

Abstract
Aim
Alport syndrome (AS) is the second most common hereditary kidney disease caused by mutations in collagen IV genes. Patients present with microhaematuria that progressively leads to proteinuria and end stage renal disease. Currently, no specific treatment exists for AS. Using mass spectrometry based proteomics, we aimed to detect early alterations in molecular pathways implicated in AS before the stage of overt proteinuria, which could be amenable to therapeutic intervention.
Methods
Kidneys were harvested from male Col4a3 −/− knock out and sex and age‐matched Col4a3 +/+ wild‐type mice at 4 weeks of age. Purified peptides were separated by liquid chromatography and analysed by high resolution mass spectrometry. The Cytoscape bioinformatics tool was used for function enrichment and pathway analysis. PPARα expression levels were evaluated by immunofluorescence and immunoblotting.
Results
Proteomic analysis identified 415 significantly differentially expressed proteins, which were mainly involved in metabolic and cellular processes, the extracellular matrix, binding and catalytic activity. Pathway enrichment analysis revealed among others, downregulation of the proteasome and PPAR pathways. PPARα protein expression levels were observed to be downregulated in Alport mice, supporting further the results of the discovery proteomics.
Conclusion
This study provides additional evidence that alterations in proteins which participate in cellular metabolism and mitochondrial homeostasis in kidney cells are early events in the development of chronic kidney disease in AS. Of note is the dysregulation of the PPAR pathway, which is amenable to therapeutic intervention and provides a new potential target for therapy in AS.
Keywords: Alport syndrome, Col4a3 knockout mice, mass spectrometry, PPARα, proteomics
SUMMARY AT A GLANCE
Alterations in proteins which participate in cellular metabolism and mitochondrial homeostasis in kidney cells, are early events in the development of CKD in Alport syndrome. In addition, pathway enrichment analysis revealed among others, dysregulation of the proteasome, protein digestion and absorption pathways as well as PPAR pathway in AS compared to control mice. Targeting the PPAR pathway could be a potential therapeutic approach in AS.
Alport syndrome (AS) is one of the major monogenic kidney diseases caused by mutations in the genes encoding for collagen IV proteins. The commonest mode of inheritance is X‐linked (∼85%) caused by mutations in the Col4a5, followed by the autosomal recessive form, caused by mutations in the Col4a3 or Col4a4 genes. The autosomal dominant form is rare. Collagen IV proteins are essential components of the Glomerular Basement Membrane (GBM) but are also found in the inner ear, lens and retina. Hence development of not only renal, but also ocular and cochlear symptoms occur in AS. 1 Patients present with early‐onset microscopic haematuria, worsening proteinuria and slow progression to end stage renal disease (ESRD). Alport syndrome accounts for more than 1% of patients on renal replacement therapy. 2
Currently, there is no specific treatment for AS. Early initiation of angiotensin‐converting enzyme inhibitors (ACE‐i), has shown to delay disease progression and increase life expectancy. 3 Based on these studies, the current recommendations for the treatment of AS include: early intervention at the stage of microalbuminuria with ACE‐i in children with a family history of early‐ESRD or severe Col4a5 mutation (deletion, nonsense or splicing mutation) and treatment for all the children with proteinuria even if normotensive. 4 The efficacy and safety of early initiation of Ramipril was recently reported in a prospective randomized placebo‐controlled trial. 5 , 6 Nonetheless, ACE‐i are not specific for AS and their renoprotective effects are mediated through their antihypertensive, antiproteinuric and other pleiotropic properties. However, the mechanisms involved in this renoprotection are poorly understood. The slow progressive clinical course of the disease provides an opportunity for interventions aiming to mitigate the deleterious effects of the ultrastructural GBM abnormalities in AS. Similarly, to ACE‐i, novel therapeutic strategies in AS could potentially be applicable to other forms of Chronic Kidney Disease (CKD), as AS mice can also serve as a model of CKD progression disease.
The aim of this study was to detect early alterations in molecular pathways, before the stage of overt proteinuria, which may cause a cascade of events leading to fibrosis and ESRD and, more importantly, identify those pathways that could be amenable to actionable therapeutic interventions. To this end, we performed untargeted comparative proteomic profiling of kidney tissues from Col4a3+/+ wild type and Col4a3 −/− Alport mice, aiming to identify significantly dysregulated proteins and pathways, affected early on in disease development.
1. MATERIALS AND METHODS
1.1. Animals and tissue harvesting
Heterozygous Col4a3 +/− transgenic mice (Jackson Laboratories, USA), bred on a 129/SvJ genetic background, were crossbred. Genotyping of the resulting F1 mice, which included heterozygous Col4a3 +/−, homozygous Col4a3 −/− knock out (Col4a3KO), and Col4a3 +/+ wild‐type mice, was carried out by polymerase chain reaction (PCR) as described before. 7 Only male mice were used in this study to minimize sex differences in kidney function, caused by hormonal variations. All animals were kept in a regular 12‐hours light‐12‐hours dark cycle under specific pathogen free conditions. Mice were anesthetized with Tribromoethanol (Avertin) (250 mg/kg) via intraperitoneal injection and subsequently exsanguinated through transcardial perfusion using cold PBS. Kidneys were then harvested at 4 weeks old mice and processed accordingly for further analysis. All animal experiments were performed in accordance to the 2010/63/EU directive. In addition, all procedures performed on mice were approved by the Cyprus Veterinary Services (License Number: CY/EXP/PR.L3/2019).
1.2. Renal function
Renal function was monitored by measuring haematuria and proteinuria in 24‐hour urine samples. Urine samples were collected from all mice at 4 weeks. Microscopic haematuria was defined as >5 red blood cells (RBCs) per high power field (HPF, 20×). Quantitative measurements for proteinuria were performed using the Bradford assay (Bio‐Rad). 8 Microscopic examination of urine, on a phase contrast microscope, was performed on urine samples that had been centrifuged for 5 minutes at 2000 rpm. The sediment was examined under high power magnification (40×).
1.3. Transmission electron microscopy (TEM)
Kidneys were fixed in 2.5% glutaraldehyde in phosphate buffer (0.1 M, pH = 7.2) for a minimum of 4 hours at 4°C. The samples were then washed in phosphate buffer (0.1 M, pH = 7.2), post fixed in 1% osmium tetroxide, dehydrated in graded ethanol, cleared in propylene oxide and embedded in an epon/araldite resin mixture and polymerized at 60°C for at least 16 hours.
Semithin sections of 1 μm thickness were cut on a Leica Reichert ultra‐microtome UCT (Vienna, Austria), stained with toluidine blue and examined in a light microscope for locating kidney glomeruli. Silver/gold interference colour ultrathin sections were cut and mounted on 200 mesh copper grids, contrasted with uranyl acetate and lead citrate, before being examined in a JEM‐JEOL1010 transmission electron microscope (JEOL, Tokyo, Japan).
1.4. GBM morphometry
The width of the GBM was determined using a method similar to previously published work. 9 In brief, assessment of GBM width was carried out on five glomeruli per mouse and measurements were taken along the GBMs, spanning peripheral glomerular capillary loops. Each measurement was taken from the fenestrated endothelium to the membrane of the podocyte. The mean of the GBM width was calculated for each animal, by taking the average of 5 measured individual points per capillary loop. Five peripheral capillary loops were measured for each glomerulus.
1.5. Untargeted LC‐MS/MS proteomics
1.5.1. Sample preparation
For each sample, 10 kidney serial cryosections of 10 μm thickness were solubilized in lysis buffer (10 mM Tris‐HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, PBS) containing proteinase inhibitors (cOmplete Mini EDTA‐free, Roche, Germany). Following overnight acetone precipitation at −20°C, the protein pellet was re‐suspended in urea buffer (8 M) and 100 μg of extracted proteins, were reduced by DTT, alkylated by iodoacetamide and digested by trypsin at a 1:50 ratio, overnight at 37°C. The enzymatic reaction was stopped by addition of TFA to a final concentration of 0.5%. Resulting peptides were further purified and desalted using solid phase extraction cartridges (Sep‐Pak tC18, Waters, Austria), dried in a vacuum centrifuge and stored in −80°C until further analysis.
1.5.2. Method
Purified peptides were re‐suspended in buffer (95% water, 5% acetonitrile, 0.1% formic acid) to a final protein concentration of 0.2 μg/μL prior LC‐MS/MS analysis, and 2 μL were loaded onto a C18 analytical column (nanoAcquity CSH C18, 75 μm ID × 250 mm, 1.7 μm, Waters, UK). Samples were eluted by a linear gradient from 3% to 40% of mobile phase B (0.1% formic acid in acetonitrile) over 145 minutes. The experiments were performed on a nanoAcquity UPLC system connected to a Synapt G2Si HDMS instrument, operated on positive ion mobility mode and using the UDMSe approach. 10
1.5.3. Data and statistical analysis
The raw MS data were interpreted using the Progenesis QI for proteomics software (version 2.0, Waters, UK) against the UniProtKB/Swiss‐Prot mouse reference proteome database (version 26/06/2017). Normalization of the data was performed by Progenesis QIp analysis software using the “normalised to all proteins” option. Peptide identifications were performed using the MSe search identification and a peptide false discovery rate (FDR) threshold of <1%. Identified proteins med the following criteria: confidence score ≥ 5, sequence length ≥ 6 and hits ≥ 2. Protein‐level relative quantitation was performed using the Hi‐N approach (N = 3) as implemented in the Progenesis QIp. Additionally, a variation of one‐factor ANOVA calculation and an FDR approach for multiple comparisons, as implemented in the Progenesis QIp software, was performed for every identified and quantified protein.
Data were subjected to functional enrichment analysis using the Cytoscape bioinformatics tool and its plugin ClueGO. 11 Principal component analysis (PCA) was carried out using the R statistics software version R 3.5.3 (R Core Team, 2019).
1.6. Immunofluorescence
Fresh kidney tissues were snap‐frozen in liquid nitrogen and stored at −80°C. Cryosections of 10 μm thickness were mildly fixed with 4% paraformaldehyde in 0.1 M phosphate buffer and blocked with 20% bovine serum albumin (BSA) in PBS at room temperature for 1 hour. The sections were then incubated with primary antibodies against Nephrin (Abcam, ab 50 339, 1:50) and anti‐Peroxisome Proliferator Activated Receptor α (PPAR‐α, Abcam, ab 215 270, 1:200), overnight at 4°C. Primary antibodies were diluted in 5% BSA and 1% Triton X in PBS. After PBS washes, secondary antibody, goat anti‐rabbit immunoglobulins/biotinylated (Daco E0432, 1:100) diluted in 5% BSA and 1% Triton X in PBS, was applied for 1 hour at room temperature. Subsequently, sections were incubated with streptavidin/AlexaFluor 488 (Invitrogen, S11223, 1:1000) diluted with PBS for 1 hour at room temperature. Finally, sections were washed with PBS, mounted with fluorescence mounting medium (DAKO, S3023), and examined in a ZEISS fluorescent microscope (Carl Zeiss Microimaging, Oberkochen, Germany).
1.7. Western blot and densitometry
Frozen kidney tissues were lysed using RIPA lysis buffer containing protein inhibitors under sonication. Kidney homogenates were separated by SDS‐PAGE and transferred onto PVDF membranes. Membranes were blocked with 5% BSA in PBS‐t for 1 hour at room temperature and then incubated with primary antibodies (PPARα, Abcam, ab 215 270, 1:500) overnight at 4°C. Following incubation with the appropriate HRP‐conjugated secondary antibody (Santa Cruz, sc‐2004, 1:5000 in PBS‐t) for 1 hour at room temperature, protein bands were visualized using Luminata Forte Western HRP Substrate (Millipore, WBLUF0100) under UVP bio‐imaging system. All immunoblotting experiments were performed in duplicate.
ImageJ was used to perform densitometry calculations, while all bands were normalized against β‐actin (Abcam, ab 8227, 1:1000) loading control. The same reference sample was used in all western blotting in order to allow cross‐gel comparisons.
1.8. Statistical analysis
The data are presented as mean (SD). Statistical analysis was performed using Student's t‐test.
2. RESULTS
2.1. Evaluation of renal function
Renal function was evaluated by assessing haematuria and proteinuria in 24 hours urine samples. Mild microscopic haematuria was present in 4 weeks Alport mice and was absent in wild type mice. No proteinuria was observed in the Alport as well as the wild type mice at 4 weeks.
2.2. Electron microscopy
EM examination was performed on kidneys from the experimental mice in order to examine ultrastructural changes of the GBM that occur at 4 weeks. Overall, the GBM of Col4a3KO mice exhibited mild structural changes compared to wild type (Figure 1A). Focal thinning and thickening as well as areas with multi‐laminated segments of the GBM were observed in Col4a3KO mice (Figure 1A,V). In addition, the width of the GBM for both mouse groups was measured. The average thickness of the GBM of Alport mice was significantly reduced, 112.2 nm (P‐value: 2.2E‐64), which is about 16% below the corresponding average width (133.3 nm) of the wild type mice (Figure 1B).
FIGURE 1.
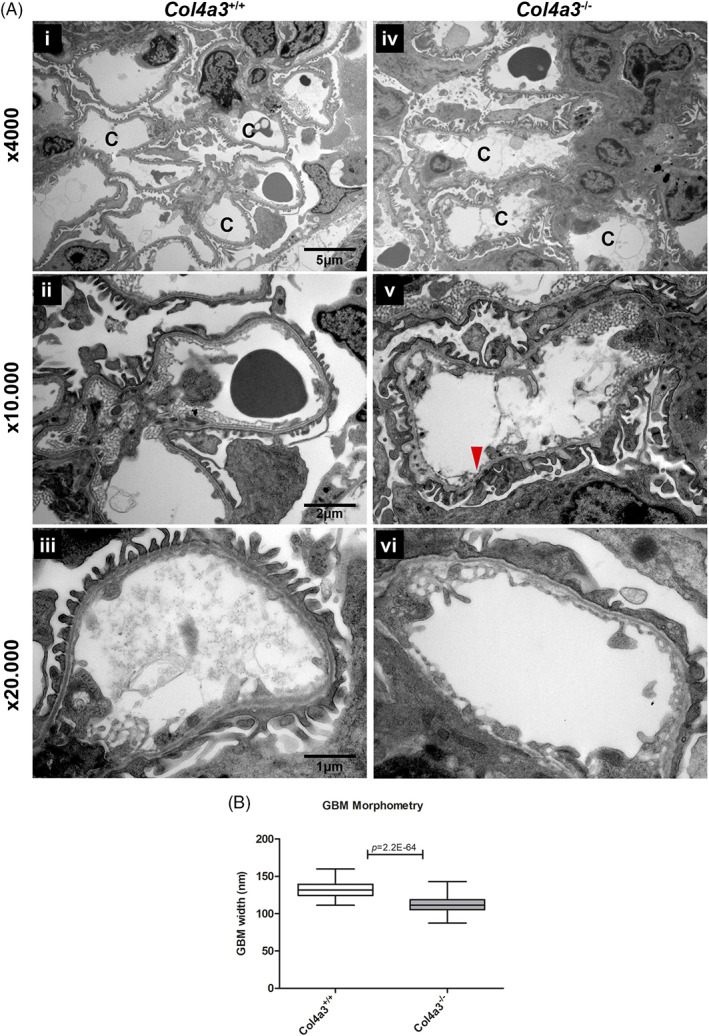
Electron microscopy and GBM measurements of 4 week mice. A, Electron micrographs of mouse kidneys Col4a3 −/− Alport (n = 4) and Col4a3 +/+ wild type mice (n = 4) at 4 weeks, at three different magnifications x4000, x10.000 and x20.000. (i) Wild type mice showing normal glomerular capillaries (C). (ii‐iii) Wild type mice showing uniform thickness of glomerular basement membranes with regular inner and outer contours. (iv) Alport mice showing normal glomerular capillaries (C). (v) Alport mice showing focal thinning (arrow heads) and focal areas with multi‐laminated segments (arrows) of GBM. (vi) Alport mice showing uniform thickness of glomerular capillaries with regular inner and outer contours. B, Box and whisker plots showing GBM width of Col4a3 −/− Alport (n = 4) and Col4a3 +/+ wild type mice (n = 4). Significant reduction in the GBM thickness (16%) was observed in Col4a3 −/− Alport mice when compared to the Col4a3 +/+ wild type mice. Col4a3 +/+ wild type mice show an average thickness of 133.3 nm with SD: 10.2 (total measurements: 122). Col4a3 −/− Alport shows an average thickness of 112.2 nm with SD: 10.2 (total measurements: 176). Statistical analysis was performed by a Student's t‐test with equal variances
2.3. Proteomic profiling using untargeted LC‐MS/MS
Comprehensive proteomic profiling was performed on kidneys from Col4a3KO (n = 4) and wild type (n = 4) mice at 4 weeks of age. Overall, 1960 unique proteins were identified, of which 415 were found to have significant changes in their expression levels with a P ≤ .05. Out of these 415 proteins, 45 proteins exhibited a difference of equal or more than 2‐fold,comprising 9 up‐regulated and 36 downregulated proteins in Alport compared to control mice. More details about the 415 dysregulated proteins are given in Table S1.1, while the 45 proteins are listed in Table 1. To enable unbiased classification of samples and detection of outliers, PCA analysis was applied on the statistically significant differentially expressed proteins. PCA score plots revealed a clear separation between disease and control samples into two distinct clusters, based on their respective protein expression profiles (Figure 2).
TABLE 1.
List of 45 significantly dysregulated proteins showing equal or more than 2fold change, identified by discovery proteomics
| Accession Number | Gene symbol | ANOVA (p) | Max FC | Highest mean condition | Lowest mean condition | Description |
|---|---|---|---|---|---|---|
| P27005 | S10A8 | 2.74E‐04 | 14.7 | MM | NN | Protein S100‐A8 |
| P20852 | CP2A5 | 1.59E‐02 | 4.0 | MM | NN | Cytochrome P450 2A5 |
| Q8JZZ0 | UD3A2 | 2.03E‐06 | 3.0 | MM | NN | UDP‐glucuronosyltransferase 3A2 |
| Q99K74 | MED24 | 1.09E‐02 | 2.5 | MM | NN | Mediator of RNA polymerase II transcription subunit 24 |
| Q9JIY7 | NAT8 | 1.86E‐02 | 2.2 | MM | NN | N‐acetyltransferase 8 |
| Q8K0H1 | S47A1 | 1.26E‐02 | 2.1 | MM | NN | Multidrug and toxin extrusion protein 1 |
| Q9D0G0 | RT30 | 1.04E‐02 | 2.1 | MM | NN | 28S ribosomal protein S30, mitochondrial |
| P02463 | CO4A1 | 6.30E‐03 | 2.0 | MM | NN | Collagen alpha‐1(IV) chain |
| Q60936 | ADCK3 | 1.23E‐02 | 2.0 | MM | NN | Chaperone activity of bc1 complex‐like, mitochondrial |
| Q9WVM8 | AADAT | 3.62E‐04 | 0.5 | NN | MM | Kynurenine/alpha‐aminoadipate aminotransferase, mitochondrial |
| Q02248 | CTNB1 | 3.90E‐02 | 0.5 | NN | MM | Catenin beta‐1 |
| P43274 | H14 | 1.82E‐04 | 0.5 | NN | MM | Histone H1.4 |
| P05201 | AATC | 1.20E‐03 | 0.5 | NN | MM | Aspartate aminotransferase, cytoplasmic |
| P17427 | AP2A2 | 5.34E‐03 | 0.5 | NN | MM | AP‐2 complex subunit alpha‐2 |
| P54869 | HMCS2 | 4.00E‐04 | 0.5 | NN | MM | Hydroxymethylglutaryl‐CoA synthase, mitochondrial |
| Q02013 | AQP1 | 8.23E‐03 | 0.5 | NN | MM | Aquaporin‐1 |
| P30412 | PPIC | 3.51E‐03 | 0.5 | NN | MM | Peptidyl‐prolyl cis‐trans isomerase C |
| P24452 | CAPG | 1.30E‐02 | 0.5 | NN | MM | Macrophage‐capping protein |
| Q8CDN6 | TXNL1 | 4.29E‐02 | 0.5 | NN | MM | Thioredoxin‐like protein 1 |
| Q64331 | MYO6 | 2.53E‐02 | 0.5 | NN | MM | Unconventional myosin‐VI |
| P05784 | K1C18 | 1.04E‐04 | 0.5 | NN | MM | Keratin, type I cytoskeletal 18 |
| P14094 | AT1B1 | 2.55E‐03 | 0.5 | NN | MM | Sodium/potassium‐transporting ATPase subunit beta‐1 |
| P43276 | H15 | 2.02E‐05 | 0.5 | NN | MM | Histone H1.5 |
| Q99JW5 | EPCAM | 6.48E‐03 | 0.5 | NN | MM | Epithelial cell adhesion molecule |
| Q8VDN2 | AT1A1 | 3.98E‐03 | 0.5 | NN | MM | Sodium/potassium‐transporting ATPase subunit alpha‐1 |
| P45591 | COF2 | 2.00E‐02 | 0.5 | NN | MM | Cofilin‐2 |
| P15626 | GSTM2 | 2.01E‐02 | 0.5 | NN | MM | Glutathione S‐transferase Mu 2 |
| P52825 | CPT2 | 4.34E‐02 | 0.5 | NN | MM | Carnitine O‐palmitoyltransferase 2, mitochondrial |
| P17809 | GTR1 | 2.83E‐03 | 0.5 | NN | MM | Solute carrier family 2, facilitated glucose transporter member 1 |
| O70133 | DHX9 | 1.61E‐03 | 0.5 | NN | MM | ATP‐dependent RNA helicase A |
| O88322 | NID2 | 4.49E‐03 | 0.5 | NN | MM | Nidogen‐2 |
| Q8CIE6 | COPA | 2.24E‐02 | 0.5 | NN | MM | Coatomer subunit alpha |
| O70456 | 1433S | 4.88E‐02 | 0.5 | NN | MM | 14–3‐3 protein sigma |
| P28666 | MUG2 | 4.21E‐02 | 0.5 | NN | MM | Murinoglobulin‐2 |
| P52480 | KPYM | 6.26E‐03 | 0.5 | NN | MM | Pyruvate kinase PKM |
| Q9CQF3 | CPSF5 | 5.22E‐04 | 0.4 | NN | MM | Cleavage and polyadenylation specificity factor subunit 5 |
| Q60604 | ADSV | 7.17E‐04 | 0.4 | NN | MM | Adseverin |
| P10922 | H10 | 2.44E‐05 | 0.4 | NN | MM | Histone H1.0 |
| P48758 | CBR1 | 4.46E‐04 | 0.4 | NN | MM | Carbonyl reductase [NADPH] 1 |
| Q91VJ1 | AIM2 | 1.49E‐02 | 0.4 | NN | MM | Interferon‐inducible protein AIM2 |
| G3X9C2 | FBX50 | 1.48E‐03 | 0.3 | NN | MM | F‐box only protein 50 |
| P45376 | ALDR | 3.16E‐03 | 0.2 | NN | MM | Aldose reductase |
| P17879 | HS71B | 6.43E‐05 | 0.2 | NN | MM | Heat shock 70 kDa protein 1B |
| P23927 | CRYAB | 7.36E‐04 | 0.2 | NN | MM | Alpha‐crystallin B chain |
| Q9CZT8 | RAB3B | 5.69E‐10 | 0.1 | NN | MM | Ras‐related protein Rab‐3B |
Notes: NN: Col4a3+/+; MM: Col4a3−/−. Red colour indicates up‐regulated proteins in disease compared to control mice, Green colour indicates down‐regulated proteins in disease compared to control mice.
Abbreviation: FC, fold change.
FIGURE 2.
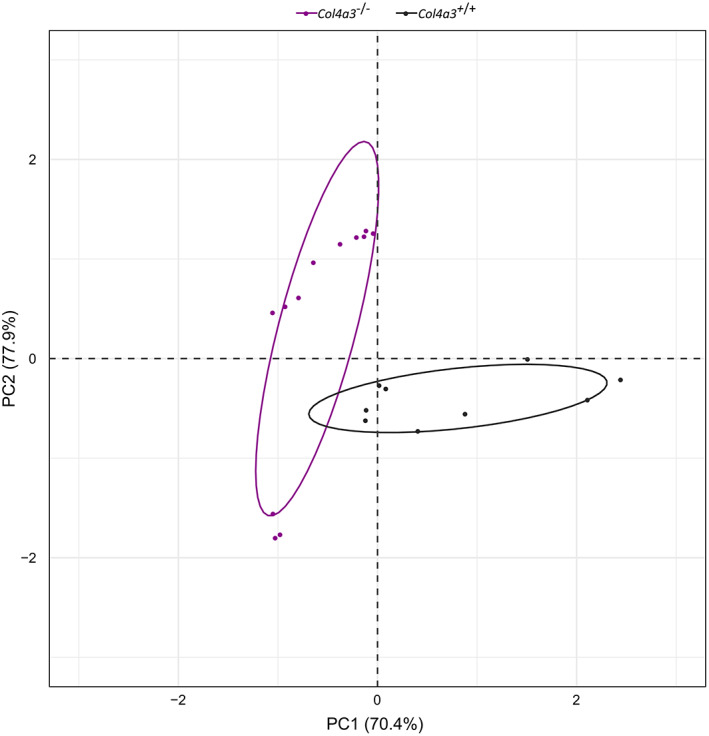
PCA analysis. PCA score plot of protein expression patterns of all proteins with P‐value ≤.05, showing a clear separation of Col4a3 −/− Alport mice (grey dots) and Col4a3 +/+ wild type mice (violet dots) into two distinct clusters. Statistical analysis was performed by one‐way ANOVA test using the Progenesis QI for proteomics software, while PCA analysis was carried out by the R statistics software version R 3.5.3 (R Core Team, 2019)
2.4. Functional annotation and enrichment analysis of protein profile
To gain valuable insights in the significant differentially expressed proteins detected by the proteomic analysis, gene ontology (GO) and pathway enrichment analyses were carried out. In total, 150, 55 and 89 statistically significant GO enriched terms were identified for biological process, molecular function, and cellular component analysis, respectively (Table S1.2, S1.3 and S1.4). The top 20 significantly enriched terms for each GO category are shown in Table 2. The biological process evaluation of the GO annotations revealed that the identified proteins are mainly involved in metabolic and cellular processes. When analysed in terms of molecular function, the identified proteins were found to be involved in the following two major categories of binding and catalytic activity. Similarly, on cellular component analysis, some proteins were found to be located in the extracellular space, while others are organelle related proteins, and yet others are part of membrane complexes.
TABLE 2.
Top 20 significantly enriched GO categories and KEGG pathways
| GO Categories/KEGG | ID | Hits | P‐value |
|---|---|---|---|
| GO: Biological Process | |||
| Organic acid metabolic process | GO:0006082 | 69 | 3.66E‐19 |
| Oxoacid metabolic process | GO:0043436 | 67 | 2.67E‐18 |
| Nucleotide metabolic process | GO:0009117 | 53 | 4.15E‐18 |
| Nucleoside phosphate metabolic process | GO:0006753 | 53 | 7.07E‐18 |
| Purine ribonucleoside monophosphate metabolic process | GO:0009167 | 34 | 1.08E‐17 |
| Purine nucleoside monophosphate metabolic process | GO:0009126 | 34 | 1.23E‐17 |
| Carboxylic acid metabolic process | GO:0019752 | 63 | 2.05E‐17 |
| Nucleobase‐containing small molecule metabolic process | GO:0055086 | 54 | 2.98E‐17 |
| Ribonucleoside monophosphate metabolic process | GO:0009161 | 34 | 4.54E‐17 |
| Nucleoside monophosphate metabolic process | GO:0009123 | 34 | 2.52E‐16 |
| Cofactor metabolic process | GO:0051186 | 38 | 3.97E‐16 |
| Purine ribonucleoside triphosphate metabolic process | GO:0009205 | 31 | 1.13E‐15 |
| Oxidoreduction coenzyme metabolic process | GO:0006733 | 25 | 2.15E‐15 |
| ATP metabolic process | GO:0046034 | 29 | 2.17E‐15 |
| Ribonucleoside triphosphate metabolic process | GO:0009199 | 31 | 2.40E‐15 |
| Purine nucleoside triphosphate metabolic process | GO:0009144 | 31 | 2.71E‐15 |
| Coenzyme metabolic process | GO:0006732 | 33 | 4.71E‐15 |
| Cellular component assembly | GO:0022607 | 115 | 1.26E‐14 |
| Macromolecular complex assembly | GO:0065003 | 84 | 1.70E‐14 |
| Nucleoside triphosphate metabolic process | GO:0009141 | 31 | 1.97E‐14 |
| GO: Molecular Function | |||
| Cadherin binding | GO:0045296 | 34 | 1.32E‐14 |
| Cell adhesion molecule binding | GO:0050839 | 40 | 1.55E‐13 |
| Identical protein binding | GO:0042802 | 89 | 1.84E‐13 |
| Protein complex binding | GO:0032403 | 53 | 1.81E‐12 |
| Coenzyme binding | GO:0050662 | 23 | 3.56E‐11 |
| Actin filament binding | GO:0051015 | 21 | 1.07E‐10 |
| RNA binding | GO:0003723 | 74 | 8.38E‐10 |
| Actin binding | GO:0003779 | 30 | 6.59E‐09 |
| Oxidoreductase activity, acting on CH‐OH group of donors | GO:0016614 | 17 | 9.11E‐09 |
| Cytoskeletal protein binding | GO:0008092 | 47 | 2.51E‐08 |
| Oxidoreductase activity, acting on the CH‐OH group of donors, NAD or NADP as acceptor | GO:0016616 | 15 | 4.87E‐08 |
| Nucleoside phosphate binding | GO:1901265 | 83 | 1.39E‐07 |
| Nucleotide binding | GO:0000166 | 83 | 1.39E‐07 |
| ATPase activity | GO:0016887 | 28 | 1.51E‐07 |
| ATPase activity, coupled | GO:0042623 | 24 | 5.66E‐07 |
| NAD binding | GO:0051287 | 10 | 1.21E‐06 |
| mRNA binding | GO:0003729 | 17 | 1.42E‐06 |
| Poly(A) binding | GO:0008143 | 6 | 1.61E‐06 |
| Carnitine O‐acyltransferase activity | GO:0016406 | 4 | 4.34E‐06 |
| Oxidoreductase activity, acting on NAD(P)H | GO:0016651 | 10 | 1.23E‐05 |
| GO: Cellular Component | |||
| Extracellular vesicle | GO:1903561 | 214 | 8.52E‐77 |
| Extracellular organelle | GO:0043230 | 214 | 1.44E‐76 |
| Extracellular exosome | GO:0070062 | 213 | 1.79E‐76 |
| Extracellular space | GO:0005615 | 220 | 2.15E‐57 |
| Vesicle | GO:0031982 | 230 | 8.64E‐57 |
| Myelin sheath | GO:0043209 | 43 | 1.82E‐28 |
| Mitochondrion | GO:0005739 | 110 | 5.28E‐25 |
| Mitochondrial part | GO:0044429 | 75 | 6.75E‐24 |
| Mitochondrial inner membrane | GO:0005743 | 48 | 2.68E‐21 |
| Mitochondrial envelope | GO:0005740 | 60 | 3.84E‐21 |
| Organelle inner membrane | GO:0019866 | 50 | 1.56E‐20 |
| Mitochondrial membrane | GO:0031966 | 57 | 1.66E‐20 |
| Organelle envelope | GO:0031967 | 70 | 1.59E‐16 |
| Envelope | GO:0031975 | 70 | 1.67E‐16 |
| Actin cytoskeleton | GO:0015629 | 38 | 1.80E‐12 |
| Inner mitochondrial membrane protein complex | GO:0098800 | 20 | 2.49E‐12 |
| Mitochondrial membrane part | GO:0044455 | 23 | 7.83E‐11 |
| Mitochondrial protein complex | GO:0098798 | 20 | 8.52E‐11 |
| Membrane protein complex | GO:0098796 | 43 | 1.78E‐10 |
| Mitochondrial respiratory chain | GO:0005746 | 13 | 7.51E‐09 |
| KEGG Pathway | |||
| Huntington's disease | KEGG:05016 | 27 | 2.50E‐09 |
| Parkinson's disease | KEGG:05012 | 22 | 3.00E‐08 |
| Oxidative phosphorylation | KEGG:00190 | 20 | 2.97E‐07 |
| Proximal tubule bicarbonate reclamation | KEGG:04964 | 7 | 2.24E‐04 |
| Bacterial invasion of epithelial cells | KEGG:05100 | 12 | 2.35E‐04 |
| Glycolysis/Gluconeogenesis | KEGG:00010 | 11 | 4.10E‐04 |
| Alzheimer's disease | KEGG:05010 | 18 | 4.98E‐04 |
| Peroxisome | KEGG:04146 | 12 | 6.64E‐04 |
| Pyruvate metabolism | KEGG:00620 | 8 | 1.20E‐03 |
| Central carbon metabolism in cancer | KEGG:05230 | 10 | 1.66E‐03 |
| Citrate cycle (TCA cycle) | KEGG:00020 | 7 | 3.12E‐03 |
| Cardiac muscle contraction | KEGG:04260 | 10 | 9.25E‐03 |
| Pentose phosphate pathway | KEGG:00030 | 6 | 2.70E‐02 |
| Protein digestion and absorption | KEGG:04974 | 10 | 2.97E‐02 |
| Proteasome | KEGG:03050 | 7 | 3.26E‐02 |
| Fructose and mannose metabolism | KEGG:00051 | 6 | 4.30E‐02 |
| Non‐alcoholic fatty liver disease (NAFLD) | KEGG:04932 | 13 | 5.04E‐02 |
| Spliceosome | KEGG:03040 | 12 | 5.23E‐02 |
| PPAR signalling pathway | KEGG:03320 | 9 | 7.63E‐02 |
| Valine, leucine and isoleucine degradation | KEGG:00280 | 7 | 1.02E‐01 |
Abbreviations: GO; gene ontology, KEGG; Kyoto Encyclopaedia of Genes and Genomes.
In order to explore the initiating signalling events that could be triggered from the structural changes in the GBM, pathway analysis was performed. In total, 52 statistically significant enriched Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways were identified to be altered in AS mice (see Table S1.5). The top 20 significantly enriched pathways are shown in Table 2. Among others, the oxidative phosphorylation, the proximal tubule bicarbonate reclamation, proteasome, protein digestion and absorption, as well as the peroxisome pathways, were identified. Of particular interest is that the majority of the proteins involved in these 20 pathways were found to be downregulated in AS mice, early on before proteinuria ensues. Proteins involved in the proteasome, protein digestion and absorption pathways as well as peroxisome‐proliferator‐activated receptors (PPAR) signalling pathways are illustrated in Tables 3, 4 and 5, respectively. In addition, a graphical illustration of PPAR pathway, highlighting the proteins identified in our study as given by DAVID bioinformatics tool, is shown in the Figure S1. Further tables demonstrating the remaining 17 pathways are given in Tables S1.6 to S1.22.
TABLE 3.
List of proteins involved in proteasome pathway
| Accession Number | Gene symbol | ANOVA (p) | Max FC | Highest mean condition | Lowest mean condition | Description |
|---|---|---|---|---|---|---|
| O70435 | Psma3 a | 1.09E‐02 | 1.5 | NN | MM | Proteasome subunit alpha type‐3 |
| Q9Z2U0 | Psma7 | 2.92E‐02 | 1.4 | NN | MM | Proteasome subunit alpha type‐7 |
| P99026 | Psmb4 a | 3.10E‐02 | 1.2 | NN | MM | Proteasome subunit beta type‐4 |
| P46471 | Psmc2 a | 2.19E‐02 | 1.3 | NN | MM | 26S protease regulatory subunit 7 |
| O88685 | Psmc3 a | 7.42E‐04 | 1.4 | NN | MM | 26S protease regulatory subunit 6A |
| Q3TXS7 | Psmd1 | 3.97E‐02 | 1.3 | NN | MM | 26S proteasome non‐ATPase regulatory subunit 1 |
| O35226 | Psmd4 | 4.51E‐02 | 1.5 | NN | MM | 26S proteasome non‐ATPase regulatory subunit 4 |
Abbreviation: FC, fold change.
Alternative gene names: Psma3 = PSA3, Psma3 = PSA7, Psmb4 = PSB4, Psmc2 = PRS7, Psmc3 = PRS6A. NN: Col4a3+/+; MM: Col4a3−/−.
TABLE 4.
List of proteins involved in protein digestion and absorption pathway
| Accession Number | Gene symbol | ANOVA (p) | Max FC | Highest mean condition | Lowest mean condition | Description |
|---|---|---|---|---|---|---|
| Q8R0I0 | Ace2 | 1.59E‐03 | 1.4 | NN | MM | Angiotensin‐converting enzyme 2 |
| Q8VDN2 | Atp1a1 | 3.98E‐03 | 2.0 | NN | MM | Sodium/potassium‐transporting ATPase subunit alpha‐1 |
| Q6PIE5 | Atp1a2 | 4.70E‐02 | 1.8 | NN | MM | Sodium/potassium‐transporting ATPase subunit alpha‐2 |
| P14094 | Atp1b1 | 2.55E‐03 | 1.9 | NN | MM | Sodium/potassium‐transporting ATPase subunit beta‐1 |
| P39061 | Col18a1 a | 2.00E‐04 | 1.5 | NN | MM | Collagen alpha‐1(XVIII) chain |
| P28825 | Mep1a | 3.35E‐03 | 1.4 | NN | MM | Meprin A subunit alpha |
| Q61847 | Mep1b | 1.46E‐04 | 1.7 | NN | MM | Meprin A subunit beta |
| Q61391 | Mme a | 4.38E‐03 | 1.6 | NN | MM | Neprilysin |
| Q9D687 | Slc6a19 | 4.69E‐03 | 1.7 | MM | NN | Sodium‐dependent neutral amino acid transporter B(0)AT1 |
| P02463 | Col4a1 | 6.30E‐03 | 2.0 | MM | NN | Collagen alpha‐1(IV) chain |
Abbreviation: FC, fold change.
Alternative gene names: Col181 = Col1, Mme = NEP. NN: Col4a3+/+; MM: Col4a3−/−.
TABLE 5.
List of proteins involved in PPAR signalling pathway
| Accession Number | Gene symbol | ANOVA (p) | Max FC | Highest mean condition | Lowest mean condition | Description |
|---|---|---|---|---|---|---|
| P51174 | Acadl | 3.52E‐02 | 1.6 | NN | MM | Long‐chain specific acyl‐CoA dehydrogenase, mitochondrial |
| Q9QXD1 | Acox2 | 1.71E‐02 | 1.2 | NN | MM | Peroxisomal acyl‐coenzyme A oxidase 2 |
| P41216 | Acsl1 | 3.61E‐02 | 1.8 | NN | MM | Long‐chain‐fatty‐acid‐CoA ligase 1 |
| P97742 | Cpt1a | 8.54E‐03 | 1.5 | NN | MM | Carnitine O‐palmitoyltransferase 1, liver isoform |
| P52825 | Cpt2 | 4.34E‐02 | 2.0 | NN | MM | Carnitine O‐palmitoyltransferase 2, mitochondrial |
| P11404 | Fabp3 a | 4.99E‐03 | 1.6 | NN | MM | Fatty acid‐binding protein, heart |
| Q05816 | Fabp5 | 1.46E‐02 | 1.3 | NN | MM | Fatty acid‐binding protein, epidermal |
| P54869 | Hmgcs2 a | 4.00E‐04 | 1.9 | NN | MM | Hydroxymethylglutaryl‐CoA synthase, mitochondrial |
| O55222 | Ilk | 4.82E‐02 | 1.7 | NN | MM | Integrin‐linked protein kinase |
Abbreviation: FC, fold change.
Alternative gene names: Fabp3 = FABPH, Hmgcs2 = HMCS2. NN: Col4a3+/+; MM: Col4a3−/−.
2.5. PPARα protein expression levels in mouse kidney tissues
PPAR pathway was one of the top 20 significantly dysregulated pathways identified in our study. Importantly, an emerging body of evidence suggests that targeting this pathway through PPARα agonists exerts renoprotective effects. 12 , 13 , 14 , 15 , 16 Because of the above and the fact that the PPARα protein was not identified by discovery proteomics, we proceeded to measure its levels using targeted methods including, immunofluorescence and immunoblot analysis. Immunofluorescence staining showed downregulation of PPARα protein in AS compared to control mice, which was found to be predominantly expressed in the renal proximal tubules; this is in agreement with previously reported data 17 , 18 (Figure 3A). In addition, these observations were confirmed by immunoblot analysis, indicating significant downregulation of PPARα protein in AS compared to control mice (P‐value < .01) (Figure 3B).
FIGURE 3.
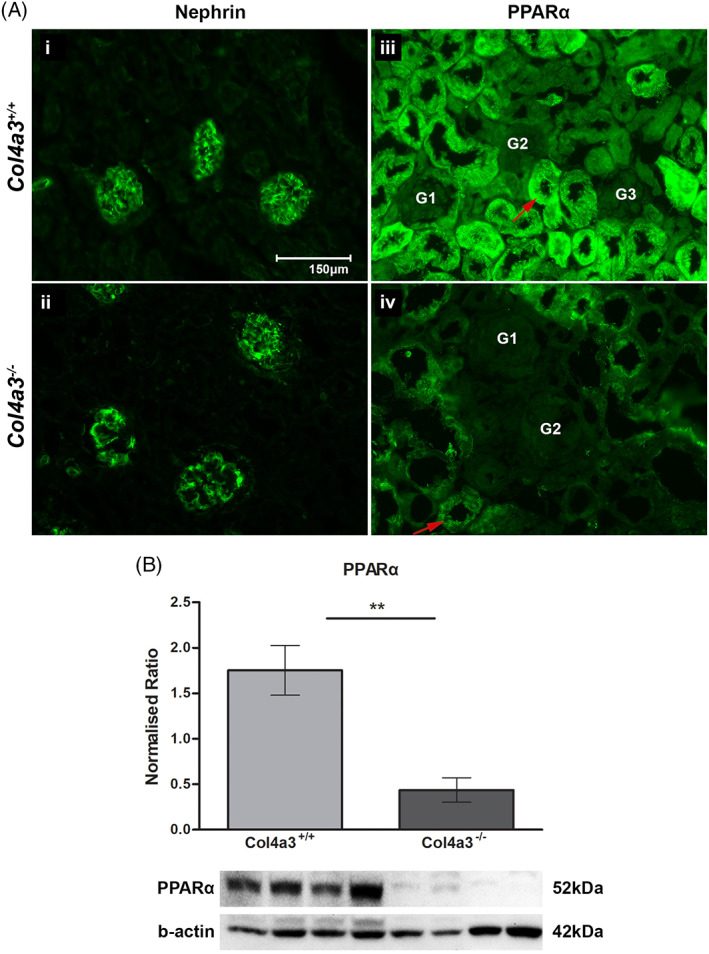
Expression of PPARα protein in mouse renal tissues. A, Representative images of immunofluorescence staining for Nephrin and PPARα proteins in renal tissues of Col4a3 −/− Alport mice and Col4a3 +/+ wild type mice at 4 weeks. (i, ii) No difference was observed in the expression levels of Nephrin protein between the two groups. In addition, Nephrin was observed to be solely expressed in the glomeruli. (iii‐iv) PPARα protein was found to be downregulated in Col4a3 −/− Alport mice (iv) compared to Col4a3 +/+ wild type mice (iii). PPARα protein was found to be predominantly expressed in proximal renal tubules (arrows). n = 4/group, Scale bar = 150 μm. B, Relative quantification of PPARα via immunoblotting. Significant decrease of PPARα expression levels was observed in Col4a3 −/− Alport mice when compared to the Col4a3 +/+ wild type mice. n = 4/group, the data are presented as mean (SD). Statistical analysis was performed using a paired Student's t test. *P < 0.05, **P < 0.01, ***P < 0.001. PPARα, Peroxisome Proliferator Activated Receptor α
3. DISCUSSION
The application of discovery proteomics to investigate kidney diseases such as AS provides the opportunity to gain a more comprehensive insight into disease pathogenesis and identify novel therapeutic targets. 19
In this study, proteomic profiling of kidney tissues from Col4a3+/+ wild type and Col4a3−/− Alport mice resulted to the detection of 1960 unique proteins. Differentially expressed proteins were mapped to molecular pathways through functional enrichment analysis in order to unravel early pathogenic mechanisms. Note that this study was designed and performed on 4 weeks old mice, a stage critical for disease progression, at which time point AS mice do not manifest proteinuria, but show mild microscopic haematuria and also exhibit mild ultrastructural GBM changes. The latter include the presence of thick and thin areas as well as the presence of focal multi‐lamellated GBM segments, resembling the typical basket weave appearance, seen in AS cases. In addition, measurement of the average thickness of the GBMs in Col4a3 −/− Alport mice, showed a small but significant reduction (16%), compared to the GBMs of wild type mice. These results are in agreement with previous published data and represent the ultrastructural changes typical of AS. 6
Of the 1960 detected proteins 415 were significantly dysregulated and of these, S100‐A8 was the protein with the highest expression in kidneys from AS compared to WT mice (max FC, 14.7). Protein S100‐A8/A9 complex is released upon tissue injury, acting as a damage‐associated molecular marker. It has multiple intracellular and extracellular biological functions, including cytoskeleton modulation of phagocytes and epithelial cells, transfer of polyunsaturated fatty acids and activation of NADPH oxidase. 20 S100‐A8 and S100‐A9 expression was shown to be increased in renal biopsies from patients with obstructive hydronephrosis and was localized to the tubulo‐interstitial compartment. In‐vitro studies indicate that S100‐A8/A9 may induce tubular epithelial cell apoptosis and lead to tubular atrophy and progressive fibrosis. 21
Pathway analysis demonstrated that dysregulated pathways related to cellular metabolism were amongst the most significantly enriched pathways. These pathways included the oxidative phosphorylation, glycolysis/glyconeogenesis, pyruvate metabolism, peroxisome and the PPAR pathways. Cellular component analysis confirmed that most of the identified proteins are related to mitochondria, whilst the biological processes analysis confirmed that proteins are mostly involved in metabolic processes, including organic acid, oxoacid and carboxylic acid metabolism. When analysed in terms of molecular function, the identified proteins were assigned into two major categories of binding and catalytic activity. The latter include carnitine‐O transferase as well as ATPase activity. The mitochondrial carnitine system is important for the transport of fatty acids into the mitochondrial matrix for β‐oxidation. 22 Upregulation of this mitochondrial system was shown to recover ATP depletion and attenuate tubular injury in animal models of ischemia/reperfusion injury. 23
The identification of a downregulated PPAR pathway in AS among the 20 significantly enriched pathways was of particular importance. Firstly, PPAR has a central role in fatty acid metabolism (β‐oxidation) with downstream effects on peroxisomal and mitochondrial function, which were highly represented in our analyses. Secondly, it can be targeted therapeutically to improve mitochondrial and peroxisomal biogenic functions in animal models of AS and CKD progression. 12 , 13 , 15 , 24 , 25 Thirdly, our results and those of previous studies show that PPAR deregulation is detected early on in the disease course, prior to histopathological changes of tubulointerstitial fibrosis (TIF) and manifestation of proteinuria, suggesting a possible pathogenic role in fibrosis and CKD progression. Lastly, as the proximal tubules contain the highest number of mitochondria than any other renal structure, the downstream effects of a deregulated PPAR pathway on mitochondria may be related to the pathogenic process in the tubulo‐interstitial compartment. In our study, immunofluorescence confirmed reduced expression of PPARα predominantly in proximal tubular cells in AS compared to wild type mice, providing further support for proximal tubules involvement early in the disease course.
It is interesting that the majority of the 415 dysregulated proteins or altered pathways are not associated with a podocyte specific function as would have been expected. It is established that changes in the GBM type IV collagen composition is the cause of AS glomerular pathology. Mechanistically, though it remains unclear how this structural alteration triggers and drives disease progression. One possible scenario is that GBM composition changes may directly influence collagen receptor signalling, contributing to podocyte dysfunction in AS. Further, it has been recently proposed that AS should not only be considered as a podocyte‐centric disorder, but changes in other types of cells may also be involved. Indeed, changes in the GBM structure may induce biomechanical strain, alter signalling that may affect all three types of cells namely, podocytes, mesangial and endothelial cells. It is of interest that although the GBM has abnormal type IV collagen composition at birth, the renal glomerulus in AS functions normally for a while, and for several years in humans. 26 Based on the above, we believe that we did not detect any abnormalities in proteins specific to podocyte function because the analysis was performed at a very early stage, before the full podocyte pathological response was elicited. Another possibility is that changes detected in proteins within mitochondria and peroxisomes, may also represent dysfunctional podocytes.
Recently, Gomez et al. demonstrated that administration of anti‐miR21 oligonucleotides to Col4a3 −/− mice, improved kidney function, proteinuria and kidney histopathology (fibrosis, inflammation, glomerulosclerosis), increasing the life span of affected mice by more than 40%. The effects of the anti‐miR21 therapy were exerted through the enhancement of the PPARα/Retinoid X Receptor pathway. 12 Administration of PPARα agonists in a CKD model of unilateral ischemia re‐perfusion injury sustained PPARα expression, reduced lipid accumulation and protected from lipotoxicity and TIF. 13 Furthermore, in the aging rat and PPARα−/− aging mouse model, Chung KW et al. have demonstrated that impaired renal PPARα signalling reduces Fatty Acid β‐Oxidation (FAO) activity and increases lipid tubular accumulation aggravating age‐associated renal fibrosis. 27 In addition, genome‐wide transcriptome human studies comparing normal and fibrotic kidney tubule samples, confirmed higher lipid accumulation in disease renal proximal tubular cells (PTCs) and marked downregulation of PPARα. 14
PPARα is a nuclear receptor transcription factor which controls lipid metabolism and plays an important role in modulating energy metabolism though direct transcriptional control of genes involved in FAO and oxidant production in peroxisomes and mitochondria. 28 , 29 Hence, PPARα is mainly expressed in organs that possess high mitochondrial and β‐oxidation activity such as the kidney, liver and heart. In the kidney, it is abundantly expressed in PTCs which have very high metabolic demands and utilize fatty acids as their main energy source. 13 Metabolism of fatty acids in PTCs requires their transport to peroxisomes and mitochondria, mediated by carnitine palmitoyl‐transferase 1, 23 where they produce the high energy yield required for PTCs' function. Consequently, dysregulation of the PPAR pathway and defective FAO in PTCs has been associated with intracellular lipid accumulation, ATP depletion and cell death. Upstream the PPAR pathway, overexpression of the transcription factor ATF6a of the unfolded protein response (UPR) under prolonged ER stress, was shown to downregulate PPARα. 13 Others have shown that miR‐21 is involved in regulating tissue repair responses after injury. It is upregulated in chronic kidney disease and renal aging and downregulates the PPARα regulated signalling pathways. Both the abnormal UPR pathway activation and miR‐21 overexpression were implicated in AS pathogenesis. 12 , 30 Collectively, these data demonstrate the potential therapeutic effects of PPARα in AS. Although the PPAR pathway was found to be downregulated in the current study, the PPARα protein was not identified among the dysregulated proteins. Therefore, targeted methods were employed to assess its expression levels in our samples, confirming downregulation in AS. The missing PPARα dysregulation in the discovery proteomics data could be related to its low abundance in combination with the protein extraction method used in the current study that did not favour protein enrichment of nuclear receptors such as PPARα. 16
Other significantly enriched pathways included the protein digestion and absorption pathway as well as the proteasome pathway. The ubiquitin‐proteasome system degrades a large number of misfolded or damaged proteins and rigorously selected regulatory proteins, maintaining proteostasis and cell integrity. Ubiquitination is a highly selective process of protein quality control which takes place in the endoplasmic reticulum (ER). Ubiquitinated proteins are transferred into the cytoplasm for degradation by the proteasome pathway. 31 Recently, the defective localization of Col4a3 chains in human podocytes and in an AS mouse model (Col4a3‐G1332E mutation) was shown to cause unfolded protein response activation and ER stress and is now considered one of the main pathogenic mechanisms in AS. 30 Zhang et al. showed that the excessive ER stress and ER stress‐related apoptosis seen in podocyte models of a truncated Col4a3 mutation could be mitigated with MG132, a potent proteasome inhibitor. 32
In conclusion, the results of our proteomic analysis are in agreement with recent breakthroughs in AS pathogenesis. In addition, we provided further evidence of the emerging concept that alterations in proteins which participate in cellular metabolism and mitochondrial homeostasis in kidney cells, PTCs in particular, are early events in the development of CKD in AS. Importantly, the PPAR pathway was amongst the significantly downregulated pathways in AS that can be therapeutically targeted through PPARα agonists. However, further studies are needed to determine whether targeting this pathway could mechanistically rescue kidney function and thus delay disease progression in AS.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
Orthodoxia Nicolaou and Andreas Kousios performed the experiments, analysed the data and prepared the manuscript. Kleitos Sokratous designed and performed the mass spectrometry experiments. Kyriacos Kyriacou and Kleitos Sokratous designed and supervised the study and critically revised the manuscript. Lola Koniali and George Neophytou performed the western blot and immunofluorescence analysis. Revekka Papacharalampous performed immunofluorescence analysis. Maria Zanti performed the PCA analysis. Kyriacos Kyriacou and Louiza Potamiti performed the electron microscopy analysis. Andreas Hadjisavvas, Theo M. Luider and Christoph Stingl provided critical feedback. All authors reviewed and approved the manuscript.
Supporting information
Data S1 Table S1.1: List of 415 significantly dysregulated proteins quantified by discovery proteomics in the kidneys of Col4a3 knock out compared to control mice at 4 weeks. Information about accession number, protein name, identification confidence score, fold‐change and P‐value of each protein are available. Accession, gene symbol, confidence score for identification, ANOVA p values for each protein, maximum fold change and the highest and lowest mean conditions are given in the table along with the full protein names.
Table S1.2: List of significantly enriched Gene Ontology terms for Biological Process. Functional enrichment analysis was performed using the Cytoscape bioinformatics tool and its plugin ClueGO. GO terms, term p values, percentage and number of associated genes along with the gene symbols of associated genes are given in the table.
Table S1.3: List of significantly enriched Gene Ontology terms for Molecular Function. Functional enrichment analysis was performed using the Cytoscape bioinformatics tool and its plugin ClueGO. GO terms, term p values, percentage and number of associated genes along with the gene symbols of associated genes are given in the table.
Table S1.4: List of significantly enriched Gene Ontology terms for Cellular Component. Functional enrichment analysis was performed using the Cytoscape bioinformatics tool and its plugin ClueGO. GO terms, term p values, percentage and number of associated genes and genes along with the gene symbols of associated genes are given in the table.
Table S1.5: List of significantly enriched KEGG pathways. Pathway analysis was performed using the Cytoscape bioinformatics tool and its plugin ClueGO. GO terms, term p values, percentage and number of associated genes along with the gene symbols of associated genes are given in the table.
Table S1.6‐S1.22: List of proteins involved in the 17 out of the 20 top significantly dysregulated pathways. Accession, gene symbol, confidence score for identification, ANOVA p values for each protein, maximum fold change and the highest and lowest mean conditions are given in the table along with the full protein names
Data S2 Figure S1: Graphical illustration of PPAR pathway as given by DAVID bioinformatics resources. Differentially expressed proteins identified by discovery proteomics with P ≤ .05 were used for this analysis. Pathway enrichment analysis was carried out using the DAVID bioinformatics tool (DAVID 6.8). Statistical significance for KEGG pathways was established at P ≤ .05. Proteins involved in the pathway are indicated with red asterisks. Note that Hmgcs2/HMCS2 protein that was identified to be involved in the PPAR pathway by Cytoscape bioinformatics resources, was not identified in this analysis.
ACKNOWLEDGEMENTS
We would like to thank Professor Oliver Gross for kindly sending us the heterozygous Col4a3 mice, for establishing the colonies of transgenic mice used in the present experiments. In addition, we would like to acknowledge the help of Andreas Stylianou, who contributed to the measurements of the GBMs in the electron micrographs.
Nicolaou O, Kousios A, Sokratous K, et al. Alport syndrome: Proteomic analysis identifies early molecular pathway alterations in Col4a3 knock out mice. Nephrology. 2020;25:937–949. 10.1111/nep.13764
Orthodoxia Nicolaou and Andreas Kousios contributed equally to this work.
REFERENCES
- 1. Kashtan C. Alport syndrome and thin basement membrane nephropathy Comprehensive Pediatric Nephrology. Elsevier Inc.; 2008:229‐237. [Google Scholar]
- 2. Gross O, Kashtan CE, Rheault MN, et al. Advances and unmet needs in genetic, basic and clinical science in Alport syndrome: report from the 2015 international workshop on Alport Syndrome. Nephrol Dial Transplant. 2017;32:916‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gross O, Licht C, Anders HJ, et al. Early angiotensin‐converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012;81:494‐501. [DOI] [PubMed] [Google Scholar]
- 4. Kashtan CE, Ding J, Gregory M, et al. Clinical practice recommendations for the treatment of Alport syndrome: a statement of the Alport Syndrome research collaborative. Pediatr Nephrol. 2013;28:5‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gross O, Friede T, Hilgers R, Gorlitz A, Gavenis K, Ahmed R, Durr U. Safety and efficacy of the ACE‐inhibitor Ramipril in Alport Syndrome: the double‐blind, randomized, placebo‐controlled, multicenter phase III EARLY PRO‐TECT Alport trial in pediatric patients. ISRN Pediatr 2012;2012:436046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gross O, Tonshoff B, Weber LT, et al. A multicenter, randomized, placebo‐controlled, double‐blind phase 3 trial with open‐arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport's syndrome. Kidney Int. 2020;97:1275‐1286. [DOI] [PubMed] [Google Scholar]
- 7. Cosgrove D, Meehan DT, Grunkemeyer JA, et al. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996;10:2981‐2992. [DOI] [PubMed] [Google Scholar]
- 8. Prakash M, Shetty JK, Dash S, Barik BK, Sarkar A, Prabhu R. Determination of urinary peptides in patients with proteinuria. Indian J Nephrol. 2008;18:150‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marquez B, Zouvani I, Karagrigoriou A, Anastasiades E, Pierides A, Kyriacou K. A simplified method for measuring the thickness of glomerular basement membranes. Ultrastruct Pathol. 2003;27:409‐416. [PubMed] [Google Scholar]
- 10. Distler U, Kuharev J, Navarro P, Levin Y, Schild H, Tenzer S. Drift time‐specific collision energies enable deep‐coverage data‐independent acquisition proteomics. Nat Methods. 2014;11:167‐170. [DOI] [PubMed] [Google Scholar]
- 11. Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug‐in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gomez IG, MacKenna DA, Johnson BG, et al. Anti‐microRNA‐21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest. 2015;125:141‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jao TM, Nangaku M, Wu CH, et al. ATF6alpha downregulation of PPARalpha promotes lipotoxicity‐induced tubulointerstitial fibrosis. Kidney Int. 2019;95:577‐589. [DOI] [PubMed] [Google Scholar]
- 14. Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li S, Mariappan N, Megyesi J, et al. Proximal tubule PPARalpha attenuates renal fibrosis and inflammation caused by unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2013;305:F618‐F627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Q, Ding C, Liu W, et al. In‐depth proteomic characterization of endogenous nuclear receptors in mouse liver. Mol Cell Proteomics. 2013;12:473‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guan Y, Breyer MD. Peroxisome proliferator‐activated receptors (PPARs): novel therapeutic targets in renal disease. Kidney Int. 2001;60:14‐30. [DOI] [PubMed] [Google Scholar]
- 18. Lin H, Cheng CF, Chen HH. Role of PPARα and its agonist in renal diseases. PPAR Res. 2010;2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nicolaou O, Sokratous K, Makowska Z, et al. Proteomic analysis in lupus mice identifies Coronin‐1A as a potential biomarker for lupus nephritis. Arthritis Res Ther. 2020;22:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang S, Song R, Wang Z, Jing Z, Ma J. S100A8/A9 in inflammation. Front Immunol. 2018;9:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tammaro A, Florquin S, Brok M, et al. S100A8/A9 promotes parenchymal damage and renal fibrosis in obstructive nephropathy. Clin Exp Immunol. 2018;193:361‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sigauke E, Rakheja D, Kitson K, Bennett MJ. Carnitine palmitoyltransferase II deficiency: a clinical, biochemical, and molecular review. Lab Invest. 2003;83:1543‐1554. [DOI] [PubMed] [Google Scholar]
- 23. Idrovo JP, Yang WL, Nicastro J, Coppa GF, Wang P. Stimulation of carnitine palmitoyltransferase 1 improves renal function and attenuates tissue damage after ischemia/reperfusion. J Surg Res. 2012;177:157‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li S, Basnakian A, Bhatt R, et al. PPAR‐alpha ligand ameliorates acute renal failure by reducing cisplatin‐induced increased expression of renal endonuclease G. Am J Physiol Renal Physiol. 2004;287:F990‐F998. [DOI] [PubMed] [Google Scholar]
- 25. Li S, Gokden N, Okusa MD, Bhatt R, Portilla D. Anti‐inflammatory effect of fibrate protects from cisplatin‐induced ARF. Am J Physiol Renal Physiol. 2005;289:F469‐F480. [DOI] [PubMed] [Google Scholar]
- 26. Cosgrove D, Liu S. Collagen IV diseases: a focus on the glomerular basement membrane in Alport syndrome. Matrix Biol. 2017;57‐58:45‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chung KW, Lee EK, Lee MK, Oh GT, Yu BP, Chung HY. Impairment of PPARalpha and the fatty acid oxidation pathway aggravates renal fibrosis during aging. J Am Soc Nephrol. 2018;29:1223‐1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest. 2006;116:571‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang YX. PPARs: diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010;20:124‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pieri M, Stefanou C, Zaravinos A, et al. Evidence for activation of the unfolded protein response in collagen IV nephropathies. J Am Soc Nephrol. 2014;25:260‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meyer‐Schwesinger C. The ubiquitin‐proteasome system in kidney physiology and disease. Nat Rev Nephrol. 2019;15:393‐411. [DOI] [PubMed] [Google Scholar]
- 32. Zhang HD, Huang JN, Liu YZ, Ren H, Xie JY, Chen N. Endoplasmic reticulum stress and proteasome pathway involvement in human podocyte injury with a truncated COL4A3 mutation. Chin Med J. 2019;132:1823‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Table S1.1: List of 415 significantly dysregulated proteins quantified by discovery proteomics in the kidneys of Col4a3 knock out compared to control mice at 4 weeks. Information about accession number, protein name, identification confidence score, fold‐change and P‐value of each protein are available. Accession, gene symbol, confidence score for identification, ANOVA p values for each protein, maximum fold change and the highest and lowest mean conditions are given in the table along with the full protein names.
Table S1.2: List of significantly enriched Gene Ontology terms for Biological Process. Functional enrichment analysis was performed using the Cytoscape bioinformatics tool and its plugin ClueGO. GO terms, term p values, percentage and number of associated genes along with the gene symbols of associated genes are given in the table.
Table S1.3: List of significantly enriched Gene Ontology terms for Molecular Function. Functional enrichment analysis was performed using the Cytoscape bioinformatics tool and its plugin ClueGO. GO terms, term p values, percentage and number of associated genes along with the gene symbols of associated genes are given in the table.
Table S1.4: List of significantly enriched Gene Ontology terms for Cellular Component. Functional enrichment analysis was performed using the Cytoscape bioinformatics tool and its plugin ClueGO. GO terms, term p values, percentage and number of associated genes and genes along with the gene symbols of associated genes are given in the table.
Table S1.5: List of significantly enriched KEGG pathways. Pathway analysis was performed using the Cytoscape bioinformatics tool and its plugin ClueGO. GO terms, term p values, percentage and number of associated genes along with the gene symbols of associated genes are given in the table.
Table S1.6‐S1.22: List of proteins involved in the 17 out of the 20 top significantly dysregulated pathways. Accession, gene symbol, confidence score for identification, ANOVA p values for each protein, maximum fold change and the highest and lowest mean conditions are given in the table along with the full protein names
Data S2 Figure S1: Graphical illustration of PPAR pathway as given by DAVID bioinformatics resources. Differentially expressed proteins identified by discovery proteomics with P ≤ .05 were used for this analysis. Pathway enrichment analysis was carried out using the DAVID bioinformatics tool (DAVID 6.8). Statistical significance for KEGG pathways was established at P ≤ .05. Proteins involved in the pathway are indicated with red asterisks. Note that Hmgcs2/HMCS2 protein that was identified to be involved in the PPAR pathway by Cytoscape bioinformatics resources, was not identified in this analysis.