.
Pipetting up and down the beads 10–20 times and incubate at room temperature (21°C–26°C) for 5 min.
-
83.
Quick spin the tube, place it back on the magnetic stand and wait for 3 min.
-
84.
Carefully transfer 50 μL of the supernatant (purified synthesized library) to a new DNase-free PCR tube.
-
85.
Add 40 μL of AMPure beads to mix with 50 μL of the supernatant (purified synthesized library). Incubate at room temperature (21°C–26°C) for 8 min.
Note: Around 90% of the RNA molecules are ribosomal RNA (rRNA) in total RNA samples. Depleting rRNA from the total RNA would save costs on sequencing. With small amount of immunoprecipitated RNA, initial rRNA depletion from total RNA is not efficient and would lose significant amount of the total RNA. In contrast, depletion of ribosome cDNA after initial cDNA amplification using specific probes is well-suited for dealing with low input library synthesis.
-
86.
Place the tube with AMPure beads and synthesized library on a magnetic stand for 5 min.
-
87.
Add 1.5 μL of the R-Probes v2 in a DNase-free PCR tube, place it on a preheated hot-lid thermal cycler and run the following program:
| Reaction conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Incubation | 72°C | 2 min | 1 |
| Cooling | 4°C | 2 min | 1 |
| Ready ∗ | 4°C | 13 min | 1 |
Note: “Ready ∗”: The R-Probes are ready for use while they are at this step. The R-Probes might not function well if they are held at 4°C for either too long (after step “Ready”) or too short (before step “Ready”).
-
88.
Remove the supernatant from the tube from step 86 while keeping the tube on the magnetic stand.
-
89.
Keep the tube on the magnetic stand, add 200 μL of freshly made 80% ethanol, await 30 s, and then discard the 80% ethanol.
-
90.
Repeat step 89 once.
-
91.
Remove the tube from the magnetic stand and quick spin the tube.
-
92.
Put the tube back on the magnetic stand for 30 s and remove the residual ethanol.
-
93.
Keep the tube open on the magnetic stand for 5 min.
-
94.
Prepare the ribosomal cDNA depletion mix by combining the following reagents in the order shown and mix well by brief vortexing:
| Reagent | Volume (μL) |
|---|---|
| Nuclease-Free Water | 16.8 |
| 10× ZapR Buffer | 2.2 |
| ZapR v2 | 1.5 |
| R-Probes v2 (72°C-incubated from step 87) | 1.5 |
-
95.
Add the mixed ribosomal cDNA depletion mix onto the bead pellets in the tube from step 93.
-
96.
Remove the tube from the magnetic stand, pipet up and down 10–20 times to mix the beads, and then incubate at room temperature (21°C–26°C) for 5 min.
-
97.
Quick spin the tube, place it back on the magnetic stand and wait for 3 min.
-
98.
Carefully transfer 20 μL of the supernatant (purified synthesized library) to a new DNase-free PCR tube.
-
99.
Place the tube in a preheated hot-lid thermal cycler and run a program as follows:
| Reaction conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Incubation | 37°C | 60 min | 1 |
| Incubation | 72°C | 10 min | 1 |
| Hold | 4°C | Forever | |
-
100.
Prepare library amplification PCR master mix as follows:
| Reagent | Volume (μL) |
|---|---|
| Nuclease-Free Water | 26 |
| SeqAmp CB PCR Buffer | 50 |
| PCR2 Primers v2 | 2 |
| SeqAmp DNA Polymerase | 2 |
-
101.
Add the library amplification PCR master mix into the PCR tube from step 94 and mix well with the ribosomal cDNA depleted library. Place the final mix on a preheated hot-lid thermal cycler and run the following PCR program:
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 94°C | 1 min | 1 |
| Denaturation | 98°C | 15 s | 10–14 cycles |
| Annealing | 55°C | 15 s | |
| Extension | 68°C | 30 s | |
| Hold | 4°C | Forever | |
-
102.
Take out AMPure beads from fridge to warm them up to room temperature (21°C–26°C).
-
103.
Prepare freshly made 80% ethanol.
-
104.
Resuspend the room temperature AMPure beads completely and add 100 μL of the AMPure beads to the 100 μL amplified library (100 μL PCR product), mix well and incubate at room temperature (21°C–26°C) for 8 min.
-
105.
Quick spin the tube and place the tube on a magnetic stand for 8 min to separate the beads and the solution.
-
106.
Remove the supernatant while keeping the tube on the magnetic stand.
-
107.
Keep the tube on the magnetic stand, add 200 μL of freshly made 80% ethanol and wait 30 s, discard the 80% ethanol.
-
108.
Repeat step 107 once.
-
109.
Remove the tube from the magnetic stand and quick spin the tube.
-
110.
Put the tube back on the magnetic stand for 30 s and remove the residual ethanol.
-
111.
Keep the tube open on the magnetic stand for 5 min.
-
112.
Add 22 μL of Nuclease-Free Water onto the beads pellet and remove the tube from the magnetic stand.
-
113.
Pipetting up and down the beads 10–20 times and incubate at room temperature (21°C–26°C) for 5 min.
-
114.
Quick spin the tube, place it back on the magnetic stand and wait for 3 min.
-
115.
Carefully transfer 20 μL of the supernatant (purified synthesized library) to a new DNase-free PCR tube.
-
116.
Proceed to library validation immediately or store at –20°C
-
117.
Evaluate library size distribution by running samples on the Agilent 2100 Bioanalyzer using the Agilent High Sensitivity DNA Kit.
-
118.
Quantify libraries with Qubit dsDNA HS kit (Thermo Fisher Scientific).
Sequencing and data analysis
In this procedure, the RNA sequencing libraries are sequenced, and initial analysis is performed.
-
119.
Library Sequencing: Read 1 matches the antisense sequence of the input RNA and for paired-end sequencing; Read 2 will correspond to the sense strand.
Note: This library synthesis kit incorporates SMART (Switching Mechanism At 5′ end of RNA Template) cDNA synthesis technology and generates Illumina-compatible libraries via PCR amplification. There is no need for adapter ligation. Besides, this strategy preserves the strand orientation of the original RNA, making it possible to obtain strand-specific sequencing data from the synthesized cDNA.
-
120.
Data analysis: From the raw reads to differentially expressed genes, we used highly compacted software package RSEM for the initial data analysis.
Expected outcomes
Studying brain functions in behavior and disease is challenged by the enormous complexity of cell types in the brain with many cell types are in low abundance and sparsely distributed. Although fluorescence activated cell sorting method has been used to isolate specific cell types from the brain (Gao et al., 2017; Shen et al., 2019), tissue dissociation has been shown to cause gene expression changes (Habib et al., 2016). Isolation of nuclei may minimize the impact of cell isolation, however, extensive processes and fine synaptic terminals of mature neurons where protein translation is active are largely lost. Therefore, RiboTag-Seq, without the need for disruptive cell or nuclei isolation, has a significant advantage by preserving gene expression in native state. In addition, the RiboTag (Rpl22HA) mice with knock-in HA tag express HA-RPL22 at the endogenous levels, which minimizes the concerns for overexpression of tagged ribosomal protein, as in the case of GFP and ribosome protein L10a fusion protein (Heiman et al., 2014). Furthermore, comparing to the original RiboTag-Seq protocol (Sanz et al., 2019; Sanz et al., 2009), this sncRiboTag-Seq protocol is significantly modified for low number and sparse cells in tissue, allowing for isolation and use of very low amount of RNA [300 pg, versus 2,000 pg by Sanz et al (Sanz et al., 2019; Sanz et al., 2009)].
We expect that sncRiboTag-Seq protocol will yield high quality and consistent data. In our published paper (Gao et al., 2020), we aimed to study gene expression changes adult new neurons that exist in low numbers in the brain. We crossed RiboTag mice (Sanz et al., 2009) with neural stem cell driver Cre mice (Nestin-CreERT2) (Lagace et al., 2007; Li et al., 2018) and performed sncRiboTag-Seq. Theoretically, 0.25 ng RNA is sufficient for our RNA-seq protocol. The numbers of cells needed to obtain this amount of RNA varies depending on cell types. In our study (Gao et al., 2020), we have obtained about 0.8 ng and 4.1 ng RNA from immature and mature adult-born hippocampal neurons of one mouse, respectively (Figure 7). Based on our published papers (Guo et al., 2011; Li et al., 2016), we estimated that the number of RiboTag-expressing cells in the hippocampus of one adult Nestin-CreERT2;RiboTag mouse induced by tamoxifen injections should be about 30,000 (7–14 days after tamoxifen injection) and 40,000 (28 days after tamoxifen injection) for immature and mature new neurons, respectively. Therefore, to obtain 0.25 ng RNA, it requires around 10,000 RiboTag-expressing immature new neurons or 2,500 RiboTag-expressing mature new neurons. We decided to use pooled tissue from multiple mice, not only to increase RNA yield therefore minimize library amplification but also to reduce the contribution of variabilities among individual animals. We must point out that these calculations are based on adult-born neuron in the hippocampus. Different cell types will likely have different yield. We recommend that you use the number of cells that yield at least 1 ng immunoprecipitated RNA for the RNA-seq library construction. The HA-ribosome bound RNA (Figures 6 and 7) and the next-generation sequencing libraries (Figure 8) produced using our method were high quality. Our sequencing results demonstrated consistency across all samples (Gao et al., 2020) (Figure 9). This method proves powerful in identifying intrinsic genome-wide molecular changes in adult new neurons and can be applied to other small number of cells in mature tissues.
Figure 7.

RNA yields and RNA quality from two batches of sncRiboTag IP
The yield (per mouse) and quality (RIN) of RNA from two different batches of sncRiboTag IP of Nestin-CreERT2;RiboTag mice. RIN, RNA integrity number.
Figure 6.

Representative results for RNA quality assessment
The quality of two representative RiboTag-isolated RNA samples (A and B) was assessed by an Agilent 2100 bioanalyzer using a sensitive Eukaryote Total RNA Pico Chip.
Figure 8.

Representative results for sncRiboTag-Seq assessment
Prior to sequencing, synthesized RiboTag libraries were analyzed by an Agilent 2100 bioanalyzer. (A) and (B) show Agilent results of two representative libraries. To date, we have synthesized 57 libraries in 5 batches from two different sparse cell types, including adult-born new neurons in the dentate gyrus of adult mouse hippocampus (Gao et al., 2020) and parvalbumin-expressing interneurons in dorsal hippocampus, ventral hippocampus, and prefrontal cortex of adult mice (unpublished). All our libraries have met the requirements for next-generation sequencing and yielded publishable data.
Figure 9.

sncRiboTag-seq yields sufficient and consistent result without batch effect
The number of detected transcripts that have FPKM over 0.1 (FPKM > 0.1, A) or 1 (FPKM > 1, B) among 3 different experiment batches. Therefore, there was no strong batch effect among all the IP samples, even though the IP and library synthesis were processed at 3 different times using different animals (Gao et al., 2020).
(A) Using our sncRiboTag-Seq protocol, we were able to detect around 17,600 transcripts (FPKM > 0.1) in each sample and the number of detected transcripts was similar among three different experimental batches, as described in our published study (Gao et al., 2020).
(B) The number of detected transcripts that have FPKM over 1 (FPKM > 1) is around 13,000 and similar number of transcripts was also found for three different experimental batches.
Limitations
There are at least three limitations of this protocol. First, the cell-type-specific RiboTag expression is heavily dependent on Cre expression patterns. Therefore, highly specific and efficient Cre-driver mouse line or promoters for viral vectors is needed to target RiboTag to specific cell type. Second, although sncRiboTag-Seq protocol allows scientists to profile translatome in specific cell-type cells, it cannot reveal the diversities of individual cells within the cell type. However, advanced bioinformatics methods can be applied to integrate sncRiboTag-Seq data with other genome-wide data at single cell and bulk cell levels to unveil novel information in biology that cannot be obtained using an individual method. Third, similar to other immunoprecipitation-based experiments, the immunoprecipitated RNA may contain non-specific contaminations. Therefore, it is important to perform preliminary experiments to optimize the IP condition. We advise that you perform preliminary experiments using two negative controls: IP from mice without Cre expression (only PRL22) and IP from RiboTag or wild-type (WT) mice using IgG instead of HA antibodies. We believe that the former is a more important negative control. To optimize this protocol, we performed IP on both PV-Cre;RiboTag and WT mice (negative control) using the same HA antibody. The IP from WT mouse yielded RNA (0.14 ng), but it was far lower than the RNA yielded from RiboTag mouse (19.46 ng) (Figure 10). In addition, the quality of RNA from WT IP was very poor (RIN=1.0, highly degraded) compared to the RNA from PV-Cre;RiboTag mouse (RIN=8.9, high quality) (Figure 10). In step 64 (Fragmentation of RNA), high quality mRNA will be fragmented into appropriate sizes, however, the low-quality background mRNA molecules are expected to be fragmented into even smaller RNA fragments, from which the smaller DNA fragments would be generated and these small DNA fragment are expected to be removed in the subsequent three rounds of purifications using AMPure beads (because the purifications using AMPure beads removes DNA fragments smaller than 250 bp) (see first round of purification: steps 71–84, second round of purification: steps 85–98, and third round of purification: steps 102–116). Therefore, even if this IP protocol yielded some background RNA, these RNA would likely to be cleaned out from the libraries before sequencing.
Figure 10.

sncRiboTag- IP of negative control tissue yields small quantity and low-quality background RNA
(A and B) sncRiboTag IP of frontal cortex tissue from parvalbumin (PV)-expressing interneurons of adult PV-Cre;RiboTag mice yields 19.46 ng high quality RNA (RIN = 8.9, A). The analysis of input RNA from the same mouse (B) is provided as a comparison.
(C and D) sncRiboTag IP of frontal cortex tissues from wild-type (WT) mice yields small amount (C, 0.14 ng) and low-quality RNA (C, RIN =1.0). The analysis of input RNA from WT mice (D) is provided as a comparison.
Troubleshooting
Problem 1
During genotyping, the negative control shows positive (step 9 in the part “Fast Genotyping Methods for Cre and/or RiboTag Mice”).
Potential solution
This is gDNA contamination. To solve this problem, re-dilute the digested tail mix, use new tubes of EmeraldAmpMaster Mix, primers, and water, and repeat the genotyping PCR.
Problem 2
Extremely low yield of RNA and the RIN values of the purified RNA below 7 (step 61).
Potential solution
There are a number of possible reasons and potential solutions:
The tissue you isolated may not have RiboTag expression. Check the RiboTag expression by staining with HA antibody to ensure that RiboTag is expressed in the tissues that you plan to use for IP. In addition, make sure your genotyping result is correct and any false positive gel bands for either RiboTag, or Cre would cause this.
For tissue processing, ensure the homogenization is efficient, if possible, check the homogenate under a microscope to make sure there is no tissue chunk, otherwise, homogenize more times.
During tissue processing and homogenization, always keep tissues cold to prevent RNA degradation.
Test the HA antibody used for immunoprecipitation to make sure your antibody works for immunoprecipitation.
Check the expire date of the RNA purification kit and ensure it is not expired when you use it.
Ensure the working area and equipment are RNase-free and use RNaseZap Wipes to clean the working bench, pipettes, tip boxes, etc.
Use filter tips or barrier tips.
It is possible that low RNA reading is caused by any mistakes during Agilent Bioanalyzer analysis. You may repeat it together with an RNA preparation as positive control.
Problem 3
Failed library synthesis (steps 117 and 118).
Potential solution
Check the input RNA used for library synthesis and make sure you used sufficient amount (>250 pg) and quality of RNA. Partially degraded RNA can work but requires optimization. Check the degradation level of your RNA and perform fragmentation with corresponding fragmentation time.
For library purification steps, make sure you use warmed-up purification beads and make sure the beads are within the expiration date.
Check whether you used reasonable number of PCR amplification, add more PCR cycles if needed.
Problem 4
Too few transcripts are detected from RNA-seq (step 120).
Potential solution
Check your sequencing depth. If too low, you might consider re-sequence to obtain more reads.
Make sure you have done at least two QC steps: (1) QC step 1: check quality of the isolated RNA; (2) QC step 2: check the quantity and profiles of your synthesized sequencing libraries.
Problem 5
Too many duplicate reads (step 120).
Potential solution
Increase the amount of input RNA. You can increase RNA yield by pooling tissues from more animals.
Perform fewer number of PCR amplifications.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xinyu Zhao (xinyu.zhao@wisc.edu).
Materials availability
No material was generated in this protocol.
Data and code availability
No data or code generated in this protocol. The data used in this protocol are from our published study (Gao et al., 2020)
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01NS105200, R56MH113146, R01MH116582, and R21NS095632 to X.Z.; U54HD090256 to the Waisman Center), UW Vilas Trust (Mid-Career Award), Wisconsin Alumni Research Foundation, and Jenni and Kyle Professorship (to X.Z.). We thank S. Kannan, J. Hoang, Y. Xing, R. Spitzer, D. Wagner, A. Meara, M. Syed, X. Lyu, S. Pham, J. Le, and Y. Choi for technical assistance during optimization of this protocol; J. Pinnow, M. Eastwood, D. Bolling, and K. Knobel at the Waisman Intellectual and Developmental Disabilities (IDD) Model Core; and S. Splinter-BonDurant at the University of Wisconsin (UW)-Madison Biotechnology Center for next-generation sequencing services.
Author contributions
X.Z. conceived the concept. Y.G. designed and performed experiments, collected data, and analyzed data. Y.G. and X.Z. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Yu Gao, Email: yu.gao@wisc.edu.
Xinyu Zhao, Email: xinyu.zhao@wisc.edu.
References
- Gao Y., Shen M., Gonzalez J.C., Dong Q., Kannan S., Hoang J.T., Eisinger B.E., Pandey J., Javadi S., Chang Q. RGS6 mediates effects of voluntary running on adult hippocampal neurogenesis. Cell Rep. 2020;32:107997. doi: 10.1016/j.celrep.2020.107997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Wang F., Eisinger B.E., Kelnhofer L.E., Jobe E.M., Zhao X. Integrative single-cell transcriptomics reveals molecular networks defining neuronal maturation during postnatal neurogenesis. Cereb. Cortex. 2017;27:2064–2077. doi: 10.1093/cercor/bhw040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Allan A.M., Zong R., Zhang L., Johnson E.B., Schaller E.G., Murthy A.C., Goggin S.L., Eisch A.J., Oostra B.A. Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat. Med. 2011;17:559–565. doi: 10.1038/nm.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N., Li Y., Heidenreich M., Swiech L., Avraham-Davidi I., Trombetta J.J., Hession C., Zhang F., Regev A. Div-Seq: single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 2016;353:925–928. doi: 10.1126/science.aad7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M., Kulicke R., Fenster R.J., Greengard P., Heintz N. Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP) Nat. Protoc. 2014;9:1282–1291. doi: 10.1038/nprot.2014.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagace D.C., Whitman M.C., Noonan M.A., Ables J.L., DeCarolis N.A., Arguello A.A., Donovan M.H., Fischer S.J., Farnbauch L.A., Beech R.D. Dynamic contribution of Nestin-expressing stem cells to adult neurogenesis. J. Neurosci. 2007;27:12623–12629. doi: 10.1523/JNEUROSCI.3812-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Stockton M.E., Bhuiyan I., Eisinger B.E., Gao Y., Miller J.L., Bhattacharyya A., Zhao X. MDM2 inhibition rescues neurogenic and cognitive deficits in a mouse model of fragile X syndrome. Sci. Transl. Med. 2016;8:336ra361. doi: 10.1126/scitranslmed.aad9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Stockton M.E., Eisinger B.E., Zhao Y., Miller J.L., Bhuiyan I., Gao Y., Wu Z., Peng J., Zhao X. Reducing histone acetylation rescues cognitive deficits in a mouse model of Fragile X syndrome. Nat. Commun. 2018;9:2494. doi: 10.1038/s41467-018-04869-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E., Bean J.C., Carey D.P., Quintana A., McKnight G.S. RiboTag: ribosomal tagging strategy to analyze cell-type-specific mRNA expression in vivo. Curr. Protoc. Neurosci. 2019;88:e77. doi: 10.1002/cpns.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E., Yang L., Su T., Morris D.R., McKnight G.S., Amieux P.S. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. U S A. 2009;106:13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M., Wang F., Li M., Sah N., Stockton M.E., Tidei J.J., Gao Y., Korabelnikov T., Kannan S., Vevea J.D. Reduced mitochondrial fusion and Huntingtin levels contribute to impaired dendritic maturation and behavioral deficits in Fmr1-mutant mice. Nat. Neurosci. 2019;22:386–400. doi: 10.1038/s41593-019-0338-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data or code generated in this protocol. The data used in this protocol are from our published study (Gao et al., 2020)