

Abstract
Purpose
In the era of precision medicine, genomic characterization of blind patients is critical. Here, we evaluate the effects of comprehensive genetic analysis on the etiologic diagnosis of potentially hereditary vision loss and its impact on clinical management.
Methods
We studied 100 non‐syndromic and syndromic Spanish patients with a clinical diagnosis of blindness caused by alterations on the retina, choroid, vitreous and/or optic nerve. We used a next‐generation sequencing (NGS) panel (OFTALMOgenics™), developed and validated within this study, including up to 362 genes previously associated with these conditions.
Results
We identified the genetic cause of blindness in 45% of patients (45/100). A total of 28.9% of genetically diagnosed cases (13/45) were syndromic and, of those, in 30.8% (4/13) extraophthalmic features had been overlooked and/or not related to visual impairment before genetic testing, including cases with Mainzer‐Saldino, Bardet‐Biedl, mucolipidosis and MLCRD syndromes. In two additional cases–syndromic blindness had been proposed before, but not specifically diagnosed, and one patient with Heimler syndrome had been misdiagnosed as an Usher case before testing. 33.3% of the genetically diagnosed patients (15/45) had causative variants in genes targeted by clinical trials exploring the curative potential of gene therapy approaches.
Conclusion
Comprehensive genomic testing provided clinically relevant insights in a large proportion of blind patients, identifying potential therapeutic opportunities or previously undiagnosed syndromes in 42.2% of the genetically diagnosed cases (19/45).
Keywords: genomic diagnostics, hereditary, inherited retinal dystrophies, next‐generation sequencing, panel sequencing, precision ophthalmology, vision loss
Introduction
The approval of Luxturna®, the first gene therapy for an inherited condition to reach the clinic in the USA and Europe, has made reality the promise of a cure for genetic blindness (Russell et al. 2017). Although only patients with visual impairment caused by a defective RPE65 gene can benefit from this therapy, advanced clinical trials involving several forms of blindness caused by mutations in other genes are under way. In them, patient’s eligibility depends on the fact that his/her disease‐causing gene matches the target of the particular therapy. Thus, by holding the key to therapeutic intervention, molecular diagnostics of genetic blindness has reached the top of clinical applicability.
Inherited retinal dystrophies (IRDs), characterized by the degeneration of photoreceptor and retinal pigment epithelial cells, are the main cause of genetic blindness. With a worldwide prevalence of about 1/4000, retinitis pigmentosa (RP) is the commonest IRD (Verbakel et al. 2018). Besides the handicapping effects of visual impairment, between 20% and 30% of patients with RP have an associated non‐ocular condition, ranging from mild morphologic changes to life‐threatening pathologies. With more than 250 genes described as responsible for syndromic and/or non‐syndromic forms of the disease, the genetic heterogeneity of IRDs is overwhelming. Phenotypic heterogeneity is also characteristic of many IRD genes (Costa et al. 2017). As a result, the clinical testing approaches used for IRD diagnosis (electroretinography, optical coherence tomography, funduscopic examination, fundus autofluorescence, dark adaptation or visual field testing) are not usually able to predict the causative gene in a given patient (Hafler 2017).
Although the features mentioned above have historically hampered the clinical genetics of IRD, molecular diagnostics has demonstrated its utility for multiple aspects of patient management long before the availability of gene therapies. Thus, in paediatric patients, genetic diagnosis may dictate visual prognosis and guide educational and/or supportive plans for the proband (Jauregui et al. 2018). Correct IRD molecular diagnosis can also uncover associations with syndromes that manifest with disabling systemic disease, leading to early interventions that facilitate improved outcomes (Werdich et al. 2014). This is especially relevant, because many syndromic patients present initially to the ophthalmologist long before they are seen by the paediatrician with systemic symptoms (Sadagopan 2017).
Therefore, since 2012, the American Academy of Ophthalmology Task Force recommends genetic testing when clinical findings suggest that a known Mendelian disorder may affect the patient (Stone et al. 2012). However, comprehensive genetic testing of IRDs is not feasible by traditional Sanger sequencing techniques, as these are not suited for analysing hundreds of genes per patient. With the advent of next‐generation sequencing (NGS), extensive IRD molecular diagnosis has become possible, even for patients whose clinical diagnosis and inheritance pattern are uncertain after ophthalmologic evaluation (Lee & Garg 2015).
Here we: (1) present the development and validation of an NGS‐based panel for the comprehensive genomic diagnosis of blind patients with IRDs and/or optical nerve disorders; (2) evaluate its clinical performance and diagnostic yield in a series of 100 Spanish patients; and (3) illustrate, with real‐life data, how the availability of a molecular diagnosis frequently reveals unnoticed syndromes/phenotypic expansions and opens the door to personalized therapeutic opportunities.
Material
Purpose of test
The performed test (OFTALMOgenics™) was aimed at detecting the molecular aetiology of individual clinical diagnoses of syndromic/non‐syndromic IRDs and/or optic nerve disorders.
Panel content design: criteria for inclusion of specific genes
Genes associated with blindness caused by alterations in the vitreous, choroid, retina and optic nerve, including both syndromic and non‐syndromic forms, were considered.
First, the Human Gene Mutation Database (HGMD) was queried using a list of phenotypes potentially related to ophthalmologic defects (Table S1) to generate a preliminary gene list. This was manually curated to identify genes fulfilling the following criteria: (I) gene defects cause alterations in any of the four ocular structures previously mentioned; (II) published evidence supports the gene‐phenotype association in at least two independent families; and (III) at least one publication demonstrates convincing cosegregation of phenotype with gene variants. Then, a tiered classification system was devised. Genes with strong/moderate association with blindness (criteria I, II and III described above) formed tier 1, while genes with weak/preliminary association (criterion I, but not criteria II and/or III) were grouped in tier 2. The panel evolved with revision of newly published literature, yielding versions v1, v2, v3 and v4 (Tables S2–S4 and Table 1, respectively). v1 and v2 were used in the research and development (R&D) phase of the study, while v3 was used both during the R&D phase and as the first version of the clinical panel. v4 is the current optimized clinical version. Each patient was tested with only one of the four different versions: 20 cases were analysed with v1, 19 with v2, 55 with v3 (16 during the R&D phase and 39 in the clinical phase) and 6 with v4 (Table S5).
Table 1.
Tier 1 and tier 2 genes included in the OFTALMOgenics™ panel (v4).
Sample types
Four millilitre of peripheral blood in conventional EDTA‐tubes, 1.5 ml of saliva in Danagen saliva collection containers (Danagen‐Bioted, S.L., Badalona, Spain) or ≥200 ng of germline genomic DNA (quantitated by a fluorimetric method) were required per patient.
Library preparation, target enrichment and sequencing
Library preparation was carried out as previously described for the OTOgenics NGS platform (Cabanillas et al. 2018), except for library capture, that was performed with the OFTALMOgenics™ probes. The OFTALMOgenics™ NGS pipeline targeted the coding exons and intron–exon junctions of 278 genes (v1) (Table S2), 290 genes (v2) (Table S3), 297 genes (v3) (Table S4) or 362 genes (v4) (Table 1).
Bioinformatics for variant identification and annotation
Next‐generation sequencing results were processed using the bioinformatics software Genome One Core (DREAMgenics, Oviedo, Spain), certified with CE/IVD‐marking. The pipeline has been adapted from those previously described as part of the ONCOgenics and OTOgenics NGS platforms (Cabanillas et al. 2017; Cabanillas et al. 2018). The workflow of bioinformatics analysis that includes the FASTQ read generation, alignment, duplicate removal, variant identification, filtering and annotation has been previously described (Cabanillas et al. 2018).
Copy‐number variants (CNVs) were systematically assessed as described in Cabanillas et al. (2018). Detection was performed with an adapted version of the exome2cnv algorithm, incorporating a combination of read depth and allelic imbalance computations. The algorithm employs a background of pooled samples processed using the same capturing protocol and sequencing technology (Valdes‐Mas et al. 2012; Cabanillas et al. 2017). To improve sensitivity for large homozygous deletions, genomic regions with no sequencing coverage in an individual sample, but showing proper coverages in the remaining samples, were identified (i.e. homoz. BBS4 exon 1 deletion).
Estimation of analytical sensitivity, specificity and accuracy
The methodology for calculation of analytical sensitivity and specificity for SNVs and Indels has been described (Cabanillas et al. 2018). Accuracy was estimated based on the same dataset and it was expressed as the average ± standard deviation of the absolute value of the difference between the expected and the observed allelic frequencies of each variant.
Multiplex Ligation‐dependent Probe Amplification (MLPA)
Multiplex Ligation‐dependent Probe Amplification analysis was performed using MRC‐Holland (Amsterdam, The Netherlands) probe sets targeting five genes EYS (cat. # P328), PRPF31, RHO and RPE65 (cat. # P235) and USH2A (cat. #’s P361 and P362) on 31 patients without causative variants, following the manufacturer’s instructions.
Variant interpretation, classification and diagnostic yield
Clinical classification of variants from v1 and v2 cases was performed as described (Cabanillas et al. 2017). For v3 and v4 cases, variants that could potentially explain the vision loss phenotype of the probands, based on zygosity of the variant, presence of additional variants and mode of inheritance, were further considered. After that, variants were clinically classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines as described (Richards et al. 2015; Cabanillas et al. 2018). Diagnostic yield was defined as the percentage of tested patients with pathogenic/likely pathogenic variants capable of explaining their IRD phenotype.
Variant validation
Pathogenic/likely pathogenic SNVs and indels were validated by PCR + Sanger sequencing. For validation and identification of the breakpoints flanking the BBS4 exon 1 deletion multiple PCR reactions were performed. Primers used in validation PCRs are described in Table S6.
Patient population
Between September 2014 and May 2019, 100 consecutive patients (47 male, 53 female) with syndromic/non‐syndromic IRDs were selected after excluding non‐genetic causes. Consent was obtained from patients or their parents. The study was approved by the Comité de Ética de la Investigación del Principado de Asturias (research project #74/14).
Results
Performance of targeted NGS
Mean coverage of tier 1 genes was 453x for v1, 383x for v2, 347x for the 16 R&D phase cases performed with v3, 910x for the 39 clinical cases performed with v3 and 1056x for v4. 99.08%, 99.58%, 98.39%, 99.86% and 99.74% of their target bases were covered by ≥20 reads, respectively. The minimum, average and maximum coverage (average read depth of all target bases of the gene) and callabilities (% of the target bases of the gene with minimum read depths of 10, 20, 50 and 100 reads per each target base of the gene) for every tier 1 and tier 2 gene on samples analysed with OFTALMOgenics™ v4 is shown in Table S7. In clinical cases, regions from tier 1 genes with less than 100% coverage ≥20 reads (DP20) and specific positions within those regions affected by such limitation were included in each individual patient’s report.
Analytical sensitivity, specificity and accuracy
A genotyped mixture of 10 lymphoblastoid cell lines was evaluated to determine the analytical sensitivities, specificities and accuracies of the clinical versions of the panel (v3 and v4). For v3, 1277/1286 variants with expected allelic frequency ≥0.1 (1208/1216 SNVs and 69/70 indels) were detected, what gives a sensitivity >0.993 (>99.3%). The v3 version of the platform did not call any SNV or indel in 936684 of the 937048 positions of the probe design region known to not be affected by these type of variants, yielding a specificity of 0.9996. (936684/937048; >99.95%). The accuracy of v3 for determining the allelic frequencies of SNVs and indels with expected allelic frequencies ≥0.1 was 0.03 ± 0.06 (Table S8). The sensitivity, specificity and accuracy of v4, calculated as described above for v3, were 0.9917, 0.9995 and 0.033 ± 0.055, respectively (Table S9).
Orthogonal validation of sequencing results
All variants considered responsible for the IRDs phenotypes of the probands (Table 2) were successfully validated by approaches alternative to NGS. These included 48 instances of SNVs or indels (validated by PCR and Sanger sequencing) and 1 CNV: 1 homozygous deletion of BBS4 exon 1 (validated by breakpoint‐specific PCR).
Table 2.
Clinical and genetic characteristics of cases with causative mutations.
| Case ID | Pretest phenotype | Previous genetic study | Pretest inheritance pattern | Age at diagnosis | Gene | Allele variant | Variant zygosity | ACMG classification |
Clasificación HGMD/ClinVar |
Fulfilled ACMG pathogenicity criteria | Gene‐associated phenotype | Gene‐associated phenotype inheritance pattern | Pretest syndrome, hidden syndrome or non‐syndromic |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OFTALMO.001 | Retinitis pigmentosa | Not reported | AR † | Old age (61 years) | DHDDS | c.124A>G, p.(Lys42Glu) | Hom. | P | DM/P | PM3_VS, PP1_S, PM2, PS3_P | Retinitis pigmentosa type 59 | AR | Non‐syndromic |
| OFTALMO.002 | Retinitis pigmentosa | Not reported | AD/ AR | Youth (26 years) | ABCA4 | c.868C>T, p.(Arg290Trp) | Hom. | LP | DM/Absent | PM3_S, PM2 |
Stargardt disease type 1 Fundus flavimaculatus Retinitis pigmentosa type 19 Cone‐rod dystrophy type 3 |
AR | Non‐syndromic |
| OFTALMO.003 | Retinitis pigmentosa | Not reported | AD/ AR | Youth (26 years) | EYS | c.5928‐2A>G | Hom. | P | DM?/P | PVS1, PM2, PM3, PP1 | Retinitis pigmentosa type 25 | AR | Non‐syndromic |
| OFTALMO.004 | Retinitis pigmentosa | Not reported | AD/ AR | Childhood (9 years) | RP1 | c.1625C>G, p.(Ser542*) | Hom. | LP | DM/Absent | PM2, PM3_VS | Retinitis pigmentosa type 1 | AR | Non‐syndromic |
| OFTALMO.005 | Retinitis pigmentosa | Not reported | AR † | Youth (24 years) | CNGB1 | c.2957A>T, p.(Asn986Ile) | Hom. | P | DM/P | PM1, PM2, PM3_VS, PP1, BS1_P | Retinitis pigmentosa type 45 | AR | Non‐syndromic |
| OFTALMO.006 | Retinitis pigmentosa | Not reported | AR † | Youth (15 years) | IFT140 | c.874G>A, p.(Val292Met) | Hom. | P | DM/P | PS3_P, PM2, PM3_VS |
Retinitis pigmentosa type 80 Mainzer–Saldino syndrome Jeune syndrome Leber congenital amaurosis |
AR | Non‐syndromic |
| OFTALMO.007 | Usher syndrome | Not reported | AR † | Youth (16 years) | USH2A | c.9304C>T, p.(Gln3102*) | Hom. | P | Absent/P | PVS1, PM2, PM3_P | Usher syndrome type 2A | AR | Pretest syndrome |
| OFTALMO.008 | Retinitis pigmentosa | Not reported | XD/ XR | Youth (28 years) | CHM | c.1514delT, p.(Leu505Trpfs*4) | Hem. | LP | Absent/Absent | PVS1, PM2 | Choroideremia | XD | Non‐syndromic |
| OFTALMO.009 | Retinitis pigmentosa | Not reported | AR | Youth (21 years) | IFT140 | c.874G>A, p.(Val292Met) | Hom. | P | DM/P | PS3_P, PM2, PM3_VS |
Retinitis pigmentosa type 80 Mainzer–Saldino syndrome Jeune syndrome Leber congenital amaurosis |
AR | Hidden syndrome |
| OFTALMO.010 | Retinitis pigmentosa | Not reported | AR | Youth (18 years) | PROM1 | c.1414delC, p.(Arg472Glufs*18) | Hom. | P | Absent/Absent | PVS1, PM2, PM3_P | Retinitis pigmentosa type 41 | AR | Non‐syndromic |
| OFTALMO.011 | Retinitis pigmentosa | Yes | AD/ AR | Adulthood (44 years) | FAM161A | c.1355_1356delCA, p.(Thr452Serfs*3) | Hom. | P | DM/P | PVS1, PM2, PM3_VS, PP1_S | Retinitis pigmentosa type 28 | AR | Non‐syndromic |
| OFTALMO.012 | Retinitis pigmentosa | Not reported | AD † | Adulthood (31 years) | RHO | c.173C>G, p.(Thr58Arg) | Hom. | LP | DM/P | PM2, PP1_S, PP3 | Retinitis pigmentosa type 4 | AD | Non‐syndromic |
| OFTALMO.013 | Retinitis pigmentosa | Not reported | AR † | Adulthood (37 years) | CNGB1 | c.2957A>T, p.(Asn986Ile) | Hom. | P | DM/P | PM1, PM2, PM3_VS, PP1, BS1_P | Retinitis pigmentosa type 45 | AR | Non‐syndromic |
| OFTALMO.014 | Retinitis pigmentosa | Not reported | AR † | Youth (18 years) | CNGB1 | c.2957A>T, p.(Asn986Ile) | Hom. | P | DM/P | PM1, PM2, PM3_VS, PP1, BS1_P | Retinitis pigmentosa type 45 | AR | Non‐syndromic |
| OFTALMO.015 | Retinitis pigmentosa | Not reported | AD/AR | Adulthood (40 years) | BBS1 | c.1169T>G, p.(Met390Arg) | Hom. | P | DM/P | PS3, PM3_VS, PP1, BS1_P | Bardet–Biedl syndrome type 1 | AR | Hidden syndrome |
| OFTALMO.016 | Retinitis pigmentosa | Not reported | AD/AR | Childhood (8 years) | MCOLN1 | c.878‐1G>A | Hom. | P | Absent/Absent | PVS1, PM2, PM3_P | Mucolipidosis type IV | AR | Hidden syndrome |
| OFTALMO.017 | Usher syndrome | Yes | AR † | Childhood (10 years) | MYO7A | c.6025delG, p.(Ala2009Profs*32) | Hom. | P | DM/P | PVS1, PM2, PM3_VS, PP1_S | Usher syndrome, type 1B | AR | Pretest syndrome |
| OFTALMO.018 | Usher syndrome | Not reported | AR | Adulthood (31 years) | MYO7A | c.397C>T, p.(His133Tyr) | Hom. | LP | DM?/LP | PM1, PM2, PM3, PM5, PP3 | Usher syndrome, type 1B | AR | Pretest syndrome |
| OFTALMO.019 | Stargardt disease | Not reported | AR | Youth (16 years) | ABCA4 | c.6449G>A, p.(Cys2150Tyr) | Hom. | P | DM/LP | PM1, PM2, PM3_VS, PM5, PP3 |
Stargardt disease type 1 Fundus flavimaculatus Retinitis pigmentosa type 19 Cone‐rod dystrophy type 3 |
AR | Non‐syndromic |
| OFTALMO.020 | Retinitis pigmentosa | Not reported | XD/ XR | Youth (15 years) | CHM | c.340G>T, p.(Glu114*) | Hem. | LP | Absent/Absent | PVS1, PM2 | Choroideremia | XD | Non‐syndromic |
| OFTALMO.021 | Maculopathy | Not reported | AR † | Youth (18 years) | ABCA4 |
c.5044_5058delGTTGCCATCTGCGTG, p.(Val1682_Val1686del) c.3943C>T, p.(Gln1315*) |
Het. Het. |
P P |
DM/np DM/Absent |
PM2, PM3_VS, PM4 PVS1, PM2, PM3_S |
Stargardt disease type 1 Fundus flavimaculatus Retinitis pigmentosa type 19 |
AR | Non‐syndromic |
| OFTALMO.022 | Retinitis pigmentosa | Not reported | AR | Adulthood (27 years) | OAT | c.416T>G, p.(Met139Arg) | Hom. | LP | Absent/Absent | PM1, PM2, PM3_P, PP3 | Gyrate atrophy of choroid and retina with or without ornithinemia | AR | Non‐syndromic |
| OFTALMO.024 | Retinitis pigmentosa | Yes | AD/ AR | Old age (57 years) | PRPH2 | c.660_665del, p.(Pro221_Cys222del) | Het. | LP | Absent/Absent | PM1, PM2, PM4 |
Macular dystrophy Retinitis pigmentosa type 7 Retinitis punctata albescens Choroidal dystrophy, central areolar type 2 |
AD | Non‐syndromic |
| OFTALMO.029 | Retinitis pigmentosa | Not reported | XR † | NA | RPGR (ORF15) | c.2284delG, p.(Glu762Lysfs*53) | Hem. | LP | Absent/Absent | PVS1, PM2 | Retinitis pigmentosa type 3 Cone‐rod dystrophy type 1 | XR | Non‐syndromic |
| OFTALMO.031 | Retinitis pigmentosa | Not reported | AD/ AR | Youth (14 years) | RP1 |
c.1625C>G, p.(Ser542*) c.227T>C, p.(Leu76Pro) |
Het. Het. |
LP LP |
DM/Absent DM/Absent |
PM2, PM3_S PM1, PM2, PM3, PP3 | Retinitis pigmentosa type 1 | AR | Non‐syndromic |
| OFTALMO.033 | Retinitis pigmentosa | Not reported | AD/ AR | Adulthood (39 years) | CERKL | c.356G>A, p.(Gly119Asp) | Hom. | LP | DM/Absent | PM1, PM2, PM3_S, PP1 | Retinitis pigmentosa type 26 Cone‐rod dystrophy | AR | Non‐syndromic |
| OFTALMO.041 |
Malformative syndrome, suspicion of Bardet– Biedl syndrome. Retinitis pigmentosa, mild mental retardation, patent foramen ovale, polydactyly in hands and feet, genital hypoplasia, renal dysplasia, obesity, hypertension. |
Yes | AR | Childhood (10 years) | BBS4 | c.‐2274_23 + 930del | Hom. | LP | Absent/Absent | PVS1, PM2, PM3_P | Bardet–Biedl syndrome | AR | Pretest syndrome |
| OFTALMO.046 | Retinitis pigmentosa | Not reported | AR † | Youth (18 years) | ARL6 |
c.36_39dupCCTG, p.(Lys14Profs*15) c.499G>A, p.(Gly167Arg) |
Het. Het. | P LP |
Absent/Absent Absent/Absent |
PVS1, PM2, PM3_S, PP1 PM1, PM2, PM3_S, PP1, PP3 | Retinitis pigmentosa type 55 | AR | Non‐syndromic |
| OFTALMO.047 | Retinitis pigmentosa | Not reported | AD/ AR | Childhood (4 years) | IMPDH1 | c.933C>G, p.(Asp311Glu) | Het. | LP | Absent/Absent | PS2_M, PM1, PM2, PP3 | Retinitis pigmentosa type 10 | AD | Non‐syndromic |
| OFTALMO.052 | Retinitis pigmentosa | Not reported | AD/ AR | Youth (29 years) | USH2A |
c.9304C>T, p.(Gln3102*) c.2276G>T, p.(Cys759Phe) |
Het. Het. | P LP |
DM/P Absent/P |
PVS1, PM2, PM3_P PM1, PM3_S, PP1, PP3, BS1_P | Retinitis pigmentosa type 39 | AR | Non‐syndromic |
| OFTALMO.054 | Usher syndrome | Not reported | AR | Adulthood (49 years) | PEX1 |
c.3077T>C, p.(Leu1026Pro) c.1548delT, p.(Leu517Cysfs*2) |
Het. Het. |
LP LP |
DM/Absent Absent/Absent | PM1, PM2, PP1, PP3 PVS1, PM2 | Heimler syndrome | AR | Pretest syndrome |
| OFTALMO.060 | Nystagmus. Complete atrophy of the optic nerve. Retinitis pigmentosa. Macular atrophy. Mental retardation | Not reported | AD/ AR | Congenital (at birth) | KIF11 | c.2548‐2A>G | Het. | P | Absent/Absent | PVS1, PM2, PP3 |
MLCRD syndrome: Microcephaly with or without chorioretinopathy, lymphedema or mental retardation |
AD | Hidden syndrome |
| OFTALMO.063 | Macular atrophy with perimacular ‘flecks’ compatible with Stargardt's disease | Not reported | AD/ AR | Childhood | ABCA4 |
c.5819T>C, p.(Leu1940Pro) c.3386G>T, p.(p.Arg1129Leu) |
Het. Het. |
P P |
DM/P DM/LP | PM2, PM3_VS, PP1, PP3 PS4, PM1, PM2, PM3_S, PM5, PP3 | Stargardt disease type 1 Fundus flavimaculatus | AR | Non‐syndromic |
| OFTALMO.068 |
Retinitis pigmentosa. Atrophy of the optic nerve. Deafness, hypertension and |
Not reported | AD/ AR † | Childhood (8 years) | USH2A |
c.2276G>T, p.(Cys759Phe) c.12093delC, p.(Tyr4031*) |
Het. Het. |
LP LP |
DM/P DM/Absent | PM1, PM3_S, PP1, PP3, BS1_P PVS1, PM2 | Usher syndrome type 2A | AR | Pretest syndrome |
| OFTALMO.072 |
Retinitis pigmentosa. Atrophy of the optic nerve. Deafness of the right ear |
Not reported | AR † | Adulthood (40 years) | ABCA4 | c.5882G>A, p.(Gly1961Glu) | Hom. | P | DM/P | PS3_P, PS4, PM3_VS, PP3, BA1 |
Stargardt disease type 1 Fundus flavimaculatus Retinitis pigmentosa type 19 Cone‐rod dystrophy type 3 |
AR | Pretest syndrome |
| OFTALMO.076 | Macular dystrophy in pattern with flecks | Not reported | AR † | Old age (67 years) | ABCA4 |
c.3056C>T, p.(Thr1019Met) c.6718A>G, p.(Thr2240Ala) |
Het. Het. |
LP LP |
DM/P DM?/Absent | PM1, PM2, PM3, PP3 PM2, PM3_S, PP3 | Stargardt disease type 1 Fundus flavimaculatus | AR | Non‐syndromic |
| OFTALMO.077 |
Retinitis pigmentosa with macular edema. Mean myopia. Bilateral moderate non‐progressive hearing loss |
Not reported | AR | Youth (15 years) | USH2A |
c.8917_8918delCT, p.(Leu2973Lysfs*79) c.9119G>A (p.Trp3040*) |
Het. Het. |
P P |
DM/Absent DM/Absent |
PVS1, PM2, PM3_S PVS1, PM2, PM3, PP1 | Usher syndrome type 2A | AR | Pretest syndrome |
| OFTALMO.081 | Retinitis pigmentosa. Tapetoretinian dystrophy | Not reported | AD/ AR | Old age (66 years) | PRPH2 | c.422delA, p.(Tyr141Serfs*12) | Het. | LP | Absent/Absent | PVS1, PM2 |
Macular dystrophy Retinitis pigmentosa type 7 Retinitis punctata albescens Choroidal dystrophy, central areolar type 2 |
AD | Non‐syndromic |
| OFTALMO.082 | Infectious choroiditis (25 years). Macular degeneration | Not reported | AR † | Old age (65 years) | PRPH2 | c.421T>C, p.(Tyr141His) | Het. | LP | DM/Absent | PM1, PM2, PM5, PP1_S, PP3 |
Macular dystrophy Retinitis pigmentosa type 7 Retinitis punctata albescens Choroidal dystrophy, central areolar type 2 |
AD | Non‐syndromic |
| OFTALMO.087 | Nystagmus and visual deficit | Yes | AR † | Congenital (3 months) | CEP290 |
c.384_387delTAGA, p.(Asp128Glufs*34) c.7341_7344dupACTT, p.(Ser2449Thrfs*8) |
Het. Het. |
P P |
DM/P Absent/Absent |
PVS1, PM3_S, BA1 PVS1, PM2, PM3 |
Leber congenital amaurosis type 10 Senior‐Loken syndrome type 6 Joubert syndrome type 5 |
AR | Non‐syndromic |
| OFTALMO.093 | Bilateral atrophy, diabetes and hypertension | Not reported | AD † | Adulthood (40 years) | PRPH2 | c.537G>A, p.(Trp179*) | Het. | LP | Absent/Absent | PVS1, PM2 |
Macular dystrophy Retinitis pigmentosa type 7 Retinitis punctata albescens Choroidal dystrophy, central areolar type 2 |
AD | Non‐syndromic |
| OFTALMO.094 | Bull's eye maculopathy. Possible macular dystrophy | Not reported | AD/ AR † | Youth (12 years) | ABCA4 |
c.982G>T, p.(Glu328*) c.5882G>A, p.(Gly1961Glu) |
Het. Het. |
P P |
DM/Absent DM/P | PVS1, PM2, PM3 PS3_P, PS4, PM3_VS, PP3, BA1 | Stargardt disease type 1 Fundus flavimaculatus | AR | Non‐syndromic |
| OFTALMO.095 | Macular atrophy with perimacular flecks | Not reported | AD/ AR | Childhood (15 years) | ABCA4 |
c.3386G>T, p.(Arg1129Leu) c.6006‐2A>G |
Het. Het. | P LP |
DM/LP Absent/Absent |
PS4, PM1, PM2, PM3_S, PM5, PP3 PVS1_S, PM2 | Stargardt disease type 1 Fundus flavimaculatus | AR | Non‐syndromic |
| OFTALMO.097 | Retinitis pigmentosa, diabetic retinopathy, deafness, epilepsy | Not reported | AD/ AR | Childhood (8 years) | PRPS1 | c.292G>A, p.(Asp98Asn) | Het. | LP | Absent/Absent | PM1, PM2, PM6_P, PP3 | Syndromic retinitis pigmentosa (ocular asymmetry, hearing loss and neurological disorders) | XD | Pretest syndrome |
| OFTALMO.098 | Optical and macular atrophy | Not reported | AD † | Youth (15 years) | OPA1 | c.1847 + 1G>T | Het. | LP | DM/LB | PVS1, PM2 | Optic atrophy 1 Optic atrophy plus syndrome | AD | Non‐syndromic |
AD = autosomal dominant; AR = autosomal recessive; Hem = Hemizygous; Het = Heterozygous; Hom = Homozygous; LB = Likely benign; LP = Likely pathogenic; NA = not available; np = not provided; P = Pathogenic; XD = X‐linked dominant.
Family history of eye disease.
Performance at challenging regions: RPGR ORF15
Certain regions in the genome represent a major challenge for short‐reading NGS technologies. Of the genes included in the OFTALMOgenics designs, RPGR poses the highest difficulties. RPGR encodes several isoforms, but only isoform C (NM_001034853), also known as RPGR ORF15, is highly expressed in the retina and, consequently, involved in the pathogenesis of retinitis pigmentosa (Vervoort et al. 2000). Several regions of RPGR ORF15 bear low sequence complexity, causing a decrease in base quality at the end of NGS reads. This, combined with the highly repetitive nature of the RPGR ORF15 reference sequence, leads to low mapping quality, limiting NGS‐based RPGR ORF15 analysis. As a result, many laboratories analyse RPGR ORF15 using traditional Sanger sequencing (Chiang et al. 2015). Being aware of these difficulties, and as we observed that 92.7% of R&D cases (51/55) showed regions with less than 100% of their bases covered by ≥20 reads, we increased sequencing coverage and dedicated a larger number of capture probes to this conflictive region in the design of the panel, reaching 100% of the RPGR target region covered by 20 or more reads in v4 cases (Table S10). Of note, one of the cases in our series had a causative variant detected by the NGS pipeline in RPGR affecting ORF15 (NM_001034853.1: c.2284delG; p.(Glu762Lysfs*53); corresponding with c.1905 + 379delG according to the main NM_000328.2 RPGR RefSeq), demonstrating the ability of the test to identify these challenging mutations.
Analysis of causative variants and diagnostic yield
Of 100 cases with low vision caused by alterations in the vitreous, choroid, retina and/or optic nerve, a genetic cause for their vision loss phenotype was found in 45 (45%) after identifying 49 different pathogenic/likely pathogenic variants in 26 genes (Fig. 1, Table 2 and Table S6). Causative mutations were found in 31.2% (5/16) of cases in which the existence of a previous genetic study, with negative results, had been reported before OFTALMOgenics testing (Table 2 and Table S11). No causative mutation could be identified in 55 patients (55%) (Table S11). These are likely to be a pool of patients without a genetic aetiology, with mutations in genes not included in the panel or with mutations in genes included in the panel but not detectable by the methodology used. The commonest causative genes in our population were ABCA4 (8 patients), PRPH2 (4 patients) and USH2A (4 patients). 1 pathogenic variant in IMPDH1 was de novo (after confirmation of maternity and paternity). The post‐test inheritance patterns of the molecularly diagnosed patients were autosomal recessive (AR) in 75.5% (34/45) of cases, autosomal dominant (AD) in 17.8% (8/45), and X‐linked in 6.5% (3/46) (Fig. 2 and Table 2).
Fig. 1.
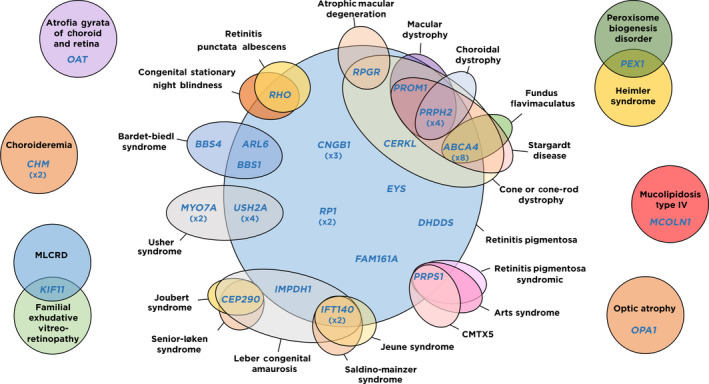
Genes considered responsible for hereditary visual impairment in our cohort and their associated phenotypes. Ellipses represent phenotypes (black characters) involving vision loss caused by alterations in the retina, vitreous, choroid and/or optic nerve and consistently associated to defects in the genes (blue characters) they contain. For genes with causative alterations affecting more than one patient in our cohort, the number of affected patients is shown in brackets below. Phenotype acronyms: CMTX5: Charcot‐Marie‐Tooth disease, X‐linked recessive, 5; MLCRD: microcephaly, lymphoedema, chorioretinal displasia syndrome.
Fig. 2.
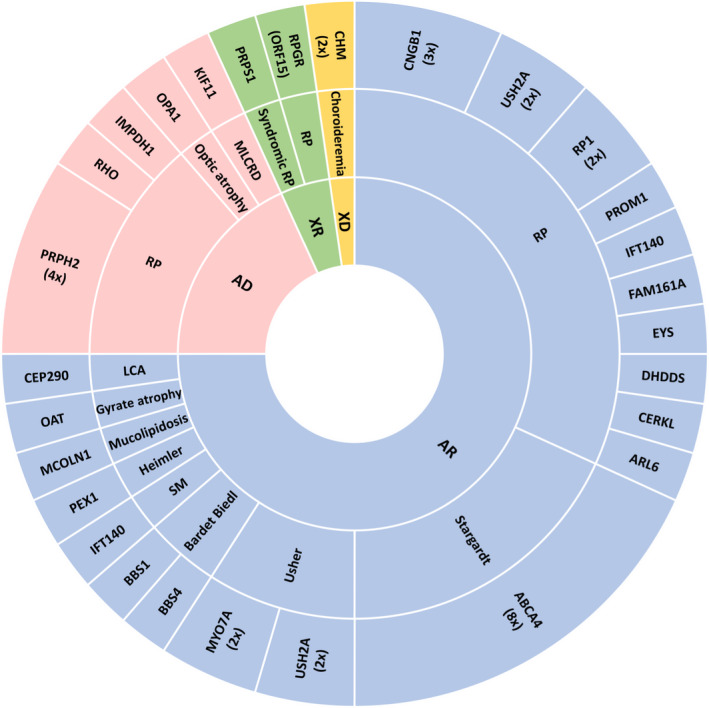
Modes of inheritance of the genetic conditions considered responsible for hereditary vision loss in our cohort. From outer to inner rings, affected genes, associated phenotypes and modes of inheritance are shown. The area of each sector is proportional to the number of cases represented within it. For genes with causative alterations affecting more than one patient, the number of affected patients is shown in brackets. AD: autosomal dominant; AR: autosomal recessive; X‐linked recessive, 5; LCA: Leber congenital amaurosis; MLCRD: microcephaly, lymphoedema, chorioretinal displasia syndrome; RP: retinitis pigmentosa; SM: Saldino–Mainzer syndrome; XD: X‐linked dominant; XR: X‐linked recessive.
CNV analysis identified a causative variant in 1 of the 45 molecularly diagnosed patients (2.2%): a homozygous BBS4 exon 1 deletion in a patient with suspected Bardet–Biedl syndrome (retinitis pigmentosa, mild mental retardation, polydactyly in hands and feet, genital hypoplasia, renal dysplasia, obesity and hypertension). As no heterozygous CNVs were detected by our NGS platform, we analysed by MLPA a group of five genes included in our panel and previously described to be affected by pathogenic deletions/duplications (including USH2A, EYS, PRPF31, RHO and RPE65) on a subset of 31 cases without a genetic diagnosis from our series. This analysis identified no homozygous or heterozygous alterations (Table S13).
Two of the 49 causative variants (ABCA4 c.5882G>A and CEP290 c.384_387delTAGA) are present in one control population each with allele frequencies above 0.005, the threshold we used for the application of the BA1 (benign‐standalone) ACMG criterion to recessive variants: 0.005054 (613/121302) in ExAC for ABCA4 c.5882G>A and 0.01074 (122/11360) in GO‐ESP for CEP290 c.384_387delTAGA. Their respective GnomAD frequencies are 0.00350 (110/31396) and 0.00003 (1/31362), below the BA1 threshold and compatible with disease incidence in the general population. Moreover, the 0.01074 (122/11360) frequency reported for CEP290 c.384_387delTAGA in GO‐ESP does not match those of the same variant in other databases mentioned by dbSNP, all with higher numbers of alleles analysed [0.000056 (14/249068) in GnomAD_exome, 0.000104 (13/125568) in TOPMED and 0.00074 (58/78700) in PAGE_STUDY] and is suspiciously high for a truncating variant in a gene in which inactivating mutations have consistently been shown to be pathogenic. Taking these data altogether and considering that, otherwise, both of these variants meet ACMG criteria for pathogenicity, we propose that they should be part of the exclusion list for application of the BA1 criterion, to avoid false negatives caused by automatic filtering based on allele frequency.
In total, 1081 variants in tier 1 genes and 119 variants in tier 2 genes were identified and evaluated in the full cohort of 100 patients. 476/1081 (44%) and 81/119 (68.1%) of the identified tier 1 and tier 2 variants, respectively, were absent from the HGMD professional and ClinVar databases (hereafter ‘clinical databases’). No tier 2 variant was considered responsible for the IRD phenotype. Eighteen of the variants absent from the clinical databases (all from tier 1) were classified as pathogenic or likely pathogenic and responsible for the IRD. Globally, those 18 variants were considered responsible for the IRD phenotype in 18 cases. As a result, 40% of the genetically diagnosed cases (18/45) were explained by variants not described in the clinical databases (Table 2). In addition, 31 non‐redundant variants were identified that explained positive cases: 30 of these were classified as pathogenic (DM) by HGMD and/ or pathogenic/ likely pathogenic by ClinVar for ophthalmological phenotypes, and 1 (ABCA4 c.6718A>G, p.(Thr2240Ala)) absent in ClinVar and ‘questionable’ (DM?) in HGMD for a phenotype of retinal dystrophy (Table 2). We found 363 variants of uncertain significance (VUS) that appeared a total of 412 instances considering all patients (Table S12). These results highlight the importance of manual interpretation and curation for the clinical classification of variants.
Increased clinical sensitivity and specificity by jointly analysing non‐syndromic and syndromic genes
For 7 of the 45 patients in which a genetic diagnosis was obtained, the phenotypes associated with the genes affected by causative variants either did not match the pretest clinical features/diagnoses or corresponded to syndromes not previously diagnosed in the proband’s personal and familial medical records (hereafter ‘hidden syndromes’; Table 2):
Two patients, male and female, both with a pretest diagnosis of non‐syndromic RP, shared the same homozygous mutation in IFT140 (c.874G>A, p.(Val292Met)). The IFT140 gene had been associated with recessive syndromic (Mainzer–Saldino and Jeune syndromes) (Perrault et al. 2012; Schmidts et al. 2013) and non‐syndromic phenotypes (RP80 and Leber Congenital Amaurosis) (Xu et al. 2015). Post‐test clinical assessment of the male patient revealed renal hypertension and two collapsed vertebrae, both sings compatible with Mainzer–Saldino syndrome (RP, renal and skeletal dysplasia). Thus, a hidden syndrome explained the phenotype. The clinical diagnosis of the female remained as non‐syndromic RP post‐test. Interestingly, in our two cases, the same variant (IFT140 c.874G>A; p.(Val292Met)) was associated with a syndromic and a non‐syndromic phenotype.
A male patient with a pretest diagnosis of RP was affected by a homozygous pathogenic variant in BBS1 (c.1169T>G, p.(Met390Arg)). BBS1 alterations have been associated with recessive non‐syndromic (Estrada‐Cuzcano et al. 2012; Sharon & Banin 2015) and syndromic RP (Bardet‐Biedl syndrome ‐RP, diabetes‐linked obesity, polydactyly, kidney abnormalities and intellectual disability‐). Upon post‐test revaluation of the case, the patient was found to have pancreas divisum and unilateral postaxial polydactyly emerging from the fifth toe (right foot). However, no intellectual disability, nor renal or hormonal alterations were detected.
A female patient with a pretest diagnosis of RP had a novel canonical splicing mutation in MCOLN1 (c.878‐1G>A) in homozygosis. MCOLN1 inactivation has only been associated with type 4 mucolipidosis, characterized by neurologic and ophthalmologic abnormalities (Boudewyn & Walkley 2019). Post‐test clinical review of the case revealed that the patient had psychomotor delay, intellectual disability and a corneal lesion with microcystic epithelial oedema, suggestive of corneal dystrophy.
A male with nystagmus, macular atrophy, typical RP, retinal ischaemia, complete optic nerve atrophy and mental retardation that had not been clinically matched to any specific syndrome pretest, had a heterozygous pathogenic KIF11 variant (c.2548‐2A>G). This gene has been associated with exudative familial vitreoretinopathy and with microcephaly with or without chorioretinopathy, lymphedema or mental retardation (MLCRD), both dominant phenotypes (Robitaille et al. 2014). Post‐test revaluation of the case confirmed that the phenotype matched MLCRD syndrome.
A male patient with a clinical diagnosis of Usher syndrome was found to be affected by two likely pathogenic compound heterozygous variants affecting the PEX1 gene (c.1548delT, p.(Leu517Cysfs*2) and c.3077T>C, p.(Leu1026 Pro)). The PEX1 gene has been associated with peroxisome biogenesis disorders types 1A and 1B, as well as with Heimler syndrome, all with recessive inheritance (Ratbi et al. 2015). Heimler syndrome combines the hearing loss and retinal dystrophy phenotypes typical of Usher syndrome with enamel hypoplasia of the secondary dentition and nail abnormalities, which may be overlooked. Therefore, genetic assessment increased the specificity of the diagnosis.
Finally, a female patient with hearing loss, diabetes and RP without a specific syndromic diagnosis pretest was affected by a missense, likely pathogenic, heterozygous PRPS1 variant (c.292G>A, p.(Asp98Asn)). Pathogenic variants in this gene have been associated with three X‐linked conditions involving blindness: Charcot‐Marie‐Tooth disease type 5, Arts syndrome and syndromic retinitis pigmentosa (ocular asymmetry, hearing loss and neurological alterations). The latter has only been described in females with missense variants and shows a dominant mode of inheritance (Fiorentino et al. 2018).
Aside from these seven cases, another female patient with a pretest diagnosis of non‐syndromic RP and a blind sister had two compound heterozygous variants affecting ARL6 [p.(Lys14Profs*15), pathogenic and p.(Gly167Arg), likely pathogenic]. This gene has been associated both with non‐syndromic RP and with Bardet–Biedl syndrome. Clinical review of the family post‐test revealed that, while the patient had no extraophthalmic manifestations, her sister was born with an extra toe that was later removed by surgery. Although the sister has not been genetically analysed so far, this likely represents another case of a single genotype causing syndromic and non‐syndromic hereditary blindness, in this case within the same family.
In total, 28.9% of the genetically diagnosed patients (13/45) had syndromic visual impairment (Table 2). Of those, 30.8% (4/13) had a previously unrecognized (hidden) syndrome: Mainzer‐Saldino (1 patient), Bardet–Biedl type 1 (1 patient), mucolipidosis type IV (1 patient) and MLCRD (1 patient). Likewise, 23.1% (3/135) had different syndromic manifestations, but they did not have a specific clinically diagnosed syndrome before the test was performed: Usher type 2A (2 patients) and syndromic retinitis pigmentosa caused by mutations in the PRPS1 gene (1 patient). Additionally, one patient with Heimler syndrome according to the test results had previously been imprecisely diagnosed as an Usher case based on clinical features (2.2%; 1/45) (Table 2). In contrast, of the 55 patients without a genetic diagnosis, only 5 (9.1%) had potentially syndromic features (Table S11). These findings demonstrate that comprehensive genomic diagnosis not only increases diagnostic performance (sensitivity and specificity), but also provides information with high clinical relevance.
Genomic diagnosis identifies potential gene therapy options for a considerable proportion of IRD patients
Thirteen gene therapy clinical trials aimed at compensating genetic defects responsible for hereditary blindness of patients genetically diagnosed by our platform were open as of November 2019 or had been open for a period of time during this study (Table 3). Of the genes targeted by these clinical trials, ABCA4 is the one altered by variants responsible for blindness in most patients of our series (8 patients), followed by USH2A (2 patients), CHM (2 patients), MYO7A (2 patients) and RPGR (1 patient) (Table 3). In total, 33.3% of the genetically diagnosed patients (15/45) and 15% of the genetically evaluated patients (15/100) could have potentially benefited from therapeutic approaches with curative potential in this time window (Table 3).
Table 3.
Gene therapy clinical trials aimed at correcting deffects responsible for hereditary blindness of patients genetically diagnosed in this study. Trials currently open or open at some point between the start of the study and November 2019 are shown.
| OFTALMO ID | Date of report | Sex | Gene | Causative mutations | Zigosity | Clinical trial title | Web links to trial details | Phase |
|---|---|---|---|---|---|---|---|---|
| OFTALMO.002 | 26/02/2016 | F | ABCA4 | c.868C>T, p.(Arg290Trp) | Homozygous | A Phase I/IIa Study of SAR422459 in Patients With Stargardt's Macular Degeneration | https://www.clinicaltrialsregister.eu/ctr‐search/trial/2010‐023111‐34/IT/ | 1, 2a |
| OFTALMO.019 | 06/04/2016 | F | c.6449 G>A, p.(Cys2150Tyr) | Homozygous | ||||
| OFTALMO.021 | 21/04/2016 | M | c.5044_5058delGTTGCCATCTGCGTG, p.(Val1682_Val1686del)/ c.3943C>T, p.(Gln1315*) | Compound heterozygous | ||||
| OFTALMO.063 | 31/01/2017 | M | c.5819T>C, p.(Leu1940Pro)/ c.3386G>T, p.(Arg1129Leu) | Compound heterozygous | ||||
| OFTALMO.072 | 19/05/2017 | F | c.5882G>A, p.(Gly1961Glu) | Homozygous | ||||
| OFTALMO.076 | 24/08/2017 | M | c.3056C>T, p.(Thr1019Met)/ c.2828G>A, p.(Thr2240Ala) | Compound heterozygous | ||||
| OFTALMO.094 | 28/03/2019 | M | c.982G>T, p.(Glu328*)/ c.5882G>A, p.(Gly1961Glu) | Compound heterozygous | ||||
| OFTALMO.095 | 08/04/2019 | F | c.3386G>T, p.(Arg1129Leu)/ c.6006‐2A>G | Compound heterozygous | ||||
| Safety and Dose Escalation Study of AAV2‐hCHM in Subjects With CHM (Choroideremia) Gene Mutations | https://clinicaltrials.gov/ct2/show/NCT02341807 | 1, 2 | ||||||
| OFTALMO.008 | 07/03/2016 | M | c.1514delT, p.(Leu505Trpfs*4) | Long‐term Safety and Efficacy Follow‐up of AAV2‐REP1 for the Treatment of Choroideremia (SOLSTICE) | https://clinicaltrials.gov/ct2/show/NCT03584165 | ? | ||
| A Safety Study of Retinal Gene Therapy for Choroideremia (GEMINI) | https://clinicaltrials.gov/ct2/show/NCT03507686 | 2 | ||||||
| CHM | Hemizygous | Choroideremia Gene Therapy Clinical Trial | https://clinicaltrials.gov/ct2/show/NCT02553135 | 2 | ||||
| OFTALMO.020 | 20/04/2016 | M | c.340G>T, p.(Glu114*) | An Open Label Clinical Trial of Retinal Gene Therapy for Choroideremia | https://clinicaltrials.gov/ct2/show/NCT02077361 | 1, 2 | ||
| Efficacy and Safety of AAV2‐REP1 for the Treatment of Choroideremia (STAR) | https://clinicaltrials.gov/ct2/show/NCT03496012 | 3 | ||||||
| OFTALMO.017 | 31/03/2016 | M | MYO7A | c.6025delG, p.(Ala2009Profs*32) | Homozygous | Study of SAR421869 in Patients With Retinitis Pigmentosa Associated With Usher Syndrome Type 1B | https://clinicaltrials.gov/ct2/show/NCT01505062 | 1, 2a |
| OFTALMO.018 | 04/04/2016 | F | c.397C>T, p.(His133Tyr) | Homozygous | ||||
| Gene Therapy for X‐linked Retinitis Pigmentosa (XLRP) Retinitis Pigmentosa GTPase Regulator (RPGR) | https://clinicaltrials.gov/ct2/show/NCT03252847 | 1, 2 | ||||||
| RPGR | A Clinical Trial of Retinal Gene Therapy for X‐linked Retinitis Pigmentosa (XIRIUS) | https://clinicaltrials.gov/ct2/show/NCT03116113 | 1, 2, 3 | |||||
| OFTALMO.029 | 27/07/2016 | M | (ORF15) | c.2284delG, p.(Glu762Lysfs*53) | Hemizygous | Safety and Efficacy of rAAV2tYF‐GRK1‐RPGR in Subjects With X‐linked Retinitis Pigmentosa Caused by RPGR‐ | https://clinicaltrials.gov/ct2/show/NCT03316560 | 1, 2 |
| Gene Therapy Trial for People with Retinitis Pigmentosa (progressive reduction in vision) due to a gene defect on | https://globalclinicaltrialdata.com/trial/GCT022016‐003967‐21 | 1, 2 | ||||||
| OFTALMO.052 | 02/11/2016 | F | USH2A | c.9304C>T,p.(Gln3102*)/ c.2276G>T, p.(Cys759Phe) | Compound heterozygous | Study to Evaluate Safety and Tolerability of QR‐421a in Subjects With RP Due to Mutations in Exon 13 of the USH2A Gene (Stellar) | https://clinicaltrials.gov/ct2/show/NCT03780257 | 1, 2 |
| OFTALMO.068 | 18/05/2017 | F | c.2276G>T, p.(Cys759Phe)/ c.12093delC, p.(Tyr4031*) | Compound heterozygous |
Discussion
Out of the different outputs of this work, the most appealing one is probably the confirmation of the utility of comprehensive genomic diagnosis of blind patients for the identification of potential therapeutic options matching their genetic profile in a relatively large proportion of them. Nonetheless, the advantages of testing hundreds of blindness genes in parallel reach far beyond therapeutic guidance. Thus, in the diagnostic setting, because the complex genetic heterogeneity of retinal dystrophies makes very hard predicting the genetic alteration based on the clinical characterization of the patient only, and mutation of a single gene may cause a broad range of different clinical manifestations, comprehensive testing is particularly useful (Hafler 2017).
In fact, the ability to perform phenotype‐agnostic testing is providing a growing body of evidence in support of the concept of phenotypic expansion, by means of the identification of disease‐causing mutations in genes not previously related to the proband’s clinical phenotype (Bowne et al. 2011; Nishiguchi et al. 2014; Consugar et al. 2015). This approach can also tackle the diagnostic dilemmas appearing when phenotypic variability is observed within the same family, a common situation in inherited retinal dystrophies (Consugar et al. 2015). Moreover, as phenotypes evolve over time, comprehensive testing reduces the possibility that the causative gene is not evaluated because the proband's symptoms do not match the phenotype associated to it at the moment of testing. Finally, as visual impairment may be the presenting symptom of a systemic syndrome, an accurate molecular diagnosis may be critical for identifying a so far not revealed and wider health problem. Apart from its informative value with regard to prognosis, because some of these syndromes can cause significant morbidity, prompt identification and referral for systemic assessment and monitorization may secure early intervention and a better outcome (Werdich et al. 2014).
A total of 15.6% (7/45) of the patients for which a genetic diagnosis was obtained in our study corresponded to cases whose clinical diagnoses pretest did not match the phenotypes associated with the affected genes at the time of reporting the results to the requesting clinician, involving the following genes: BBS1, IFT140 (2x), KIF11, MCOLN1, PEX1 and PRPS1. Post‐test reassessment revealed that four of these cases represented hidden syndromes [cases with BBS1, IFT140 (1 of the 2 cases), KIF11 and MCOLN1 causative variants]. Of the remaining three, one of them is a case of phenotypic expansion (the second IFT140 case), other had been diagnosed as syndromic pre‐test, but without a specific diagnosis (the PRPS1 case) and the remaining one corresponded to misdiagnosis of a Heimler syndrome (caused by a PEX1 variant) as an Usher syndrome. These results reinforce the utility of using phenotype‐agnostic panels for the genomic diagnosis of vision loss.
The two IFT140 cases from our series, caused by the same mutation, showed very different phenotypes: the male had a phenotype matching Mainzer–Saldino syndrome whereas the female had non‐syndromic RP. Similarly, the non‐syndromic patient with two causative variants affecting ARL6 had a sister (not genetically tested) with polydactyly, which is strongly suggestive of Bardet–Biedl syndrome. These are clear examples of phenotypic heterogeneity that further reinforce the difficulty to predict the affected genes from clinical assessment and, as a result, the convenience of using comprehensive gene panels.
So far, the clinical application of NGS to the molecular diagnosis of hereditary blindness has followed two main paths: exome sequencing and panel sequencing. While exome sequencing has the advantage of being able to identify variants in any gene (thus not depending on gene selection and being able to discover novel genes not previously related to vision loss), this advantage is actually more useful in the research setting than in the clinical setting, where a solid molecular diagnosis, based on existing published gene‐phenotype associations and able to guide patient management, is required. In fact, this advantage is deemed to be less and less relevant with time, as a recent study shows that the discovery of novel genes associated to recessive IRD approaches saturation (Patel et al. 2018). As a result, up‐to‐date panels including both genes with strong/moderate association with inherited blindness and recently discovered candidate genes, as the one presented herein, should minimize this theoretical advantage of exome sequencing.
Consugar et al. concluded that focusing the sequencing and interpretation efforts in a panel with a limited subset of genes consistently associated with blindness phenotypes outperforms exome‐based sequencing strategies (Consugar et al. 2015). This outperformance was based on improved detection rates (attributable to a probe design better tailored to genes associated with IRD, as probes for aprox. 10% of the regions targeted by their panel were missing from commercially available exome capture kits) and reduced turnaround times. Time reduction does not only apply to laboratory geneticists, but also to clinicians, as exome sequencing demands from them additional time for the informed consent process (including discussions about incidental secondary findings), for interpreting laboratory reports and for reviewing results (many of which refer to conditions they are likely not familiar with) with patients (Biesecker & Green 2014; Gillespie et al. 2014). A more recent study, aimed at evaluating the performance of exome sequencing in the clinical context, concludes that the current standard of 120x average read depth may be insufficient for consistent breadth of coverage across all relevant genes (Kong et al. 2018).
Our test includes genes with all modes of inheritance in a single gene panel. An advantage of this is the possibility of detecting dominant de novo or incomplete penetrance mutations in families with apparent recessive patterns of disease transmission. In total, in our series, 6 of the 45 cases (13.3%) with a genetic diagnosis have causative variants in genes associated with dominant phenotypes and, however, do not have a family history of IRDs (Table 2). Of note, a recessive IRD panel would not have detected these dominant causative variants.
As any diagnostic technique, panel sequencing has its difficulties and limitations. The ability of our panel to detect SNVs and indels with high analytical sensitivity and specificity has been validated in this study. However, certain variant types, such as trinucleotide expansions and CNVs, as well as those affecting highly homologous or repetitive regions, can be missed. Nonetheless, these limitations are not exclusive of panel sequencing, but extensive to other NGS approaches, such as exome or, to a lower extent, genome sequencing (Bamshad et al. 2011; Schrijver et al. 2012). One relevant IRD gene affected by such limitations is RPGR, specifically in its ORF15 variant isoform (Audo et al. 2012). Although we initially observed difficulties in the capture, sequencing and/or alignment of sequences from this region, optimization of our capture probe has allowed us to overcome them in the current clinical version of the panel (v4) (Table S10).
The ability of our methodology to detect CNVs from panel data has previously been evaluated for a panel of 199 deafness‐related genes (Cabanillas et al. 2018). In that case, our NGS pipeline was able to correctly detect all homozygous and heterozygous whole gene CNVs previously found by qPCR and MLPA affecting STRC (a particularly challenging gene because of its highly homologous STRCP1 pseudogene). However, detection of partial gene deletions/duplications by NGS is even more challenging. The lack of homozygous or heterozygous deletions/duplications in the five genes we evaluated by MLPA in a subset of 31 patients without a genetic diagnosis from our current blindness series is encouraging. However, we do not rule out the possibility that these and other patients from this study without a genetic diagnosis have causative variants in genes that were investigated, either because of the aforementioned limitations or because the regions affected by the variants were not included in the design and, thus, were not captured and sequenced.
It is also possible that some of the patients from the current study tested with the earlier versions of the probe have causative variants in genes included in the later versions. These are not likely to represent a large fraction of the cases, though, as all causative variants in our series affected genes present in all panel versions. Nonetheless, a limitation of the panel approach is that, if no causative variants are detected in the patient and a new disease‐associated gene is discovered, that gene cannot be retrospectively evaluated from the available sequence. As mentioned before, exome data could provide this information with no extra sequencing.
The gene most frequently found to be responsible for the visual impairment phenotype in our series is ABCA4 (8/46 cases, 17.4%) followed by USH2A and PRPH2 (4/46 cases, 8.7%, each), CNGB1 (3/46, 6.5%) and CHM, IFT140, MYO7A and RP1 (2/46, 4.3%, each). The remaining 18 cases (40%) were each caused by variants in one of 18 different genes (Figs 1 and 2). The high prevalence of ABCA4, USH2A and CHM variants is in agreement with a recent and large retrospective analysis done by Motta et al. that evaluated 549 patients with IRD genetic testing in Brazil (Motta et al. 2018). Of the other four genes highlighted by Motta and collaborators as very prevalent disease‐causing genes (CEP290, CRB1, RPGR and CHM), three of them (CEP290, RPGR and CHM) are also affected by causative mutations in our series, although not recurrently (Figs 1 and 2).
Of the 100 cases evaluated in our series, 15 (15%) could potentially benefit from clinical trials aimed at correcting their genetic defects, including those affected by mutations in ABCA4 (8 cases), USH2A (2 cases), CHM (2 cases), MYO7A (2 cases) and RPGR (1 case). Thus, our results suggest that potential eligibility from a gene‐corrective clinical trial with curative purposes is not a rare possibility for IRD patients. Moreover, this data, combined with the four cases in which a hidden syndrome was revealed, indicate that the proportion of genetically diagnosed patients in which comprehensive genetic testing identified clinically relevant insights was at least 42.2% (19/45).
Genetic diagnosis is the key eligibility criterion for clinical trials and treatments based on gene therapy. With the advent of CRISPR/Cas9 technologies the possibility to correct not just the mutated genes but also their specific pathogenic variants has become a reality. This is the case of the CEP290 c.2991 + 1655A>G intronic mutation, targeted by the EDIT‐101 investigational new drug for type 10 Leber congenital amaurosis (Maeder et al. 2019). In this scenario, comprehensive genomic characterization should soon be a must in the clinical management of blind patients. Hopefully, a substantial proportion of the ongoing clinical trials will boost the approval of more therapies that, combined with appropriate genomic testing, will provide life‐changing options for those affected by such highly disabling conditions.
Supporting information
Table S1. List of phenotypes potentially related to visual impairment used as keywords for initial query on HGMD professional.
Table S2. Tier 1 and tier 2 genes included in v1 of the panel.
Table S3. Tier 1 and tier 2 genes included in v2 of the panel.
Table S4. Tier 1 and tier 2 genes included in v3 of the panel.
Table S5. Cases analyzed with each version of the panel.
Table S6. Primers used to validate causative variants (pathogenic and likely pathogenic variants considered responsible for the ophthalmologic phenotype).
Table S7. Coverages¥ and callabilities# for every tier 1 and tier 2 gene on v4 samples (n = 6).
Table S8. SNV and indel sensitivity, specificity and accuracy of the OFTALMOgenics v3 platform.
Table S9. SNV and indel sensitivity, specificity and accuracy of the OFTALMOgenics v4 platform.
Table S10. RPGR target regions with <100% bases covered by ≥10 reads or ≥20 reads in R&D, clinical v3 and clinical v4 cases.
Table S11. Clinical characteristics of cases without causative mutations.
Table S12. Variants of uncertain significance identified in this study.
Table S13. Results from MLPA analysis on 31 cases from this study with no causative variants identified.
Acknowledgements: We are grateful to all participating patients and their families. Work performed at IMOMA has been supported by Fundación María Cristina Masaveu Peterson.
Conflict of interest: The following authors are or have been employed by IMOMA, the owner of the OFTALMOgenicsTM platform: MD (Clinical Molecular Geneticist), RaC (Molecular Biologist), GAC (Biotechnologist), AO (Genetic Counsellor), AS (Bioinformatitian), NSD (Lab. Technician), RA (Lab Technician), RuC (Director of the Area of Precision Medicine) and JC (Director of the Laboratory of Molecular Medicine). The authors declare no other conflicts of interests.
Ethics approval and consent to participate: The study was approved by the Comité de Ética de Investigación del Principado de Asturias (research project #74/14). Consent was obtained from all patients or their parents.
Author’s contributions: MD performed most of the experimental work and contributed to results analysis and interpretation and writing of the manuscript. RaC contributed to results interpretation and data analysis. GAC contributed to experimental work and results interpretation. BF‐V, EV, AO, MV‐D, IH and AF‐V provided patient samples and associated clinical data. AS contributed to results analysis. PCP, DC and GRO performed bioinformatics analyses. NSD, RA and CGL contributed to experimental work. RuC and JC participated in study design, interpretation of results and writing of the manuscript. All authors commented on the manuscript and approved the submitted version.
Data Availability Statement: The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
[Correction added on 10 June 2020, after first publication: the acknowledgements section has been updated with funding information in this version.]
Contributor Information
Rubén Cabanillas, Email: rcabanillas@imoma.es.
Juan Cadiñanos, Email: jcb@imoma.es, Email: rcabanillas@imoma.es.
References
- Audo I, Bujakowska KM, Leveillard T et al. (2012): Development and application of a next‐generation‐sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis 7: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA & Shendure J (2011): Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12: 745–755. [DOI] [PubMed] [Google Scholar]
- Biesecker LG & Green RC (2014): Diagnostic clinical genome and exome sequencing. N Engl J Med 370: 2418–2425. [DOI] [PubMed] [Google Scholar]
- Boudewyn LC & Walkley SU (2019): Current concepts in the neuropathogenesis of mucolipidosis type IV. J Neurochem.148, 669–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne SJ, Humphries MM, Sullivan LS et al. (2011): A dominant mutation in RPE65 identified by whole‐exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet 19: 1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanillas R, Dineiro M, Castillo D et al. (2017): A novel molecular diagnostics platform for somatic and germline precision oncology. Mol Genet Genomic Med 5: 336–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanillas R, Dineiro M, Cifuentes GA et al. (2018): Comprehensive genomic diagnosis of non‐syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med Genomics 11: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JP, Lamey T, McLaren T, Thompson JA, Montgomery H & De Roach J (2015): Progress and prospects of next‐generation sequencing testing for inherited retinal dystrophy. Expert Rev Mol Diagn 15: 1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consugar MB, Navarro‐Gomez D, Place EM et al. (2015): Panel‐based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med 17: 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa KA, Salles MV, Whitebirch C, Chiang J & Sallum JMF (2017): Gene panel sequencing in Brazilian patients with retinitis pigmentosa. Int J Retina Vitreous 3: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada‐Cuzcano A, Koenekoop RK, Senechal A et al. (2012): BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet‐Biedl syndrome. Arch Ophthalmol 130: 1425–1432. [DOI] [PubMed] [Google Scholar]
- Fiorentino A, Fujinami K, Arno G et al. (2018): Missense variants in the X‐linked gene PRPS1 cause retinal degeneration in females. Hum Mutat 39: 80–91. [DOI] [PubMed] [Google Scholar]
- Gillespie RL, Hall G & Black GC (2014): Genetic testing for inherited ocular disease: delivering on the promise at last? Clin Exp Ophthalmol 42: 65–77. [DOI] [PubMed] [Google Scholar]
- Hafler BP (2017): Clinical progress in inherited retinal degenerations: gene therapy clinical trials and advances in genetic sequencing. Retina 37: 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauregui R, Cho GY, Takahashi VKL, Takiuti JT, Bassuk AG, Mahajan VB & Tsang SH (2018): Caring for hereditary childhood retinal blindness. Asia Pac J Ophthalmol (Phila) 7: 183–191. [DOI] [PubMed] [Google Scholar]
- Kong SW, Lee IH, Liu X, Hirschhorn JN & Mandl KD (2018): Measuring coverage and accuracy of whole‐exome sequencing in clinical context. Genet Med 20: 1617–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K & Garg S (2015): Navigating the current landscape of clinical genetic testing for inherited retinal dystrophies. Genet Med 17: 245–252. [DOI] [PubMed] [Google Scholar]
- Maeder ML, Stefanidakis M, Wilson CJ et al. (2019): Development of a gene‐editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 25: 229–233. [DOI] [PubMed] [Google Scholar]
- Motta FL, Martin RP, Filippelli‐Silva R, Salles MV & Sallum JMF (2018): Relative frequency of inherited retinal dystrophies in Brazil. Sci Rep 8: 15939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi KM, Avila‐Fernandez A, van Huet RA et al. (2014): Exome sequencing extends the phenotypic spectrum for ABHD12 mutations: from syndromic to nonsyndromic retinal degeneration. Ophthalmology 121: 1620–1627. [DOI] [PubMed] [Google Scholar]
- Patel N, Alkuraya H, Alzahrani SS et al. (2018): Mutations in known disease genes account for the majority of autosomal recessive retinal dystrophies. Clin Genet 94: 554–563. [DOI] [PubMed] [Google Scholar]
- Perrault I, Saunier S, Hanein S et al. (2012): Mainzer‐Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet 90: 864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratbi I, Falkenberg KD, Sommen M et al. (2015): Heimler syndrome is caused by hypomorphic mutations in the peroxisome‐biogenesis genes PEX1 and PEX6. Am J Hum Genet 97: 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S et al. (2015): Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille JM, Gillett RM, LeBlanc MA et al. (2014): Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol 132: 1393–1399. [DOI] [PubMed] [Google Scholar]
- Russell S, Bennett J, Wellman JA et al. (2017): Efficacy and safety of voretigene neparvovec (AAV2‐hRPE65v2) in patients with RPE65‐mediated inherited retinal dystrophy: a randomised, controlled, open‐label, phase 3 trial. Lancet 390: 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagopan KA (2017): Practical approach to syndromic pediatric retinal dystrophies. Curr Opin Ophthalmol 28: 416–429. [DOI] [PubMed] [Google Scholar]
- Schmidts M, Frank V, Eisenberger T et al. (2013): Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney disease. Hum Mutat 34: 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijver I, Aziz N, Farkas DH et al. (2012): Opportunities and challenges associated with clinical diagnostic genome sequencing: a report of the Association for Molecular Pathology. J Mol Diagn 14: 525–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon D & Banin E (2015): Nonsyndromic retinitis pigmentosa is highly prevalent in the Jerusalem region with a high frequency of founder mutations. Mol Vis 21: 783–792. [PMC free article] [PubMed] [Google Scholar]
- Stone EM, Aldave AJ, Drack AV, Maccumber MW, Sheffield VC, Traboulsi E & Weleber RG (2012): Recommendations for genetic testing of inherited eye diseases: report of the American Academy of Ophthalmology task force on genetic testing. Ophthalmology 119: 2408–2410. [DOI] [PubMed] [Google Scholar]
- Valdes‐Mas R, Bea S, Puente DA, Lopez‐Otin C & Puente XS (2012): Estimation of copy number alterations from exome sequencing data. PLoS ONE 7: e51422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbakel SK, van Huet RAC, Boon CJF et al. (2018): Non‐syndromic retinitis pigmentosa. Prog Retin Eye Res 66: 157–186. [DOI] [PubMed] [Google Scholar]
- Vervoort R, Lennon A, Bird AC et al. (2000): Mutational hot spot within a new RPGR exon in X‐linked retinitis pigmentosa. Nat Genet 25: 462–466. [DOI] [PubMed] [Google Scholar]
- Werdich XQ, Place EM & Pierce EA (2014): Systemic diseases associated with retinal dystrophies. Semin Ophthalmol 29: 319–328. [DOI] [PubMed] [Google Scholar]
- Xu M, Yang L, Wang F et al. (2015): Mutations in human IFT140 cause non‐syndromic retinal degeneration. Hum Genet 134: 1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of phenotypes potentially related to visual impairment used as keywords for initial query on HGMD professional.
Table S2. Tier 1 and tier 2 genes included in v1 of the panel.
Table S3. Tier 1 and tier 2 genes included in v2 of the panel.
Table S4. Tier 1 and tier 2 genes included in v3 of the panel.
Table S5. Cases analyzed with each version of the panel.
Table S6. Primers used to validate causative variants (pathogenic and likely pathogenic variants considered responsible for the ophthalmologic phenotype).
Table S7. Coverages¥ and callabilities# for every tier 1 and tier 2 gene on v4 samples (n = 6).
Table S8. SNV and indel sensitivity, specificity and accuracy of the OFTALMOgenics v3 platform.
Table S9. SNV and indel sensitivity, specificity and accuracy of the OFTALMOgenics v4 platform.
Table S10. RPGR target regions with <100% bases covered by ≥10 reads or ≥20 reads in R&D, clinical v3 and clinical v4 cases.
Table S11. Clinical characteristics of cases without causative mutations.
Table S12. Variants of uncertain significance identified in this study.
Table S13. Results from MLPA analysis on 31 cases from this study with no causative variants identified.