

Abstract
To effectively apply microwell array cell delivery devices their biodegradation rate must be tailored towards their intended use and implantation location. Two microwell array devices with distinct degradation profiles, either suitable for the fabrication of retrievable systems in the case of slow degradation, or cell delivery systems capable of extensive remodeling using a fast degrading polymer, were compared in this study. Thin films of a poly(ethylene glycol)‐poly(butylene terephthalate) (PEOT‐PBT) and a poly(ester urethane) were evaluated for their in vitro degradation profiles over 34 weeks incubation in PBS at different pH values. The PEOT‐PBT films showed minimal in vitro degradation over time, while the poly(ester urethane) films showed extensive degradation and fragmentation over time. Subsequently, microwell array cell delivery devices were fabricated from these polymers and intraperitoneally implanted in Albino Oxford rats to study their biocompatibility over a 12‐week period. The PEOT‐PBT implants shown to be capable to maintain the microwell structure over time. Implants provoked a foreign body response resulting in multilayer fibrosis that integrated into the surrounding tissue. The poly(ester urethane) implants showed a loss of the microwell structures over time, as well as a fibrotic response until the onset of fragmentation, at least 4 weeks post implantation. It was concluded that the PEOT‐PBT implants could be used as retrievable cell delivery devices while the poly(ester urethane) implants could be used for cell delivery devices that require remodeling within a 4–12 week period.
Keywords: biodegradation, cell–material interactions, foreign body reactions (response), implant design, regenerative medicine
1. INTRODUCTION
A key feature of cell delivery devices is that they are composed of biomaterials which elicit no or minimal foreign body response. Depending on the clinical application these biomaterials can be selected in such a way that their biodegradation matches the intended tissue or implant location. For instance, delivery devices that should be retrievable within the first couple of months should display high polymer stability with minimal tissue interactions to ensure that the whole device can be easily removed or replaced at the end of the experiment or during the clinical treatment. In other cases, extensive engraftment and tissue regeneration might be required and, in those instances, degradation should be synchronous with new tissue formation. A very clear example of retrievable devices can be found in the field of islet transplantation, where insulin‐producing β‐cells or pancreatic donor islets are combined with a so‐called islet delivery device to treat type 1 diabetes (Buitinga et al., 2017; Skrzypek et al., 2017). The general consensus is that these devices should consist of nonbiodegradable biomaterials to allow for retrieval in case a severe inflammatory response against the allogenic cells might occur or if over time insulin‐producing cells have to be replenished to obtain sufficient insulin independence. However, it is also crucial that insulin‐producing cells are sufficiently supplied with nutrients and oxygen to maintain their survival and glucose responsiveness (Lau, Henriksnäs, Svensson, & Carlsson, 2009). This requires proper engraftment which is better supported by degradable materials that elicit a minimal foreign body response than by nondegradable materials. On the other hand, extensive engraftment is for instance required for the embedding of organoids, such as kidney organoids generated from human pluripotent stem cells. Proper implant engraftment in the surrounding tissue and vasculature ingrowth is a major concern during implant design, which is most optimal in a biocompatible, fast degrading implant in which the implant is gradually replaced by regenerated tissue.
We have chosen two distinct polymers which can both be used to create a thin film microwell array islet delivery device with very different degradation properties (Deschamps et al., 2002; Hadavi et al., 2018; Hadavi et al., 2019). The slow degradable polymer used in this study belongs to a family of block copolymers composed of poly(ethylene oxide terephthalate) and poly(butylene terephthalate) (PEOT–PBT) which is a thermoplastic elastomer used in various compositions for cartilage repair (Barron et al., 2015), nerve tissue engineering (Santos, Wieringa, Moroni, Navarro, & Valle, 2017), pancreatic islet delivery devices (Buitinga et al., 2013; Buitinga et al., 2017; Hadavi et al., 2019), clinically applied bone fillers (Bartha et al., 2013), and dermal substitutes (Mensik, Lamme, Riesle, & Brychta, 2002). We report on a specific composition namely 4000PEOT30PBT70, with 4000 being the molecular weight of the poly(ethylene oxide) group, and the 30 and 70 denoting the mass ratio percentage between the “soft” PEOT and “hard” PBT, that is also previously used in a diabetic mouse study (Buitinga et al., 2017). In comparison we also report on a poly(ester urethane) polymer that has been used in biomedical research for meniscal inserts (van Tienen et al., 2002), nerve guides for nerve regeneration (Chiono et al., 2011), and vascular tissue engineering (Jovanovic et al., 2010). Clinical trials have been performed with a medical device called C‐seal, made from the same poly(ester urethane) to support stapled colorectal anastomoses (Bakker et al., 2017; Kolkert, Havenga, ten Cate Hoedemaker, Zuidema, & Ploeg, 2011; Morks et al., 2013) while in another form it was clinically used as so‐called NasoPore nasal dressing foam (Romano et al., 2017; Wang, Cai, & Wang, 2014). The poly(ester urethane) was prepared using a diisocyanate, butanediol, and a dihydroxy end functionalized random copolyester of lactide and ε‐caprolactone, into a linear segmented copolymer, consisting of alternating “hard” and “soft” segments (Guelcher, 2008).
Both biomaterials are thermoplastic polymers that allow reshaping of the material by microthermoforming into a thin film microwell array cell delivery device similar to the one described by Hadavi et al. (2019). Such a delivery device consists of an array of micrometer‐sized wells to retain cells aggregates, organoids, or pancreatic islets, thereby effectively preventing clustering of cells. The implant was designed to be a thin as possible with a large surface area to maximize the survival of encapsulated cells in the device. On the other hand, this design makes the biomaterial prone to degradation/disintegration. Observing the degradation/disintegration characteristics of biopolymers in their final implant shape is therefore essential to predict the effectiveness of future cell delivery studies. The aim of this study is to evaluate the in vitro degradation profile and in vivo biomaterial–tissue interactions of microwell array cell delivery devices that should pave the way for retrievable implants and implants involved in extensive tissue remodeling.
2. MATERIALS AND METHODS
2.1. Materials
Similar sized thin films of the 4000PEOT30PBT70 and poly(ester urethane) were made by solvent casting as described previously (Hadavi et al., 2019). The 4000PEOT30PBT70 (PolyActive™, produced by Polyvation BV, Groningen, the Netherlands) is composed of poly(ethylene oxide) with a molecular weight (Mw) of 4000, and the weight percentage (wt%) of PEOT and PBT blocks was 30 wt% and 70 wt%, respectively (Sutherland, Mahoney 2nd, Coury, & Eaton, 1993). The poly(ester urethane) was provided by Polyganics BV (Groningen, the Netherlands). For preparation of polymer solutions chloroform (Merck, Darmstadt, Germany) and 1,1,1,3,3,3‐hexafluoro‐2‐isopropanol (Biosolve, Valkenswaard, the Netherlands) were used as solvents (Bartha et al., 2013; Mensik et al., 2002).
2.2. Polymer film fabrication
Thin films with a thickness of 40 μm were fabricated using solvent casting. The 4000PEOT30PBT70 was dissolved in a 65:35 (wt/wt) mixture of chloroform and 1,1,1,3,3,3‐hexafluoro‐2‐isopropanol at a concentration of 15 wt% and casted on a 100‐mm diameter silicon wafer (Okmetic) at room temperature. The polymer films were dried under a continuous nitrogen stream for 4 hr followed by immersion in ethanol to remove residual solvent and easily peeling from the substrate. Subsequently, the polymer films were dried in a vacuum oven (Heraeus, Hanau, Germany) at 30°C for 3 days. Poly(ester urethane) films with a thickness of 40 μm were fabricated in a similar way from a 5 wt% polymer solution in chloroform.
2.3. Mechanical and thermal properties
The mechanical properties of dry polymer films were determined using a universal tensile testing device (Zwick Z020, Germany). Samples with a thickness of 40 μm were cut according to American Society for Testing and Materials (ASTM) specifications (50 mm × 4 mm). Tensile tests were carried out at room temperature using a 0.01 N pre‐load, a crosshead speed of 50 mm/min and an initial grip to grip separation of 30 mm. Thermal properties were determined with differential scanning calorimetry (DSC). 4000PEOT30PBT70 samples were heated from −50 to 235°C at a rate of 20°C.min−1 and quenched to −50°C. After holding the temperature at −50°C for 6 min the samples were heated to 235°C at a rate of 20°C. min−1. The thermal data reported were from the second heating run. Similar experiments were performed on poly(ester urethane) samples except that the upper temperature was 125°C.
2.4. In vitro degradation
Controlled in vitro hydrolytic degradation experiments were carried out by immersing 20 mg polymer films in phosphate‐buffered saline (PBS) solution at pH 5, 7.4, or 9 at 37°C and were gently shaken using a table top orbital shaker at 150 rpm. Solutions with different pH were used to determine the relative polymer stability at physiological pH compared to artificially‐stimulated biomaterial degradation, which are unlikely for biomaterials to be exposed to in the human body. Three samples were retrieved for every pH at every time point [week 0, 1, 3, 5, 7, 12 and 34 for 4000PEOT30PBT70 and week 0, 1, 3, 5, 12, 15, and 34 for poly(ester urethane)]. Polymer mass was determined after drying, and the average was calculated. Subsequently, FTIR spectra, as well as 1H NMR spectra of dissolved samples in DMSO‐d6 or CDCl3 were taken. The changes in surface morphology of degraded polymer films were examined using scanning electron microscopy (JEOL JSM‐6400 SEM, Japan). Before scanning electron microscopy samples were gold coated using a Cressington 108 auto sputter coater (Cressington, Watford UK).
2.5. Fabrication of microwell scaffolds
Thin films holding 4,630 microwells were made using the method described by Hadavi et al. (2019) by means of micro‐thermoforming (Truckenmüller et al., 2011). The 4000PEOT30PBT70 microwell scaffolds were produced by pressing the polymer films in between a metal mold (Lightmotif, Enschede, the Netherlands) and a 560 μm thick polyethylene film functioning as backing material (Figure 1a,c). The construct was placed in a hydraulic press (Atlas Manual Hydraulic press, Specac), upon which the mold pressure was set to 40 kN at 30°C. The temperature was subsequently increased to 85°C while the pressure was kept at 40 kN. As soon as the temperature reached 85°C, the pressure was increased to 65 kN and the temperature was set to 20°C. When the press was cooled to 20°C, the pressure was released. The mold was then submerged in 70% ethanol for 15 min at room temperature to ease separating the polymer film from the mold.
FIGURE 1.
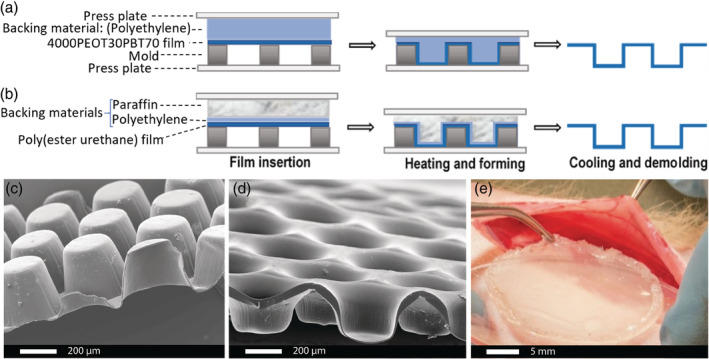
Microthermoforming of polymer films. Micro‐thermoforming steps to manufacture microwell array devices either from (a) 4000PEOT30PBT70 or (b) poly(ester urethane). The construct is assembled, upon which heat and pressure is gradually increased to form the microwells. The construct is then cooled to fix the architecture and subsequently demolded. SEM images of the side view of thermoformed (c) 4000PEOT30PBT70 and (d) poly(ester urethane) microwell scaffold. (e) The microwell array delivery device is closed with a thin film made from the same biomaterial serving as a lid. The lid is annealed to the thermoformed bottom by a tailor‐made hot sealing system
Poly(ester urethane) microwell scaffolds were fabricated by pressing a 20‐μm thick polyethylene film in between the metal mold and a 540‐μm thick paraffin film functioning as backing material (Figure 1b,d). The construct was placed in the hydraulic press, clamped in between the two pressure plates and heated to 41°C. During heating the pressure was gradually increased. When the temperature increased to 25°C, 30°C, 33°C, and 41°C the pressure was simultaneously increased to 30 kN, 40 kN, 50 kN, and 60 kN, respectively. As soon as the temperature reached 41°C, the pressure was released and samples were submerged in ice‐cold ethanol for 15 min in order to separate the scaffold from the mold. Subsequently, the microwells were incubated in xylene for 1 min to remove any residual traces of paraffin. Xylene was exchanged for ethanol by immersion in ethanol for five times. After washing with ethanol, the microwell scaffold were thoroughly rinsed in distilled water.
The microwell‐containing films were then covered with a thin film made from the same biomaterial serving as a lid and annealed by a tailor‐made hot sealing system to finally form the microwell array cell delivery device (Figure 1e). The constructs were then disinfected overnight by incubation in 70% ethanol, drying and subsequent washing with water.
2.6. Implantation
The 30 mm diameter disc‐shaped 4000PEOT30PBT70 and poly(ester urethane) microwell films were implanted intraperitoneally in male Albino Oxford rats, weighing ~200–250 g, to evaluate biomaterial–tissue interactions and in vivo degradation at 1, 4, and 12 weeks post implantation (n = 3 for each time point and each biomaterial). The study was approved by the Dutch Central Authority for Scientific Procedures on Animals (CCD) and the intervention protocols were approved (3006AA) by the local animal care committee of the University of Groningen, the Netherlands.
2.7. Histological evaluation
To assess the biocompatibility of the implanted microwell films, 4000PEOT30PBT70 samples were embedded in paraffin with a HistoStar™ embedding workstation (ThermoFisher Scientific). Subsequently, 5 μm sections were prepared with a microtome (Microm HM 355S, Thermo Scientific). Since the melting point of the poly(ester urethane) scaffolds is lower than that of paraffin, these scaffolds were embedded in Tissue Tek (OCT, Sakura, catalogue# 4583) and sectioned into 5 μm thick sections with a cryotome (Leica, CM3050). All samples were stained with haematoxylin (VWR, VWRK4085‐9002) and eosin (Sigma Aldrich, E4009) to evaluate the tissue–biomaterial interactions, or with trichrome (Sigma Aldrich HT15) for the visualization of collagen deposition and fibrous tissue. Histological slides were imaged using a Nikon Eclipse Ti inverted microscope and analyzed using ImageJ software (http://rsb.info.nih.gov.ij).
3. RESULTS AND DISCUSSION
3.1. In vitro degradation
The 4000PEOT30PBT70 and poly(ester urethane) (Figure 2) were commercially available materials for biomedical research. The microstructure of both polymers are given in the Supporting information (Figure S1–S5 and accompanying text). in vitro degradation of the poly(ester urethane) and 4000PEOT30PBT70 was followed by incubation of polymer films (~20 mg) in PBS buffer at a pH of 5, 7.4, or 9 over 34 weeks. At every time point, polymer samples were removed from the incubation buffer and rinsed with water and air dried. Sample weight was determined, FTIR spectra and 1H NMR spectra were recorded, and macroscopic changes were assessed by scanning electron microscopy of each sample.
FIGURE 2.
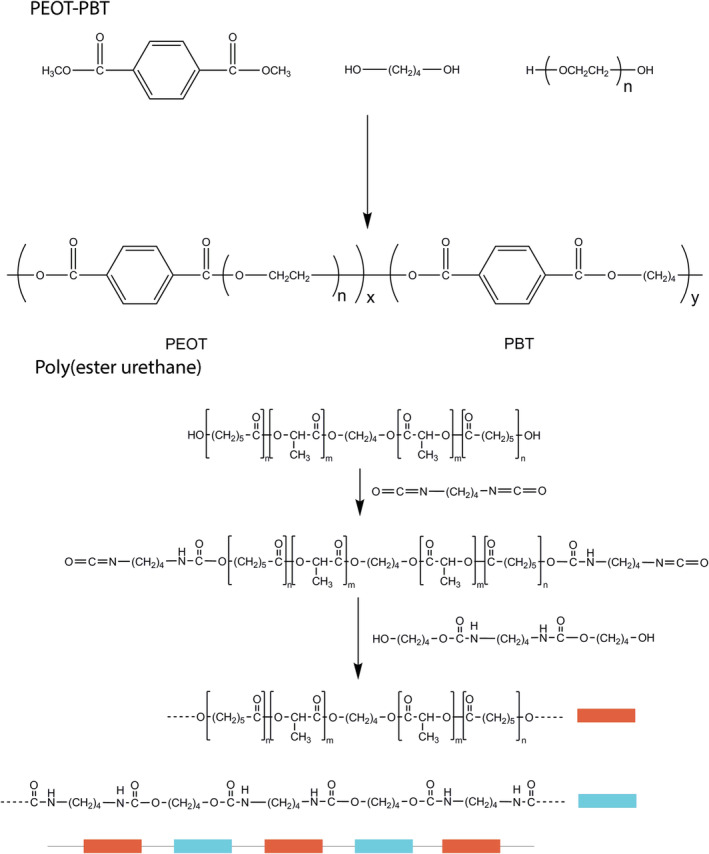
General synthesis and chemical structure of the PEOT‐PBT and poly(ester urethane)
Macroscopically, all 4000PEOT30PBT70 films, up to 34 weeks, showed no visible signs of deterioration independent of incubation at the different pH values. High magnification electron micrographs of the films revealed some surface irregularities and some cracks at 34 weeks incubation (Figure 3a–c). On the other hand, the poly(ester urethane) films showed cracks after 5 weeks and were fully fragmented after 34 weeks of incubation (Figure 3e,f).
FIGURE 3.
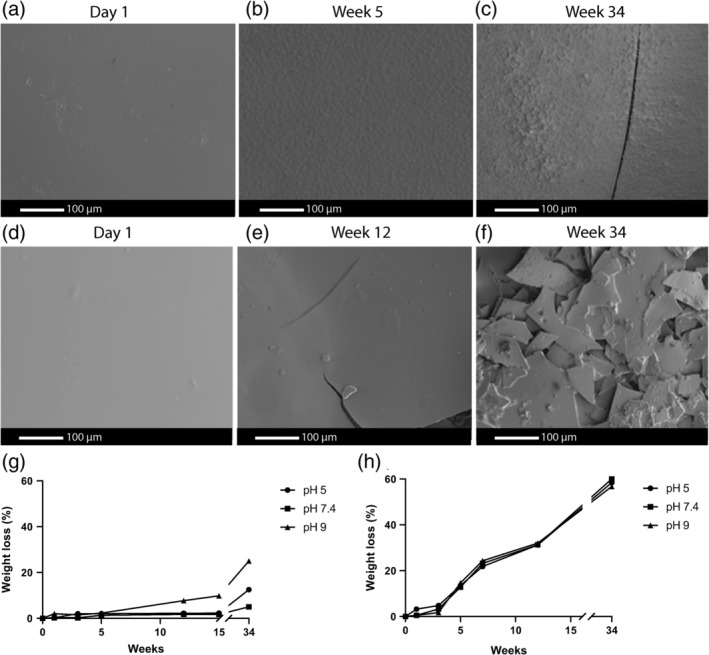
In vitro surface degradation of 4000PEOT30PBT70 and poly(ester urethane films over 34 weeks. SEM images of the surface of the pH 9 PBS‐treated 4000PEOT30PBT70 (a)–(c) and poly(ester urethane) (d)–(f) films at different time points. Weight loss over time (n = 3) of the (g) 4000PEOT30PBT70 films at week 0, 1, 3, 5, 6, 12 and 34 and (h) poly(ester urethane) films at week 0, 1, 3, 5, 12, 15 and 34 by incubation in PBS at pH 5, 7.4 or 9
The weight loss of 4000PEOT30PBT70 films over time (week 0, 1, 3, 5, 7, 12 and 34) indicated that the degradation of the polymer started earlier at a pH of 9 compared to samples incubated at a pH of 5 or 7.4 (Figure 3g). At a pH of 5 and 7.4, after 34 weeks, the weight loss was approximately 5%, while at a pH of 9 around 25% weight loss was observed. In Figure 3g, the lines drawn can only be considered as trend lines because no standard deviations were calculated. The observed linear degradation profile has previously also been described for other PEOT‐PBT polymers (Kellomäki, Paasimaa, Grijpma, Kolppo, & Törmälä, 2002). The amount of mass loss of PEOTPBT polymers has previously been described to depend on its PBT content, with mass loss ≤10% in PBS at pH 7.4 once the PBT mass ratio exceeds 50% (Deschamps et al., 2004; Zhang, Feng, & Xie, 2009). Hydrolysis of the ester bonds between the PEO and terephthalate units has been described to be most susceptible to hydrolysis within PEOT‐PBT block copolymers (Deschamps et al., 2004; Gaharwar et al., 2014). The minor changes in the surface texture of the films are most likely due to the low number of these particular ester bonds and the relatively high amount of the “hard” PBT blocks in 4000PEOT30PBT70. Moreover, both blocks in these copolymers are phase separated into low, PEOT, and high crystalline PBT domains. An uneven degradation throughout the polymer's microstructure leads to cracks and ultimately fragmentation, which becomes visible after 34 weeks of accelerated in vitro degradation at a pH of 9 (Deschamps et al., 2004; Radder et al., 1995). in vitro degradation of poly(ester urethane) showed a 60% weight loss over time (determined at week 0, 1, 3, 5, 12, 15, and 34), independent of pH (Figure 3h). Degradation of the poly(ester urethane) revealed the polymer to easily disintegrate making the retrieval of all polymer mass difficult. Although three samples were used for every time point and at every pH, the lines shown in Figure 3h should be regarded trend lines. Other poly(ester urethane) shows a similar S‐shaped degradation profile, with differences in degradation properties being linked to differences in hydrophilicity and hard segment content (Umare & Chandure, 2008).
These findings indicated that extensive bulk degradation of the poly(ester urethane) takes place, in strong contrast to 4000PEOT30PBT70, which only shows signs of some surface erosion after 34 weeks (Umare & Chandure, 2008; Van Blitterswijk, Leenders, & Baaker, 1993). Degradation of the 4000PEOT30PBT70 and poly(ester urethane) polymers proceeds through hydrolysis of the ester groups, leading to polymer segments with hydroxyl and carboxylic acid functional groups (Brown, Lowry, & Smith, 1980; Lee & Gardella, 2001; Schoonover et al., 2001). The carboxylic acid groups may accelerate the hydrolysis of the polyester segments in an autocatalytic process (Lelah & Cooper, 1986; Stokes, McVenes, & Anderson, 1995).
In vitro degradation of the polymer films was also monitored by 1H NMR and FTIR spectroscopy. Only minor changes were observed for the 4000PEOT30PBT70 upon incubation independent of pH. A clear difference can be seen for the poly(ester urethane) films before and after 12 weeks incubation as shown by characteristic 1H NMR spectra of the poly(ester urethane) before and after degradation at pH 5 after 12 weeks (Figure 4a). The spectral data show the presence of lactyl and caprolactoyl end groups at 3.55 ppm, revealing hydrolysis of ester groups has taken place. Polymer fragments recovered at later time points, independent of pH, became difficult to dissolve in deuterated chloroform. However, the recovered material could be dissolved in deuterated DMSO, and the 1H NMR data revealed a decrease in the signal of lactyl methine and caprolactoyl CH2‐C(O) methylene protons compared to the 3.1 ppm CH2NHC(O) proton signal. FTIR spectroscopy revealed only minor changes for the 4000PEOT30PBT70 upon degradation (Figure 4b). For the poly(ester urethane) films, hydrolysis of ester bonds was clearly observed (Figure 4c). The ratio between the carbonyl absorption bands of ester bonds at 1728 cm−1 and carbamate bonds at 1680 cm−1 clearly changes as a result of ester group hydrolysis. The appearance of a broad absorption peak at 3700 cm−1 revealed the presence of hydroxyl and carboxylic acid groups as a result of ester hydrolysis.
FIGURE 4.
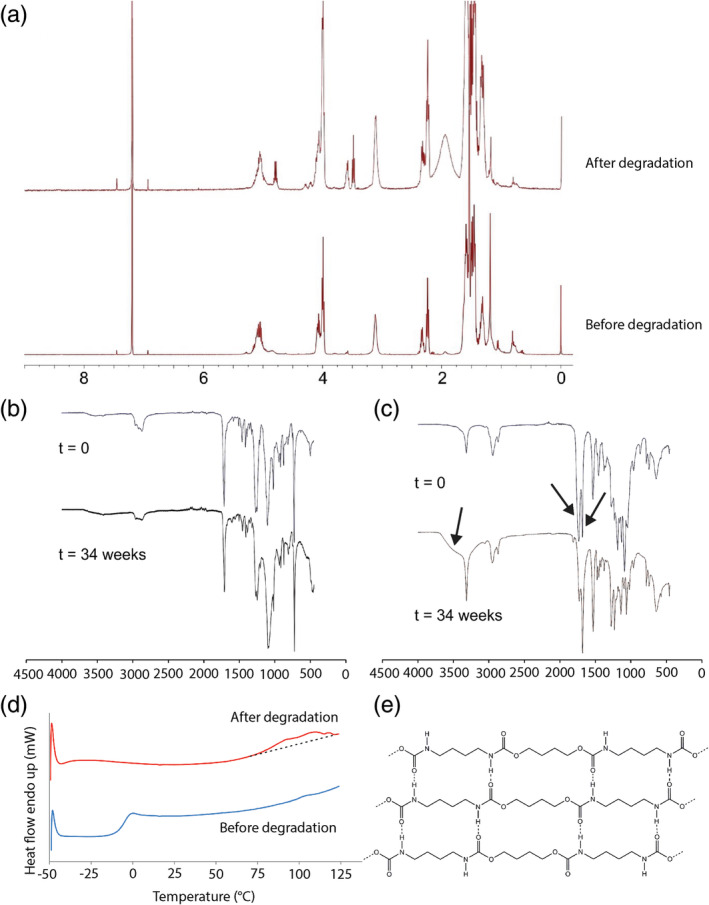
1H NMR spectrum, FTIR spectra and DSC thermograms of in vitro degradation of polymer films. (a) 1H NMR of non‐degraded poly(ester urethane) (bottom) and after degradation for 12 weeks at pH 5 (top). The polymer was dissolved in CDCl3. FTIR spectra of polymer films at day 0 and 34 weeks after degradation in PBS at pH 9 for (b) 4000PEOT30PBT70 and (c) poly(ester urethane. (d) DSC thermograms of the poly(ester urethane) before (bottom) and after 7 weeks incubation in PBS, pH 9 and at 37°C. (b) Representation of hydrogen bonded tetra‐carbamate segments. Oligoester moieties present on both ends are not depicted
3.2. Thermal and mechanical properties
DSC revealed that the 4000PEOT30PBT70 is a semi‐crystalline material with a glass transition temperature of approximately −45°C and a melting transition temperature at 218°C. No changes in these values were observed after prolonged incubation in PBS at different pH values during 34 weeks. The thermal properties of the amorphous poly(ester urethane) revealed a glass transition temperature at −6.1°C with a ΔCp value of 0.47 J g−1 °C−1. At 105°C a very small transition was observed, which can likely be explained by the molecular organization in the polymer due to hydrogen bonding of the carbamate linkages. Upon degradation the Tg fully disappeared showing hydrolysis in the polyester segments. The transition at ~100°C becomes more defined but still it is very broad (Figure 4d). The observed increase in crystallinity of the poly(ester urethane) may be due to the formation of tetra‐carbamate hydrogen bonded segments with short oligoester moieties on the ends as presented in Figure 4e.
The Youngs modulus of the poly(ester urethane) (32 MPa) was eight times lower than that of the 4000PEOT30PBT70 (260 MPa), corresponding to the semicrystaline nature of the polyester (Figure S6). Moreover, the peak stress of 4000PEOT30PBT70 (11.1 MPa) was higher compared to the poly(ester urethane) (7.2 MPa). Both polymers show a similar failure strain around 225%. A previous study of Deschamps, Grijpma, and Feijen (2001) on the tensile behavior of PEOT‐PBT polymers revealed that an increase in the weight percentage of PEOT soft segments reduces the polymer strength and stiffness, accompanied by an increase in elasticity of the material.
3.3. Histology
Both poly(ester urethane) and 4000PEOT30PBT70 films were shaped by microthermoforming into microwell scaffolds comparable to previously published research on β‐cell delivery devices (Hadavi et al., 2019). These scaffolds were implanted in Albino Oxford rats in the intraperitoneal space to assess their engraftment and tissue interaction.
Implants prepared from 4000PEOT30PBT70 showed minimal morphological changes upon implantation (Figure 5a–c) with formation of granulation/fibrous tissue after 4 weeks of implantation (Figure 5d–f) and finally formation of a clear fibrous capsule after 12 weeks of implantation (Figure 5g–l). Even though foreign body giant cells (FBGCs) were present as of week 4 of implantation (Figure 5e), their prevalence was low. Microwell structures of 4000PEOT30PBT70 implants were slightly deformed over time, but most importantly maintained the same well volume. On average, the microwells were 300–350 μm deep and showed an inner diameter around 250 μm wide during the first 4 weeks of implantation, whilst implants showed an average depth of 250 μm and inner diameter of 350 μm after 12 weeks of implantation. This altered microstructure of the device could be caused by the foreign body reaction, resulting in multilayer fibrosis that integrated into the surrounding tissue (Figure 5j–l). Here, failed phagocytosis leads to fusing of macrophages towards FBGCs and also stimulates maturation of granulation tissue towards a fibrous capsule in an attempt to isolate and prevent outspread of the foreign body to other body regions (Klopfleisch & Jung, 2017). Some of the fibroblasts differentiate towards myofibroblasts during fibrous capsule formation which could lead to capsular contracture. These myofibroblasts provide a contractile force while the collagen matrix remodels and stabilizes the contracture, leading to implant deformation and mechanical stress (Headon, Kasem, & Mokbel, 2015). Remarkably, a thicker fibrous layer (ranging between 50 and 150 μm) was observed at the open side of the 4000PEOT30PBT70 implants (Figure 5j–l), while the bottom side of the microwells only showed a fibrous layer with a thickness of 14 ± 4 μm. It has previously been described that a thicker polymer layer is associated with a thicker fibrous layer (Ward, Slobodzian, Tiekotter, & Wood, 2002). The presence of the two polymer layers in close proximity to one another may thus well explain the thick fibrous layer and the resulting deformation. This also stresses the importance of limiting device dimensions in future cell delivery applications.
FIGURE 5.
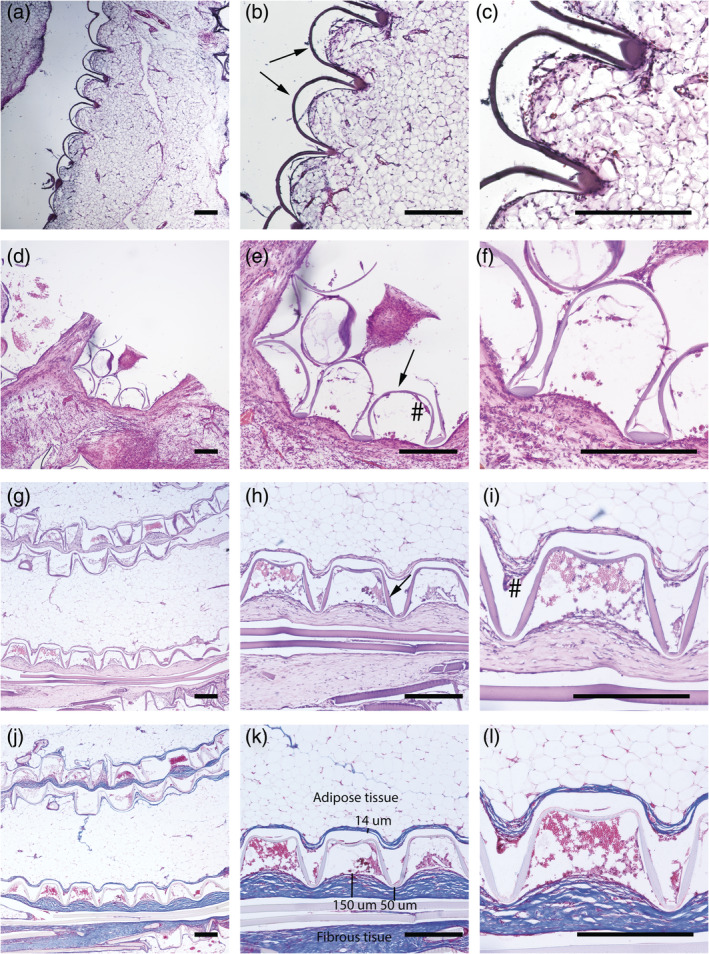
Hematoxylin eosin staining of intraperitoneally implanted 4000PEOT30PBT70 rats after week 1 (a)–(c), week 4 (d)–(f) and week 12 (g)–(i), and trichrome staining of week 12 samples (j)–(l) with blue stain for collagen, red/pink for cellular cytoplasm and black for nuclei. The presence of foreign body giant cells (#) is limited. Scale bar in all light micrographs indicates 300 μm
The macrostructure of the 4000PEOT30PBT70 implants is sometimes altered due to folding (Figure 5g,j). Although this folding does not seem to influence the mechanical stability of the implant, it may be a concern for future application of the implant. Folding of the implant can be prevented by the use of nickel–titanium (nitinol) support rings as this materials possess shape memory, as well as excellent biocompatibility properties that have made nitinol a common biomaterial for multiple biomedical devices, including vascular stents (Stoeckel, Pelton, & Duerig, 2004) and orthopaedics (Zakaria, Madi, & Kasugai, 2019). Given that the degradation of PEOT‐PBT polymers is dictated by (a) hydrolysis of the PEO and terephthalate units and (b) loss of PEOT blocks due to oxidation, the relative low frequency of PEOT units in the 4000PEOT30PBT70 polymer composition could well explain the sobered polymer mass loss over time (Deschamps et al., 2002; Deschamps et al., 2004; Gaharwar et al., 2014).
On the other hand, poly(ester urethane) implants did not engraft very well with the surrounding tissue, impeding histological analysis of the host's tissue response, especially visible for week 1 samples (Figure 6a–c). In contrast to implants made from 4000PEOT30PBT70, poly(ester urethane) implants deformed to a large extend already after 4 weeks of implantation (Figure 6d–f). The average well depth decreased over time. The well depth was 210 μm at week 1 and decreased to 80–150 μm at 4 weeks. Further loss of the microwell structure and fragmentation of the poly(ester urethane) scaffold was observed after 12 weeks of implantation (Figure 6g–i). While poly(ester urethane) implants evoked only few FBGCs after 4 weeks, there was an increased recruitment of FBGCs after 12 weeks of implantation (Figure 6i). This could be explained by the fact that degradation particles from the polymer are phagocytized by macrophages (particles with diameter < 10 μm) and FBGCs (particles with diameter of 10–100 μm) and cleared by intracellular digestion (Xia & Triffitt, 2006). If biomaterial particles are larger, biodegradation is initiated by macrophages and FBGCs via bulk digestion through the release of reactive oxygen species, enzymes, and pH lowering mechanism (Sheikh et al., 2015). Poly(ester urethane)s are known to undergo oxygen‐dependent degradation regulated by phagocytes such as macrophages and FBGCs (Sgrott et al., 2018; Sutherland et al., 1993).
FIGURE 6.
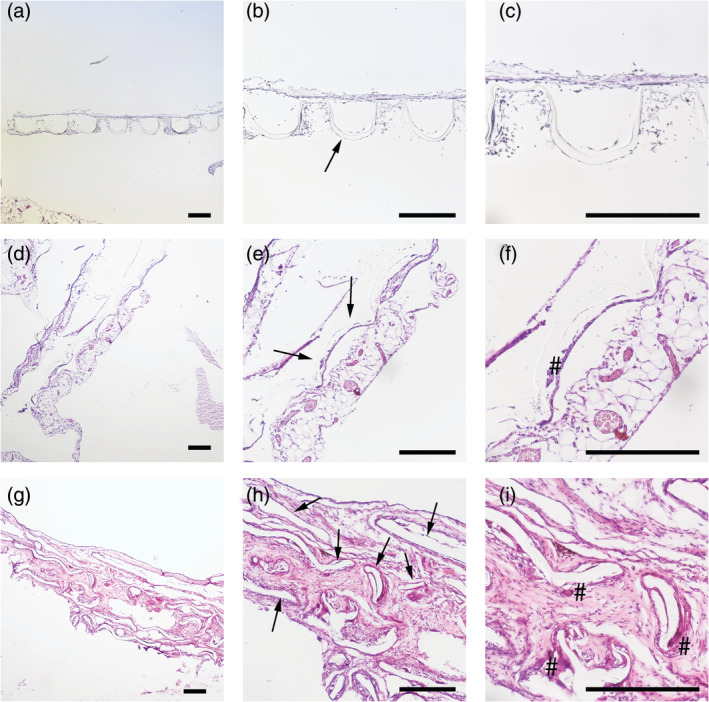
HE stain of poly(ester urethane)‐implanted rats after week 1 (a)–(c), week 4 (d)–(f) and week 12 (g)–(ii). Polymer structure is indicated with an arrow (b, e, h). Degradation of poly(ester urethane) is observed as early as 4 weeks of implantation and disrupted polymer structures are observed at week 12. Foreign body giant cells (#) are present after 12 weeks of implantation (i). Scale bar in all light micrographs indicates 300 μm
The high polymer stability over time makes the 4000PEOT30PBT70 implants suitable for long term implantation and allow retrieving of the implant. Implant retrieval could be considered a valuable characteristic, for instance in the case of induced‐pluripotent stem cells which hold a tumorigenic potential and therefore may be dangerous to the implant recipient (Lee, Tang, Rao, Weissman, & Wu, 2013). Incorporating the stem cells within a retrievable delivery device would therefore be optimal, with the device acting as a physical barrier preventing the unwanted roaming of cells throughout the body. The relative low polymer stability over time of poly(ester urethane) cell delivery devices opens ways for local cell delivery as a temporary or short term solution, and may be used as a drug delivery device, for instance for the co‐delivery of cell clusters and antibiotics (Admane et al., 2017) or oxygen‐generating biomaterials (Coronel, Liang, Li, & Stabler, 2019).
4. CONCLUSIONS
Structural and physical analysis, weight loss measurements and SEM analysis all showed minimal in vitro degradation of 4000PEOT30PBT films over 34 weeks, while poly(ester urethane) films showed severe fragmentation over 34 weeks. Similar results were obtained during histology on microwell array devices that were intraperitoneally implanted for 12 weeks in Albino Oxford rats. The 4000PEOT30PBT70 implants were capable to maintain their microwell structure and elucidate a multilayer fibrotic response that integrated into the surrounding tissue, and could therefore be used as a retrievable cell delivery device. On the other hand, poly(ester urethane) implants showed loss of microwell structures over time and a minimal fibrotic response until the onset of fragmentation 4 weeks post implantation, indicating that it could be used for cell delivery devices in need of local cell environment remodeling within a 4–12 week period. Microwell array cell delivery devices can be used to deliver cell clusters towards the human body for regenerative medicinal purposes. Possible applications include the delivery of aggregated stem cells and organoids or function as an alternative delivery site for pancreatic islets in clinical islet transplantation for type 1 diabetes.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Figure S1. 1H NMR spectrum of the 4000PEOT30PBT70. Solvent CDCl3.
Figure S2. Structure of the pre‐polyester prepared by the bulk ring opening polymerization of DL‐lactide and ε‐caprolactone using stannous octoate as a catalyst and 1,4‐butanediol as an initiator.
Figure S3. Stannous alkoxide formation (a), and ring opening polymerization mechanism of lactones (b).
Figure S4. 1H NMR spectrum of the pre‐polyester with a Mn of 2000. Sequences of lactyl (L) and caprolactoyl (CL) are indicated (CDCl3).
Figure S5. 13C NMR spectrum of the poly(ester urethane) with in the insert the assignment of lactyl (L) caprolactoyl (C) sequences in the carbonyl region.
Figure S6. Stress strain curves of the PEOT‐PBT and poly(ester urethane). The measurements were performed on dry specimens.
ACKNOWLEDGMENTS
This project was financially supported by Juvenile Diabetes Research Institute Foundation (Grant 17‐2013‐303) and the Diabetes Cell Therapy Initiative (DCTI) FES 2009 program LSH‐DCTI including the Dutch Diabetes Research Foundation.
Hadavi E, de Vries RHW, Smink AM, et al. In vitro degradation profiles and in vivo biomaterial–tissue interactions of microwell array delivery devices. J Biomed Mater Res. 2021;109B:117–127. 10.1002/jbm.b.34686
Funding information Diabetes Cell Therapy Initiative (DCTI) FES 2009 program LSH‐DCTI including the Dutch Diabetes Research Foundation; Juvenile Diabetes Research Institute Foundation, Grant/Award Number: 17‐2013‐303
REFERENCES
- Admane, P. , Gupta, J. , Ancy, I. , Kumar, R. , & Panda, A. (2017). Design and evaluation of antibiotic releasing self‐ assembled scaffolds at room temperature using biodegradable polymer particles. International Journal of Pharmaceutics, 520(1), 284–296. [DOI] [PubMed] [Google Scholar]
- Bakker, I. S. , Morks, A. N. , ten Cate Hoedemaker, H. O. , Burgerhof, J. G. M. , Leuvenink, H. G. , van Praagh, J. B. , … the Collaborative C‐seal Study Group . (2017). Randomized clinical trial of biodegradeable intraluminal sheath to prevent anastomotic leak after stapled colorectal anastomosis. The British Journal of Surgery, 104(8), 1010–1019. [DOI] [PubMed] [Google Scholar]
- Barron, V. , Merghani, K. , Shaw, G. , Coleman, C. M. , Hayes, J. S. , Ansboro, S. , … Barry, F. (2015). Evaluation of cartilage repair by mesenchymal stem cells seeded on a PEOT/PBT scaffold in an osteochondral defect. Annals of Biomedical Engineering, 43(9), 2069–2082. [DOI] [PubMed] [Google Scholar]
- Bartha, L. , Hamann, D. , Pieper, J. , Péters, F. , Riesle, J. , Vajda, A. , … Hangody, L. (2013). A clinical feasibility study to evaluate the safety and efficacy of PEOT/PBT implants for human donor site filling during mosaicplasty. European Journal of Orthopaedic Surgery & Traumatology, 23(1), 81–91. [DOI] [PubMed] [Google Scholar]
- Brown, D. W. , Lowry, R. E. , & Smith, L. E. (1980). Kinetics of hydrolytic aging of polyester urethane elastomers. Macromolecules, 13(2), 248–252. [Google Scholar]
- Buitinga, M. , Assen, F. , Hanegraaf, M. , Wieringa, P. , Hilderink, J. , Moroni, L. , … van Apeldoorn, A. (2017). Micro‐fabricated scaffolds lead to efficient remission of diabetes in mice. Biomaterials, 135, 10–22. [DOI] [PubMed] [Google Scholar]
- Buitinga, M. , Truckenmüller, R. , Engelse, M. A. , Moroni, L. , ten Hoopen, H. W. M. , van Blitterswijk, C. A. , … Karperien, M. (2013). Microwell scaffolds for the extrahepatic transplantation of islets of Langerhans. PLoS One, 8(5), e64772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiono, V. , Sartori, S. , Rechichi, A. , Tonda‐Turo, C. , Vozzi, G. , Vozzi, F. , … Ciardelli, G. (2011). Poly(ester urethane) guides for peripheral nerve regeneration. Macromolecular Bioscience, 11(2), 245–256. [DOI] [PubMed] [Google Scholar]
- Coronel, M. M. , Liang, J. P. , Li, Y. , & Stabler, C. L. (2019). Oxygen generating biomaterial improves the function and efficacy of beta cells within a macroencapsulation device. Biomaterials, 210, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps, A. A. , Claase, M. B. , Sleijster, W. J. , de Bruijn, J. D. , Grijpma, D. W. , & Feijen, J. (2002). Design of segmented poly(ether ester) materials and structures for the tissue engineering of bone. Journal of Controlled Release, 78(1), 175–186. [DOI] [PubMed] [Google Scholar]
- Deschamps, A. A. , Grijpma, D. W. , & Feijen, J. (2001). Poly (ethylene oxide)/poly (butylene terephthalate) segmented block copolymers: The effect of copolymer composition on physical properties and degradation behavior. Polymer, 42(23), 9335–9345. [Google Scholar]
- Deschamps, A. A. , van Apeldoorn, A. A. , Hayen, H. , de Bruijn, J. D. , Karst, U. , Grijpma, D. W. , & Feijen, J. (2004). In vivo and in vitro degradation of poly(ether ester) block copolymers based on poly(ethylene glycol) and poly(butylene terephthalate). Biomaterials, 25(2), 247–258. [DOI] [PubMed] [Google Scholar]
- Gaharwar, A. K. , Mihaila, S. M. , Kulkarni, A. A. , Patel, A. , di Luca, A. , Reis, R. L. , … Khademhosseini, A. (2014). Amphiphilic beads as depots for sustained drug release integrated into fibrillar scaffolds. Journal of Controlled Release, 187, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelcher, S. A. (2008). Biodegradable polyurethanes: Synthesis and applications in regenerative medicine. Tissue Engineering Part B: Reviews, 14(1), 3–17. [DOI] [PubMed] [Google Scholar]
- Hadavi, E. , Leijten, J. , Brinkmann, J. , Jonkheijm, P. , Karperien, M. , & van Apeldoorn, A. (2018). Fibronectin and collagen IV microcontact printing improves insulin secretion by INS1E cells. Tissue Engineering Part C: Methods, 24(11), 628–636. [DOI] [PubMed] [Google Scholar]
- Hadavi, E. , Leijten, J. , Engelse, M. , de Koning, E. , Jonkheijm, P. , Karperien, M. , & van Apeldoorn, A. (2019). Microwell scaffolds using collagen‐IV and Laminin‐111 Lead to improved insulin secretion of human islets. Tissue Engineering. Part C, Methods, 25(2), 71–81. [DOI] [PubMed] [Google Scholar]
- Headon, H. , Kasem, A. , & Mokbel, K. (2015). Capsular contracture after breast augmentation: An update for clinical practice. Archives of Plastic Surgery, 42(5), 532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic, D. , Engels, G. E. , Plantinga, J. A. , Bruinsma, M. , van Oeveren, W. , Schouten, A. J. , … Harmsen, M. C. (2010). Novel polyurethanes with interconnected porous structure induce in vivo tissue remodeling and accompanied vascularization. Journal of Biomedical Materials Research. Part A, 95(1), 198–208. [DOI] [PubMed] [Google Scholar]
- Kellomäki, M. , Paasimaa, S. , Grijpma, D. W. , Kolppo, K. , & Törmälä, P. (2002). In vitro degradation of Polyactive® 1000PEOT70PBT30 devices. Biomaterials, 23(1), 283–295. [DOI] [PubMed] [Google Scholar]
- Klopfleisch, R. , & Jung, F. (2017). The pathology of the foreign body reaction against biomaterials. Journal of Biomedical Materials Research Part A, 105(3), 927–940. [DOI] [PubMed] [Google Scholar]
- Kolkert, J. L. , Havenga, K. , ten Cate Hoedemaker, H. O. , Zuidema, J. , & Ploeg, R. J. (2011). Protection of stapled colorectal anastomoses with a biodegradable device: The C‐seal feasibility study. American Journal of Surgery, 201(6), 754–758. [DOI] [PubMed] [Google Scholar]
- Lau, J. , Henriksnäs, J. , Svensson, J. , & Carlsson, P. O. (2009). Oxygenation of islets and its role in transplantation. Current Opinion in Organ Transplantation, 14(6), 688–693. [DOI] [PubMed] [Google Scholar]
- Lee, A. S. , Tang, C. , Rao, M. S. , Weissman, I. L. , & Wu, J. C. (2013). Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nature Medicine, 19(8), 998–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.‐W. , & Gardella, J. A. (2001). In vitro hydrolytic surface degradation of poly (glycolic acid): Role of the surface segregated amorphous region in the induction period of bulk erosion. Macromolecules, 34(12), 3928–3937. [Google Scholar]
- Lelah, M. D. , & Cooper, S. L. (1986). Polyurethanes in medicine, Florida, USA: CRC Press. [Google Scholar]
- Mensik, I. , Lamme, E. N. , Riesle, J. , & Brychta, P. (2002). Effectiveness and safety of the PEGT/PBT copolymer scaffold as dermal substitute in scar reconstruction wounds (feasibility trial). Cell and Tissue Banking, 3(4), 245–253. [DOI] [PubMed] [Google Scholar]
- Morks, A. N. , Havenga, K. , ten Cate Hoedemaker, H. O. , Leijtens, J. W. A. , & Ploeg, R. J. (2013). Thirty‐seven patients treated with the C‐seal: Protection of stapled colorectal anastomoses with a biodegradable sheath. International Journal of Colorectal Disease, 28(10), 1433–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radder, A. M. , van Loon, J. A. , Puppels, G. J. , & van Blitterswijk, C. A. (1995). Degradation and calcification of a PEO/PBT copolymer series. Journal of Materials Science: Materials in Medicine, 6(9), 510–517. [Google Scholar]
- Romano, A. , Salzano, G. , Dell'Aversana Orabona, G. , Cama, A. , Petrocelli, M. , Piombino, P. , … Califano, L. (2017). Comparative study between biodegradable nasopore (BNP) and Merocel hemox 10 cm after septo‐turbinoplasty procedure. European Review for Medical and Pharmacological Sciences, 21(4), 669–673. [PubMed] [Google Scholar]
- Santos, D. , Wieringa, P. , Moroni, L. , Navarro, X. , & Valle, J. D. (2017). PEOT/PBT guides enhance nerve regeneration in long gap defects. Advanced Healthcare Materials, 6(3). [DOI] [PubMed] [Google Scholar]
- Schoonover, J. R. , Thompson, D. G. , Osborn, J. C. , Orler, E. B. , Wrobleski, D. A. , Marsh, A. L. , … Palmer, R. A. (2001). Infrared linear dichroism study of a hydrolytically degraded poly (ester urethane). Polymer Degradation and Stability, 74(1), 87–96. [Google Scholar]
- Sgrott, S. M. , Neves, R. D. , D'Acampora, A. J. , Bernardes, G. J. S. , Belmonte, L. , Martins, T. C. , … Piovezan, A. P. (2018). Early fragmentation of polyester urethane sheet neither causes persistent oxidative stress nor alters the outcome of normal tissue healing in rat skin. Annals of the Brazilian Academy of Sciences, 90(2 suppl 1), 2211–2222. [DOI] [PubMed] [Google Scholar]
- Sheikh, Z. , Brooks, P. J. , Barzilay, O. , Fine, N. , & Glogauer, M. (2015). Macrophages, foreign body giant cells and their response to implantable biomaterials. Materials (Basel, Switzerland), 8(9), 5671–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrzypek, K. , Groot Nibbelink, M. , van Lente, J. , Buitinga, M. , Engels, M. A. , & Stamatialis, D. (2017). Pancreatic islet macroencapsulation using microwell porous membranes. Scientific Reports, 7(1), 9186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckel, D. , Pelton, A. , & Duerig, T. (2004). Self‐expanding nitinol stents: Material and design considerations. European Radiology, 14(2), 292–301. [DOI] [PubMed] [Google Scholar]
- Stokes, K. , McVenes, R. , & Anderson, J. M. (1995). Polyurethane elastomer biostability. Journal of Biomaterials Applications, 9(4), 321–354. [DOI] [PubMed] [Google Scholar]
- Sutherland, K. , Mahoney, J. R., 2nd , Coury, A. J. , & Eaton, J. W. (1993). Degradation of biomaterials by phagocyte‐derived oxidants. The Journal of Clinical Investigation, 92(5), 2360–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truckenmüller, R. , Giselbrecht, S. , Rivron, N. , Gottwald, E. , Saile, V. , van den Berg, A. , … van Blitterswijk, C. (2011). Thermoforming of film‐based biomedical microdevices. Advanced Materials, 23(11), 1311–1329. [DOI] [PubMed] [Google Scholar]
- Umare, S. S. , & Chandure, A. S. (2008). Synthesis, characterization and biodegradation studies of poly (ester urethane) s. Chemical Engineering Journal, 142(1), 65–77. [Google Scholar]
- Van Blitterswijk, C. H. , Leenders, H. , & Baaker, D. (1993). The effect of PEO ratio on degradation, calcification and bone bonding of PEO/PBT copolymer (PolyActive). Cells & Materials, 3(1), 23–36. [Google Scholar]
- van Tienen, T. G. , Heijkants, R. G. J. C. , Buma, P. , de Groot, J. H. , Pennings, A. J. , & Veth, R. P. H. (2002). Tissue ingrowth and degradation of two biodegradable porous polymers with different porosities and pore sizes. Biomaterials, 23(8), 1731–1738. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Cai, C. , & Wang, S. (2014). Merocel versus Nasopore for nasal packing: A meta‐analysis of randomized controlled trials. PLoS One, 9(4), e93959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, W. K. , Slobodzian, E. P. , Tiekotter, K. L. , & Wood, M. D. (2002). The effect of microgeometry, implant thickness and polyurethane chemistry on the foreign body response to subcutaneous implants. Biomaterials, 23(21), 4185–4192. [DOI] [PubMed] [Google Scholar]
- Xia, Z. , & Triffitt, J. T. (2006). A review on macrophage responses to biomaterials. Biomedical Materials, 1(1), R1–R9. [DOI] [PubMed] [Google Scholar]
- Zakaria, O. , Madi, M. , & Kasugai, S. (2019). Introduction of a novel guided bone regeneration memory shape based device. Journal of Biomedical Materials Research. Part B, Applied Biomaterials, 108(2), 460–467. [DOI] [PubMed] [Google Scholar]
- Zhang, A. , Feng, Z. , & Xie, Z. (2009). Long‐term investigation on hydrolytic degradation and morphology of poly(ethylene glycol terephthalate)‐b‐poly(butylene terephthalate) copolymer films. Journal of Applied Polymer Science, 111(3), 1462–1470. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. 1H NMR spectrum of the 4000PEOT30PBT70. Solvent CDCl3.
Figure S2. Structure of the pre‐polyester prepared by the bulk ring opening polymerization of DL‐lactide and ε‐caprolactone using stannous octoate as a catalyst and 1,4‐butanediol as an initiator.
Figure S3. Stannous alkoxide formation (a), and ring opening polymerization mechanism of lactones (b).
Figure S4. 1H NMR spectrum of the pre‐polyester with a Mn of 2000. Sequences of lactyl (L) and caprolactoyl (CL) are indicated (CDCl3).
Figure S5. 13C NMR spectrum of the poly(ester urethane) with in the insert the assignment of lactyl (L) caprolactoyl (C) sequences in the carbonyl region.
Figure S6. Stress strain curves of the PEOT‐PBT and poly(ester urethane). The measurements were performed on dry specimens.