

Abstract
Farnesoid X receptor (FXR, encoded by NR1H4), a bile acid‐activated nuclear receptor, is widely implicated in human tumorigenesis. The FXR agonist obeticholic acid (OCA) has preliminarily displayed tumour suppressor potential. However, the anticancer effects of this agent on colorectal cancer (CRC) remain unclear. In this study, the treatment of colon cancer cells with OCA inhibited cell proliferation and invasion in vitro, retarded tumour growth in vivo and prevented the G0/G1 to S phase transition. Moreover, the expression of active caspase‐3, p21 and E‐cadherin was up‐regulated and the expression of cyclin D1, c‐Myc, vimentin, N‐cadherin and MMP9 was down‐regulated in OCA‐treated colon cancer cells. Mechanistic studies indicated that OCA treatment suppressed the activity of JAK2/STAT3 pathway by up‐regulating SOCS3 expression. Colivelin, an agonist of JAK2/STAT3 pathway, antagonized the tumour‐suppressive effect of OCA on colon cancer cells. Dual‐luciferase reporter and quantitative chromatin immunoprecipitation (qChIP) assays further confirmed that OCA promoted SOCS3 transcription by enhancing the binding of FXR to the FXRE/IR9 of the SOCS3 promoter. In conclusion, our study demonstrates that targeting FXR and improving its function might be a promising strategy for CRC treatment.
Keywords: colorectal cancer, Farnesoid X receptor, JAK2/STAT3 signalling, obeticholic acid, SOCS3
1. INTRODUCTION
Colorectal cancer (CRC) is the second most common cause of cancer‐related death. 1 Globally, approximately 1 800 000 new cases are diagnosed as CRC every year. Due to distant metastasis and relapse, most advanced‐stage CRC indicates a poor prognosis. 2 , 3 The 5‐year survival rate of stage I CRC in patients exceeds 90%; however, the rate of stage IV CRC in patients is slightly higher than 10%. 4 There has recently been substantial progress in multimodality therapy for CRC. Chemotherapy is the second major treatment type for CRC after surgical treatment. However, there are many problems with chemotherapy, such as drug resistance and side effects. 5 Thus, more effective strategies and novel targets for chemotherapy in this malignancy are urgently required.
Epidemiological studies and animal studies have suggested that prolonged exposure to faecal bile acids contributes to the occurrence of inflammatory bowel disease (IBD) and gastrointestinal cancer. 6 Increased amounts of bile acids generate an excess of detrimental injury on colonic mucosa including ROS‐induced DNA damage, 7 inflammation 8 and hyperproliferation. 9 Farnesoid X receptor (FXR, encoded by NR1H4), a bile acid‐activated nuclear receptor, regulates metabolism homeostasis of lipid, cholesterol and glucose. 10 Mounting evidence supports a pivotal role for FXR in human tumorigenesis. FXR exerts tumour‐promoting effect in oesophageal 11 and pancreatic carcinomas. 12 However, some studies conclude the opposite. FXR‐deficient mice are susceptible to developing hepatocellular carcinoma (HCC). 13 Diminished FXR correlates with late tumour stage and often predicts a poor prognosis. 14 The deficiency of FXR promotes intestinal inflammation and colon tumorigenesis. 15 Conversely, activation of intestinal FXR can suppress abnormal cell growth and curtail CRC progression. 16 Thus, targeting FXR and restoring its function might be an attractive tactics for CRC treatment.
Obeticholic acid (OCA) is a novel FXR agonist, a derivative of chenodeoxycholic acid (CDCA), and shows almost 10‐fold greater potency than CDCA. 17 Importantly, OCA has been approved by the US Food and Drug Administration (FDA) for the treatment of primary biliary cholangitis. 18 Recent studies indicate that OCA inhibits the malignant potential of cholangiocarcinoma 19 and HCC. 20 However, the anticancer effect of this agent on colon cancer cells remains to be elucidated. Herein, we evaluated the impact of OCA on the aggressive phenotype of colon cancer cells and further investigated the underlying mechanisms.
2. MATERIALS AND METHODS
2.1. Cell cultures
Colon cancer cells HT‐29 and Caco‐2 (Shanghai Institute of Cell Biology, Chinese Academy of Sciences) were all routinely cultured in DMEM supplemented with 10% foetal bovine serum at 5% CO2 at 37°C. Once the cells reached 70% confluence, they were pre‐treated with colivelin (0.5 µM) or interleukin‐6 (IL‐6, 10 ng/mL) for 6 hours and then were cultured with various doses of OCA (0, 1, 2 and 4 µM) for 48 hours.
2.2. Lentiviral vectors and transfection
The phU6‐EGFP‐shRNA‐SOCS3 lentiviral vectors and their control vectors were constructed and prepared by GeneChem Co., Ltd. All transfections were performed according to the manufacturer's instructions.
2.3. Cell growth curve, CCK8, colony formation, cell cycle and apoptosis assays
Cell growth curve and CCK8 assays were performed as described previously. 21 For colony formation assay, three hundred cells were seeded and cultured for 14 days. Colonies (≥50 cells/colony) were counted. Cell cycle distributions were evaluated by flow cytometry as previously described. 21 For apoptosis assay, cells were labelled with Annexin V PE/7‐AAD (BD Biosciences) according to the manufacturer's protocol as previously described. 22 Each experiment was performed in triplicate.
2.4. Wound‐healing assays
Cells were cultured in 6‐well plates until confluent. Then, 3 artificial vertical lines were created with pipette tips (10 µL) in each well. The wells were washed with phosphate‐buffered saline (PBS) to remove cell debris. The cells were then cultured for an additional 48 hours. The scratch lines were imaged under a microscope, and the scratch distances were measured. Each experiment was performed in triplicate.
2.5. Transwell assays
Cell migration and invasion were measured by using Transwell plates (Corning) with or without Matrigel (BD). In both assays, the lower chamber was filled with 600 μL of RPMI 1640 medium containing 20% FBS. For the migration assay, the cells were trypsinized and plated at 5 × 104 cells per upper chamber. The cells were incubated at 37°C for 24 hours. For the invasion assay, the upper chamber filters were pre‐coated with 50 µL of Matrigel and plated at 10 × 104 cells per upper chamber. The cells were incubated at 37°C for 48 hours. After incubation, non‐migratory cells on the upper surface of the Transwell inserts were removed by washing with fresh PBS. The migratory or invading cells on the underside of the membrane were fixed with 4% paraformaldehyde and stained with 1% crystal violet. The number of cells was counted in three randomly selected fields of fixed cells under an inverted microscope. Each experiment was performed in triplicate.
2.6. Nude mouse xenograft assay
All animal experiments were performed in accordance with the institutional guidelines and were approved by the Laboratory Animal Center of Xi'an Jiaotong University. The 5‐week‐old female BALB/c‐nude mice were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). The mice were injected with 5 × 106 HT‐29 and Caco‐2 cells into the right flanks to establish xenograft tumour model. Once the palatable xenograft tumours were established, the nude mice were treated by oral gavage of OCA (10 mg/kg/d) or DMSO solution. Tumour size was monitored using callipers every 3 days, and the tumour volume was calculated according to the formula (length × width2 × 0.5). At the end of the experiment, the mice were killed and the xenograft tumours were isolated and weighted.
2.7. RNA isolation and real‐time PCR
RNA isolation, complementary DNA (cDNA) synthesized and real‐time PCR were performed as described previously. 21 The sequences of primers were summarized in Supplementary Table S1. Each experiment was performed in triplicate.
2.8. Immunohistochemistry
The protocol was performed as previously described. 21 The extent of Ki67‐ and active caspase‐3‐stained cells was divided into 4 score ranks: 0%‐5% (0), 6% to 25% (1), 26% to 50% (2), 51% to 75% (3) and 76%‐100% (4). The staining intensity was divided into 4 score ranks: negative (0), light brown (1), brown (2) and dark brown (3). The immunoreactivity scores (IRSs) = extent score × intensity score. An IRS of ≤3 was defined as negative, and a score of >3 was defined as positive.
2.9. Total Protein extraction and Western blot
The detailed protocol was performed as described previously. 21 The antibody information was presented in Supplementary Table S2. Each experiment was performed in triplicate.
2.10. Immunofluorescence and immunocytochemistry
For Immunofluorescence (IF), the cells were fixed with 4% paraformaldehyde for 20 minutes and permeabilized with 0.2% Triton X‐100 for 10 minutes. After blocking with 5% bovine serum albumin (BSA) for 30 minutes at room temperature, the cells were incubated at 4°C overnight with primary antibodies against E‐cadherin, vimentin and STAT3 (1:100 dilution). The dishes were washed three times with PBS for 10 minutes each and then incubated with Alexa Fluor 594‐conjugated secondary antibodies (1:400 dilution; Invitrogen) for 1 hour at room temperature. The nuclei were stained with DAPI (10 μg/mL) for 10 minutes. The samples were examined via microscopy (Leica Microsystems) to analyse the expression of E‐cadherin and vimentin and the nuclear translocation of STAT3.
For immunocytochemistry (ICC), the cells were fixed with 4% paraformaldehyde for 20 minutes and permeabilized with 0.2% Triton X‐100 for 10 minutes and then incubated with the primary antibodies against E‐cadherin and vimentin. The following protocol is the same as the Immunohistochemistry (IHC) assay.
2.11. Luciferase reporter assay
For promoter analyses, a fragment of the SOCS3 5′‐flanking sequence (from −3182 bp to + 425 bp) and other truncated fragments were cloned into the luciferase reporter vector pGL3.0 Basic Vector (Promega) to generate a SOCS3 full promoter reporter construct and the truncated ones. The primers for plasmid constructs were presented in Supplementary Table S1. The detailed protocol was performed as described previously. 21 Each experiment was performed in triplicate.
2.12. Quantitative chromatin immunoprecipitation
Cells were subjected to ChIP using the EZ‐ChIP Kit (Millipore).
The detailed protocol was performed as described previously. 21 Real‐time PCR was conducted to amplify the regions of DNA fragments by using special primers (Supplementary Table S1). Each experiment was performed in triplicate.
2.13. Statistical analysis
The differences among the groups were compared by Student's t‐test or one‐way ANOVA. All statistical analyses were performed using the SPSS statistical package (SPSS Inc, Chicago, IL, USA). P < .05 was considered statistically significant.
3. RESULTS
3.1. FXR activation inhibits the proliferation of colon cancer cells in vitro
The effect of FXR activation on tumour cell viability and growth was assessed by cell growth curve and CCK8 assays. The results showed that OCA treatment dose dependently inhibited the viability and growth of HT‐29 and Caco‐2 cells in relation to the control (Figure 1A,B). This inhibitory effect was further confirmed by the colony formation assay, in which OCA treatment dose‐dependent reduced the colony formation (Figure 1C,D).
Figure 1.
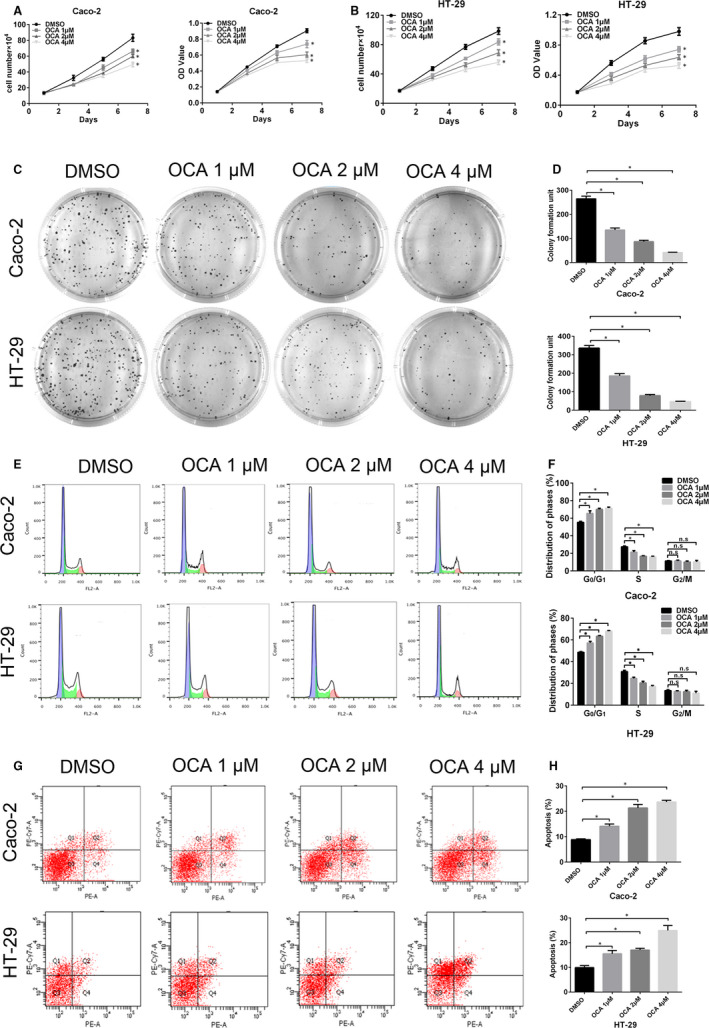
Farnesoid X receptor (FXR) activation inhibits the proliferation of colon cancer cells by inducing cell cycle arrest and apoptosis. A and B, Effect of different doses of obeticholic acid (OCA) on the growth and viability of Caco‐2 (A) and HT‐29 (B) cells detected by cell growth and CCK8 assays. C, Results of colony formation assays for Caco‐2 and HT‐29 cells after OCA exposure for 14 d. D, Quantification of the cell colonies. E, FACS analysis of the cell cycle distribution of Caco‐2 and HT‐29 cells after OCA exposure. F, Comparison of the cell cycle distribution. G, FACS analysis of PE/APC staining for apoptosis in Caco‐2 and HT‐29 cells after OCA exposure. H, Quantification of apoptotic cells. *P < .05
As cell proliferation inhibition was observed after OCA treatment, cell cycle distribution and apoptotic state were assessed by flow cytometry assay. OCA treatment resulted in a dose‐dependent accumulation of cells in the G0/G1 phase with a decrease in cells in the S phase (Figure 1E,F). However, OCA had no significant impact on the accumulation of cells in the G2 phase. Moreover, exposure to OCA dose dependently increased the apoptotic rate of colon cancer cells (Figure 1G,H).
We then examined the impact of OCA treatment on the key proteins associated with G1/S transition and apoptosis. Western blot analysis showed that OCA treatment dose dependently elevated the levels of p21 and active caspase‐3 and reduced the levels of cyclin D1 and c‐Myc (Figure S1A‐D). Altogether, these data suggested that FXR activation retards colon cancer cell proliferation by preventing G1/S transition and inducing apoptosis.
3.2. FXR activation retards the tumour growth in nude mice
Next, xenograft mouse model was conducted to assess the effect of FXR activation on tumour growth in vivo. The xenograft tumours in the OCA‐treated group showed a decline in growth rate in relative to the DMSO‐treated group (Figure 2A,B,D,E). Moreover, mean weight of xenograft tumours in the OCA‐treated group is lighter than that in the DMSO‐treated group (Figure 2C,F). Ki67 and active caspase‐3 are well‐known markers evaluating cellular proliferation and apoptosis. Thus, we detected Ki67 and active caspase‐3 expression in xenograft tumours by using IHC staining. The xenograft tumour tissues in the OCA‐treated group displayed weaker Ki67 (Figure 2G,H) and stronger caspase‐3 staining intensity (Figure 2I,J) than those in the DMSO‐treated group. Collectively, our data indicated that FXR activation retards the tumour growth in nude mice.
Figure 2.
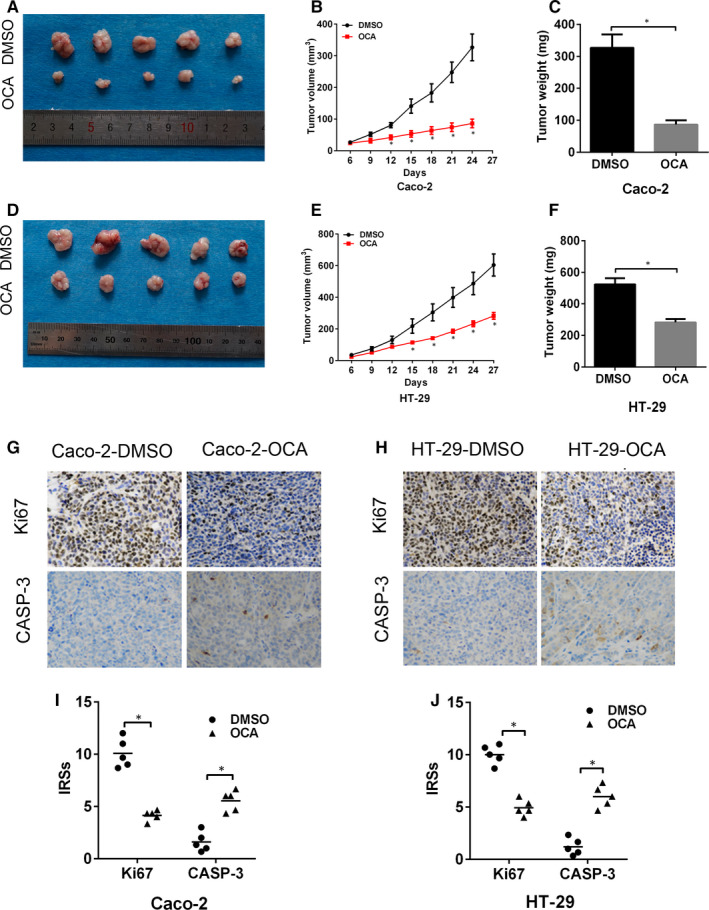
Farnesoid X receptor (FXR) activation retards the tumour growth in nude mice. A, Image of xenograft tumours formed by Caco‐2 cells after obeticholic acid (OCA) treatment. B and C, Tumour growth curves (B) and tumour weights (C) for Caco‐2 cells after OCA treatment. D, Image of xenograft tumours formed by HT‐29 cells after OCA treatment E and F, Tumour growth curves (E) and tumour weights (F) for HT‐29 cells after OCA treatment. G and H, Immunohistochemical staining for Ki67 (G) and active caspase‐3 (H) in tumour xenografts of Caco‐2 (B) cells. I and J, Immunohistochemical staining for Ki67 (I) and active caspase‐3 (J) in tumour xenografts of HT‐29 cells. *P < .05
3.3. FXR activation inhibits the invasion and migration of colon cancer cells by arresting EMT
Considering that tumour metastasis is the leading cause of cancer‐related death in CRC, we thus aimed to evaluate the impact of FXR activation on the invasive and migratory abilities of colon cancer cells. The wound‐healing assay showed that OCA treatment resulted in a decreased rate of wound healing (Figure 3A,B). The Transwell assays showed that the number of invasive and migrating cells in the OCA‐treated group was less than that in the DMSO‐treated group (Figure 3C,D). Epithelial‐mesenchymal transition (EMT) has been linked to the mobility and dissemination of CRC by conferring increased invasiveness and metastatic potential to cells. 23 , 24 Our study further assessed the levels of EMT‐related genes by using real‐time PCR and Western blotting analysis. OCA treatment increased the mRNA and protein levels of E‐cadherin and reduced vimentin, N‐cadherin and MMP9 levels (Figure S2A‐F). Furthermore, ICC (Figure S3A,B) and IF analyses (Figure S3C‐F) showed that OCA‐treated cells had a stronger E‐cadherin staining and a weaker vimentin staining than DMSO‐treated cells. Collectively, our data indicated that FXR activation inhibits the invasive and migratory ability of colon cancer cells by arresting EMT.
Figure 3.
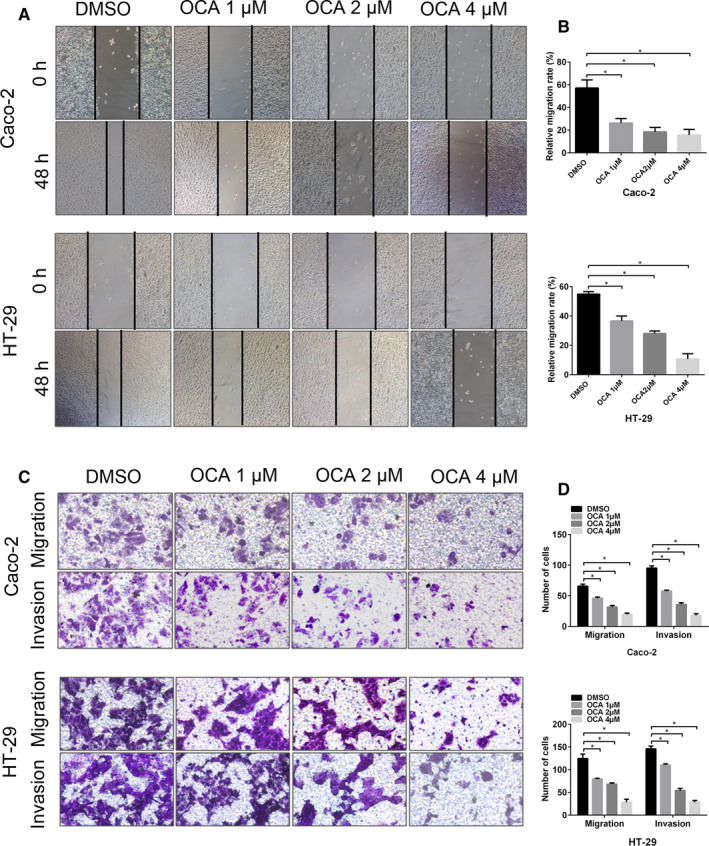
Farnesoid X receptor (FXR) activation inhibits the invasion and migration of colon cancer cells in vitro. A, Wound‐healing assay in Caco‐2 and HT‐29 cells after obeticholic acid (OCA) exposure measured at 48 h. B, The percentage of wound healing in Caco‐2 and HT‐29 cells. C, Invasion and migration assays in Caco‐2 and HT‐29 cells after OCA exposure. D, The number of invasive and migrating cells in Caco‐2 and HT‐29 cells. *P < .05
3.4. FXR activation suppresses JAK2/STAT3 pathway by up‐regulating SOCS3 expression in colon cancer cells
The Janus tyrosine kinases/signal transducer and activator of transcription (JAK/STAT) pathway, which is negatively regulated by the SOCS family, plays a crucial role in cellular responses by regulating survival, proliferation, invasion and differentiation. 25 , 26 Our study attempted to detect the impact of FXR activation on the activity of JAK/STAT signalling. OCA treatment inhibited the protein expression of p‐STAT3 and p‐JAK2 (Figure 4A,B). However, OCA treatment had no significant impact on phosphorylated JAK1 expression (Figure 4A,B). Furthermore, we found that OCA treatment resulted in an up‐regulation of SOCS3 expression at both mRNA and protein levels (Figure 4A,B). To further validate the involvement of SOCS3 in OCA‐mediated inhibition of JAK/STAT signalling, we knocked down SOCS3 in colon cancer cells. Depleting SOCS3 expression restored the activity of JAK/STAT signalling in OCA‐treated cells (Figure 4D,E).
Figure 4.
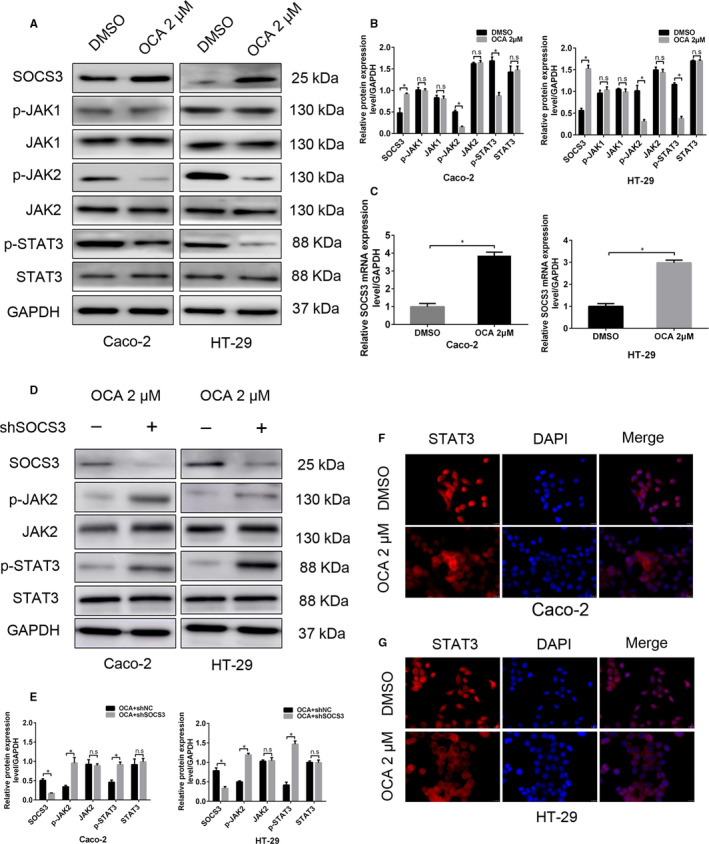
Farnesoid X receptor (FXR) activation suppresses JAK2/STAT3 pathway by up‐regulating SOCS3 expression in colon cancer cells. A, Western blot bands of the critical proteins of JAK/STAT pathway in Caco‐2 and HT‐29 cells after obeticholic acid (OCA) exposure. B, Quantitative analysis of the critical proteins of JAK/STAT pathway in Caco‐2 and HT‐29 cells. C, Real‐time PCR of SOCS3 levels in Caco‐2 and HT‐29 cells after OCA exposure. D, Western blot bands of the critical proteins of JAK/STAT pathway in OCA‐treated Caco‐2 and HT‐29 cells upon SOCS3 knockdown. E, Quantitative analysis of the critical proteins of JAK/STAT pathway in Caco‐2 and HT‐29 cells. F and G, Immunofluorescence staining of STAT3 in Caco‐2 (F) and HT‐29 (G) after OCA exposure. *P < .05
Phosphorylated STATs dimerize and translate into the nucleus to regulate target gene transcription. 27 Thus, we attempted to assess the localization of STAT3 protein by using IF staining. Exposure to OCA resulted in increased STAT3 protein accumulation in the cell cytoplasm (Figure 4F,G). Taken together, these results suggest that FXR activation might suppress JAK2/STAT3 pathway in colon cancer cells by up‐regulating SOCS3 expression.
It has been demonstrated that FXR usually modulates target genes transcription by specifically binding to FXR response element (FXRE). 28 We employed the dual‐luciferase reporter assay to assess the impact of FXR activation on SOCS3 transcription. FXR‐activating cells transfected with the full‐length SOCS3 promoter displayed a high luciferase intensity in relative to the control cells (Figure 5A,B). However, OCA treatment did not result in the change in the luciferase intensity of the other truncated fragments compared with the control, suggesting that FXR activation could transcriptionally activate SOCS3 expression through binding to the −3182 bp to −2834 bp region of the SOCS3 promoter in colon cancer cells.
Figure 5.
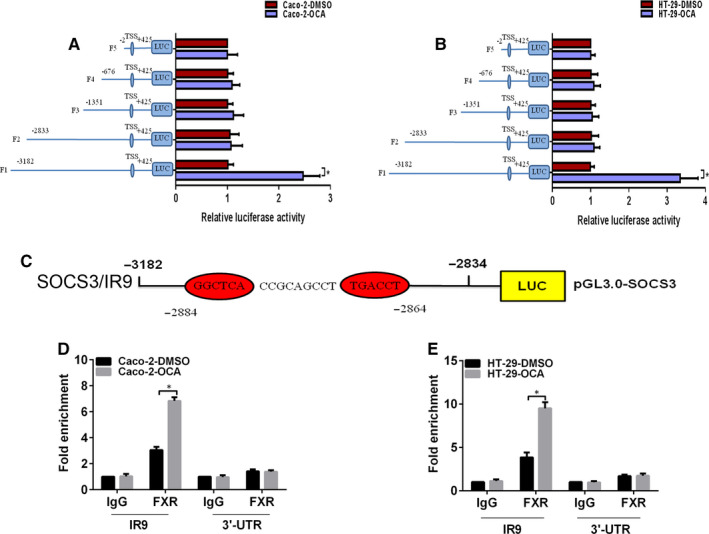
Farnesoid X receptor (FXR) transactivates the expression of SOCS3 by binding to the FXRE/IR9 in the SOCS3 promoter in colon cancer cells. A and B, The activities of the full SOCS3 promoter reporter construct and the truncated ones in Caco‐2 (A) and HT‐29 (B) cells after obeticholic acid (OCA) exposure using the dual‐luciferase assay. C, Schematic representation of a putative FXRE in the human SOCS3 promoter region (SOCS3/IR9). Enrichment level of FXR binding to the putative FXRE in the SOCS3 promoter region in Caco‐2 (D) and HT‐29 (E) cells determined by the qChIP assay. *P < .05
Next, we attempted to search for the putative binding site of FXR in the SOCS3 promoter by using JASPAR database. A potential FXRE/IR9 (an inverted repeat spaced by nine nucleotides, −2884 bp to −2864 bp) was identified in the −3182 bp to −2834 bp region of the SOCS3 promoter (Figure 5C). We conducted the Quantitative chromatin immunoprecipitation (qChIP) assay to determine whether OCA treatment facilitates the binding of FXR to the FXRE/IR9 of the SOCS3 promoter. As expected, an enrichment of FXR binding to the FXRE/IR9 was observed in OCA‐treated cells in relative to the control cells (Figure 5D,E). All of these results indicate that FXR activation inhibits JAK2/STAT3 pathway by regulating SOCS3 transcription in colon cancer cells.
3.5. Activation of JAK2/STAT3 signalling rescues the tumour‐suppressive effect of FXR activation
In order to determine that JAK2/STAT3 signalling is indispensable for the tumour‐suppressive effect of FXR activation on colon cancer cells, colivelin and IL‐6, the agonists of JAK/STAT3 pathway, 29 , 30 were adopted to treat FXR‐activated cells. IL‐6 is a classic STAT3 activator, while colivelin, a potent humanin (HN) derivative, has recently been proved to activate JAK2/STAT3 signalling via binding to cell surface receptors involving CNTFRα, WSX‐1 and gp130. 30 In the current study, colivelin and IL‐6 rescued the proliferative and invasive abilities inhibited by FXR activation (Figure 6A‐F, Figure 7A,B). Consistent with the observations above, colivelin and IL‐6 up‐regulated the protein levels of p‐STAT3, p‐JAK2, vimentin, c‐Myc and cyclin D1 but reduced the level of p21, active CASP‐3 and E‐cadherin (Figure 7C‐F). Taken together, these results demonstrate that JAK2/STAT3 signalling participates in OCA‐mediated tumour inhibition.
Figure 6.
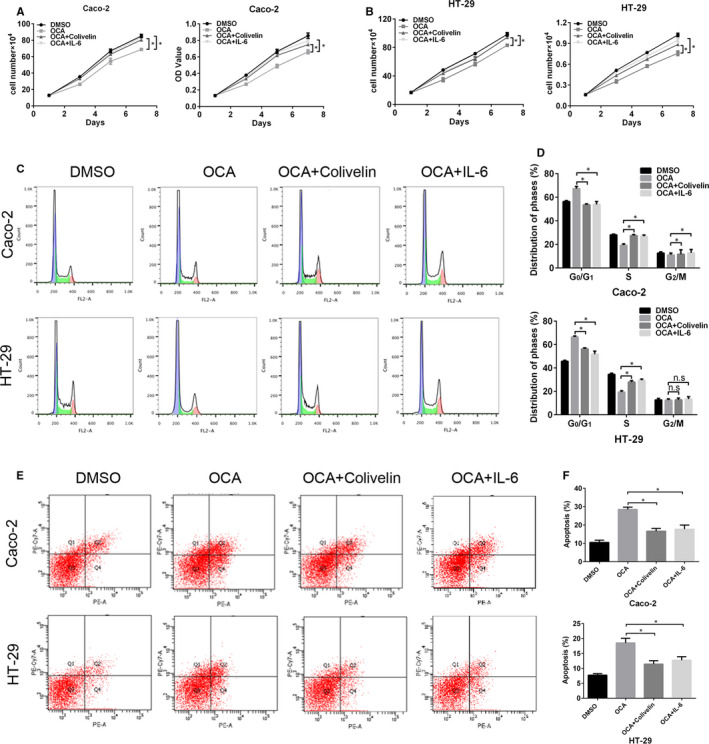
Activation of the JAK2/STAT3 pathway rescues the inhibitory effect of Farnesoid X receptor (FXR) activation on the proliferation of colon cancer cells. A and B, The effect of colivelin and IL‐6 on the growth and viability of FXR‐activated Caco‐2 (A) and HT‐29 (B) cells detected by cell growth and CCK8 assays. C, FACS analysis of the cell cycle distribution of FXR‐activated Caco‐2 and HT‐29 cells after colivelin and IL‐6 exposure. D, Comparison of the cell cycle distribution. E, FACS analysis of PE/APC staining for apoptosis in FXR‐activated Caco‐2 and HT‐29 cells after colivelin and IL‐6 exposure. F, Quantification of apoptotic cells. *P < .05
Figure 7.
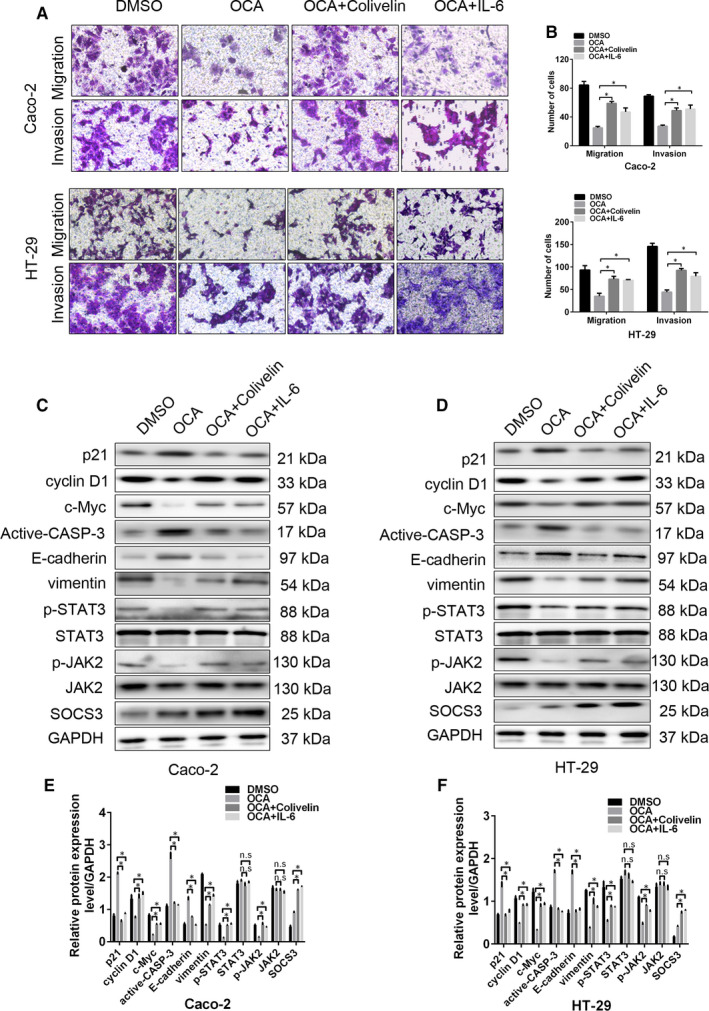
Activation of the JAK2/STAT3 pathway rescues the inhibitory effect of Farnesoid X receptor (FXR) activation on the growth‐, apoptosis‐ and EMT‐related proteins of colon cancer cells. A, The effect of colivelin and IL‐6 on the invasion of FXR‐activated Caco‐2 and HT‐29 detected by invasion and migration assays. B, The number of invasive and migrating cells in Caco‐2 and HT‐29 cells. C and D, Western blot bands of the growth‐, apoptosis‐ and EMT‐related proteins in FXR‐activated Caco‐2 (C) and HT‐29 (D) cells after colivelin and IL‐6 exposure. E and F, Quantitative analysis of the growth‐, apoptosis‐ and EMT‐related proteins in Caco‐2 (E) and HT‐29 (F) cells. *P < .05
4. DISCUSSION
Recently, due to the limited effectiveness of conventional therapeutic strategies, there has been a strong interest in the field of novel antitumour drugs. These novel drugs characterized by the good markers available are the major goals of molecular oncology. The tumour‐suppressive role of FXR in colorectal tumorigenesis has inspired us to restore FXR activity as a novel therapeutical strategy. Our study indicated that FXR activation by OCA inhibited the proliferative potential of colon cancer cells by arresting G1/S transition and inducing apoptosis. In support of these data, treatment with OCA significantly altered the levels of growth‐ and apoptosis‐related proteins. FXR agonist GW4064 has been reported to trigger apoptosis and retard tumour growth by activating its target SHP. 31 Moreover, restoring FXR expression inhibited cyclin D1 and c‐Myc levels by repressing the Wnt/β‐catenin signalling. 32
Epithelial‐mesenchymal transition is a biological process that involves the morphological transformation of epithelial cells, with the cells acquiring mesenchymal‐like and motile phenotypes. 33 OCA treatment impaired the invasive and migratory potential of colon cancer cells by arresting EMT. Inconsistent with the result of our work, Kainuma M et al reported that treatment of HCC with OCA treatment promoted EMT phenotypes, 34 although FXR may act as a tumour suppressor at the early stage of HCC through the regulation of hepatic inflammatory responses. 20 Collectively, our study for the first time reported the tumour‐suppressive effect of OCA on CRC.
The highly malignant behaviour of colon cancer cells is closely associated with the activity of JAK/STAT pathway. 35 Our data indicated that OCA treatment repressed the activation of JAK2/STAT3 pathway in colon cancer cells, which was further confirmed by IF staining, showing increased levels of STAT3 protein located in the cell cytoplasm. In addition, restoration of JAK2/STAT3 signalling by colivelin and IL‐6 abolished OCA‐mediated proliferation and invasion inhibition. Intriguingly, the concentration of colivelin used in CRC and oesophageal cancer 36 in vitro is much higher than that used in neurodegenerative disease, which may reflect that the regulatory mechanism antagonizing JAK/STAT signalling exists in different disease models. FXR deficiency in HCC results in a high activity of STAT3. 37 Following this activation step, phosphorylated STATs dimerize and translate into the nucleus to regulate the transcription of target genes related to CRC progression.
SOCS3, an important member of the SOCS family, negatively regulates JAK/STAT pathway. 38 SOCS3 expression is reduced in CRC tissues due to promoter methylation. 39 Lack of SOCS3 contributes to accelerative intestinal crypt growth and facilitates tumour growth. 40 In the current study, OCA treatment increased the mRNA and protein levels of SOCS3. Similarly, restoration of SOCS3 by FXR agonist GW4064 is observed in hepatocellular inflammation and HCC, 41 , 42 indicating that FXR‐SOCS3 axis may serve as a new potential target for the prevention and treatment of HCC. Furthermore, our study revealed that OCA treatment could promote SOCS3 transcription by binding to the −3182 bp to −2834 bp of the SOCS3 promoter, as determined by the dual‐luciferase reporter assay. Nuclear hormone receptors regulate gene transcription by recognizing the motif consisting of repeats with the core element AGGTCA aparted by one or more nucleotides. 43 We predicted a potential FXRE/IR9 (−2884 bp to −2864 bp) in the −3182 bp to −2834 bp of the SOCS3 promoter by using the JASPAR database. The qChIP assay demonstrated that FXR specifically and directly binds to the FXRE/IR9 of SOCS3 promoter. Some pharmacological compounds have been shown to repress STAT3 activity by restoring SOCS3 expression. 44 , 45 , 46 Manipulation of SOCS3 expression via the regulatory role of FXR might be expected to be a promising therapeutic option for cancer chemotherapy. Intriguingly, the activation of FXR by GW4064 decreased the expression of HER2, the upstream of JAK/STAT3 pathway. 47 Besides JAK/STAT signalling, OCA might exert tumour suppressor role by directly regulating FXR target genes SHP 31 and MMP7 48 or suppressing Wnt/β‐catenin 49 and EGFR/ERK signalling. 50 Hence, OCA exerts its tumour suppressor functions via multiple molecular mechanisms.
In the present study, the FXR agonist OCA inhibited colon cancer cell proliferation and invasion by repressing JAK2/STAT3 pathway via regulating SOCS3 transcription. Our study provided the first evidence that FXR activation by OCA may represent a potential novel therapeutic strategy for CRC treatment.
CONFLICT OF INTEREST
All authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Shan Li: Project administration (equal); Writing‐original draft (lead); Writing‐review & editing (equal). Zhengshui Xu: Project administration (equal); Software (lead); Writing‐review & editing (equal). Jing Guo: Methodology (equal); Project administration (equal); Software (lead); Validation (lead). Jianbao Zheng: Funding acquisition (equal); Resources (equal); Writing‐review & editing (equal). Xuejun Sun: Conceptualization (equal); Funding acquisition (lead); Supervision (lead). Junhui Yu: Conceptualization (lead); Methodology (lead); Project administration (equal); Supervision (equal); Writing‐original draft (equal); Writing‐review & editing (lead).
Supporting information
Supplementary Material
Figure S1
Figure S2
Figure S3
ACKNOWLEDGEMENTS
This work was funded by a grant from the National Natural Science Foundation of China (Grant Serial Numbers: 81972720, 81101874, 81172362), the Coordinative and Innovative Plan Projects of the Science and Technology Program in Shaanxi Province (Grant Serial Numbers: 2013KTCQ03‐08), The Science and Technology Project of Shaanxi Province (Grant serial number: 2016SF‐015, 2019SF‐065) and The Fundamental Research Funds for the Central Universities (Grant serial number: xjj2018123).
Li S, Xu Z, Guo J, Zheng J, Sun X, Yu J. Farnesoid X receptor activation induces antitumour activity in colorectal cancer by suppressing JAK2/STAT3 signalling via transactivation of SOCS3 gene. J Cell Mol Med. 2020;24:14549–14560. 10.1111/jcmm.16083
Contributor Information
Xuejun Sun, Email: sunxy@mail.xjtu.edu.cn.
Junhui Yu, Email: sunxy@mail.xjtu.edu.cn, Email: 13571850623@163.com.
DATA AVAILABILITY STATEMENT
The data used to support the findings of this study are included within the article.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Coppede F, Lopomo A, Spisni R, Migliore L. Genetic and epigenetic biomarkers for diagnosis, prognosis and treatment of colorectal cancer. World J Gastroenterol. 2014;20:943‐956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mei Z, Liu Y, Liu C, et al. Tumour‐infiltrating inflammation and prognosis in colorectal cancer: systematic review and meta‐analysis. Br J Cancer. 2014;110:1595‐1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490‐1502. [DOI] [PubMed] [Google Scholar]
- 5. Gerhards NM, Rottenberg S. New tools for old drugs: functional genetic screens to optimize current chemotherapy. Drug Resist Updat. 2018;36:30‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jia W, Xie G, Jia W. Bile acid‐microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol. 2018;15:111‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rosignoli P, Fabiani R, De Bartolomeo A, Fuccelli R, Pelli MA, Morozzi G. Genotoxic effect of bile acids on human normal and tumour colon cells and protection by dietary antioxidants and butyrate. Eur J Nutr. 2008;47:301‐309. [DOI] [PubMed] [Google Scholar]
- 8. Chen ML, Takeda K, Sundrud MS. Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol. 2019;12:851‐861. [DOI] [PubMed] [Google Scholar]
- 9. Moschetta A, Portincasa P, van Erpecum KJ, Debellis L, Vanberge‐Henegouwen GP, Palasciano G. Sphingomyelin protects against apoptosis and hyperproliferation induced by deoxycholate: potential implications for colon cancer. Dig Dis Sci. 2003;48:1094‐1101. [DOI] [PubMed] [Google Scholar]
- 10. Chavez‐Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, Type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. 2017;152(7):1679‐1694.e3. [DOI] [PubMed] [Google Scholar]
- 11. Guan B, Li H, Yang Z, Hoque A, Xu X. Inhibition of farnesoid X receptor controls esophageal cancer cell growth in vitro and in nude mouse xenografts. Cancer. 2013;119:1321‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Macha MA, Rachagani S, Gupta S, et al. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling. Cancer Lett. 2013;341:166‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu N, Meng Z, Lou G, et al. Hepatocarcinogenesis in FXR‐/‐ mice mimics human HCC progression that operates through HNF1alpha regulation of FXR expression. Mol Endocrinol. 2012;26:775‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Selmin OI, Fang C, Lyon AM, et al. Inactivation of adenomatous polyposis coli reduces Bile Acid/Farnesoid X receptor expression through Fxr gene CpG methylation in mouse colon tumors and human colon cancer cells. J Nutr. 2016;146:236‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maran RR, Thomas A, Roth M, et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther. 2009;328:469‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fu T, Coulter S, Yoshihara E, et al. FXR regulates intestinal cancer stem cell proliferation. Cell. 2019;176(5):1098‐1112.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Markham A, Keam SJ. Obeticholic acid: first global approval. Drugs. 2016;76:1221‐1226. [DOI] [PubMed] [Google Scholar]
- 18. Ali AH, Lindor KD. Obeticholic acid for the treatment of primary biliary cholangitis. Expert Opin Pharmacother. 2016;17:1809‐1815. [DOI] [PubMed] [Google Scholar]
- 19. Erice O, Labiano I, Arbelaiz A, et al. Differential effects of FXR or TGR5 activation in cholangiocarcinoma progression. Biochim Biophys Acta. 2018;1864:1335‐1344. [DOI] [PubMed] [Google Scholar]
- 20. Attia YM, Tawfiq RA, Ali AA, Elmazar MM. The FXR agonist, obeticholic acid, suppresses HCC proliferation & metastasis: role of IL‐6/STAT3 signalling pathway. Sci Rep. 2017;7:12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu J, Liu D, Sun X, et al. CDX2 inhibits the proliferation and tumor formation of colon cancer cells by suppressing Wnt/beta‐catenin signaling via transactivation of GSK‐3beta and Axin2 expression. Cell Death Dis. 2019;10:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu J, Li S, Qi J, et al. Cleavage of GSDME by caspase‐3 determines lobaplatin‐induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qi L, Sun B, Liu Z, Cheng R, Li Y, Zhao X. Wnt3a expression is associated with epithelial‐mesenchymal transition and promotes colon cancer progression. J Exp Clin Cancer Res: CR. 2014;33:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang JX, Mai SJ, Huang XX, et al. MiR‐29c mediates epithelial‐to‐mesenchymal transition in human colorectal carcinoma metastasis via PTP4A and GNA13 regulation of beta‐catenin signaling. Ann Oncol. 2014;25:2196‐2204. [DOI] [PubMed] [Google Scholar]
- 25. Chang R, Song L, Xu Y, et al. Loss of Wwox drives metastasis in triple‐negative breast cancer by JAK2/STAT3 axis. Nat Commun. 2018;9:3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin L, Cao L, Liu Y, et al. B7–H3 promotes multiple myeloma cell survival and proliferation by ROS‐dependent activation of Src/STAT3 and c‐Cbl‐mediated degradation of SOCS3. Leukemia. 2019;33:1475‐1486. [DOI] [PubMed] [Google Scholar]
- 27. Abroun S, Saki N, Ahmadvand M, Asghari F, Salari F, Rahim F. STATs: an old story. Yet mesmerizing. Cell J. 2015;17:395‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Plass JR, Mol O, Heegsma J, et al. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology (Baltimore, MD). 2002;35:589‐596. [DOI] [PubMed] [Google Scholar]
- 29. Zhao H, Feng Y, Wei C, et al. Colivelin rescues ischemic neuron and axons involving JAK/STAT3 signaling pathway. Neuroscience. 2019;416:198‐206. [DOI] [PubMed] [Google Scholar]
- 30. Chiba T, Yamada M, Sasabe J, et al. Amyloid‐beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol Psychiatry. 2009;14:206‐222. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y, Soto J, Park K, et al. Nuclear receptor SHP, a death receptor that targets mitochondria, induces apoptosis and inhibits tumor growth. Mol Cell Biol. 2010;30:1341‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U. Increased activation of the Wnt/beta‐catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther. 2011;338:12‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial‐mesenchymal transitions in development and disease. Cell. 2009;139:871‐890. [DOI] [PubMed] [Google Scholar]
- 34. Kainuma M, Takada I, Makishima M, Sano K. Farnesoid X receptor activation enhances transforming growth factor β‐induced epithelial‐mesenchymal transition in hepatocellular carcinoma cells. Int J Mol Sci. 2018;19:1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ren R, Sun H, Ma C, Liu J, Wang H. Colon cancer cells secrete exosomes to promote self‐proliferation by shortening mitosis duration and activation of STAT3 in a hypoxic environment. Cell Biosci. 2019;9:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chiu WC, Lee YC, Su YH, et al. The synthetic β‐Nitrostyrene derivative CYT‐Rx20 inhibits esophageal tumor growth and metastasis via PI3K/AKT and STAT3 pathways. PLoS One. 2016;11:e0166453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li G, Zhu Y, Tawfik O, et al. Mechanisms of STAT3 activation in the liver of FXR knockout mice. Am J Physiol Gastrointest Liver Physiol. 2013;305:G829‐G837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Inagaki‐Ohara K, Kondo T, Ito M, Yoshimura A. SOCS, inflammation, and cancer. Jak‐stat. 2013;2:e24053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Y, Deuring J, Peppelenbosch MP, Kuipers EJ, de Haar C, van der Woude CJ. IL‐6‐induced DNMT1 activity mediates SOCS3 promoter hypermethylation in ulcerative colitis‐related colorectal cancer. Carcinogenesis. 2012;33:1889‐1896. [DOI] [PubMed] [Google Scholar]
- 40. Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage‐induced crypt hyper‐proliferation and inflammation‐associated tumorigenesis in the colon. Oncogene. 2007;26:4833‐4841. [DOI] [PubMed] [Google Scholar]
- 41. Xu Z, Huang G, Gong W, et al. FXR ligands protect against hepatocellular inflammation via SOCS3 induction. Cell Signal. 2012;24:1658‐1664. [DOI] [PubMed] [Google Scholar]
- 42. Guo F, Xu Z, Zhang Y, et al. FXR induces SOCS3 and suppresses hepatocellular carcinoma. Oncotarget. 2015;6:34606‐34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269‐1304. [DOI] [PubMed] [Google Scholar]
- 44. Liang P, Cheng SH, Cheng CK, et al. Platelet factor 4 induces cell apoptosis by inhibition of STAT3 via up‐regulation of SOCS3 expression in multiple myeloma. Haematologica. 2013;98:288‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu JH, Kim KH, Kim H. SOCS 3 and PPAR‐gamma ligands inhibit the expression of IL‐6 and TGF‐beta1 by regulating JAK2/STAT3 signaling in pancreas. Int J Biochem Cell Biol. 2008;40:677‐688. [DOI] [PubMed] [Google Scholar]
- 46. Patel K, Kollory A, Takashima A, Sarkar S, Faller DV, Ghosh SK. MicroRNA let‐7 downregulates STAT3 phosphorylation in pancreatic cancer cells by increasing SOCS3 expression. Cancer Lett. 2014;347:54‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Giordano C, Catalano S, Panza S, et al. Farnesoid X receptor inhibits tamoxifen‐resistant MCF‐7 breast cancer cell growth through downregulation of HER2 expression. Oncogene. 2011;30:4129‐4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Peng Z, Chen J, Drachenberg CB, Raufman JP, Xie G. Farnesoid X receptor represses matrix metalloproteinase 7 expression, revealing this regulatory axis as a promising therapeutic target in colon cancer. J Biol Chem. 2019;294:8529‐8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu J, Li S, Guo J, Xu Z, Zheng J, Sun X. Farnesoid X receptor antagonizes Wnt/β‐catenin signaling in colorectal tumorigenesis. Cell Death Dis. 2020;11:640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peng Z, Raufman JP, Xie G. Src‐mediated cross‐talk between farnesoid X and epidermal growth factor receptors inhibits human intestinal cell proliferation and tumorigenesis. PLoS One. 2012;7:e48461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Figure S1
Figure S2
Figure S3
Data Availability Statement
The data used to support the findings of this study are included within the article.