

Abstract
Injury of the skin from exposure to toxic chemicals leads to the release of inflammatory mediators and the recruitment of immune cells. Nitrogen mustard (NM) and other alkylating agents cause severe cutaneous damage for which there are limited treatment options. Here, we show that combined treatment of vitamin D3 (VD3) and spironolactone (SP), a mineralocorticoid receptor antagonist, significantly improves the resolution of inflammation and accelerates wound healing after NM exposure. SP enhanced the inhibitory effect of VD3 on nuclear factor‐kB activity. Combined treatment of NM‐exposed mice with VD3 and SP synergistically inhibited the expression of iNOS in the skin and decreased the expression of matrix metallopeptidase‐9, C‐C motif chemokine ligand 2, interleukin (IL)‐1α, and IL‐1β. The combined treatment decreased the number of local proinflammatory M1 macrophages resulting in an increase in the M2/M1 ratio in the wound microenvironment. Apoptosis was also decreased in the skin after combined treatment. Together, this creates a proresolution state, resulting in more rapid wound closure. Combined VD3 and SP treatment is effective in modulating the immune response and activating anti‐inflammatory pathways in macrophages to facilitate tissue repair. Altogether, these data demonstrate that VD3 and SP may constitute an effective treatment regimen to improve wound healing after NM or other skin chemical injury.
Keywords: vitamin D, spironolactone, nitrogen mustard, skin injury
Our results demonstrate that vitamin D3 in combination with spironolactone treatment improves wound healing after nitrogen mustard (NM) exposure by modulating the immune response and activating anti‐inflammatory pathways. In addition to supportive treatment, this may expand the currently limited treatment options to improve clinical outcomes for NM injury.
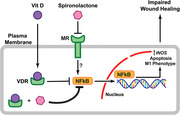
Introduction
Nitrogen mustard (NM) is an alkylating agent used as a chemotherapy drug for the treatment of skin lymphoma. 1 NM is closely related to sulfur mustard, a chemical warfare agent used during World War I and in the modern era. 2 Toxic exposure to mustard leads to irreversible DNA alkylation and cell death, resulting in skin and mucous membrane blistering, as well as pulmonary injury and airway edema. 3 , 4 As there are no effective therapies for mustard‐induced injury, more efficacious treatments addressing the underlying cutaneous inflammation causing morbidity and mortality after mustard exposure are of significant clinical relevance. 5 , 6
Our laboratory has previously shown that treatment with vitamin D (VD) significantly decreases skin inflammation caused by exposure to NM or ultraviolet (UV) radiation. 7 , 8 , 9 , 10 , 11 Vitamin D3 (VD3), the predominant form of VD in humans, can be produced in the skin after UV exposure or be derived from animal‐based foods. Activation of VD is tightly regulated and requires two separate enzymatic reactions, first by the liver to produce 25‐hydroxyvitamin D (25(OH)D) and the second by the kidney to produce 1,25‐dihydroxycholecalciferol (1,25(OH)2D or calcitriol), the biologically active form of VD.
The VD receptor (VDR) is ubiquitously expressed throughout the body. Traditionally, the role of VD has been described in governing mineral metabolism and bone health, but recent research had demonstrated the importance of VD in a wide variety of physiological processes, including cell proliferation and differentiation, cardiovascular disease, and modulation of immune response. 12 , 13 , 14 This is supported by the findings that in addition to the kidney, extrarenal cells of myeloid origin are also capable of producing active VD. 13 The response evoked by VD in immune cells relies, at least in part, on the inhibition of the nuclear factor‐κB (NF‐κB) pathway. 15 , 16 , 17 In our previous studies, we found that VD suppresses macrophage‐mediated inducible nitric oxide synthase (iNOS) production and enhances autophagy in anti‐inflammatory M2 macrophages. 7 , 11 This leads to a pronounced anti‐inflammatory response that improves wound healing and facilitates tissue repair.
In this study, we investigated whether the addition of spironolactone (SP) to VD leads to greater skin wound repair, given its purported anti‐inflammatory role in other organ systems. Drugs, such as SP and eplerenone (EP), are mineralocorticoid receptor (MR) antagonists and have been demonstrated to inhibit inflammation by decreasing inflammatory cytokines, regulating ion channels expression and activity, and reducing tissue edema. 18 , 19 , 20 The use of SP for acute injury and wound repair in the skin has not been well characterized. Recently, topical inhibition of MR by SP was shown to rescue delayed reepithelialization and impaired healing in a model induced by the glucocorticoid agonist clobetasol. 21 , 22
Given their anti‐inflammatory properties, we hypothesized that coadministration of VD and SP could synergistically reduce inflammation and improve wound healing after NM exposure. Our findings demonstrate that the combined action of VD and SP significantly improves skin repair by modulating the immune response, decreasing proinflammatory cytokines expression, and decreasing apoptosis.
Materials and methods
RAW‐DualTM cell culture and treatment
RAW‐DualTM (IRF‐Lucia/KI‐[MIP‐2]SEAP) murine macrophage reporter cells were commercially purchased from InvivoGen and cultured according to the suppliers protocol in Dulbecco's modified Eagle's medium (DMEM, containing high glucose, 110 mg/L sodium pyruvate, and 584.0 mg/L l‐glutamine) (Gibco) supplemented with 10% heat‐inactivated fetal bovine serum (FBS) (Gibco), 1% Pen‐Strep (HyClone), and 100 μg/mL NormocinTM (InvivoGen). RAW 264.7 reporter cells were plated and incubated overnight in a humidified incubator at 37 °C and 5% CO2 to allow for attachment. The following day, cells were rinsed with phosphate‐buffered saline (PBS; Corning, without calcium and magnesium) and treated with 100 ng/mL LPS (Sigma‐Aldrich, L2630) and 20 ng/mL mouse interferon‐gamma (IFN‐γ) (R&D Systems, 485‐MI/CF) in DMEM with 1% Pen‐Strep (without FBS and NormocinTM). After 1 h of treatment with LPS/IFN‐γ, calcitriol (Cayman Chemical) or 25(OH)D (Sigma‐Aldrich, H4014) in the presence/absence of SP (Sigma‐Aldrich) or EP (Cayman Chemical, 15616) was added to the media for 24 hours. Calcitriol, SP, EP, and 25(OH)D were dissolved in pure ethyl alcohol (Sigma‐Aldrich, E7023).
Cell line reporter assay
RAW 264.7 culture media was collected 24 h after treatment for secreted alkaline phosphatase reporter assay. Collected media was added to a 96‐well plate (20 μL) in duplicate, exposed to 180 μL QUANTI‐Blue™ Solution (InvivoGen), and incubated at 37 °C and 5% CO2 for 1 hour. Secreted alkaline phosphatase levels were quantified using a VICTOR X5 (PerkinElmer) microplate reader set to measure absorbance at 630 nanometers.
Animals
All animal studies have been approved by the Northwestern University IACUC. Six‐ to eight‐week‐old C57BL/6J female mice (stock no: 000664) were purchased from Jackson Laboratories.
NM skin injury model and treatment
The dorsal area of mice was shaved and chemically depilated using Nair® hair removal product 48 h before skin injury induction. Before injury, the skin was carefully examined for signs of inflammation (redness, swelling, etc.) and animals exhibiting any sign of inflammation were excluded from the study. Mechlorethamine hydrochloride (NM) (Sigma, 122564) was weighed under a chemical fume hood and diluted to a 0.75% solution in 1.5% DMSO‐PBS immediately before the application. Anesthesia was induced by intraperitoneal (i.p.) injections of freshly prepared, sterile‐filtered 1.25% Avertin (Sigma, T48402) solution (pH = 7.0–7.4). Doses were given within the recommended range of 125–250 mg/kg body weight. 23 , 24 Mice were placed on a heat pad under a chemical fume hood. A 12‐mm diameter filter paper disk (Fisher, 0980455) saturated with 40 μL of NM solution was placed on the depilated area for 30 minutes. Animals were kept under a chemical fume hood for 2 h as per safety procedures.
Five nanograms of 25(OH)D (Sigma H4014) in 300 μL mineral oil (Sigma, M8410) per mouse was injected i.p. 1 h after NM exposure. 25(OH)D was given once. SP (Sigma, S3375) was administered via oral gavage in water at a dose of 200 mg/kg daily, for up to 5 days for animals that were monitored longer than 5 days. The first dose of SP was administered 1 h after NM exposure. The maximum volume of gavage was 1% of body weight. Control animals received vehicle (water) by gavage and i.p. injections of corresponding vehicle (mineral oil). The experimental animals were daily monitored for signs of distress.
Monitoring skin injury and measurement of wound healing
Mice were followed‐up noninvasively after induction of skin injury. Mice were briefly anesthetized with isoflurane using an induction chamber connected to a gas scavenging system. Monitoring was performed daily, starting at the day of skin injury, for the first 7 days, and weekly for up to 30 days. The bifold skin thickness of the injured area was measured, photographs of the injured area were taken, and animals were weighed. A caliper (Mitutoyo, PK0505CPX) was used for skin thickness measurements. Photographs of the wounds were traced, analyzed, and quantified using ImageJ (the National Institutes of Health, Bethesda, MD) software.
Quantitative real‐time PCR
Skin injury and wound healing–related gene expression profiling was measured with real‐time quantitative PCR. Total RNA was extracted from mouse skin tissue using TRIzol® reagent (Invitrogen, 15596026), according to the manufacturer's protocol. TaqMan® Gene Expression assays were used for measuring the relative expression levels of IL‐1α (Mm00439620_m1), IL‐1β (Mm00434228_m1), tumor necrosis factor‐alpha (TNF‐α; Mm99999068_m1), matrix metallopeptidase 9 (MMP9; Mm00442991_m1), iNOS (Mm01309902_m1), and C‐C motif chemokine ligand 2 (CCL2; Mm00441242_m1). 18s (Hs99999901_s1) was used as the control housekeeping gene. One hundred nanograms of RNA was amplified using each assay and TaqMan RNA‐to Ct 1‐Step Kit (ABI, 4392938). The relative expression fold change was calculated using the ddCt method.
Flow cytometry
Mouse skin samples were mechanically disrupted followed by digestion using Liberase TL (Sigma, 1 h incubation at 37 °C) at concentration 0.25 mg/mL dissolved in RPMI‐1640 with l‐glutamine (Corning), supplemented with 1 mM sodium pyruvate, MEM nonessential amino acids, and 20 mM HEPES (all from Gibco). Following digestion, the cell suspension was filtered twice consequently using 70‐ and 40‐μm strainers (Greiner), and washed and stained for analysis in flow buffer containing 2% bovine serum albumin (Sigma). The following anti‐mouse antibodies were used: F4/80 APC, CD45 eFluor® 450, Ly6c Alexa Fluor® 488, CD206 PE‐Cy7, and Fixable Viability Dye eFluor 506 (all from Invitrogen and Thermo Fisher Scientific). Appropriate isotype controls were also purchased from Invitrogen, Thermo Fisher Scientific, and TruStain FcX, and Fc blocking antibodies were from Biolegend. Data were collected using BD FACSCanto® II cytometer and analyzed with FlowJo v10.5.3 software (BD).
Immunohistochemistry
The skin samples from euthanized mice were taken and fixed in 10% formalin diluted in PBS (Fisher Scientific, 23 245684). Samples were then embedded in paraffin, sectioned (8‐μm thickness), and stained. Caspase‐3 staining was performed using the DAB‐HRP protocol. The sections were incubated with the primary antibody against cleaved caspase‐3 (Asp175) (Cell Signaling, #9661) and HRP rabbit secondary antibody (Cell Signaling, #8114), and stained with the DAB substrate kit (Cell Signaling, #8059).
Statistical analysis
GraphPad® Prism V.8.3.0 software (San Diego, CA) was used to create visual graphics and calculate statistical significance. One‐way ANOVA was used to calculate P values. ImageJ software was used to calculate the progress of the wound healing process.
Results
SP enhances VD inhibition of NF‐κB activity in mouse macrophages
It is widely accepted that NF‐κB signaling plays a pivotal role in a multitude of immunological transcriptional responses. 25 Since VD has been previously shown to inhibit factors downstream of NF‐κB activation after injury in vivo, 11 we wanted to determine NF‐κB activity in LPS/IFN‐γ–stimulated macrophages in vitro followed by intervention with VD alone or in combination with SP. For our assays, we utilized a murine RAW 264.7 macrophage cell line (RAW‐DualTM) stably expressing secreted embryonic alkaline phosphatase under an endogenous MIP‐2 promoter, as well as the Lucia luciferase gene under control of an ISG54 minimal promoter in conjunction with five IFN‐stimulated response elements. We confirmed that our reporter cell line expressed MR using western blotting (data not shown). Supernatants were assayed for the cumulative effect of NF‐κB and IFN activation at 24 hours. In our in vitro assays, we used calcitriol, the active form of VD.
We found that calcitriol alone decreased NF‐κB activation at a concentration of 500 nM but not at lower concentrations (Fig. 1A). Unlike previous studies in which SP pretreatment altered NF‐κB activity, SP administered 1 h after LPS/IFN‐γ stimulation had no measurable effect on NF‐κB activity when used over the concentration range from 10 nM to 10 μM (Fig. S1, online only). However, SP in combination with calcitriol had an additive effect on modulating NF‐κB activity. A dose of 10 μM of SP significantly decreased NF‐κB activity at all examined concentrations of calcitriol (20, 100, and 500 nM) (Fig. 1B). The activity of the Ifna promoter did not yield statistically significant differences between all tested conditions (data not shown).
Figure 1.
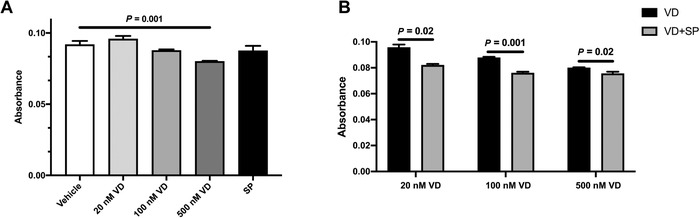
SP enhances vitamin D–dependent inhibition of NF‐κB activity. RAW 264.7 reporter cells were activated with 100 ng/mL LPS and 20 ng/mL IFN‐γ. One hour later, cells were treated with VD (calcitriol) in the absence/presence of SP as indicated. Absorbance was read after 24‐h incubation. (A) NF‐κB activity reduced by 500 nM VD (calcitriol) treatment. (B) Combined treatment of the cells with SP and VD (calcitriol) further decreased NF‐κB activity. SP was used at 10 μM concentration, and Et‐OH served as a vehicle. The average ± SEM; n = 4. VD, calcitriol; SP, spironolactone.
Next, we employed a selective MR inhibitor EP to confirm that the observed NF‐κB inhibition was due to blocking of MR and not due to off‐target effects of SP. Similar to SP, EP alone at 0.1 or 1 μM did not inhibit NF‐κB activity of LPS/IFN‐γ–stimulated cells (Fig. S2, online only). However, the addition of EP led to decreased NF‐κB activity at lower doses of calcitriol. Taken together, these experiments confirm the role of MR antagonism.
To verify that calcitriol inhibition of NF‐κB is mediated by VDR binding, we next performed the experiments with 25(OH)D, the prehormone form of calcitriol. Although 25(OH)D has a very high affinity and selectivity for the VDR, it lacks the biological activity of calcitriol. As shown in Figure S3 (online only), no inhibition of NF‐κB activity was observed in LPS/IFN‐γ–stimulated cells treated with 25(OH)D.
VD and SP treatment decreases the expression of proinflammatory cytokines and tissue remodeling factors in mouse skin after NM exposure
On the basis of the results of our in vitro experiments, we further investigated whether VD and SP treatment, either alone or in combination, affected inflammatory signaling in the skin using an established NM skin injury model. 7 In all animal experiments, VD was given in the prehormone form of 25(OH)D, which undergoes enzymatic activation in vivo. Skin wounds were harvested on days 1, 2, and 5 posttreatment. We focused our analysis on a panel of hallmark proinflammatory factors involved in tissue damage (interleukin (IL)‐1α, IL‐1β, TNF‐α, CCL2, iNOS, and MMP9) and their level of expression, as compared with NM exposure alone.
Analysis by quantitative real‐time PCR data revealed gradual increased expression levels of all inflammatory factors following the application of NM, beginning on days 1 and 2 (data not shown) with significant increases on day 5 (Fig. 2).
Figure 2.
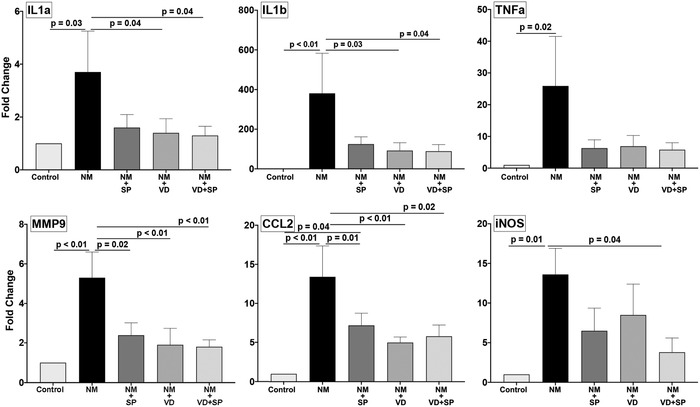
VD and SP treatment regulates the expression of proinflammatory and tissue remodeling factors in the skin. C57BL/6 mice were exposed to NM and treated as indicated. At day 5 post‐application, the wounds were harvested and gene expression was analyzed by qPCR. The average ± SEM; n = 5 mice per group. Control, animals not exposed to NM; NM, nitrogen mustard; VD, 25(OH)D; SP, spironolactone.
VD treatment alone compared with NM demonstrated the decreased expression of proinflammatory cytokines IL‐1α and IL‐1β in addition to the tissue remodeling, MMP9, and chemokine CCL2. SP alone demonstrated trends toward decreased expression in all the factors tested but was only statistically significant for MMP9 and CCL2. Only iNOS was significantly downregulated by the combination of VD and SP. The levels of TNF‐α were reduced by all treatments, but the data did not reach statistical significance threshold.
VD and SP treatment increases M2/M1 macrophages ratio in mouse skin after NM exposure
The degree of inflammation resulting from the insult is determined by several factors, including the ratio of pro and anti‐inflammatory signaling in damaged tissue. To investigate the effect of VD and SP treatment on the immunological response in the skin, we analyzed whole‐skin cell isolates from mice exposed to NM and treated with either VD, SP, or the combination of the two in vivo. Since we had previously reported that VD strongly regulates inflammatory response in UV‐damaged skin by increasing M2 repair in macrophages, 11 for this study, we focused the analysis mainly on the subsets of macrophages in the skin.
Flow cytometric analysis revealed that in mice that were not exposed to NM, the vast majority of live F4/80+ CD45+ macrophages are Ly6c low, indicating the absence of the proinflammatory M1 (defined as Ly6chi CD206low) subtype and the presence of the anti‐inflammatory Ly6clow CD206hi M2 subtype (Fig. 3A). Following NM treatment, we observed marked increase in the total number of macrophages (Fig. 3B). This increase was mostly due to a surge of Ly6chi CD206low M1 population (Fig. 3C). As expected, NM exposure resulted in a dramatic increase in proinflammatory M1 macrophages. By contrast, animals treated with the combination of VD and SP had the lowest number of proinflammatory M1 cells (2.6 ± 0.4% versus 6.3 ± 0.9% in the NM group). The number of M1 macrophages in the VD and SP combination group was statistically different from both NM and VD groups, but not from the SP group. There was no statistically significant differences in anti‐inflammatory M2 populations between all groups (Fig. 3D). Thus, we found that treatment of animals with the combination of VD and SP changed the distribution pattern of infiltrating macrophages, resulting in an increased ratio of anti‐inflammatory F4/80+ CD45+ Ly6Clow CD206hi M2 macrophages relative to proinflammatory F4/80+ CD45+ Ly6Chi CD206low M1 macrophages. The M2/M1 ratio in animals treated with combination of drugs was significantly higher than in animals treated with either VD or SP alone (P < 0.05), indicating decreased inflammatory response and potentially improved ability to heal cutaneous wounds (Fig. 3E).
Figure 3.
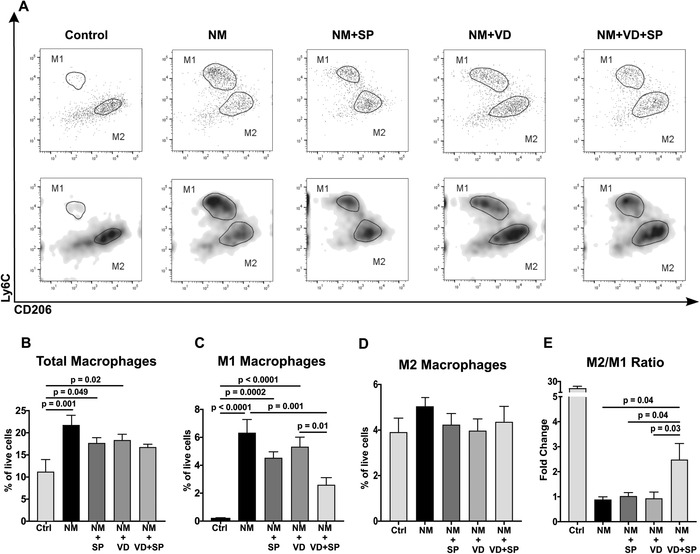
VD and SP treatment triggers an anti‐inflammatory immune response. Skin samples harvested from mice subjected to NM injury and treated as indicated were investigated by flow cytometry 48 h after injury. After initial selection, M1 macrophages were defined as F4/80+ CD45+ Ly6chigh CD206low cells, and M2 macrophages as F4/80+ CD45+ Ly6clow CD206high cells. (A) Representative images showing M1 and M2 populations in animals from different treatment groups. Top panels show dot plots, and bottom panels show density plots of the same sample; (B) total number of macrophages; (C) M1 macrophages; (D) M2 macrophages; and (E) ratios of M2/M1 macrophage populations. The average ± SEM; n = 7–9 mice per group. Control animals were sham treated and not exposed to NM. NM, nitrogen mustard; VD, 25(OH)D; SP, spironolactone.
VD and SP treatment facilitates skin wound healing in vivo
We next inquired whether the combined treatment of VD and SP can improve skin wound healing longitudinally. In our experiments, 6–8 weeks old C57BL/6J female mice were followed for 21 days after initial NM exposure and treatment. Representative images of skin wound development and healing are shown in Figure 4A. While NM application leads to uniform injury, intervention with SP, VD, or combination partially mitigated hemorrhagic crust formation on days 1–3. By days 5–7, the protective effects are most pronounced in the combination treatment group and to a lesser extent in the VD group.
Figure 4.
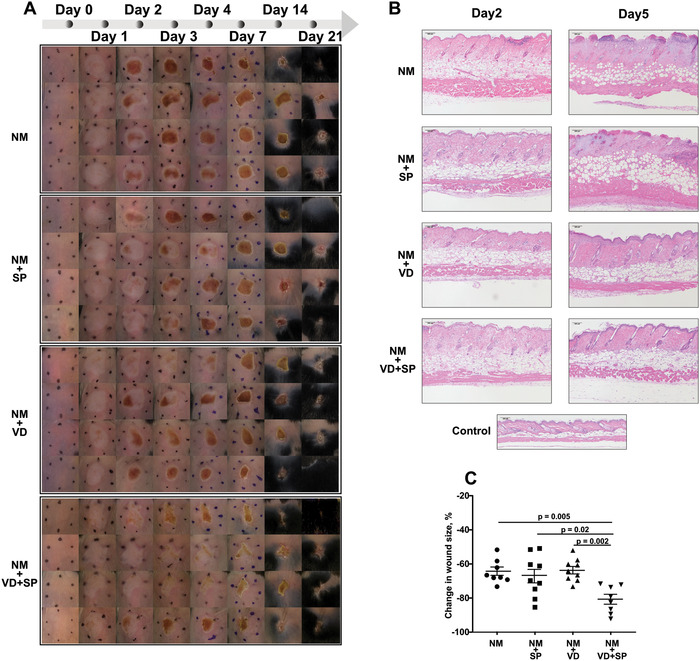
Combination treatment of VD and SP accelerates healing from chemical injury. C57BL/6 mice were exposed to NM and treated as shown. At indicated time points, images and measurements were taken. (A) Representative wound images at indicated days post‐application (dpa); (B) representative images of H&E staining of mouse skin samples, 10× magnification, scale bar = 100 μm; and (C) changes in the wound size from dpa 7 to 21. Wound areas were traced and measured using ImageJ. The data were calculated as a percent of the decrease in the wound area at given dpa relative to the corresponding wound area at dpa 1. The average ± SEM; n = 8–9 mice per group. NM, nitrogen mustard; VD, 25(OH)D; SP, spironolactone.
Histological evaluation of the wounds at day 5 post‐NM application (Fig. 4B) showed that NM exposure resulted in massive skin damage accompanied by full‐thickness necrosis. The histologic sections reveal edema with dense inflammatory infiltrates compared with control naive skin. At day 5, SP treatment alone did not mitigate the damage caused by NM exposure. However, VD alone or in combination with SP demonstrated reduced inflammatory infiltrates with focal necrosis in the epidermis and superficial dermis. The treatments also decreased skin edema that is further supported by daily noninvasive bifold skin thickness (Fig. S4, online only).
We used wound area measurements to quantify skin healing. Though the initial damage at day 1 post‐NM application looked similar across all groups, mice treated with the combination of VD and SP healed at the fastest rate. The treatment intervention with the combination of VD and SP delayed and lessened the formation of skin necrosis and hemorrhagic crust formation (Fig. 4A) with the most pronounced rate of healing than in any other group (Fig. 4C).
Overall, the wound repair was significantly enhanced by a combined VD and SP treatment. The combination treatment proved to be superior to VD or SP alone.
VD and SP treatment inhibits apoptosis in mouse skin after NM exposure
To determine if the treatment with VD and/or SP protects the cells in NM‐damaged skin from apoptosis, mouse skin sections were stained for active caspase‐3 (Fig. 5). NM induced an injury that resulted in a strong apoptotic response in the skin adjacent to the wound. This response was significantly reduced by SP treatment. VD treatment alone did not decrease the number of active caspase‐3–positive cells. However, we observed that the combination of VD and SP had reduced the number of caspase‐3–positive cells.
Figure 5.
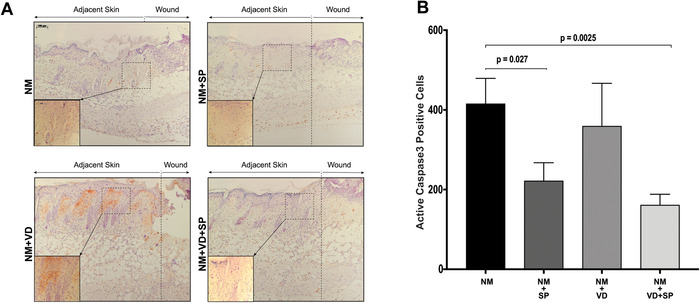
Combination treatment of VD and SP inhibits apoptosis in NM‐injured skin. Skin samples of mice subjected to NM injury and treated as indicated were immunostained for activated caspase‐3 at dpa 5. (A) Representative images of mouse skin samples (10× magnification, inlets 40× magnification, scale bar = 100 μm); apoptotic cells are seen as brown‐stained cells. (B) Quantification of caspase‐3–positive cells. Two equal size sections were selected from the left and right side of the wound from each slide. The area was chosen to include a portion of both the wound and adjacent skin. Stained cells were counted using ImageJ software. The average ± SEM; n = 4 mice per group. NM, nitrogen mustard; VD, 25(OH)D; SP, spironolactone.
Discussion
NM exposure leads to severe acute skin injury that is characterized by blistering and robust inflammatory response. In this study, we demonstrate that administration of VD together with SP decreases NF‐κB activity, reduces the expression of proinflammatory cytokines, shifts the ratio of M2/M1 macrophages, and accelerates skin repair (Fig. 6).
Figure 6.
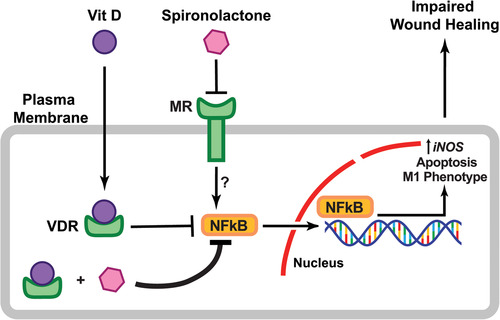
Schematic representation for a model of VD and SP effects on skin wound healing.
An inflammatory response plays a crucial role in skin wound healing. 26 During this process, macrophages protect the skin from infection, clear cellular debris, and secrete cytokines that stimulate the proliferation of keratinocytes, fibroblasts, and epithelial cells. Under normal physiological conditions, inflammation resolves as environmental cues trigger polarization of macrophages into an anti‐inflammatory M2 phenotype. However, the imbalance of pro and anti‐inflammatory signals can alternatively prolong and amplify the inflammatory phase, resulting in a further tissue damage and the development of chronic wounds. 26 , 27 Therefore, timely regulation of anti‐inflammatory signaling is crucial for proper wound healing.
The NF‐κB signaling pathway is a central mediator of inflammation. 28 VD, upon binding to the VDR, can regulate NF‐κB activity in different tissues and cell types. 16 , 17 , 29 , 30 , 31 Multiple mechanisms of NF‐κB activity inhibition by VD have been reported, including increasing expression of IκB‐alpha, 29 arresting p65 nuclear translocation, suppressing transcription of RelB, 32 or through direct VDR binding to IKK‐beta. 17
Our in vitro experiments using murine macrophages found decreased NF‐κB activity in response to VD treatment. SP significantly enhanced VD‐dependent inhibition, even at the doses of VD that did not elicit a statistically significant response when given alone (at 20 and 100 nM). SP alone at concentrations up to 10 μM did not affect LPS‐induced NF‐κB signaling. This observation agrees with the data published by Kato et al., 33 where authors observed inhibition of NF‐κB in RAW 264.7 macrophages only at 100 μM concentration of SP. At a high dose of calcitriol (500 nM), the additional effect of SP on NF‐κB activity appeared to decrease, though it remained statistically significant. This observation could be due to the fact that at high concentrations, VD already reached the inhibition plateau, and, therefore, the additional effect of SP is less pronounced.
In general, the read‐outs in the NF‐κB reporter assay can be explained by the experimental setup. While in other publications, cells were pretreated with SP for several hours before stimulation with LPS, 33 , 34 we chose to treat our cells with VD and/or SP 1 h after LPS stimulation. This allowed us to examine whether our treatment regimen could decrease NF‐κB activity in a situation that more closely resembles clinical settings, where treatment would be given as an intervention after skin injury rather than as a prophylaxis.
Data using EP, a highly selective MR antagonist, suggest that the effects of SP on NF‐κB activity are indeed MR dependent. The role of direct VD–VDR interaction in inhibiting NF‐κB activity was also confirmed by using 25(OH)D, a prohormone form of VD that significantly lacks its biological activity, while retaining affinity and selectivity for VDR. 35
We next investigated the effects of combined VD and SP treatment on proinflammatory gene expression after NM exposure. Treatment with VD alone or in combination with SP significantly decreased the expression of proinflammatory cytokines IL‐1α and IL‐1β. This is consistent with the inhibition of IL‐1β by VD that has been observed in cultured human macrophages 36 and monocyte‐derived macrophages. 37 While the reduction in TNF‐α expression was not statistically significant, there was a trend toward reduced expression in all treatment groups. This supports published data showing that VD both regulates TNF‐α levels in human macrophages 37 and reduces TNF‐α expression in the skin following UV‐induced damage. 11 MMP9 and CCL2 levels were significantly inhibited in all SP and VD treatment conditions. This again validates our previous findings that MMP9 can be inhibited by VD. 7 , 11 Similarly, MR inhibition was shown to decrease MMP9 expression in the cerebral vascular wall of rats, 38 and CCL2 plays an important role in MR‐mediated development of cardiac fibrosis. 39 Interestingly, only the combined treatment group was able to significantly inhibit the levels of iNOS transcription in this injury model. We had previously shown that VD reduces M1 macrophage–specific iNOS production. 7 , 9 The role of SP in iNOS regulation appears to be complex and context dependent. In IL‐1–treated cardiomyocytes, SP upregulated iNOS production. 40 On the other hand, SP did not affect iNOS expression in the kidneys of l‐NAME–treated rats, 41 but rather inhibited iNOS levels in rat aortic smooth muscle cells. 42 Therefore, it is possible that VD and SP affect different iNOS regulation pathways, and combined treatment results in synergistic reduction of iNOS production.
Ultimately, our results suggest that both VD and SP treatments affect the anti‐inflammatory response individually and synergistically.
This is highlighted by data demonstrating that VD and SP treatment after NM exposure significantly increased the ratio of anti‐inflammatory M2 macrophages to proinflammatory M1 macrophages in the skin wounds. Under baseline conditions, there is an abundance of M2 macrophages over M1 macrophages. Restoring the relative ratio where there are more M2 macrophages present compared with M1 macrophages in the wound microenvironment can facilitate tissue repair and return to homeostasis. NM‐induced injury leads to a dramatic increase in the number of M1 macrophages. Treatment of NM‐injured mice with the combination of VD and SP greatly reduced the number of M1 macrophages, but did not significantly affect M2 macrophages. Taken together, this leads to an increase in the M2/M1 ratio and improved wound repair.
By contrast, UV‐induced skin injury affects M2 macrophage numbers more than M1 macrophage numbers. 11 These findings suggest that the modulation of macrophage polarization by VD can vary depending on the skin injury model, likely owing to different stimulation signals from the wound microenvironment. In addition to VD, MR can also regulate macrophage polarization. Studies have shown that activation of MR by aldosterone in rats enhanced expression of M1 markers while not affecting M2 markers, 43 and aldosterone also stimulated M1 markers in isolated mice peritoneal macrophages. 44 In the model of bleomycin‐induced pulmonary fibrosis, SP had reduced the number of CD11b+ Ly6Chi monocytes, but had no effect on F4/80+ CD206+ macrophages. 45
The combination treatment of VD and SP also delayed skin necrosis progression, hemorrhagic crust development, and reduced tissue edema after NM application. This further corroborates our previous findings where VD significantly improved survival and wound healing in animals by regulating macrophage iNOS production. 7 Since SP was administered systemically, it can bind to aldosterone receptors in the distal tubule and collecting duct of the kidney and function as a mild diuretic. Therefore, it was surprising that SP alone did not significantly decrease skin swelling. This may be due to SP being a weak diuretic that is better suited to treat milder chronic conditions and not an acute injury to the skin. However, the combination of VD and SP led to improved healing of NM‐induced wounds, suggesting that SP has anti‐inflammatory treatment effects beyond just water balance.
In addition to anti‐inflammatory effects, the combined VD and SP treatment also resulted in a significant inhibition of apoptosis in the NM‐injured mouse skin. This finding is supported by the reports of SP‐mediated inhibition of apoptosis in the cardiovascular system. 46 , 47 Though VD can decrease apoptosis after UV‐induced skin damage, 11 treatment with VD alone in NM‐induced skin injury did not significantly reduce apoptosis. Although NF‐κB signaling is usually regarded as antiapoptotic, in some instances, NF‐κB can promote apoptosis. 48 , 49 Whether the inhibition of NF‐κB by VD and SP is responsible for the antiapoptotic effects we observed in the skin wounds is a subject of further research.
Wound healing is a complex process that requires a coordinated response from various cell types. Since SP was administered for up to 5 consecutive days in vivo, it is possible that several tissues involved in a wound healing process could be affected. For instance, SP can reduce inflammation in the endothelium and vascular smooth muscle cells, 50 , 51 , 52 regulate other immune cells, such as Treg cells, TH17 helper cells, and glial cells. 53 , 54 In addition, SP inhibited fibrosis in cardiac and kidney injury. 55 , 56 All of these effects could potentially add to the benefits we observed in our skin healing model. However, MR epidermal knockout mice showed increased susceptibility to local skin damage. Similarly, NF‐κB activity was actually increased in MR‐deficient keratinocytes. 57 This suggests that improvements in wound healing observed in our study are indeed likely due to the effect of SP‐induced MR inhibition in immune cells rather than keratinocytes.
Our study demonstrates that VD in combination with SP reduces inflammation and improves wound healing after NM exposure. However, it should be noted that SP may exert some effects independently of MR signaling. 19 For example, other possible mechanisms may include SP‐induced suppression of NF‐κB signaling via degradation of the XPB subunit of the transcription factor II human. 58 , 59 SP has also been shown to activate the NKG2D ligand in colon carcinoma cells in an MR‐independent manner, apparently through the retinoid X receptor gamma (RXR‐gamma). 60 Since VDR forms dimers with the RXR‐gamma, 61 , 62 it cannot be ruled out that SP can modulate VD activity through MR‐independent mechanisms. This possibility requires further investigation.
Overall, our results demonstrate that VD in combination with SP treatment improves wound healing after NM exposure by modulating the immune response and activating anti‐inflammatory pathways. In addition to supportive treatment, this may expand the currently limited treatment options to improve clinical outcomes for NM injury.
Author contributions
K.Q.L., D.B., and U.V.O. designed the experiments. D.B., U.V.O., M.D., and S.E. performed the experiments, acquired the data, did statistical analysis, and analyzed the data. J‐M.T. analyzed the data and edited the manuscript. D.B. wrote, and P.D. and K.Q.L. edited the manuscript. All authors reviewed the manuscript.
Competing interests
The authors declare no competing interests.
Supporting information
Figure S1. Spironolactone at concentrations up to 10 μM does not inhibit NF‐κB activity in RAW 264.7 cells.
Figure S2. Eplerenone enhances vitamin D (calcitriol)–dependent inhibition of NF‐κB activity in RAW 264.7 cells.
Figure S3. Eplerenone and the prehormone form of vitamin D 25(OH)D does not affect NF‐κB activity in RAW 264.7 cells.
Figure S4. Skin thickness measurements after NM exposure.
Supporting information
Acknowledgments
This work was supported by NIH research Grants AR064144 and AR071168 (to K.Q.L.). The NU‐SBDRC Skin Tissue Engineering and Morphology Core facility assisted in the morphologic analysis. The Skin Biology & Diseases Resource‐Based Center (NU‐SBDRC) is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Grant AR075049. This work was supported by the Northwestern University Interdepartmental ImmunoBiology Flow Cytometry Core Facility. The authors wish to thank Dr. R.M. Lavker for critical reading of the manuscript.
[Correction added on September 10, 2020 after online publication: The author name “Ummiye V. Onay” was corrected.]
References
- 1. Lessin, S.R. et al 2013. Topical chemotherapy in cutaneous T cell lymphoma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.02%, gel in mycosis fungoides. JAMA Dermatol. 149: 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jenner, J. 2016. Toxicology of vessicants In Chemical Warfare Toxicology: Volume 1: Fundamental Aspects. Worek F., Jenner J. & Thierman H., Eds.: 29–80. The Royal Society of Chemistry. [Google Scholar]
- 3. Shakarjian, M.P. et al 2009. Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol. Sci. 114: 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tewari‐Singh, N. & Agarwal R.. 2016. Mustard vesicating agent‐induced toxicity in the skin tissue and silibinin as a potential countermeasure. Ann. N.Y. Acad. Sci. 1374: 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen, Y. , Jia Y., Song W. & Zhang L.. 2018. Therapeutic potential of nitrogen mustard based hybrid molecules. Front. Pharmacol. 9 10.3389/fphar.2018.01453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singh, R.K. , Kumar S., Prasad D.N. & Bhardwaj T.R.. 2018. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: historic to future perspectives. Eur. J. Med. Chem. 151: 401–433. [DOI] [PubMed] [Google Scholar]
- 7. Au, L. et al 2015. Suppression of hyperactive immune responses protects against nitrogen mustard injury. J. Invest. Dermatol. 135: 2971–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scott, J.F. & Lu K.Q.. 2017. Vitamin D as a therapeutic option for sunburn: clinical and biologic implications. DNA Cell Biol. 36: 879–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scott, J.F. et al 2017. Oral vitamin D rapidly attenuates inflammation from sunburn: an interventional study. J. Invest. Dermatol. 137: 2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Das, L.M. , Binko A.M., Traylor Z.P., et al 2018. Defining the timing of 25(OH)D rescue following nitrogen mustard exposure. Cutan. Ocul. Toxicol. 37: 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Das, L.M. , Binko A.M., Traylor Z.P., et al 2019. Vitamin D improves sunburns by increasing autophagy in M2 macrophages. Autophagy 15: 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gil, A. , Plaza‐Diaz J. & Mesa M.D.. 2018. Vitamin D: classic and novel actions. Ann. Nutr. Metab. 72: 87–95. [DOI] [PubMed] [Google Scholar]
- 13. Bikle, D.D. 2014. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 21: 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Christakos, S. , Dhawan P., Verstuyf A., et al 2016. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol. Rev. 96: 365–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun, J. et al 2006. Increased NF‐kappaB activity in fibroblasts lacking the vitamin D receptor. Am. J. Physiol. Endocrinol. Metab. 291: E315–E322. [DOI] [PubMed] [Google Scholar]
- 16. Deb, D.K. et al 2009. 1,25‐dihydroxyvitamin D3 suppresses high glucose‐induced angiotensinogen expression in kidney cells by blocking the NF‐{kappa}B pathway. Am. J. Physiol. Renal Physiol. 296: F1212–F1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen, Y. et al 2013. Vitamin D receptor inhibits nuclear factor kappaB activation by interacting with IkappaB kinase β protein. J. Biol. Chem. 288: 19450–19458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gilbert, K.C. & Brown N.J.. 2010. Aldosterone and inflammation. Curr. Opin. Endocrinol. Diabetes Obes. 17: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jaisser, F. & Farman N.. 2016. Emerging roles of the mineralocorticoid receptor in pathology: toward new paradigms in clinical pharmacology. Pharmacol. Rev. 68: 49–75. [DOI] [PubMed] [Google Scholar]
- 20. van der Heijden, C. , Deinum J., Joosten L.A.B., et al 2018. The mineralocorticoid receptor as a modulator of innate immunity and atherosclerosis. Cardiovasc. Res. 114: 944–953. [DOI] [PubMed] [Google Scholar]
- 21. Nguyen, V.T. et al 2016. Re‐epithelialization of pathological cutaneous wounds is improved by local mineralocorticoid receptor antagonism. J. Invest. Dermatol. 136: 2080–2089. [DOI] [PubMed] [Google Scholar]
- 22. Stojadinovic, O. , Lindley L.E., Jozic I. & Tomic‐Canic M.. 2016. Mineralocorticoid receptor antagonists—a new sprinkle of salt and youth. J. Invest. Dermatol. 136: 1938–1941. [DOI] [PubMed] [Google Scholar]
- 23. Lieggi, C.C. et al 2005. Efficacy and safety of stored and newly prepared tribromoethanol in ICR mice. Contemp. Top. Lab. Anim. Sci. 44: 17–22. [PubMed] [Google Scholar]
- 24. Lieggi, C.C. et al 2005. An evaluation of preparation methods and storage conditions of tribromoethanol. Contemp. Top. Lab. Anim. Sci. 44: 11–16. [PubMed] [Google Scholar]
- 25. Dorrington, M.G. & Fraser I.D.C.. 2019. NF‐kappaB signaling in macrophages: dynamics, crosstalk, and signal integration. Front. Immunol. 10 10.3389/fimmu.2019.00705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Larouche, J. , Sheoran S., Maruyama K. & Martino M.M.. 2018. Immune regulation of skin wound healing: mechanisms and novel therapeutic targets. Adv. Wound Care (New Rochelle) 7: 209–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Laskin, D.L. & Laskin J.D.. 1996. Macrophages, inflammatory mediators, and lung injury. Methods 10: 61–70. [DOI] [PubMed] [Google Scholar]
- 28. Lawrence, T. 2009. The nuclear factor NF‐kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1: a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Riis, J.L. et al 2004. 1alpha,25(OH)(2)D(3) regulates NF‐kappaB DNA binding activity in cultured normal human keratinocytes through an increase in IkappaBalpha expression. Arch. Dermatol. Res. 296: 195–202. [DOI] [PubMed] [Google Scholar]
- 30. Zhang, Z. et al 2007. 1,25‐dihydroxyvitamin D3 targeting of NF‐kappaB suppresses high glucose‐induced MCP‐1 expression in mesangial cells. Kidney Int. 72: 193–201. [DOI] [PubMed] [Google Scholar]
- 31. Chen, Y. et al 2013. 1,25‐dihydroxyvitamin D promotes negative feedback regulation of TLR signaling via targeting microRNA‐155‐SOCS1 in macrophages. J. Immunol. 190: 3687–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dong, X. et al 2003. Direct transcriptional regulation of RelB by 1alpha,25‐dihydroxyvitamin D3 and its analogs: physiologic and therapeutic implications for dendritic cell function. J. Biol. Chem. 278: 49378–49385. [DOI] [PubMed] [Google Scholar]
- 33. Kato, Y. et al 2014. Spironolactone inhibits production of proinflammatory mediators in response to lipopolysaccharide via inactivation of nuclear factor‐kappaB. Immunopharmacol. Immunotoxicol. 36: 237–241. [DOI] [PubMed] [Google Scholar]
- 34. Sønder, S.U. , Woetmann A., Odum N. & Bendtzen K.. 2006. Spironolactone induces apoptosis and inhibits NF‐kappaB independent of the mineralocorticoid receptor. Apoptosis 11: 2159–2165. [DOI] [PubMed] [Google Scholar]
- 35. Lou, Y.R. et al 2010. 25‐hydroxyvitamin D(3) is an agonistic vitamin D receptor ligand. J. Steroid Biochem. Mol. Biol. 118: 162–170. [DOI] [PubMed] [Google Scholar]
- 36. Villaggio, B. , Soldano S. & Cutolo M.. 2012. 1,25‐dihydroxyvitamin D3 downregulates aromatase expression and inflammatory cytokines in human macrophages. Clin. Exp. Rheumatol. 30: 934–938. [PubMed] [Google Scholar]
- 37. Neve, A. , Corrado A. & Cantatore F.P.. 2014. Immunomodulatory effects of vitamin D in peripheral blood monocyte‐derived macrophages from patients with rheumatoid arthritis. Clin. Exp. Med. 14: 275–283. [DOI] [PubMed] [Google Scholar]
- 38. Tada, Y. et al 2009. Role of mineralocorticoid receptor on experimental cerebral aneurysms in rats. Hypertension (Dallas, Tex.:1979) 54: 552–557. [DOI] [PubMed] [Google Scholar]
- 39. Shen, J.Z. , Morgan J., Tesch G.H., et al 2014. CCL2‐dependent macrophage recruitment is critical for mineralocorticoid receptor‐mediated cardiac fibrosis, inflammation, and blood pressure responses in male mice. Endocrinology 155: 1057–1066. [DOI] [PubMed] [Google Scholar]
- 40. Chun, T.Y. , Bloem L.J. & Pratt J.H.. 2003. Aldosterone inhibits inducible nitric oxide synthase in neonatal rat cardiomyocytes. Endocrinology 144: 1712–1717. [DOI] [PubMed] [Google Scholar]
- 41. Pechanova, O. et al 2006. Effect of spironolactone and captopril on nitric oxide and S‐nitrosothiol formation in kidney of L‐NAME‐treated rats. Kidney Int. 70: 170–176. [DOI] [PubMed] [Google Scholar]
- 42. Godfrey, V. , Martin A.L., Struthers A.D. & Lyles G.A.. 2011. Effects of aldosterone and related steroids on LPS‐induced increased expression of inducible NOS in rat aortic smooth muscle cells. Br. J. Pharmacol. 164: 2003–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin‐Fernandez, B. et al 2016. Aldosterone induces renal fibrosis and inflammatory M1‐macrophage subtype via mineralocorticoid receptor in rats. PLoS One 11: e0145946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Usher, M.G. et al 2010. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J. Clin. Invest. 120: 3350–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ji, W.J. et al 2013. Spironolactone attenuates bleomycin‐induced pulmonary injury partially via modulating mononuclear phagocyte phenotype switching in circulating and alveolar compartments. PLoS One 8: e81090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Williams, T.A. , Verhovez A., Milan A., et al 2006. Protective effect of spironolactone on endothelial cell apoptosis. Endocrinology 147: 2496–2505. [DOI] [PubMed] [Google Scholar]
- 47. Loan Le, T.Y. et al 2012. Low‐dose spironolactone prevents apoptosis repressor with caspase recruitment domain degradation during myocardial infarction. Hypertension (Dallas, Tex.:1979) 59: 1164–1169. [DOI] [PubMed] [Google Scholar]
- 48. Khandelwal, N. et al 2011. Nucleolar NF‐kappaB/RelA mediates apoptosis by causing cytoplasmic relocalization of nucleophosmin. Cell Death Differ. 18: 1889–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Strozyk, E. , Poppelmann B., Schwarz T. & Kulms D.. 2006. Differential effects of NF‐kappaB on apoptosis induced by DNA‐damaging agents: the type of DNA damage determines the final outcome. Oncogene 25: 6239–6251. [DOI] [PubMed] [Google Scholar]
- 50. Zhu, C.J. et al 2012. The mineralocorticoid receptor‐p38MAPK‐NFkappaB or ERK‐Sp1 signal pathways mediate aldosterone‐stimulated inflammatory and profibrotic responses in rat vascular smooth muscle cells. Acta Pharmacol. Sin. 33: 873–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang, X. et al 2014. Aldosterone induces C‐reactive protein expression via MR‐ROS‐MAPK‐NF‐kappaB signal pathway in rat vascular smooth muscle cells. Mol. Cell. Endocrinol. 395: 61–68. [DOI] [PubMed] [Google Scholar]
- 52. Xiao, H.B. , Lu X.Y., Liu Z.K. & Luo Z.F.. 2016. Kaempferol inhibits the production of ROS to modulate OPN‐alphavbeta3 integrin pathway in HUVECs. J. Physiol. Biochem. 72: 303–313. [DOI] [PubMed] [Google Scholar]
- 53. Chantong, B. , Kratschmar D.V., Nashev L.G., et al 2012. Mineralocorticoid and glucocorticoid receptors differentially regulate NF‐kappaB activity and pro‐inflammatory cytokine production in murine BV‐2 microglial cells. J. Neuroinflammation 9 10.1186/1742-2094-9-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Amador, C.A. et al 2014. Spironolactone decreases DOCA‐salt‐induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension (Dallas, Tex.:1979) 63: 797–803. [DOI] [PubMed] [Google Scholar]
- 55. Johar, S. , Cave A.C., Narayanapanicker A., et al 2006. Aldosterone mediates angiotensin II‐induced interstitial cardiac fibrosis via a Nox2‐containing NADPH oxidase. FASEB J. 20: 1546–1548. [DOI] [PubMed] [Google Scholar]
- 56. Fukuda, S. et al 2011. Aldosterone‐induced kidney injury is mediated by NFkappaB activation. Clin. Exp. Nephrol. 15: 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boix, J. , Sevilla L.M., Saez Z., et al 2016. Epidermal mineralocorticoid receptor plays beneficial and adverse effects in skin and mediates glucocorticoid responses. J. Invest. Dermatol. 136: 2417–2426. [DOI] [PubMed] [Google Scholar]
- 58. Alekseev, S. et al 2014. A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the chemosensitization of tumor cells to platinum. Chem. Biol. 21: 398–407. [DOI] [PubMed] [Google Scholar]
- 59. Elinoff, J.M. et al 2018. Spironolactone‐induced degradation of the TFIIH core complex XPB subunit suppresses NF‐kappaB and AP‐1 signalling. Cardiovasc. Res. 114: 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Leung, W.H. et al 2013. Modulation of NKG2D ligand expression and metastasis in tumors by spironolactone via RXRgamma activation. J. Exp. Med. 210: 2675–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. de la Fuente, A.G. et al 2015. Vitamin D receptor‐retinoid X receptor heterodimer signaling regulates oligodendrocyte progenitor cell differentiation. J. Cell Biol. 211: 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kephart, D.D. , Walfish P.G., DeLuca H. & Butt T.R.. 1996. Retinoid X receptor isotype identity directs human vitamin D receptor heterodimer transactivation from the 24‐hydroxylase vitamin D response elements in yeast. Mol. Endocrinol. 10: 408–419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Spironolactone at concentrations up to 10 μM does not inhibit NF‐κB activity in RAW 264.7 cells.
Figure S2. Eplerenone enhances vitamin D (calcitriol)–dependent inhibition of NF‐κB activity in RAW 264.7 cells.
Figure S3. Eplerenone and the prehormone form of vitamin D 25(OH)D does not affect NF‐κB activity in RAW 264.7 cells.
Figure S4. Skin thickness measurements after NM exposure.
Supporting information