

Abstract
Germline mutation in the PTEN gene is the genetic basis of PTEN hamartoma tumor syndrome with the affected individuals harboring features of autism spectrum disorders. Characterizing a panel of 14 autism‐associated PTEN missense mutations revealed reduced protein stability, catalytic activity, and subcellular distribution. Nine out of 14 (64%) PTEN missense mutants had reduced protein expression with most mutations confined to the C2 domain. Selected mutants displayed enhanced polyubiquitination and shortened protein half‐life, but that did not appear to involve the polyubiquitination sites at lysine residues at codon 13 or 289. Analyzing their intrinsic lipid phosphatase activities revealed that 78% (11 out of 14) of these mutants had twofold to 10‐fold reduction in catalytic activity toward phosphatidylinositol phosphate substrates. Analyzing the subcellular localization of the PTEN missense mutants showed that 64% (nine out of 14) had altered nuclear‐to‐cytosol ratios with four mutants (G44D, H123Q, E157G, and D326N) showing greater nuclear localization. The E157G mutant was knocked‐in to an induced pluripotent stem cell line and recapitulated a similar nuclear targeting preference. Furthermore, iPSCs expressing the E157G mutant were more proliferative at the neural progenitor cell stage but exhibited more extensive dendritic outgrowth. In summary, the combination of biological changes in PTEN is expected to contribute to the behavioral and cellular features of this neurodevelopmental disorder.
Keywords: autism spectrum disorders, macrocephaly, neurodevelopmental disorders, PTEN, PTEN hamartoma tumor syndrome
Mutation of the PTEN gene has been linked to autism spectrum disorders (ASDs) and other disorders with autism‐like features. Here, Andrew Chen and co‐authors characterized a panel of PTEN autism‐associated mutants, unearthing a E157G PTEN mutant that displays preferential localization to the cell nucleus. The E157G mutant was knocked‐in to an iPSC line that recapitulated a similar nuclear‐targeting preference. In addition, iPSCs expressing the E157G mutant were more proliferative at the neural progenitor cell stage and exhibited more extensive dendritic outgrowth when induced to undergo neuronal differentiation, hinting at the contribution of PTEN to the cellular features of ASDs.
Abbreviations
- ASDs
autism spectrum disorders
- PHTS
PTEN hamartoma tumor syndrome
- PI3‐K
phosphoinositide 3‐kinase
- PTEN
phosphatase and tensin homolog deleted on chromosome 10
Introduction
Phosphatase and tensin homolog deleted on chromosome 10 hamartoma tumor syndrome (PHTS) represents a collection of four overgrowth syndromes including Cowden, Bannayan–Riley–Ruvalcaba, Proteus, and Proteus‐like, with germline PTEN mutation detected at variable frequency from 7% to 85% [1]. While having distinct diagnostic criteria, these overgrowth syndromes share common clinical features of multiple hamartomatous growth and increased risk for developing breast and thyroid cancers. In individuals with PTEN germline mutation, macrocephaly is diagnosed in 94% [2], and 22% of PHTS subjects have features of autism spectrum disorders (ASDs) [3]. ASD is a collection of neurodevelopmental disorders including autistic disorder, Asperger syndrome, pervasive developmental disorder not otherwise specified (PDD‐NOS), and Rett syndrome [4]. The core symptoms are social and communication deficits, and sensory hypersensitivity. Affected individuals also display repetitive behavior, epileptic episodes, and mental retardation [5]. The early onset (< 3 years) reflects aberrant neurogenesis at a stage when sensory inputs are shaping excitatory and inhibitory synapses. Altered anatomy, function, and connectivity have been demonstrated in ASD brains. Anatomically, ASD is characterized by overgrowth early in life (2–4 years old) in the amygdala and frontal cortex (reviewed in Ref. [6]). These changes coincide with the developmental stage of intense acquisitions of language and social skills.
Autism spectrum disorders has a strong genetic component. Almost 80% of ASD cases are sporadic with the remaining 20% being syndromic [7]. Syndromic ASD patients have autistic phenotypes characterized by the pathologies of some well‐established neurodevelopmental syndromes such as fragile X (FMR1), Rett syndrome (MECP2), Angelman syndrome (UBE3A), Phelan–McDermid syndrome (SHANK3), and PHTS (PTEN).
PTEN was originally isolated as a tumor suppressor gene inactivated in 30% of human cancers (reviewed in Ref. [8]). It encodes a lipid phosphatase and dephosphorylates phosphatidylinositol (3,4,5)‐triphosphate (PIP3), a second messenger that activates the phosphoinositide 3‐kinase (PI3‐K). Thus, PTEN loss hyperactivates PI3‐K and a host of downstream effectors. Structurally, PTEN is composed of a catalytic domain in the amino (N) terminus and a lipid‐binding C2 domain in the carboxyl (C) terminus (Fig. 1). This is followed by a flexible ‘tail’ region composed of multiple phosphorylation sites, which ends in a postsynaptic density/Dlg1/ZO‐1‐binding motif (PDZ‐BM). PTEN is mainly localized to microtubule‐associated vesicles in the cytoplasm [9]. It is also localized in the nucleus where it plays a role in genome stability [10, 11]. Post‐translational modifications involving ubiquitination at Lys13 and Lys289 or sumoylation at Lys254 and Lys266 have been shown to regulate PTEN proteolysis and nuclear translocation, respectively (reviewed in Ref. [12]).
Fig. 1.
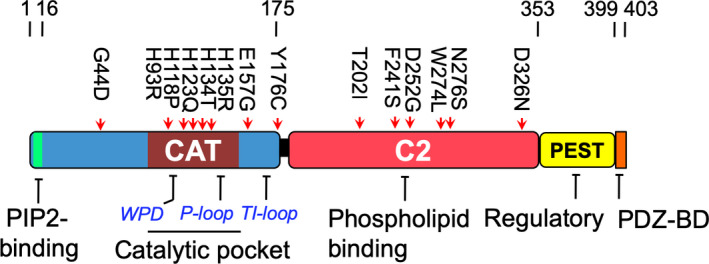
PTEN mutations in PHTS patients with autistic features. Schematic representation of the PTEN mutants used in this study: G44D, H93R, H118P, H123Q, M134T, I135R, E157G, and Y176C are located in the catalytic (CAT) domain; T202I, F241S, D252G, W274L, N276S, and D326N are located in the C2 domain.
Independently, knockout of Pten gene in differentiated neurons results in viable mice with macrocephaly and abnormal social interaction and learning [13]. Mice that are germline heterozygous for PTEN resemble those with PHTS, harboring macrocephaly, impaired social interactions, and repetitive behaviors [14, 15]. In this report, we comprehensively characterized a list of PTEN missense mutants associated with ASD and identified diverse changes in biochemical properties that may perturb the physiological functions of PTEN.
Results
ASD‐associated PTEN missense mutants possess reduced protein expression
To investigate the ASD‐associated functional defects in PTEN mutants, a total of 14 missense mutations were identified based on a literature search for PHTS patients with autistic features (Fig. 1). Eight mutants were located in the N‐terminal half of PTEN where the catalytic domain resides, while six were in the C2 domain, with none reported in the PEST domain and PDZ‐BM. As a control, the PTEN R130Q mutant frequently detected in human endometrial carcinoma was also included [16]. To examine the properties of these PTEN missense mutants, similar amounts of expression plasmids were transfected into three different mammalian cell lines. Specifically, 293T cells expressed wild‐type (WT) PTEN, while both U87MG and PC3 were PTEN‐null. Western blotting analysis clearly revealed two distinct groups of mutants. The first group was composed of five mutants (H93R, H123Q, I135R, E157G, and Y176C) that had protein expression levels very similar to that of WT PTEN (Fig. 2). The second group was composed of nine mutants that had a greater than threefold decrease in the 57‐kDa protein species of PTEN (Fig. 2). This drastic reduction in PTEN protein expression was observed in all of the C2 domain mutants, while only three out of eight N‐terminal domain mutants (H118P, M134T, and I135R) were affected (Fig. 2A). Interestingly, the catalytic domain mutant, H118P, had the lowest expression among all of the missense mutants. Very similar observations were also detected in 293T and U87MG cells (Fig. 2B,C). In addition, there were subtle differences in the molecular mass with the H93R mutant showing a band of faster mobility, while the I135R mutant had reduced mobility compared with the WT (Fig. 2A–C). These differences are likely to be mutation‐specific rather than cell type‐specific as similar changes were observed in all three cell lines tested.
Fig. 2.
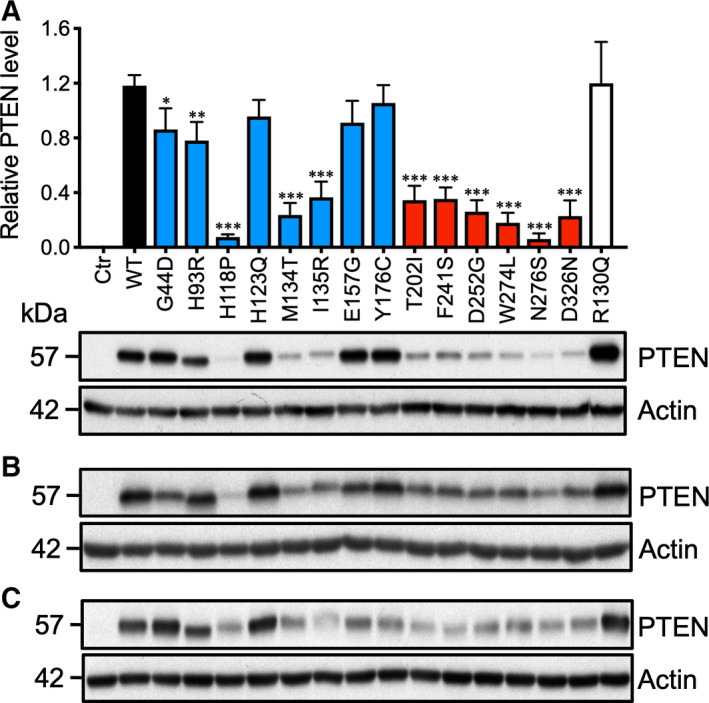
Expression analysis of ASD‐associated PTEN mutants. (A) The indicated expression plasmids were transfected into PTEN‐null PC3 cells. After 48 h, the cells were solubilized with Laemmli buffer. Western blotting analysis was carried out using anti‐PTEN and anti‐actin antibodies. Similar experiments were conducted in (B) U87MG and (C) 293T cells with comparable results being obtained. The results from three independent experiments were analyzed by one‐way ANOVA with Dunnett's post‐tests, and the significance of the data is relative to the WT. Bars, SD. *P < 0.05; **P < 0.01; ***P < 0.001.
Reduced protein half‐life and enhanced polyubiquitination of ASD‐associated PTEN mutants
Phosphatase and tensin homolog deleted on chromosome 10 is a relatively stable protein with an average half‐life of ~ 9 h [17]. To test whether the reduced protein levels in the PTEN missense mutants were due to reduced protein half‐life, protein expression was evaluated following cycloheximide treatment. We selected two low‐PTEN expression mutants each from the catalytic domain (H118P, I135R) and C2 domain (D252G, N276S). As a control, we selected the E157G mutant, which has comparable protein expression to the WT. The WT PTEN expressed in 293T cells had an average half‐life of ~ 6 h (Fig. 3). All four mutants, H118P, I135R, D252G, and N276S, with reduced PTEN protein levels had shorter half‐lives of 3.0–4.0 h (Fig. 3). As expected, the E157G mutant, which retained 80% PTEN protein expression, had a half‐life resembling that of the WT.
Fig. 3.

Reduced protein stability of ASD‐associated PTEN mutants. 293T cells were transfected with the indicated expression plasmids. The cells were treated with cycloheximide (80 μg·mL−1) for the indicated durations. The cells were solubilized in Laemmli buffer, and the levels of PTEN were determined by western blotting analysis and normalized to the actin levels. The results from three independent experiments were quantified. Bars, SD.
It is well established that PTEN protein turnover is regulated by ubiquitin‐mediated proteolysis events. To test whether missense mutants with shortened protein half‐life had greater ubiquitination, cells expressing these mutants were first treated with a proteasome inhibitor, MG132, for 6 h. The WT PTEN showed almost undetectable polyubiquitination, and ubiquitination only became observable following MG132 treatment (Fig. 4A). All of the missense mutants except E157G had readily detectable basal polyubiquitinated protein species, and polyubiquitination was greatly potentiated by MG132 (Fig. 4A,B). Among the missense mutants tested, I135R had the greatest polyubiquitination, followed by D252G, H1118P, and N276S (Fig. 4B). To elucidate the polyubiquitination sites responsible, two known PTEN ubiquitination sites at K13 and K289 were mutated in the two unstable mutants, I135R and N276S, and in the relatively stable E157G mutant. In addition, because PTEN sumoylation and ubiquitination events have previously been shown to display crosstalk [18], we also eliminated the two known sumoylation sites at K254 and K266 through substitutions. Unexpectedly, none of the ubiquitination‐ and sumoylation‐site mutants could rescue the protein expression levels of either the I135R or N276S mutants (Fig. 5A). In contrast, the expression levels of the more stable E157G mutant could be elevated by mutating the K13 but not the K289 ubiquitination site (Fig. 5B). Furthermore, mutations in the K254 but not the K266 sumoylation site significantly reduced the expression levels of the E157G mutant (Fig. 5B). Therefore, individual ASD‐associated PTEN missense mutants are differentially affected by specific ubiquitination and sumoylation events, which affect their steady‐state protein expression.
Fig. 4.
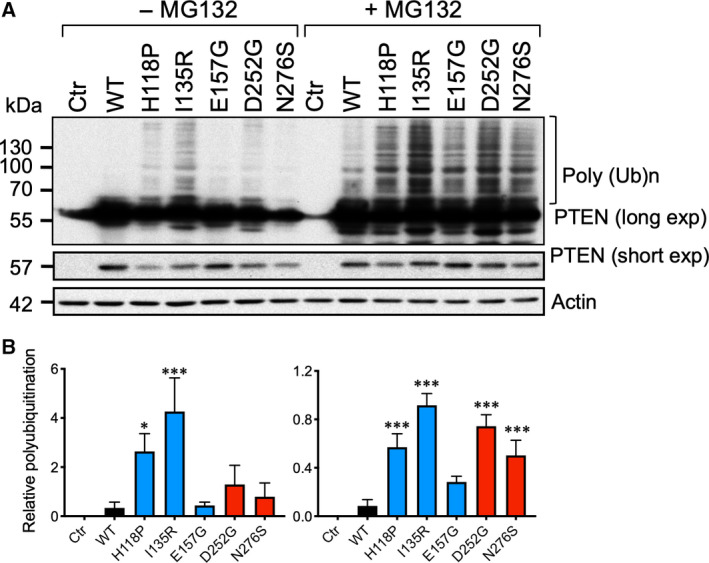
Enhanced polyubiquitination of ASD‐associated PTEN mutants. (A) Western blotting analysis of whole‐cell extracts of 293T transfected with the indicated expression plasmids. The cells were treated with (+) or without (−) 10 μm of MG132 for 6 h. The intensity of the polyubiquitinated (PolyUb) PTEN protein species (bracketed) was quantified from a film with long exposure and normalized to the levels of the unmodified PTEN using a film with short exposure. (B) The results from three independent experiments were analyzed by one‐way ANOVA with Dunnett's post‐tests, and the significance of the data is relative to the WT. Bars, SD. *P < 0.05; ***P < 0.001.
Fig. 5.

Effects of mutating ubiquitination or sumoylation sites of ASD‐associated PTEN mutants. (A) Western blotting analysis of whole‐cell extracts of PC3 transfected with I135R and N276S or (B) E157G mutants harboring substitutions at ubiquitination or sumoylation sites. The relative expression levels of the PTEN mutants were quantified and normalized to actin. The results from three independent experiments were analyzed by one‐way ANOVA with Dunnett's post‐tests, and the significance of the data is relative to I135R (@), N276S (*), or E157G (*). Bars, SD. *P < 0.05; **P < 0.01; @@@/***P < 0.001.
Previous studies also demonstrated that phosphorylation at the C‐terminal PEST domain could confer protein stability. In particular, the cluster of serine and threonine residues at S362, T366, S379, S380, T382, T383, and S385 has been shown to be phosphorylated by glycogen synthase kinase 3‐β, polo‐like kinase 3, and casein kinase II (CKII) [19]. We examined the phosphorylation states of the CKII phosphorylation cluster of S380/T382/T383 and the PTEN auto‐dephosphorylation site at T366 with phospho‐specific antibodies. However, no significant differences were observed among all of the PTEN missense mutants (Fig. 6).
Fig. 6.
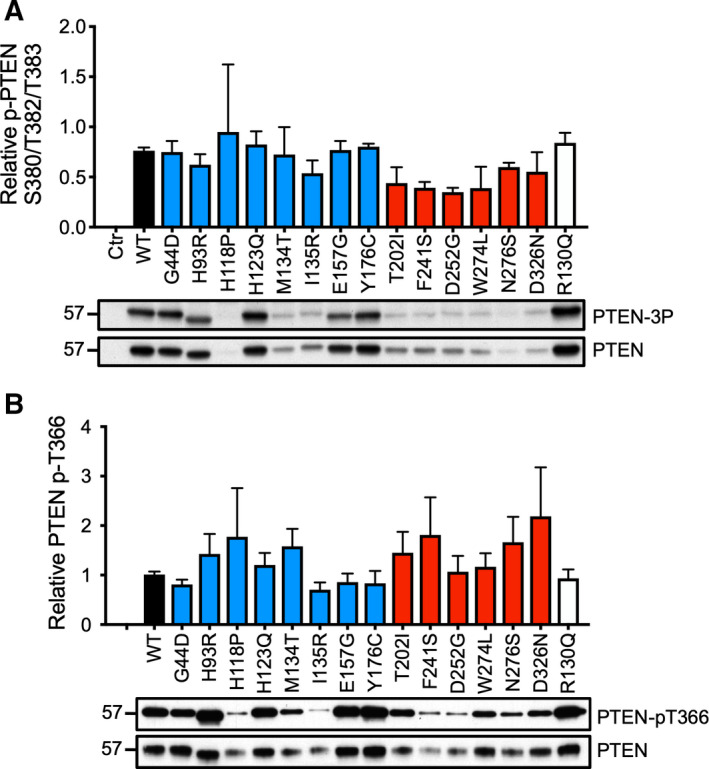
Phosphorylation states of ASD‐associated PTEN mutants. (A) Total cell lysates were resolved by SDS/PAGE, and western blotting analysis was carried out using anti‐p‐PTEN S380/T382/T383 (p‐3P) or anti‐PTEN antibodies. (B) Total cell lysates from parallel cultures were immunoprecipitated with an anti‐AU5 antibody, and western blotting analysis was carried out with anti‐p‐PTEN T366 or anti‐PTEN antibodies. The specific phosphorylation levels of each mutant were quantified. The results were derived from three independent experiments. Bars, SD.
Partial inactivation of lipid phosphatase activity of PTEN missense mutants
For mutants with comparable protein half‐lives as WT PTEN, attempts were made to unravel the biological properties that were disrupted. PTEN is a lipid phosphatase with specificity toward PIP3, and the catalytic pocket of PTEN is located in the N‐terminal portion of the protein [20]. To test the relative ability of the PTEN missense mutants in suppressing the PI3‐K signaling pathway, similar amounts of expression plasmids were transfected into the PTEN‐null cell line U87MG, which harbors appreciable levels of p‐Akt. The WT PTEN strongly suppressed p‐Akt by 80% (Fig. 7A). While almost all of the N‐terminal PTEN missense mutants, except I135R, retained > 80% of the WT's activity, only half of the C‐terminal mutants retained some capacity to suppress Akt phosphorylation. As expected, the catalytic‐dead mutant R130Q was completely devoid of suppressive activity (Fig. 7A). These data were, however, confounded by the variable expression levels of individual PTEN mutants expressed in U87MG cells.
Fig. 7.
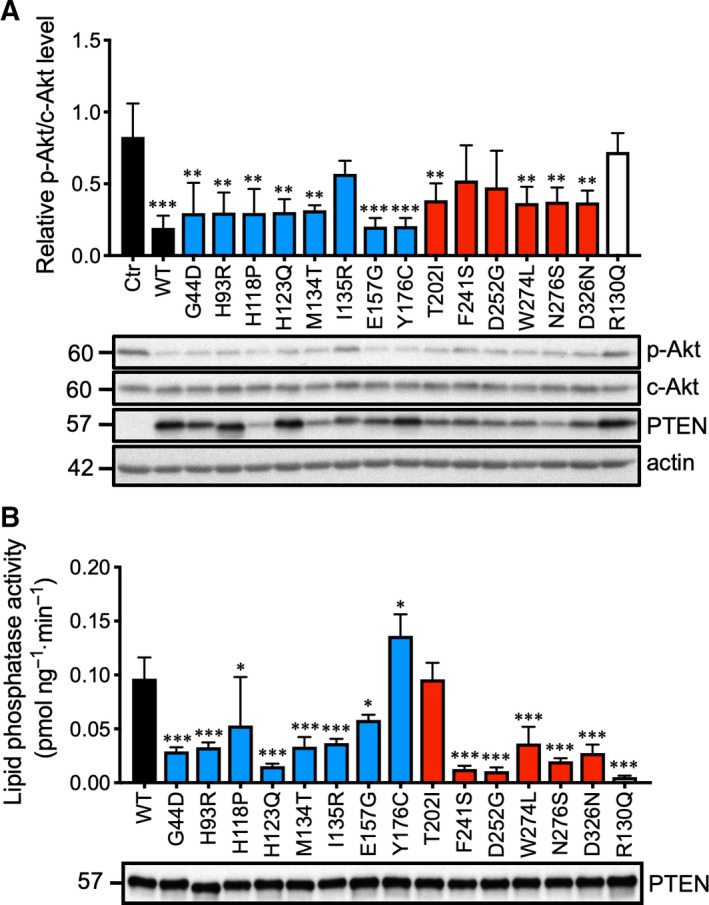
Lipid phosphatase activity of ASD‐associated PTEN mutants. (A) The control (Ctr) and the indicated expression plasmids were transfected into U87MG cell lines. The levels of p‐Akt S473 (p‐Akt) and total Akt (c‐Akt) were examined by western blotting analysis. The specific p‐Akt levels were calculated from three individual experiments, and the results were analyzed by one‐way ANOVA with Dunnett's post‐tests. The significance of the data is relative to Ctr. Bars, SD. **P < 0.01; ***P < 0.001. (B) The indicated plasmids were transfected into 293T cells. PTEN was immunoprecipitated by an anti‐AU5 antibody and probed with an anti‐PTEN antibody (lower panel). The PTEN protein levels were normalized, and a lipid phosphatase assay was carried out with PIP3 as substrates with the released phosphate measured. Lipid phosphatase activities from three independent experiments were quantified, and the results were analyzed by one‐way ANOVA with Dunnett's post‐tests. The significance of the data is relative to the WT. Bars, SD. *P < 0.05; ***P < 0.001.
To clearly define the intrinsic phosphatase activity of these missense mutants, AU5 epitope‐tagged mutants were transfected into 293T cells and similar amounts of PTEN proteins were immunoprecipitated with an anti‐AU5 antibody. Lipid phosphatase reaction was initiated by the addition of PIP3 to PTEN proteins immobilized on sepharose beads followed by incubation for 10 min. The levels of phosphate released were measured by the standard malachite green assay. As shown in Fig. 7B, all of the PTEN missense mutants with the exception of H118P, E157G, Y176C, and T202I had a 60–80% decrease in phosphatase activity compared with the WT. In contrast, the cancer‐associated catalytic‐dead mutant R130Q was almost completely inactivated. Thus, among the five stable missense mutants, only E157G, and Y176C, retains significant lipid phosphatase activity.
Altered subcellular distribution of PTEN missense mutants
To explore what further defects could be associated with the E157G and Y176C mutants, we examined their subcellular localization. PTEN has been shown to be localized to various subcellular compartments including plasma membrane, cell–cell junctions, microvesicles in the cytosol, and mitotic spindles of the nucleus [21]. To investigate whether the PTEN missense mutants had altered subcellular localization, expression plasmids were transfected into 293T cells and immunofluorescence was analyzed using an anti‐AU5 antibody. To minimize the confounding effects of cells in different phases of the cell cycle, the cultures were synchronized in the G1/S phase by double thymidine blocking. The majority of cells in the G1/S phase had flat morphology with the cytosolic and nuclear compartments clearly demarcated. As previously reported, WT PTEN had a more cytosolic rather than nuclear localization (Fig. 8A). Six missense mutants, H118P, M134T, I135R, F241S, D252G, and N276S, had significant reduction in nuclear localization compared with the WT (Fig. 8A,B). Four mutants, G44D, H123Q, E157G, and D326N, displayed greater nuclear localization compared with the WT. They were mostly confined to the nucleoplasm and absent in nucleoli with the exception of the D326N mutant, which also populated the nucleoli (Fig. 8A). Furthermore, in four mutants, H93R, E157G, Y176C, and R130Q, readily detectable membrane extensions resembling pseudopodia or membrane ruffles were observed (Fig. 8A).
Fig. 8.
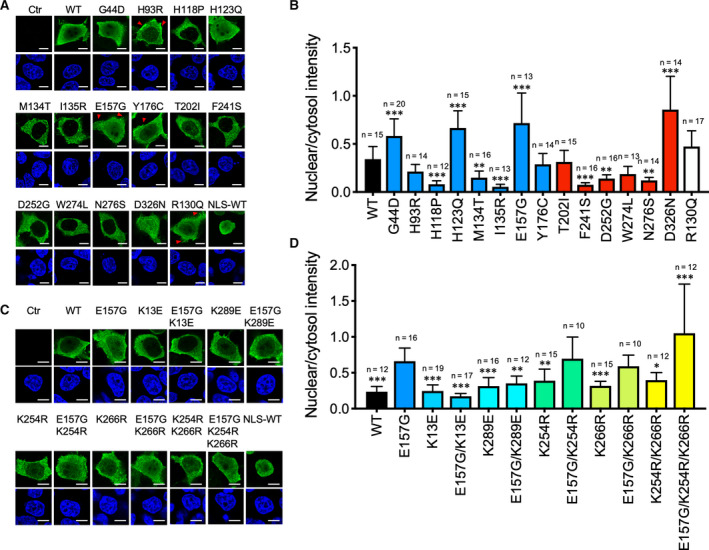
Nuclear localization of ASD‐associated PTEN mutants. (A) Immunofluorescence analysis of AU5‐tagged ASD‐associated PTEN mutants using an anti‐AU5 antibody and detected with an anti‐mouse IgG antibody conjugated with the Alexa 488 fluorophore. Cell nuclei were stained with DAPI. Positive control for nuclear staining was provided by a PTEN tagged with a NLS‐WT. Fluorescent signals were captured by confocal microscopy, and representative images are shown. Scale bars, 10 μm. (B) Fluorescent intensity was quantified for the cytosolic and nuclear compartments by the imagej software and expressed as the corresponding ratio. Results were analyzed by one‐way ANOVA with Dunnett's post‐tests, and the significance of the data is relative to the WT. The number of cells analyzed (n=) is shown. Data from a single experiment, which has been repeated once. Bars, SD. *P < 0.05; **P < 0.01; ***P < 0.001. (C) Similar experiments were carried out with E157G mutants with ubiquitination or sumoylation sites substituted. Representative images are shown. Scale bars, 10 μm. (D) Fluorescent intensity was quantified for the cytosolic and nuclear compartments and expressed as the corresponding ratio. The results were analyzed by one‐way ANOVA with Dunnett's post‐tests, and the significance of the data is relative to E157G. The number of cells analyzed (n=) is shown. Data from a single experiment, which has been repeated once. Bars, SD. *P < 0.05; **P < 0.01; ***P < 0.001.
To delineate the underlying mechanism responsible for the preferential nuclear distribution of the E157G mutant, we generated additional mutations in this missense mutant. Of note, an E157K mutant that carried an opposing polar group also displayed greater nuclear localization, suggesting the importance of this position, rather than the charge, being a critical determinant (unpublished data). Because both ubiquitination and sumoylation have been implicated in PTEN nuclear localization, we tested a panel of E157G mutants carrying mutations in lysine residues that mediated these two post‐translational events. As shown in Fig. 8C,D, mutating either of the two sumoylation sites at K254 and K266 alone failed to disrupt the nuclear targeting capacity of the E157G mutant. Unexpectedly, however, mutating both sumoylation sites increased the E157 nuclear targeting. More importantly, mutating the K13 and K289 ubiquitination sites greatly diminished the nuclear‐to‐cytosol ratio of the E157G mutants (Fig. 8C,D). We further examined the overall sumoylation state of PTEN missense mutants by a pull‐down assay. E157G mutant had a similar sumoylation level compared to WT PTEN protein (Fig. 9). Another mutant, H123Q, which had greater nuclear localization, displayed a slight reduction in sumoylation level. However, out of the 6 mutants with reduced nuclear localization, H118P, M134T, I135R, F241S, D252G, and N276S, all have elevated sumoylation levels (Fig. 9). Thus, there appears to be an inverse relationship between nuclear localization and sumoylation level in certain PTEN mutants. From these analyses, we assigned a tentative defect for the E157G mutant in displaying a preferential localization in the cell nucleus, which may involve ubiquitination events at K13 and K289.
Fig. 9.
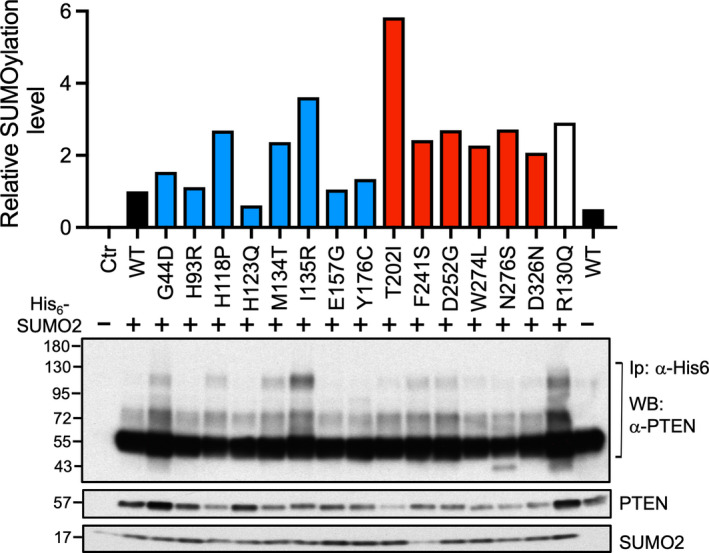
Sumoylation states of PTEN missense mutants. Sumo pull‐down assay was conducted in 293T cells by cotransfecting His6‐SUMO2 and PTEN missense mutant plasmids. Bound sumoylated PTEN was eluted from nickel beads and subjected to western blotting analysis using the antibodies indicated. Data from a single experiment was quantified (upper panel).
Endogenous E157G mutation displayed greater nuclear localization
To examine how the E157G mutation could impact on neurogenesis, this mutation was knocked‐in to a human iPSC cell line, ASE‐9203, using CRISPR/Cas9. Successful homozygous knock‐in in both alleles was confirmed by sequencing analysis (data not shown), and the identity was authenticated by short tandem repeat analysis (data not shown). Morphologically, the WT and E157G iPSCs were very similar (Fig. 10A). Next, neural induction was initiated in the iPSCs using neural induction medium and the neural progenitor cells (NPCs) were expanded in neural progenitor medium. Neuronal precursors were generated from the NPC and matured for 1–2 weeks. As expected, the cells acquired neurite‐like extensions following differentiation and maturation processes (Fig. 10A). However, more extensive and robust neurite extensions were observed in E157G cells compared with WT cells. Also, E157G at the NPC stage proliferated at a much greater rate than the WT. Western blotting analysis revealed upregulation of PTEN in both the WT and the E157G, both in NPC and during neuronal differentiation (Fig. 10B). This was correlated with a corresponding downregulation of p‐Akt. However, the levels of WT and E157G PTEN were very similar as we expected based on our results in PC3 cells (Fig. 1B). As expected, high levels of the reprogramming factors NANOG, SOX2, and OCT4 were observed in the iPSCs and their expression was downregulated upon neural induction and differentiation (Fig. 10B). However, the levels of SOX2 remained detectable in the E157G cells. More strikingly, the levels of PAX6 were upregulated following neural induction and were silenced following the induction of neuronal differentiation. Interestingly, PAX6 was more downregulated in E157G than in the WT (Fig. 10B,C). For the neuronal‐specific marker, TuJ, its expression was higher in E157G than in WT cells during the entire differentiation process. Immunofluorescence analysis also revealed more MAP2‐positive staining of microtubule structures in E157G when compared to WT in matured neurons (Fig. 10D). Finally, immunofluorescence analysis of NPC using an anti‐PTEN antibody revealed a fourfold increase in nuclear‐to‐cytosol ratio for the E157G mutant compared with the WT (Fig. 10E,F). Thus, the E157G protein expressed in the iPSC line has properties very similar to those expressed in PC3 cells.
Fig. 10.
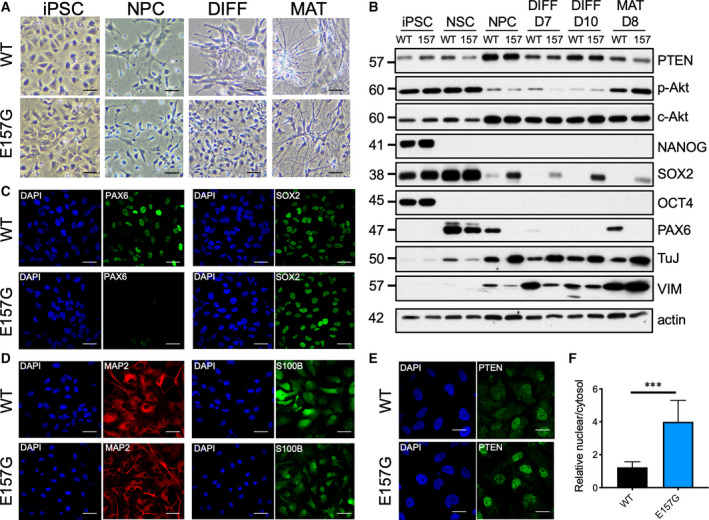
Aberrant neuronal differentiation in PTEN E157G iPSc. (A) Morphology of iPSC and different stages of neuronal development. Note the higher neurite outgrowth in E157G. Bars, 60 μm. A similar experiment was repeated once. (B) Western blotting analysis of lineage‐specific markers at different stages of neurogenesis. Immunofluorescence (IF) analysis of lineage‐specific markers in (C) NPC and (D) maturation stages. Bars, 30 μm. (E) IF of PTEN in NPC. Bars, 15 μm. Similar results were obtained in two additional experiments using iPSC cells. (F) The extent of nuclear localization of PTEN was quantified from a single experiment. Bars, SD. n = 13. ***P < 0.001. Student's t‐test, unpaired. DIFF, differentiation; MAT, maturation; NSC, neural stem cells.
Discussion
Spinelli et al. [22] had previously characterized seven PTEN mutants associated with ASD. The present study expanded on this panel by adding an additional seven mutants. Overall, the present study also observed high fraction (> 60%) of PTEN missense mutation would lead to protein instability. Our study also uncovered aberrant subcellular distribution of PTEN missense mutants that was not investigated in previous studies [22, 23]. Furthermore, the present study expanded on the previous studies by offering mechanistic insights into specific post‐translational modifications to explain the defects associated with individual missense mutations. The overall findings of the present study are summarized in Fig. 11A. Two recent studies had investigated genotype–phenotype relationship of PTEN missense mutants using saturated mutagenesis in yeast cells followed by massively parallel sequencing [24, 25]. In terms of catalytic activity, the data by Mighell et al. [24] showed striking consistency with our data with the exception of the H93R and Y176C (Fig. 11B). In terms of protein level, the report by Matreyek et al. [25] also showed high consistency with our data (Fig. 11C). Indeed, these two studies revealed that the E157G mutant has WT‐like catalytic activity and protein abundance.
Fig. 11.
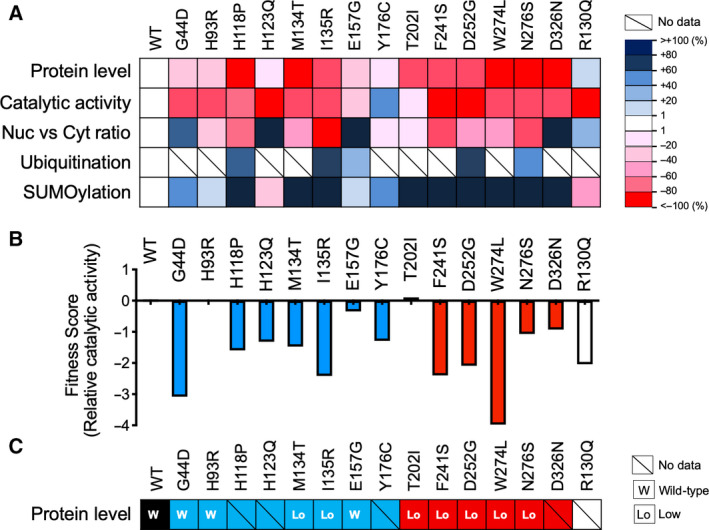
Biological properties of PTEN missense mutants. (A) Results obtained in this study are summarized by using indicated color codes. The relative levels of biological readout for individual mutants were calculated based on the mean values from the respective assays and compared with WT protein. (B) Fitness scores extracted from the study by Mighell et al. [24] for the indicated mutants are shown. (C) Protein‐level classification based on data from the study by Matreyek et al. [25] for the indicated mutant is shown.
Expression analysis of PTEN mutants in three mammalian cell lines revealed that reduced level of this protein was the most prominent defect of these ASD mutants. This was particularly striking for C2 mutants. Because the same amount of DNA was transfected in each case, it is unlikely that the observed result was due to reduced transcriptional or translational events. Both MG132 treatment and cycloheximide chase experiments indicated that reduced protein stability was the major culprit for the lowered expression levels of ASD‐associated PTEN mutants. One possibility is that some of the mutations cause conformational changes to PTEN and hence lead to structural instability. Another observation is that only three out of eight phosphatase domain mutants were unstable, while all six of the C2 domain mutants had drastic reductions in protein expression levels. This suggests that PTEN stability may be more sensitive to C2 domain mutations. Indeed, four out of six C2 domain mutants, D252, W274, N276, and D326, are located along the interphase between the C2 and phosphatase domain, with some partaking in hydrogen bond formation [26]. Mutations in these residues may disrupt these intramolecular interactions and confer structural instability. Our results resemble those reported by Spinelli et al.[22], who also characterized unstable ASD‐associated PTEN mutants. Our present study identified two mutants, H118P, and N276S, with the most reduced protein expression. The heightened polyubiquitination in the unstable mutants may explain their shortened half‐lives. Unexpectedly, however, the two major ubiquitination sites at K13 and K289 were not responsible for the half‐life shortening, at least for the unstable I135R and N276S mutants. It is possible that cryptic polyubiquitination sites are present. In contrast, the E157G mutant protein is relatively stable and retains ~ 77% of WT protein expression. This decrease in protein level was rescued by mutating the K13 ubiquitination site suggesting protein degradation mediated by K13 polyubiquitination may be responsible. The reduced expression of E157G in the K254R and K254R/K266R mutants may be related to an increase in polyubiquitination following the elimination of the K254 site. Similar crosstalk has been reported previously [18].
Previous studies conducted in Saccharomyces cerevisiae have shown that ASD‐associated PTEN mutants suffered partial loss of PTEN activity compared with tumor‐associated mutants [23]. In the present study, we performed both in vivo and in vitro assays to take into account the protein instability of PTEN mutants. Our in vivo assay also showed that most ASD‐associated PTEN mutants had partial loss of lipid phosphatase activity in U87MG. This is expected as mutations within the N‐terminal phosphatase domain are predicted to disrupt catalysis, while mutations in the C2 domain may decrease membrane binding or perturb structural integrity. Also, in most of the mutants with partial loss of in vivo phosphatase activity, the loss was not caused by their low protein expression because their in vitro lipid phosphatase activities using purified proteins were clearly compromised. Thus, the extent of loss of PTEN activities in PHTS patients reflects a combination of defects in catalytic activity, membrane‐binding ability, and protein stability.
The only two mutants with significant lipid phosphatase activity in vivo are E157G and Y176C. One possibility is that these two residues are not located in the core of the phosphatase and C2 domains. In addition, both have comparable expression levels to that of the WT. Our findings are consistent with a previous study reporting the thermostability of PTEN variants in ASD [27]. These results imply that autistic features found in patients harboring E157G and Y176C mutation may be due to defects in PTEN biochemical functions independent of lipid phosphatase activity and expression level. It is uncertain whether ‘less‐defective’ PTEN mutants are associated with milder symptoms. A patient with the Y176C mutation had difficulties in social interaction and delayed speech and was diagnosed by the Childhood Autism Rating Scale as severe [28]. The E157G mutation was discovered in a male subject diagnosed with PDD‐NOS, and his identical twin was also diagnosed with ASD [29]. These diagnostic features are not remarkably different from those PTEN mutants that have relatively significant decreases in lipid phosphatase activity and protein expression level. Attempts to link specific PTEN defects to disease diagnosis are currently hampered by the rarity of PHTS cases.
The role of nuclear PTEN in ASD is not clear. Disruption of PTEN nuclear translocation has been shown to cause autism in a transgenic mouse model [30]. Mingo et al. [31] had previously reported a Q17E germline PTEN mutant with preferential nuclear localization derived from a PHTS patient with ASD. The present study represents a more comprehensive analysis of the subcellular distribution of ASD‐associated PTEN mutants. We showed that the ASD‐associated PTEN mutants varied in their abilities in nuclear targeting, but this variation was not obviously associated with their enzymatic activities or protein expression levels. Aberrantly increased or reduced nuclear localization was observed in the E157G and I135R mutants, respectively, which highlights the importance of balanced PTEN levels in cytosolic and nuclear compartments. Mechanistically, ubiquitination events at K13 and K289 are involved in enhancing E157G nuclear localization. Indeed, K13 [32] and K289 [11] were previously suggested to be involved in nuclear translocation of PTEN. Furthermore, we observed an increase in ubiquitination of the E157G mutant (Fig. 4). We are currently testing whether the E157G mutant can interact with PTEN‐specific ubiquitin ligase.
The I135R mutant, in contrast, was excluded from the cell nucleus. Thus, it is not surprising that disrupting all of the known ubiquitination and sumoylation sites failed to restore nuclear targeting capacity. As discussed above, the I135R mutant is heavily polyubiquitinated, possibly on cryptic sites. This may disrupt ubiquitination and sumoylation events that are normally required for nuclear targeting.
Our iPSC data imply that the E157G mutation promotes proliferation of NPC but confers greater neuronal differentiation. This is consistent with the long‐held notion that loss of PTEN function increases dendritic arborization and hypertrophic growth. Indeed, hyperactivation of the mTOR pathway is directly linked to increased dendritic spine density in layer V pyramidal neurons in postmortem ASD temporal lobes [33]. The higher SOX2 expression in E157G NPC may explain the higher rate of proliferation. The persistence of SOX2 expression in E157G may also suggest that a subpopulation of neural stem cells persists following differentiation. It is currently unclear whether the enhanced neurite outgrowth seen in E157G is due to reduced cytosolic or enhanced nuclear PTEN functions. Whether the E157G mutant can directly regulate gene expression in the nucleus is not clear. Future studies to resolve these issues may yield new insights into the molecular pathogenesis of ASD.
Materials and methods
Chemicals
All chemicals used in this study were purchased from Sigma‐Aldrich (St. Louis, MO, USA) unless otherwise indicated.
Antibody reagents
Antibodies against PTEN (#9552, #9559), p‐Akt‐S473 (#3271), c‐Akt (#9272), PTEN‐pS380/pT382/pT383 (#9554), Nanog (#4903), Pax6 (#60433), Oct4 (#2750), Sox2 (#3579), GFAP (#3670), GAPDH (#5174), and horseradish peroxidase (HRP)‐conjugated anti‐rabbit or anti‐mouse IgG (#7074, #7076) were from Cell Signaling (Danvers, MA, USA). The anti‐AU5 antibody (#ab130113) and anti‐Vimentin (#ab8978) were from Abcam (Cambridge, UK), and anti‐actin antibody (#I‐19) was from Santa Cruz Inc. (Dallas, TX, USA) An antibody directed against PTEN‐pT366 was a generous gift provided by Nick Leslie (Heriot‐Watt University, UK) [34].
Compilation of PTEN missense mutants
A PubMed search was conducted using the search terms—PTEN, autism, and mutation. A list of germline PTEN missense mutations found in PHTS patients with ASD was compiled. The source references used were as follows: G44D [29], H93R [3], H118P [28], H123Q [35], M134T [35], I135R [36], E157G [29], Y176C [28], T202I [29], F241S [3], D252G [3], W274L [35], N276S [28], and D326N [37].
Expression plasmids
All PTEN missense mutants were generated by PCR‐based site‐directed mutagenesis techniques. All cDNAs generated were cloned into pEF‐AU5 mammalian expression vector and their authenticity confirmed by sequencing analysis as described previously [38].
Cell cultures
The HEK293T, PC3, and U87MG cell lines were from the cell bank at the National Cancer Institute, USA. Cultures were maintained in DMEM supplemented with 10% FBS and penicillin–streptomycin (pen–strep; Thermo Fisher Scientific, Waltham, MA, USA). For lipofection, ~ 1 × 105 cells were plated in a well of a six‐well culture dish. Samples of 0.1–3.0 μg of DNA constructs were mixed with 2 μL of Lipofectamine 2000 in 0.2 mL of Opti‐MEM (Thermo Fisher Scientific). This mixture was added to 2 mL of DMEM + 5% FBS medium without pen–strep and incubated for 16 h. The cells were analyzed 1–2 days later.
Lipid phosphatase assay
An immunoprecipitation‐based lipid phosphatase assay was conducted as previously described [39]. Around 3 × 105 293T cells in 100‐mm plates were transfected by lipofection using 10 μg of various PTEN expression constructs. After 36 h, the cells were washed with cold TBS (25 mm Tris/HCl pH 8.0, 150 mm NaCl) and resuspended in 550 μL of lysis buffer (25 mm Tris/HCl pH 8.0, 150 mm NaCl, 1% Triton X‐100, 2 mm DTT, and a protease inhibitor cocktail). PTEN was immunoprecipitated with 1 μg of an anti‐AU5 epitope antibody for 3 h at 4 °C followed by the addition of 35 μL of GammaBind G sepharose beads (Millipore, Billerica, MA, USA) for 1 h. Phosphatase reactions were initiated with the addition of 30 μL of 25 mm Tris/HCl pH 8.0, 10 mm DTT, 50 μm PIP3 (Echelon, Salt Lake City, UT, USA). After incubating at 37 °C for 10 min, 100 μL of malachite green (Echelon) was added to each reaction. The absorbance at OD 620 nm was measured in a SpectraMax i3X microplate reader (Molecular Devices, San Jose, CA, USA). The enzymatic activity of PTEN was presented as pmol pyrophosphate (PPi) released·ng−1·min−1.
Western blotting analysis
Cells were solubilized in 100 µL of either 2 × Laemmli buffer or RIPA buffer composed of 50 mm Tris/HCl pH 7.5, 1% Triton X‐100, 0.5% sodium deoxycholate, 0.1% SDS, 150 mm NaCl, 10 mm MgCl2, and protease and phosphatase inhibitors. Around 5–20 µg of total cell lysates were resolved on 10–15% SDS/PAGE gels, transferred onto nitrocellulose membranes, and probed with selected primary antibodies. Bound antibodies were detected with either HRP‐conjugated anti‐mouse or anti‐rabbit secondary antibodies. The expression levels of protein were visualized by the standard ECL method (Life Technologies, Carlsbad, CA, USA) followed by exposure to X‐ray films. Band intensity was quantified by the GS‐900 Calibrated Densitometer System (Bio‐Rad, Hercules, CA, USA).
Immunofluorescence analysis
Around 1 × 104 cells were seeded on a glass coverslip coated with 0.5% Matrigel®Matrix (CORNING #356231, Corning, NY, USA) in a 12‐well plate. The cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.2% of Triton X‐100 in PBS for 12 min at room temperature. The cells were then incubated with antibodies (1 : 100) at 4 °C overnight. They were washed with PBS and incubated with Alexa Fluor® 488‐conjugated donkey anti‐mouse IgG (H + L) antibody (1 : 200; Thermo Fisher Scientific) for 1 h. They were mounted with 300 nm DAPI on glass slides. They were then imaged with a confocal microscope (Olympus FV1200, Tokyo, Japan) using the metamorph imaging software (San Jose, CA, USA), and the fluorescence intensity was quantified by the imagej software (NIH, MD, USA). The nuclear‐to‐cytosol ratios were calculated from 15 to 20 cells using the mid‐sections of the z‐stacks of each cell.
SUMO pull‐down assay
HEK293T cells were plated at a density of 1 × 106 on a 100‐mm dish and transfected with 7 μg of PTEN expressing plasmids, 5 μg of His6‐SUMO2 plasmid, 3 μg of UBC9 plasmid, and 12 μL of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for 16 h. After 48 h, cells were lysed by denaturing cell lysis buffer (6 m guanidinium‐HCl, 10 mm Tris, 100 mm sodium phosphate buffer pH 8.0, and 5 mm β‐mercaptoethanol) and imidazole to 5 mm and then sonicated briefly. After centrifugation at 3000 g for 15 min, the supernatant was incubated with 50 μL prewashed Ni2+ NTA sepharose for 4 h at 4 °C. Beads were washed once with 5 mL cell lysis buffer supplemented with 0.1% Triton X‐100, once with 4 mL of pH 8.0 wash buffer (8 m Urea, 10 mm Tris, 100 mm sodium phosphate buffer pH 8.0, 5 mm β‐mercaptoethanol, 0.1% Triton X‐100), and three times with 4 mL of pH 6.3 wash buffer (8 m Urea, 10 mm Tris, 100 mm sodium phosphate buffer pH 6.3, 5 mm β‐mercaptoethanol, 0.1% Triton X‐100). SUMO‐conjugated proteins were eluted with an elution buffer (200 mm imidazole, 5% SDS, 150 mm Tris/HCl pH 6.7, 30% glycerol, 720 mm β‐mercaptoethanol, 0.0025% bromophenol blue).
Knock‐in K157G mutation in iPSC
A knocked‐in PTEN K157G iPSC line was generated using an Applied StemCell kit (Milpitas, CA, USA). Briefly, ASE‐9203 human iPS cells were electroporated with the following gRNA: 5′‐TAGATTTCTATGGGGAAGTAAGG‐3′ in the cloning vector, pBT‐U6‐Cas9‐2A‐GFP. The CRISPR‐Cas9 positive clone was confirmed by Sanger sequencing analysis. The iPSC cells were cultured and differentiated according to the manufacturer's instructions (STEMCELL Technologies, Vancouver, BC, Canada).
Statistical analysis
All values are expressed as means ± SD. Statistical significance was analyzed using a two‐tailed Student's t‐test for two‐group comparisons. For more than two groups, analysis of variance (ANOVA) with Dunnett's post‐tests was used. Statistical significance was considered to be P < 0.05.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
CWW, YW, TL, LL, KKC, and PMO conducted the experiments; CWW, YW, and AMC planned, conducted the experiment, and wrote the manuscript; ASC, KWC, JPHB, BF, RCCC, and AMC provided expertise in planning key experiments reported in this study.
Acknowledgements
We thank the invaluable technical assistance of the core facilities at the School of Biomedical Sciences of the Chinese University of Hong Kong. 6xHis‐tag‐SUMO2 plasmid was a generous gift from Prof. Ron Hay (University of Dundee). This work was supported by Lo Kwee‐Seong Biomedical Research Seed Fund 6903806, Hong Kong University Grants Committee, Collaborative Research Fund C4014‐14G, and Brain and Mind Institute of The Chinese University of Hong Kong Pilot Project Fund 4930742. Yubing Wang was supported by Research Grants Council of Hong Kong, Hong Kong PhD Fellowship Scheme PF12‐13876.
Chi Wai Wong and Yubing Wang contributed equally to this work.
References
- 1. Yehia L, Ngeow J & Eng C (2019) PTEN‐opathies: from biological insights to evidence‐based precision medicine. J Clin Invest 129, 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mester JL, Tilot AK, Rybicki LA, Frazier TW 2nd & Eng C (2011) Analysis of prevalence and degree of macrocephaly in patients with germline PTEN mutations and of brain weight in Pten knock‐in murine model. Eur J Hum Genet 19, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R et al (2005) Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 42, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kleijer KT, Schmeisser MJ, Krueger DD, Boeckers TM, Scheiffele P, Bourgeron T, Brose N & Burbach JP (2014) Neurobiology of autism gene products: towards pathogenesis and drug targets. Psychopharmacology 231, 1037–1062. [DOI] [PubMed] [Google Scholar]
- 5. Ebert DH & Greenberg ME (2013) Activity‐dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Courchesne E, Pierce K, Schumann CM, Redcay E, Buckwalter JA, Kennedy DP & Morgan J (2007) Mapping early brain development in autism. Neuron 56, 399–413. [DOI] [PubMed] [Google Scholar]
- 7. Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J et al (2014) Most genetic risk for autism resides with common variation. Nat Genet 46, 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parsons R (2004) Human cancer, PTEN and the PI‐3 kinase pathway. Semin Cell Dev Biol 15, 171–176. [DOI] [PubMed] [Google Scholar]
- 9. Naguib A, Bencze G, Cho H, Zheng W, Tocilj A, Elkayam E, Faehnle CR, Jaber N, Pratt CP, Chen M et al (2015) PTEN functions by recruitment to cytoplasmic vesicles. Mol Cell 58, 255–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP & Yin Y (2007) Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128, 157–170. [DOI] [PubMed] [Google Scholar]
- 11. Trotman LC, Wang X, Alimonti A, Chen Z, Teruya‐Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon‐Cardo C, Erdjument‐Bromage H et al (2007) Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kreis P, Leondaritis G, Lieberam I & Eickholt BJ (2014) Subcellular targeting and dynamic regulation of PTEN: implications for neuronal cells and neurological disorders. Front Mol Neurosci 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ & Parada LF (2006) Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clipperton‐Allen AE & Page DT (2014) Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism‐relevant behavioral tests. Hum Mol Genet 23, 3490–3505. [DOI] [PubMed] [Google Scholar]
- 15. Clipperton‐Allen AE & Page DT (2015) Decreased aggression and increased repetitive behavior in Pten haploinsufficient mice. Genes Brain Behav 14, 145–157. [DOI] [PubMed] [Google Scholar]
- 16. Minaguchi T, Yoshikawa H, Oda K, Ishino T, Yasugi T, Onda T, Nakagawa S, Matsumoto K, Kawana K & Taketani Y (2001) PTEN mutation located only outside exons 5, 6, and 7 is an independent predictor of favorable survival in endometrial carcinomas. Clin Cancer Res 7, 2636–2642. [PubMed] [Google Scholar]
- 17. Birle D, Bottini N, Williams S, Huynh H, deBelle I, Adamson E & Mustelin T (2002) Negative feedback regulation of the tumor suppressor PTEN by phosphoinositide‐induced serine phosphorylation. J Immunol 169, 286–291. [DOI] [PubMed] [Google Scholar]
- 18. Gonzalez‐Santamaria J, Campagna M, Ortega‐Molina A, Marcos‐Villar L, de la Cruz‐Herrera CF, Gonzalez D, Gallego P, Lopitz‐Otsoa F, Esteban M, Rodriguez MS et al (2012) Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis 3, e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Singh G & Chan AM (2011) Post‐translational modifications of PTEN and their potential therapeutic implications. Curr Cancer Drug Targets 11, 536–547. [DOI] [PubMed] [Google Scholar]
- 20. Hodakoski C, Fine B, Hopkins B & Parsons R (2015) Analysis of intracellular PTEN signaling and secretion. Methods 77–78, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leslie NR, Kriplani N, Hermida MA, Alvarez‐Garcia V & Wise HM (2016) The PTEN protein: cellular localization and post‐translational regulation. Biochem Soc Trans 44, 273–278. [DOI] [PubMed] [Google Scholar]
- 22. Spinelli L, Black FM, Berg JN, Eickholt BJ & Leslie NR (2015) Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J Med Genet 52, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rodriguez‐Escudero I, Oliver MD, Andres‐Pons A, Molina M, Cid VJ & Pulido R (2011) A comprehensive functional analysis of PTEN mutations: implications in tumor‐ and autism‐related syndromes. Hum Mol Genet 20, 4132–4142. [DOI] [PubMed] [Google Scholar]
- 24. Mighell TL, Evans‐Dutson S & O'Roak BJ (2018) A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype‐phenotype relationships. Am J Hum Genet 102, 943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, Kircher M, Khechaduri A, Dines JN, Hause RJ et al (2018) Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat Genet 50, 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P & Pavletich NP (1999) Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–334. [DOI] [PubMed] [Google Scholar]
- 27. Johnston SB & Raines RT (2015) Conformational stability and catalytic activity of PTEN variants linked to cancers and autism spectrum disorders. Biochemistry 54, 1576–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Orrico A, Galli L, Buoni S, Orsi A, Vonella G & Sorrentino V (2009) Novel PTEN mutations in neurodevelopmental disorders and macrocephaly. Clin Genet 75, 195–198. [DOI] [PubMed] [Google Scholar]
- 29. Varga EA, Pastore M, Prior T, Herman GE & McBride KL (2009) The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med 11, 111–117. [DOI] [PubMed] [Google Scholar]
- 30. Tilot AK, Gaugler MK, Yu Q, Romigh T, Yu W, Miller RH, Frazier TW 2nd & Eng C (2014) Germline disruption of Pten localization causes enhanced sex‐dependent social motivation and increased glial production. Hum Mol Genet 23, 3212–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mingo J, Rodriguez‐Escudero I, Luna S, Fernandez‐Acero T, Amo L, Jonasson AR, Zori RT, Lopez JI, Molina M, Cid VJ et al (2018) A pathogenic role for germline PTEN variants which accumulate into the nucleus. Eur J Hum Genet 26, 1180–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu F, Wagner S, Campbell RB, Nickerson JA, Schiffer CA & Ross AH (2005) PTEN enters the nucleus by diffusion. J Cell Biochem 96, 221–234. [DOI] [PubMed] [Google Scholar]
- 33. Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A et al (2014) Loss of mTOR‐dependent macroautophagy causes autistic‐like synaptic pruning deficits. Neuron 83, 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tibarewal P, Zilidis G, Spinelli L, Schurch N, Maccario H, Gray A, Perera NM, Davidson L, Barton GJ & Leslie NR (2012) PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor‐suppressing phenotype, but not with AKT activity. Sci Signal 5, ra18. [DOI] [PubMed] [Google Scholar]
- 35. McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF & Herman GE (2010) Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res 3, 137–141. [DOI] [PubMed] [Google Scholar]
- 36. Boccone L, Dessi V, Zappu A, Piga S, Piludu MB, Rais M, Massidda C, De Virgiliis S, Cao A & Loudianos G (2006) Bannayan‐Riley‐Ruvalcaba syndrome with reactive nodular lymphoid hyperplasia and autism and a PTEN mutation. Am J Med Genet A 140, 1965–1969. [DOI] [PubMed] [Google Scholar]
- 37. Buxbaum JD, Cai G, Chaste P, Nygren G, Goldsmith J, Reichert J, Anckarsater H, Rastam M, Smith CJ, Silverman JM et al (2007) Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet 144B, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kimmelman AC, Nunez Rodriguez N & Chan AM (2002) R‐Ras3/M‐Ras induces neuronal differentiation of PC12 cells through cell‐type‐specific activation of the mitogen‐activated protein kinase cascade. Mol Cell Biol 22, 5946–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wong CW, Or PMY, Wang Y, Li L, Li J, Yan M, Cao Y, Luk HM, Tong TMF, Leslie NR et al (2018) Identification of a PTEN mutation with reduced protein stability, phosphatase activity, and nuclear localization in Hong Kong patients with autistic features, neurodevelopmental delays, and macrocephaly. Autism Res 11, 1098–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]