

Abstract
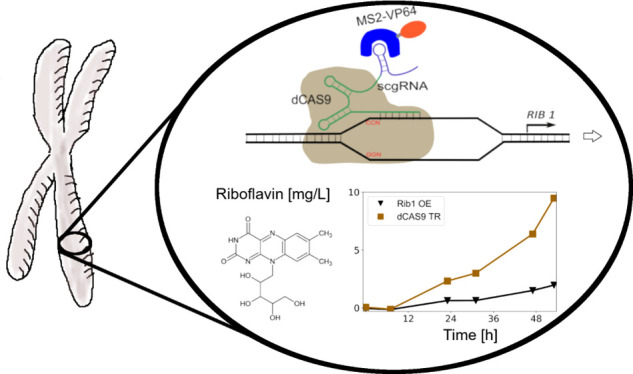
For metabolic engineering approaches, fast and reliable tools are required to precisely manipulate the expression of target genes. dCas9 can be fused via RNA scaffolds to trans-activator domains and thus regulate the gene expression when targeted to the promoter region of a gene. In this work we show that this strategy can be successfully implemented for the methylotrophic yeast Pichia pastoris. It is shown that the thiamine repressible promoter of THI11 can be activated under repression conditions using a scgRNA/dCas9 construct. Furthermore, the RIB1 gene required for riboflavin production was activated, leading to increased riboflavin production exceeding the riboflavin titers of a conventional RIB1 overexpression with a pGAP promoter.
Keywords: dCas9, CRISPR-associated RNA scaffolds, Komagataella, yeast, riboflavin, thiamine
Pichia pastoris (syn. Komagataella phaffii) is a well-known host for recombinant protein production1 and for metabolic engineering endeavors.2 Metabolic engineering approaches require flexible promoter systems to fine-tune the transcription of the desired target genes. For P. pastoris a high number of methanol inducible promoters with different expression strengths are available,3 and by a combinatorial fine-tuning of the carotenoid pathway β-carotene yields can vary up to 10-fold.4
An efficient plasmid generation system GoldenPiCS5,6 and different CRISPR/Cas9 pipelines7,8 are available for P. pastoris genetic engineering experiments. Nuclease deficient Cas9 (dCas9)9 can be linked to regulatory domains capable of activation or repression and thus can be used to regulate gene expression by linking the activation domain to the dCas9 protein.10 Further improvements have been made by using RNA scaffolds connecting the activator domain with the dCas9.11 An advantage of this strategy is that the proteins (activators/repressors) can be fused to dCas9 via a scaffold guide RNA (scgRNA), which can be more easily exchanged and enable a modular cloning strategy.
Here, we present a system to engineer transcriptional regulation using dCas9 and RNA scaffolds in P. pastoris. First, it was tested if a repressed promoter (pTHI11) can be activated by this approach overruling the strong repression by thiamine.12 Second, with RIB1, a metabolic engineering target was chosen to demonstrate the impact on riboflavin production. In this case, the scgRNA/dCas9 system is compared to a classic overexpression approach of RIB1. For both experiments both a glucose and a methanol compatible system was evaluated.
Results and Discussion
The experimental design for a scgRNA/dCas9 assisted transcription regulation system in Pichia pastoris is outlined in Figure 1(A). RNA scaffolds are used to link dCas9 with a regulatory activator domain by adopting a system reported for S. cerevisiae and HEK293T cells for P. pastoris.11,13 The system to enable transcriptional regulation of a target gene consists of 4 parts: the target DNA with a PAM sequence (protospacer adjacent motif), dCas9, a MS2 coat protein fused to VP64 (further referred as MS2-VP64) in case of gene activation (Figure 1(A)) or the MS2 coat protein alone as control further referred as no-activation control (NAC) (Figure 1(B)) and a scgRNA (scaffold guide RNA) involving 20 nucleotides complementary to the target sequence at its 5′ end and a MS2 stem loop fused to its 3′ end, which is bound by the MS2 coat fusion protein.11 To test the system a cassette expressing eGFP under the control of the THI11 promoter (pTHI11) was used as a target. pTHI11 is a P. pastoris specific promoter system, which can be used for recombinant protein production, is efficiently repressed by thiamine, and shows a constitutive expression in thiamine-free medium.12,14 Using the scgRNA/dCas9 transcriptional regulation system it was tested to which extent pTHI11 mediated transcription can be influenced both under repressing (presence of thiamine) or de-repressing conditions (absence of thiamine).
Figure 1.

Experimental design for a scgRNA/dCas9 assisted transcription regulation in Pichia pastoris. (A) dCas9 based transcription regulation is mediated by a target specific gRNA fused to a MS2 loop (M) further referred to as scaffold gRNA (scgRNA) which directs dCAS9 to the desired target and recruits an MS2-VP64 fusion protein responsible for gene activation. (B) The same setup without the activation domain is used to evaluate the effect of the activation domain (no activation (NAC) control). (C) Overview of the tested target sequences (T1–T5) on the THI11 promoter to influence eGFP expression. Arrowheads indicate the positions of the PAM sequences and the positions of the last nucleotide of the PAM sequences in respect of the transcription start site are indicated. (D) Vectors used for scgRNA/dCas9 assisted transcription, linearized and integrated into the RGI2 locus. All constructs contained a dCAS9 gene fused to a nuclear localization signal (NLS) under the control of the constitutive TEF promoter, an MS2 coat protein gene under the control of the POR1 promoter fused to an NLS and in case of gene activation purposes (I and III) fused to the VP64 activation domain. As a control the MS2 coat protein expressed without the VP64 activation domain (II and IV) was created (no activation control-NAC). The gRNA is fused to 2 self-splicing ribozymes (hammerhead (HH) at the 5′ end and HDV at the 3′ end) and the MS2 loop and is expressed under the control of either the methanol inducible ALD4 promoter (I and II) or the strong constitutive GAP promoter (III and IV).
Five target sequences for scgRNA were selected in the pTHI11 ranging from −14 to −317 bp in respect to the transcription start site (TSS) of THI11(15) (Figure 1(C)). In addition, a scgRNA was designed, which has no binding sequence within the genome of the CBS 7435 wild type strain and serves as a no-targeting control (NTC) in the subsequent experiments. All genetic constructs were assembled using a Golden Gate system established for the use in Pichia pastoris(5) and have been submitted to Addgene (Table S1).
The expression of dCas9 and the MS2 coat protein constructs was placed under control of the constitutive promoters pTEF and pPOR1, respectively. P. pastoris is frequently cultivated on different carbon sources and the system shall work both under methanol/glucose limited or glucose surplus conditions. Thus, for gRNA expression, a weak MeOH inducible promoter, pALD4 (Figure 1(D) I and II), and a strong constitutive, pGAP (Figure 1(D) III and IV), were selected based on previously reported promoter strengths.5 Every gRNA promoter combination was tested with the MS2-VP64 fusion protein and the non-fused MS2 coat protein (NAC).
Strains containing the activation domain (MS2-VP64) (Figure 2(A,B)) were compared against the NACs (Figure 2(C,D)). Both were cultivated in the presence or absence of 100 μM thiamine in a methanol and glucose limited medium. Under derepressing conditions (in the absence of thiamine), a strong decrease of eGFP production was observed when sequences T1 and T2 were targeted by the scgRNA/dCas9. About 9 to 10 times less fluorescence was measured for these two targets compared to the NTC. The effect was independent from the presence of the activation domain, as the NAC strains showed a comparable behavior. Strains expressing scgRNA with the other target sequences (T3, T4, and T5) showed a fluorescence comparable to the NTC under derepressing conditions (Figure 2(A)). Without the activation domain (NAC strains), scgRNA/dCas9 targeting T3 and T4 resulted in reduced fluorescence, while no change was observed for T5 targeting (Figure 2(C)).
Figure 2.

Targeting a pTHI11::eGFP reporter system with the dCas9 system expressing scgRNAs that target T1–T5 under the control of the methanol inducible pALD4 promoter. Log2 fold changes (fc) of eGFP fluorescence compared to the NTC. (A) Strains containing the activation domain VP64 fused to MS2 in the absence of thiamine, (B) the same strains as in A cultivated in the presence of 100 μM thiamine, (C) strains without the activation domain (NAC) in the absence of thiamine, and (D) the same strains as in C cultivated in the presence of 100 μM thiamine. Error bars indicate the sum of squared errors of the respective target sequence compared to the NTC. The number of clones is indicated above each bar chart.
Under repressing conditions, scgRNA targeting T1, T2, and T5 showed comparable eGFP fluorescence as the NTC, but significantly increased fluorescence was observed for the target sequences T3 (10-fold) and T4 (2-fold) (Figure 2(B)). For all target sequences in the NAC control strains a comparable low fluorescence compared to the NTC was detected under repressing conditions (Figure 2(D)). These results confirm that the dCas9 fusion protein that is functionally linked to the transactivator domain VP64, effectively activates expression of eGFP under repressing conditions. However, the effect is dependent on the targeted sequence. The highest activation (about 7.5-fold) was seen for scgRNA targeting T3, with a binding site 70 bp upstream of TSS. A closer position to the TSS (target sequences T1 and T2) led to no activation under repressing conditions and even a significant repression under derepressing conditions. A CRISPRi effect was observed, when the scgRNA/dCas9 is targeted to T1 to T4 (Figure 2(C)), which gets less pronounced when the target is more distant from the TSS.10 In accordance with previous results for VP64 based systems in S. cerevisiae(16) the activation effect is dependent on genomic localization of the target.
Different scgRNA expression levels were created by using the pGAP promoter under methanol/glucose limiting or glucose surplus conditions. The pGAP promoter is commonly known for its strong constitutive expression but shows nearly 4-fold lower expression under methanol/glucose limited conditions compared to glucose surplus conditions.17 By applying this setup, the impact of scgRNA expression levels on the performance of the scgRNA/dCas9 system was evaluated. Comparing the levels of eGFP fluorescence between Figure 2(A–C) and Figure 3(A–C), a similar performance of both systems can be observed showing that the scgRNA expression is sufficient when using either pALD4 or pGAP as promoters for scgRNA expression. However, slightly higher activation levels were observed for the pGAP constructs at T3 and T4 both under THI11 inducing (Figure 3(A)) and repressing conditions (Figure 3(B)).
Figure 3.

Targeting a pTHI11::eGFP reporter system with the scgRNA/dCas9 system expressing the scgRNA under the control of the strong constitutive pGAP promoter. Log2 fold changes of eGFP fluorescence compared to the NTC. (A) Strains containing the activation domain VP64 fused to MS2 in the absence of thiamine, (B) the same strains as in A cultivated in the presence of 100 μM thiamine, (C) strains without the activation domain (NAC) in the absence of thiamine. (D–F) the same strains as in (A–C) but cultivated under glucose surplus conditions. Error bars indicate the sum of squared errors of the respective target sequence and the related negative control. Number of clones is indicated on top.
The success of a scgRNA/dCas9 assisted transcription regulation seems to be dependent on the position of the target sequence. For targets T1 and T2 the repression power of dCas9 was higher as the activation power of the VP64 domain. Both target sequences are located close to the TSS and dCas9 binding probably interferes with the core promoter, which typically occupies a region of ±50 bp around the TSS in eukaryotic cells.18 Such repression effects of scgRNA binding sites close to the TSS are in accordance with previous experiments done in Yarrowia lipolytica(19) and S. cerevisiae.11 The repression strength of target sequences typically decreases with increased distance to the TSS and to the core promoter. For the THI11 promoter, repression effects targeting up to 160 bp upstream of the TSS were observed.
With scgRNA/dCas9 assisted transcription regulation it is possible to activate pTHI11 in a repressed state. With increased scgRNA expression levels (glucose surplus conditions) the activation strength was slightly increased (Figure 3(D,E)), which can be explained by the effect that the THI11 promoter yields lower expression levels on glucose surplus conditions than on glucose limited/methanol conditions.17 When the THI11 promoter is activated by the scgRNA/dCas9 system a comparable expression level was reached as for the glucose limited/methanol condition (data not shown). In summary the level of activation depends on the status of the promoter. In case the promoter is repressed, the scgRNA/dCas9 system can lead to a re-activation establishing the expression levels of the induced state. For the induced THI11 system (depleted thiamine) no further induction above its natural level was observed. This shows that this regulation approach is applicable if a fine-tuning of a gene expression system is required.
The pTHI11 system showed how a repressed promoter can be derepressed with the scgRNA/dCas9 system. In the second part a gene shall be directly induced, and therefore, the riboflavin biosynthesis pathway was targeted. The gene RIB1 (PP7435_Chr2-0540), encoding for a GTP-cyclohydrolase II, the first enzyme of the riboflavin biosynthesis pathway, was previously reported to yield an increased riboflavin production when overexpressed.21 Four target sequences (R1–R4) positioned in the range of −9 to −156 bp in respect to the TSS were tested (Figure 4(A)) using the pGAP scgRNA design (Figure 1(D) III and IV).
Figure 4.

Targeting the RIB1 gene with the scgRNA/dCas9 system. (A) Overview of the tested target sequences (R1–R4). Arrowheads indicate the position of the PAM sequence and the positions of the last nucleotide of the PAM sequences in respect to the TSS. Riboflavin productivities after 23 and 53 h of (B) MS2 VP64 and (C) NAC strains cultivated using glucose surplus conditions. (D) Log2 fold changes of relative transcript levels of the RIB1 gene compared to the wt measured after 23 h of cultivation using MeOH conditions. Riboflavin productivities measured after 23 and 53 h of (E) MS2 VP64 and (F) NAC strains cultivated using MeOH conditions. The number of biological replicates is indicated on top of each bar. Error bars indicate the standard deviation of the biological replicates for B, C, E, and F and the sum of squared errors of the Ct values of the target gene and the housekeeping gene of all technical replicates (4 per biological replicate) for D.
The selected target sequences were tested both in glucose surplus and methanol/glucose limiting conditions. As a positive control a previously described RIB1 overexpression strain (X-33 PRC RIB1)21 was used. Riboflavin productivities are presented after 23 and 53 h for MS2-VP64 strains (Figure 4(B)) and NAC strains (Figure 4(C)). The wildtype resulted only in a low signal, whereas, as expected, the RIB1 overexpression strain showed a significantly increased riboflavin production. In the presence of the activation domain targets R1, R2, and R3 showed an increased riboflavin production compared to the wildtype control. Productivities of R1, R2, and R3 were on the same level as the positive control and remaining quite stable throughout the cultivation period. With 6 mg/L riboflavin R1 even outperformed the positive control (Figure S1(A)). R4 showed no increase in riboflavin concentration compared to the wild type strain. Without the activation domain all target sequences behaved similarly as the wildtype.
To evaluate the robustness of this system, the same target sequences were tested using methanol/glucose limiting conditions. Strains targeting R2 to R4 performed comparably as under the glucose surplus condition: R2/R3 showed an increased riboflavin production whereas R4 had no improvement compared to the wildtype strain. Interestingly, the strain using target sequence R1 reached an unexpected high increase in riboflavin concentration, which was even three times higher compared to the positive control (Figure 4(E) and Figure S1(C)). Without the activation domain, R1 resulted in a decreased riboflavin production of about 50% whereas all the other targets behaved similar compared to the wildtype (Figure 4(F)).
To verify scgRNA/dCas9-mediated expression fine-tuning on transcript level, mRNA was isolated and the transcript levels of the RIB1 gene were determined for the methanol/glucose limiting conditions (Figure 4(D)). Interestingly, the RIB1 overexpression control strain showed the highest RIB1 upregulation compared to the wildtype followed with some distance by the strain with the scgRNA/dCas9 construct targeting R1 with activation domain. As expected, the strains with the constructs targeting R2, R3 showed only slightly increased transcript levels, and for R4 showed no significant change at all compared to the wildtype. A strain without the activation domain for the R1 construct (NAC) showed the highest reduction in transcript level of about 20% of the wildtype, followed by less pronounced transcript level reductions for NAC constructs targeting R2, R3, and R4. Taken together, the transcript data do not explain why condition R1 showed such a significant increase in riboflavin concentrations. However, it might be that a rather moderate upregulation of the RIB1 gene by the scgRNA/dCas9 system with the target sequence R1 yields a more optimal transcript level of RIB1 and thereby provides an optimal flux through the riboflavin biosynthesis pathway.
scgRNA/dCas9 assisted transcription regulation can be used to impact metabolite production, in this case, to increase riboflavin levels when targeting the RIB1 gene. And the strains even outperformed a previously reported strong riboflavin production strain.21 The efficiency of the system was shown to be very much dependent on the relative position of the targeted sequences to the TSS. Targets close or even overlapping with the TSS resulted in a repression effect that was not compensated by the presence of an activation domain. These results are in accordance with CRISPRi experiments.19,22 However, differences in the promoter architecture preclude to specify a universal rule, for which distance from the TSS still promotes a repression effect. Target sequences further upstream (−30 to −150 bp) can lead to activation when VP64 is bound via a scaffold structure to dCas9. Applying this system to the riboflavin biosynthesis pathway shows that fine-tuning of gene expression can have a surprising and significant impact on the productivity of microbial cell factories.
In conclusion, the presented scgRNA/dCas9 system is a promising tool for fast and reliable fine tuning of transcript levels in P. pastoris. Two different versions of the system are available, optimized for the frequently applied carbon sources, glucose or methanol. It can be used for gene activation and is even capable to overcome a tightly repressed promoter. In the case of RIB1 engineering, it outcompetes a classic overexpression experiment. However, without an activation domain the scgRNA/dCas9 tool can be also used for gene repression and is thus a flexible and versatile tool for metabolic engineering approaches.
Materials and Methods
Plasmid Assembly
Plasmids were created using GoldenPiCS as described previously5 and transformed into chemically competent E. coli DH10B (Invitrogen)23 and are made available at Addgene (Table S1).
Strain Generation
Plasmids were linearized using AscI, cleaned up and transformed into electrocompetent cells,24 and cassettes were genomically integrated into the RGI2 locus. All constructs containing THI11 target sequences were transformed into the CBS 2612 strain carrying an eGFP expression cassette under the control of the THI11 promoter integrated into the AOX1 terminator.12 All constructs containing RIB1 target sequences were transformed into CBS7435 wildtype strain. Transformations were plated on YPD plates with the appropriate selection marker (zeocin 50 mg L–1, nourseothricin 100 mg L–1)
Screening in 24 Deep Well Plates
For the evaluation of the different target sequences clones were cultivated in 24 deep well plates on a rotary shaker at 280 rpm at 25°C. These screenings were performed in minimal media (per liter: 6.3 g (NH4)2HPO4, 0.8 g (NH4)2SO4, 0.49 g MgSO4·7H2O, 2.64 g KCl, 0.0535 g CaCl·2H2O, 22g Citric acid monohydrate, 1.47 mL PTM1 (without biotin), 20 mL NH4OH (25%), 0.0004 g biotin and the pH was adjusted to 6.4 using KOH) under glucose limiting conditions using an enzyme based glucose release system with methanol shots. A preculture was performed in selective YPD media for 24 h. A main culture with an OD600 of 2 was started in 2 mL minimal media with the addition of 30 g L–1 EnPump 200 polysaccharide and 0.23% Reagent A enzyme solution (both from Enpresso, Germany). The main cultures were fed with 1% MeOH after 3 and 15 h and cultures were analyzed for eGFP production after 24 h. For screenings in 2% glucose, cells were inoculated with an OD of 2 in minimal media supplemented with 2% glucose and analyzed for eGFP production after 24 h. GFP production was normalized to the parental strain cultivated on the same plate. The analyzed strains had a comparable growth rate and thus no normalization for OD was done.
Shake Flask Cultivations
Strains were tested for riboflavin production using shake flask cultures incubated at 180 rpm and 25 °C. From a preculture (YPD media) shake flask cultures were inoculated with an OD600 of 1 in minimal media (see above). For cultivations using glucose surplus conditions, the minimal media was supplemented with 2% glucose and cultures were fed with 1.5% glucose after 23, 31, and 47 h. For cultivations using glucose limiting conditions with MeOH the minimal media was prepared as mentioned above, and the cultures were fed with MeOH after 3 (0.5 %), 6 (0.5%), 23 (1%), 31 (1%), and 47 (1 %) h.
Flow Cytometry
eGFP expression was determined using flow cytometry. Cultures were diluted with PBS to a final OD600 of 0.4 and analyzed using a CytoFLEX machine (Beckmann Coulter). The 488 nm laser and the optical filter of 525/40 BP (referred as FL1) were used. For each sample 1 × 104 events were recorded, and cell debris was excluded by setting appropriate gates. Geometric mean of height of FL1 and FSC were used for evaluation of the samples. Calculations of the specific fluorescence levels related to cell sizes were performed by dividing the FL1 signal by the forward-scatter taken to the power of 1.5.25
Quantification of Riboflavin Using Fluorescence Measurements
200 μL of 1:4 diluted culture supernatants were quantified using riboflavin standards with the following concentrations: 8, 6, 4, 2, 1, 0.75, 0.5, and 0.25 mg·L–1. Measurements were performed on an Infinite M200 (Tecan) using 488 nm as excitation wavelength and 530 nm as detection wavelength. Quantification was done by using quadratic regression. Riboflavin productivities were calculated by dividing the concentrations by the OD600 values.
RNA Extraction and Transcript Level Determination
1 mL of the shake flask cultures were taken after 23 h and total RNA was isolated as described previously.26 Integrity of the RNA was checked by gel electrophoresis. cDNA synthesis was performed using the Biozym cDNA synthesis kit using the oligo d(T)23 VN primer (NEB). Transcript levels were determined by quantitative PCR using the Biozym Blue ŚGreen qPCR kit and a Rotor-Gene Q machine (Qiagen). Quantification was performed using the ΔΔCT method27 and ACT1 (PP7435_Chr3–0993) as the housekeeping gene.
Data Visualization
Figures 2–4 were created using the python package matplotlib v.3.1.3.28 The TOC graphic was created using ACD/ChemSketch, version 2020.1.2, Advanced Chemistry Development, Inc., Toronto, ON, Canada, www.acdlabs.com, 2020.
Acknowledgments
This work is supported by the Austrian Science Fund (FWF): Project W1224—Doctoral Program on Biomolecular Technology of Proteins—BioToP). The COMET center: acib: Next Generation Bioproduction is funded by BMK, BMDW, SFG, Standortagentur Tirol, Government of Lower Austria und Vienna Business Agency in the framework of COMET—Competence Centers for Excellent Technologies. The COMET Funding Program is managed by the Austrian Research Promotion Agency FFG.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.0c00214.
Figure S1: Riboflavin concentrations [mg/mL] in the supernatants after 23 and 53 h; Table S1: Summary of plasmids submitted to Addgene (PDF)
Author Present Address
∥ (R.P.) VTU Engineering GmBH, 1110 Vienna, Austria.
Author Contributions
MB, MGS, DM, and RP designed experiments. MB carried out the experiments. MB and MGS performed data analysis and prepared the manuscript. All authors read and approved the final manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Gasser B.; Prielhofer R.; Marx H.; Maurer M.; Nocon J.; Steiger M.; Puxbaum V.; Sauer M.; Mattanovich D. (2013) Pichia pastoris: protein production host and model organism for biomedical research. Future Microbiol. 8 (2), 191–208. 10.2217/fmb.12.133. [DOI] [PubMed] [Google Scholar]
- Peña D. A.; Gasser B.; Zanghellini J.; Steiger M. G.; Mattanovich D. (2018) Metabolic engineering of Pichia pastoris. Metab. Eng. 50, 2–15. 10.1016/j.ymben.2018.04.017. [DOI] [PubMed] [Google Scholar]
- Gasser B.; Steiger M. G.; Mattanovich D. (2015) Methanol regulated yeast promoters: production vehicles and toolbox for synthetic biology. Microb. Cell Fact. 14 (1), 15–17. 10.1186/s12934-015-0387-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl T.; Sturmberger L.; Kickenweiz T.; Wasmayer R.; Schmid C.; Hatzl A. M.; Gerstmann M. A.; Pitzer J.; Wagner M.; Thallinger G. G.; et al. (2016) A toolbox of diverse promoters related to methanol utilization: functionally verified parts for heterologous pathway expression in Pichia pastoris. ACS Synth. Biol. 5 (2), 172–186. 10.1021/acssynbio.5b00199. [DOI] [PubMed] [Google Scholar]
- Prielhofer R.; Barrero J. J.; Steuer S.; Gassler T.; Zahrl R.; Baumann K.; Sauer M.; Mattanovich D.; Gasser B.; Marx H. (2017) GoldenPiCS: A golden gate-derived modular cloning system for applied synthetic biology in the yeast Pichia pastoris. BMC Syst. Biol. 11 (1), 1–14. 10.1186/s12918-017-0492-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkari P.; Marx H.; Blumhoff M. L.; Mattanovich D.; Sauer M.; Steiger M. G. (2017) An efficient tool for metabolic pathway construction and gene integration for Aspergillus niger. Bioresour. Technol. 245, 1327–1333. 10.1016/j.biortech.2017.05.004. [DOI] [PubMed] [Google Scholar]
- Gassler T.; Heistinger L.; Mattanovich D.; Gasser B.; Prielhofer R. (2019) CRISPR/Cas9-mediated homology-directed genome editing in Pichia pastoris. Methods Mol. Biol. 1923, 211–225. 10.1007/978-1-4939-9024-5_9. [DOI] [PubMed] [Google Scholar]
- Dalvie N. C.; Leal J.; Whittaker C. A.; Yang Y.; Brady J. R.; Love K. R.; Love J. C. (2020) J. Host-informed expression of CRISPR guide RNA for genomic engineering in Komagataella phaffii. ACS Synth. Biol. 9 (1), 26–35. 10.1021/acssynbio.9b00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M.; Chylinski K.; Fonfara I.; Hauer M.; Doudna J. A.; Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (Washington, DC, U. S.) 337 (6096), 816–821. 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L. A.; Larson M. H.; Morsut L.; Liu Z.; Brar G. A.; Torres S. E.; Stern-Ginossar N.; Brandman O.; Whitehead E. H.; Doudna J. A.; et al. (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154 (2), 442. 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalatan J. G.; Lee M. E.; Almeida R.; Gilbert L. A.; Whitehead E. H.; La Russa M.; Tsai J. C.; Weissman J. S.; Dueber J. E.; Qi L. S.; et al. (2015) Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 160 (1–2), 339–350. 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadlmayr G.; Mecklenbräuker A.; Rothmüller M.; Maurer M.; Sauer M.; Mattanovich D.; Gasser B. (2010) Identification and characterisation of novel Pichia pastoris promoters for heterologous protein production. J. Biotechnol. 150 (4), 519–529. 10.1016/j.jbiotec.2010.09.957. [DOI] [PubMed] [Google Scholar]
- Jensen E. D.; Ferreira R.; Jakočiunas T.; Arsovska D.; Zhang J.; Ding L.; Smith J. D.; David F.; Nielsen J.; Jensen M. K.; et al. (2017) Transcriptional reprogramming in yeast using dCas9 and combinatorial gRNA strategies. Microb. Cell Fact. 16 (1), 1–16. 10.1186/s12934-017-0664-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landes N.; Gasser B.; Vorauer-Uhl K.; Lhota G.; Mattanovich D.; Maurer M. (2016) The vitamin-sensitive promoter PTHI11 enables pre-defined autonomous induction of recombinant protein production in Pichia pastoris. Biotechnol. Bioeng. 113 (12), 2633–2643. 10.1002/bit.26041. [DOI] [PubMed] [Google Scholar]
- Valli M.; Tatto N. E.; Peymann A.; Gruber C.; Landes N.; Ekker H.; Thallinger G. G.; Mattanovich D.; Gasser B.; Graf A. B. (2016) Curation of the genome annotation of Pichia pastoris (Komagataella phaffii) CBS7435 from gene level to protein function. FEMS Yeast Res. 16 (6), 1–12. 10.1093/femsyr/fow051. [DOI] [PubMed] [Google Scholar]
- Raschmanová H.; Weninger A.; Glieder A.; Kovar K.; Vogl T. (2018) Implementing CRISPR-Cas technologies in conventional and non-conventional yeasts: current state and future prospects. Biotechnol. Adv. 36 (3), 641–665. 10.1016/j.biotechadv.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Prielhofer R.; Cartwright S. P.; Graf A. B.; Valli M.; Bill R. M.; Mattanovich D.; Gasser B. (2015) Pichia pastoris regulates its gene-specific response to different carbon sources at the transcriptional, rather than the translational, level. BMC Genomics 16 (1), 1–17. 10.1186/s12864-015-1393-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle V.; Stark A. (2018) Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 19 (10), 621–637. 10.1038/s41580-018-0028-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz C.; Frogue K.; Ramesh A.; Misa J.; Wheeldon I. (2017) CRISPRi repression of nonhomologous end-joining for enhanced genome engineering via homologous recombination in Yarrowia lipolytica. Biotechnol. Bioeng. 114 (12), 2896–2906. 10.1002/bit.26404. [DOI] [PubMed] [Google Scholar]
- Marx H.; Mattanovich D.; Sauer M. (2008) Overexpression of the riboflavin biosynthetic pathway in Pichia pastoris. Microb. Cell Fact. 7 (1), 23. 10.1186/1475-2859-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzisheuskaya A.; Shlyueva D.; Müller I.; Helin K. (2016) Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9-mediated transcriptional repression. Nucleic Acids Res. 44 (18), e141. 10.1093/nar/gkw583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R.; Rogers E. J. (2013) Transformation of chemically competent. Methods Enzymol. 529, 329–336. 10.1016/B978-0-12-418687-3.00028-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Letchworth G. J. (2004) High efficiency transformation by electroporation of Pichia pastoris pretreated with lithium acetate and dithiothreitol. BioTechniques 36 (1), 152–154. 10.2144/04361DD02. [DOI] [PubMed] [Google Scholar]
- Hohenblum H.; Borth N.; Mattanovich D. (2003) Assessing viability and cell-associated product of recombinant protein producing Pichia pastoris with flow cytometry. J. Biotechnol. 102 (3), 281–290. 10.1016/S0168-1656(03)00049-X. [DOI] [PubMed] [Google Scholar]
- Heistinger L.; Gasser B.; Mattanovich D. (2018) Creation of stable heterothallic strains of Komagataella phaffii enables dissection of mating gene regulation. Mol. Cell. Biol. 38 (2), 1–16. 10.1128/MCB.00398-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M. W. A (2001) New mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 29 (9), 16–21. 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter J. D. (2007) Matplotlib: A 2D Graphics environment. Comput. Sci. Eng. 9 (3), 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.