

Abstract
We have discovered the sirtuin‐rearranging ligands (SirReals) as a novel class of highly potent and selective inhibitors of the NAD+‐dependent lysine deacetylase sirtuin 2 (Sirt2). In previous studies, conjugation of a SirReal with a ligand for the E3 ubiquitin ligase cereblon to form a so‐called proteolysis‐targeting chimera (PROTAC) enabled small‐molecule‐induced degradation of Sirt2. Herein, we report the structure‐based development of a chloroalkylated SirReal that induces the degradation of Sirt2 mediated by Halo‐tagged E3 ubiquitin ligases. Using this orthogonal approach for Sirt2 degradation, we show that other E3 ligases than cereblon, such as the E3 ubiquitin ligase parkin, can also be harnessed for small‐molecule‐induced Sirt2 degradation, thereby emphasizing the great potential of parkin to be used as an E3 ligase for new PROTACs approaches. Thus, our study provides new insights into targeted protein degradation in general and Sirt2 degradation in particular.
Keywords: epigenetics, HDACs, PROTAC, protein degradation, sirtuins
It's degrading: We report the structure‐based development of a chloroalkylated sirtuin rearranging ligand (SirReal) that induces Sirt2 degradation mediated by Halo‐tagged E3 ubiquitin ligases. Using this approach, we show that besides cereblon, other E3 ligases such as parkin, can be harnessed for small‐molecule‐induced Sirt2 degradation.
Introduction
Eighteen specific lysine deacetylases (KDACs) have been identified in the human genome, and have been grouped into four classes according to their sequence homology. [1] The seven mammalian sirtuin isotypes (Sirt1–7), which constitute the class III KDACs, share an NAD+‐dependent catalytic mechanism. The isotype Sirt2, predominantly localized in the cytoplasm, deacetylates a variety of substrates, such as α‐tubulin, [2] NFκB, [3] BubR1, [4] and p53. [5] However, Sirt2 also acts as a nuclear deacetylase. An important nuclear function of Sirt2 is the global deacetylation of H4 K16Ac during mitosis. [6] Sirt2‐dependent deacetylation has a major impact on cell‐cycle regulation, [2] autophagy, [7] peripheral myelination, [8] and immune and inflammatory response. [9] A dysregulation of Sirt2‐mediated deacylation has been associated with several disease states, including bacterial infections, type II diabetes, neurodegenerative diseases, and cancer. [10] This highlights Sirt2 as a promising target for pharmaceutical intervention. The need for suitable tool compounds to further elucidate the cellular effects of Sirt2‐catalyzed deacylation and validate Sirt2 as a drug target provoked the discovery of several drug‐like Sirt2‐selective small‐molecule inhibitors, which have been reviewed elsewhere. [10]
In 2015, we discovered a new class of highly Sirt2‐selective inhibitors (Figure 1A). [11] These compounds inhibit Sirt2 in the low‐micromolar to nanomolar range, whereas no detectable inhibition (IC50>100 μM) can be observed for their close homologues Sirt1 and Sirt3.[ 11 , 12 ] The co‐crystal structures of Sirt2 in complex with 1 or 2 (Figure 1B), which were the first crystal structures of Sirt2 complexed with Sirt2‐selective drug‐like inhibitors, [11a] revealed a unique mode of inhibition that is characterized by a major rearrangement of the active site of Sirt2 upon ligand binding. Thus, inhibitors of this class were referred to as sirtuin rearranging ligands (SirReals) and the formed induced‐fit binding pocket was termed the selectivity pocket, as it was identified to be the key to Sirt2 selectivity. [11a] Very soon after we had reported the existence of the selectivity pocket, it was shown that this pocket accommodates the long‐chain fatty acid of a myristoyl substrate. [13] In the meantime, other Sirt2 inhibitors were also shown to gain their isotype selectivity by targeting the selectivity pocket. [14] Through conjugation of the propargylated SirReal analogue 3 with different azido‐functionalized building blocks, we generated the triazole‐SirReals 4 a and b. These compounds showed increased Sirt2 affinity, which has been rationalized by X‐ray co‐crystallography to an additional H‐bond interaction of the triazole moiety with Arg97 of the cofactor binding loop (Figure 1B). [12]
Figure 1.
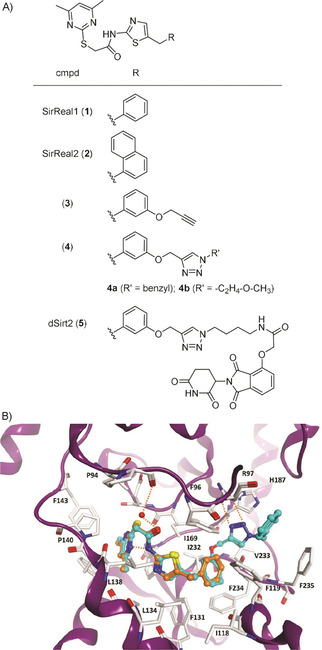
A) Chemical structures of selected SirReals (1–4), and the Sirt2‐targeted PROTAC dSirt2 (5). B) Comparison of the binding mode of SirReal1 (1, PDB ID: 4RMI, orange) and the triazole‐based SirReal 4 a (PDB ID: 5DY5, cyan) at human Sirt2 (PDB ID: 5DY5). Hydrogen bonds are shown as dashed orange lines. The conserved water molecule bridging the interaction of the SirReals is shown as a red sphere.
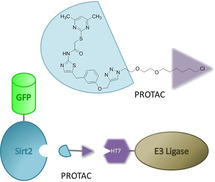
Besides an increase in affinity, the incorporation of a triazole led to improved water solubility and gave the opportunity to use the triazole as a linker moiety in order to conjugate the SirReal core with various azido‐functionalized labels by means of CuI‐catalyzed Huisgen cycloaddition. [15] Thus, we took advantage of the triazole‐SirReals as an ideal template for the straightforward development of small‐molecule tools to probe Sirt2. Fluorescently labelled SirReals were utilized for screening purposes and as unprecedented tools to study binding to the selectivity pocket. [16] A biotinylated SirReal was used in combination with biolayer interferometry for kinetic measurements that revealed the long residence time of the SirReal‐Sirt2 interaction.[ 12 , 17 ] By means of the phthalimide‐tagged SirReal analogue (5), a so‐called proteolysis targeting chimera (PROTAC), we were able to induce the proteasomal degradation of Sirt2. [18] In general, PROTACs are capable of hijacking the cellular quality control by recruiting the protein of interest (POI) to an E3 ubiquitin ligases for polyubiquitination and thus to induce its proteasomal degradation. [19] Currently, ligands for only a few of over 600 E3 ligases have been harnessed for PROTAC development. [20] Most commonly employed are phthalimide analogues leading to recruitment of cereblon, or ligands for the von Hippel‐Lindau protein (VHL) leading to its recruitment. Besides VHL and cereblon, the E3 ligases cIAP and MDM2 belong to the most frequently used ubiquitin ligases for PROTAC approaches. Since its first description by Crews and co‐workers in the year 2001, the PROTAC concept has been widely applied to induce the degradation of various proteins, such as kinases, [21] transcription factors, [22] and epigenetic reader proteins. [23] The PROTAC approach has attracted much attention in the recent years, as it comes along with several benefits compared to standard small‐molecule‐based inhibition. From a basic research point of view, PROTACs can be employed as tools to prove cellular target engagement. [24] As PROTACs induce the degradation of the whole protein, they can be used for the identification of yet unknown protein functions, e. g. protein‐protein‐interactions, which are not related to the actual binding site of the ligand/PROTAC. For example, the Sirt2‐directed PROTAC (5) enabled the discovery of the interplay between Sirt2 and the tubulin polymerase TPPP/p25 that is not dependent on the catalytic activity of Sirt2. [25] From a therapeutic perspective, PROTACs often show an improved cellular efficacy compared to occupancy‐based inhibition, which is attributed to their catalytic mode of action as well as the sustained inhibition and disruption of protein‐protein interactions as a consequence of target degradation. [26] The first PROTAC drugs (ARV‐110, ARV‐471) have recently even entered phase I clinical trials for treating patients with metastatic castration resistant prostate cancer and metastatic breast cancer, respectively, [27] which points out the therapeutic potential of the PROTAC approach.
However, first cellular mechanisms of resistance for both cereblon‐ and VHL‐engaging degraders have recently been discovered. The mechanism of resistance has been attributed to a loss of the E3 ligase itself or the downregulation of specific interaction partners of the respective E3 ligase. [28] Therefore, two major questions arise from this discovery. First, can other E3 ligases, beyond cereblon, cIAP, MDM2, and VHL, be utilized for targeted protein degradation? Second, how can new E3 ligases be identified as the potential mediators of chemically induced protein degradation, in the absence of suitable ligands enabling their recruitment to the targeted protein?
In order to address these questions, Ottis et al. studied the suitability of different E3 ligases for small‐molecule‐induced protein degradation by fusing different E3 ligases to HaloTag 7 (HT7). [29] Thereby, they replaced the native substrate binding domain of the respective E3 ligase with HT7, which is an engineered bacterial dehalogenase capable of forming a covalent bond with a chloroalkane under in vitro and in vivo conditions. [30] This system was used to present chloroalkylated ligands for the protein of interest (POI) on the surface of E3 ligase‐HT7 fusion proteins, thereby enabling the recruitment of the POI to the HT7‐tagged E3 ligase. Due to this chemically induced interaction, the ubiquitination and subsequent proteasomal degradation of the POI can be initiated, if the HT7‐tagged E3 ligase is suitable for chemically induced target degradation. By this elegant approach the authors bypassed the need for previously identified ligands for the native substrate binding site of the respective E3 ligase. In the course of their study, Ottis et al. identified the E3 ligase parkin to have the broadest efficacy for the degradation of endogenous target proteins (ABL1, SRC, YES, and EphA2). Moreover, the HT7‐parkin construct showed the broadest capability to accommodate diverse linker lengths and linker compositions. [29] In order to investigate the potential of parkin as an E3 ligase that can be broadly utilized for targeted protein degradation and to provide an unprecedented molecular tool to probe Sirt2 biology, we have developed a chloroalkylated SirReal and studied its efficacy for degrading Sirt2 in presence of different HT7‐tagged E3 ligases. In addition, molecular modeling of the ternary complex of Sirt2, HT7, and the SirReal‐probe proposes a binding model of the interaction of the bifunctional ligand with both binding pockets.
Results
For the design of the chloroalkylated SirReal, we combined the structural features of the Sirt2‐selective and highly potent triazole‐based SirReals (see Figure 1) with 2‐((6‐chlorohexyl)oxy)ethyl, a reactive linker that has been previously developed for the HaloTag labeling technology. [30a] Based on our work on a SirReal‐based affinity probe,[ 12 , 31 ] we already had valid information on where to place the linker without losing affinity to Sirt2. In addition, docking studies and molecular dynamics simulations of Sirt2‐HT7‐probe ternary complexes were carried out, in order to design suitable linkers between the triazole‐SirReal and the chloroalkyl group. To model the ternary complex of Sirt2‐HT7‐triazole‐SirReal probe, the crystal structures of human Sirt2 complexed with a triazole‐SirReal 4 a [12] and the HT7 crystal structure from Rhodococcus rhodochrous [32] with a covalently bound chloroalkane‐based ligand were used. The protein‐protein docking was carried out using program HADDOCK [33] as described in detail in the experimental section (Supporting Information, Figure S1). We predicted a 2‐(2‐((6‐chlorohexyl)oxy)ethoxy)ethyl linker to be suitable to recruit Sirt2 to HT7 with an adequate proximity that allows the formation of the covalent bond between the probe and Asp105. The obtained binary Sirt2‐HT7 complex was subsequently used to dock the designed probe assuming the formation of a covalent bond between the terminal chloroalkane and Asp105. The docking results show that the triazole‐SirReal and the 2‐(2‐((hexyl)oxy)ethoxy)ethyl group are interacting in a similar way as the co‐crystallized molecules in Sirt2 (PDB ID: 5DY5) and HT7 (PDB ID: 5VNP; Figures 2, S2, and S3).
Figure 2.
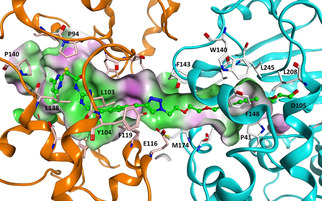
Model of the ternary complex of Sirt2‐HT7 with covalently bound chloroalkylated SirReal probe 11 (ball and stick mode, colored green). Sirt2 is shown as orange coloured ribbon and HT7 as cyan coloured ribbon. Probe surrounding amino acids are displayed as well as the molecular surface of the resulting binding pocket (color‐coded according to the hydrophobicity, hydrophobic region: green, polar region: magenta).
To analyze the stability of the obtained ternary Sirt2‐HT7‐probe complex, we carried out a 50 ns molecular dynamics (MD) simulation (see Supporting Information for details). The complex as well as the individual proteins remained stable during the simulation time with Cα‐atom RMSD values around 2 Å (Figure S4). Also the ligand changed its binding conformation only slightly (RMSD values of heavy atoms between 2 and 3 Å), esp. the SirReal part of the probe showed the same interaction with the Sirt2 protein as the unmodified SirReal inhibitor in the crystal structure. Only the flexible linker region including the triazole group, located at the interface of both proteins, showed higher flexibility during the MD simulation (the superimposition of six different complex conformations at 0, 10, 20, 30, 40, 50 ns is shown in Figure S5.
In order to synthesize the envisaged chloroalkylated SirReal (11), we set up the synthesis route depicted in Scheme 1. While planning this synthesis route, we focused our efforts on i) generating the chloroalkylated SirReal 11, capable of inducing Sirt2 degradation ii) providing a “click‐able” azido‐chloroalkyl‐conjugate as a versatile HaloTag substrate ready for conjugation with other alkynylated ligands.
Scheme 1.
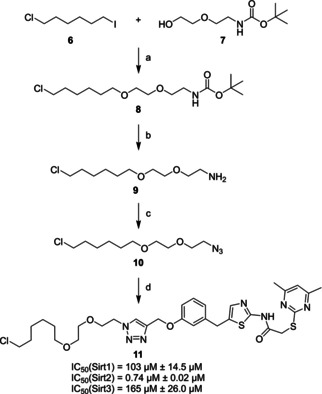
Synthesis of the chloroalkylated SirReal (11). a) NaH, THF/CH2Cl2 (2 : 1), 0 °C–RT, 12 h, 33 % yield; b) TFA, CH2Cl2, 0 °C–RT, 2 h,≥99 % yield (crude); c) 1H‐imidazole‐1‐sulfonyl azide hydrochloride, CuSO4, K2CO3, methanol, RT, 2 h, 47 % yield; d) 3, sodium ascorbate, CuSO4, water/tert‐BuOH (1 : 1), RT, 16 h, yield 79 % yield. IC50 values are indicated as mean value±standard deviation (n=3).
In the first step of our synthesis route, 1‐chloro‐6‐iodohexane (6) was used for the alkylation of the primary alcohol (7) to form the ether (8). The amine (9) was generated by removing the tert‐butyloxycarbonyl protecting group by the addition of trifluoroacetic acid. A diazotransfer reaction was used to form the “click‐able” azido‐chloroalkyl‐conjugate (10). Finally, compound 10 was conjugated with the propargylated SirReal analogue 3 through a CuI‐catalyzed Huisgen cycloaddition [15] to yield the chloroalkylated SirReal (11).
The chloroalkylated SirReal (11) was evaluated for its in vitro inhibition of Sirt2 in a fluorescence‐based deacetylase activity assay as previously described. [34] As expected, a potent inhibition of Sirt2 with an IC50 value of 0.74±0.02 μM was observed, whereas Sirt1 and Sirt3, the closest homologues of Sirt2, were only affected at much higher concentrations (Scheme 1).
To assess the suitability of the chloroalkylated SirReal (11) for chemically induced Sirt2 degradation, we transfected HeLa cells with plasmids coding for a small panel of engineered HT7‐tagged E3 ligases, covering the three major classes of E3 ligases: U‐box, HECT, and RING. The plasmids for the engineered HT7‐tagged E3 ligases HT7‐NEDD4L, MARCH5‐HT7, SIAH1‐HT7, HT7‐parkin, βTrCP‐HT7, and HT7‐CHIP have been previously developed by Ottis et al. and were a generous gift from the Crews laboratory. [29] By immunofluorescence microscopy and Western blot experiments, we were able to show the expression of the HT7‐E3 constructs in human HeLa cells; however, the extent of the expression seemed to be highly dependent on the nature of the constructs as well as the approach used for the detection (Figures 3 and S6). The immunofluorescence microscopic visualization has shown unambiguous immunopositivity of the engineered HT7‐parkin, βTrCP‐HT7, and HT7‐CHIP plasmids, comparable staining with the control (empty plasmid) (Figure 3). The Western blot analyses provided additional evidence for the significant expression of these three constructs, although their immunopositivities appear to be dependent on the antibodies specific for the HaloTag or HA‐tag of the constructs (Figure S6).
Figure 3.
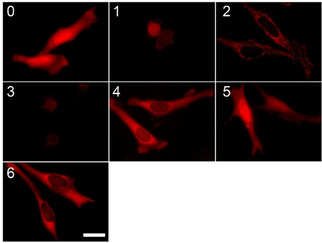
The expression of HaloTag E3 ligase constructs in transiently transfected HeLa cells. 0: empty plasmid (no E3), 1: HT7‐NEDD4L, 2: MARCH‐HT7, 3: SIAH‐HT7, 4: HT7‐parkin, 5: βTrCP‐HT7, and 6: HT7‐Chip, respectively. Transfected cells are stained by specific fluorescent HaloTag TMR Ligand (HT7‐L, 50 nM) and detected as red signal by immunofluorescence microscopy. Scale bar: 10 μm.
To investigate whether the three well‐expressed HaloTag E3 ligase constructs (HT7‐parkin, βTrCP‐HT7, and HT7‐CHIP) can mediate Sirt2 degradation induced by 11, the intracellular level of the tubulin deacetylase was quantified in HeLa cells by Western blot using a Sirt2‐specific antibody. As shown in Figure 4A, the engineered constructs HT7‐parkin, βTrCP‐HT7, and HT7‐CHIP display an effect on the Sirt2 level, as compared to the control experiments (empty vector). The quantification of the concentration‐dependent effect of the fusion constructs reveals that the most effective construct is HT7‐parkin (Figure 4B). It already induces marked Sirt2 degradation at a concentration of 20 nM of the chloroalkylated SirReal (11), whereas tenfold higher concentrations of our previously reported cereblon‐mediated PROTAC (5) were required to provoke similar effects. [18] Moreover, we were able to show that the effect of 11 on Sirt2 degradation can be counteracted either by the proteasome inhibitor MG132 [35] or by competition with the Sirt2 inhibitor 4 b [12] and the HaloTag competitor HT7‐L, respectively (Figure 4C). Even though MG132 could only reduce but not fully counteract the Sirt2 degradation, these findings indicate that observed reduction of Sirt2 levels can be attributed to chemically induced protein degradation by our chloroalkylated SirReal (11). To investigate the functional consequences of Sirt2 degradation by means of 11, we probed the acetylation levels of tubulin, a well‐known Sirt2 substrate, [2] via immunofluorescence microscopy using a specific anti‐acetylated‐tubulin antibody (Figure 4D). In cells expressing HT7‐parkin (red), degradation of Sirt2 due to treatment with 11 resulted in a more pronounced acetylation of the microtubule network as compared to cells where 11 can only act as a Sirt2 inhibitor due to the absence of HT7‐parkin. The observation that treatment with our chloroalkylated SirReal (11) in combination with HT7‐parkin expression results in a more pronounced hyperacetylation of the tubulin network, as compared to our cereblon‐mediated PROTAC (5), is consistent with the high activity of 11 detected by means of our western blot experiments (Figure 4A−B). However, when comparing the effects of 11 and 5, it should be taken into account that the effects of 11 are mediated by an artificially expressed fusion protein (HT7‐parkin), whereas Sirt2 degradation induced by the thalidomide‐labelled PROTAC (5) is mediated by endogenous expression of the E3 ligase cereblon.
Figure 4.
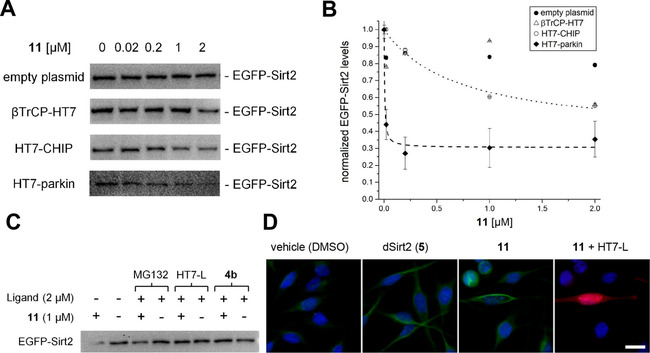
The chloroalkylated SirReal (11) induces the proteasomal degradation of Sirt2 in HeLa cells transiently co‐transfected with EGFP‐Sirt2 and different Halo‐tagged E3 ligase constructs. A) Representative Western blots showing the effect of E3‐HaloTag constructs on the intracellular EGFP‐Sirt2 level in the presence of 11 (0.02 μM – 2 μM, exposure time=4 h). B) Western blot was detected by using a Sirt2 specific antibody and quantified by densitometry. The values are normalized with respect to 0 μM of 11 for each plasmid. The obtained values are indicated as mean value ± standard deviation (n=3). Curves for HT7‐CHIP and HT7‐parkin were fitted using OriginPro 2018 software by non‐linear curve fitting applying a one‐phase decay model. C) Representative Western blot image detected by Sirt2‐specific antibody shows the effect of 11 on the EGFP‐Sirt2 level in the absence and presence of the proteasome inhibitor MG132, the HaloTag blocker HT7‐L, and the Sirt2 inhibitor 4 b (n=4). D) Immunofluorescence microscopic images of HeLa cells stained for acetyl‐tubulin immunopositivity (green). In cultured HeLa cells expressing HT7‐parkin (red, labelled with 50 nM fluorescent HaloTag TMR Ligand (HT7‐L)), depletion of Sirt2 due to treatment with 11 results in a more pronounced acetylation of the microtubule network (green), as compared to cereblon‐mediated Sirt2 degradation by 5 or sole enzymatic inhibition by 11 in the absence of HT7‐parkin. Samples were pre‐incubated with compounds at 1 μM concentration. Nuclei were DAPI stained (blue). Scale bar: 10 μm.
Discussion and Conclusion
In this study, we report the structure‐based development of a chloroalkylated sirtuin rearranging ligand (SirReal) that induces the degradation of Sirt2 mediated by Halo‐tagged E3 ligases. While the approach of targeted protein degradation mediated by Halo‐tagged E3 ligases has been established for kinases and binding proteins, [29] this is the first example of a chloroalkylated PROTAC targeting an amidohydrolase or any histone modifying enzyme. On the one hand, our study shows the versatility of this approach developed by Ottis et al., [29] on the other hand, we were able to expand our SirReal‐based chemical biology toolset, thereby highlighting the SirReals as an ideal template for the development of further Sirt2‐targeted molecular tools. Moreover, with our azido‐chloroalkyl‐conjugate (10) we provide a versatile HaloTag ligand building block, ready to be “clicked” to alkynylated ligands targeting other proteins. Given the broad functional group tolerance of this type of chemistry [36] and the availability of alkynylated ligands for many targets that have been used in a large variety of chemical biology studies, this technique will be amenable to a plethora of different targets and inhibitors. Most importantly, by means of our chloroalkylated SirReal (11) we were able to validate the broad substrate spectrum of HT7‐parkin, thereby emphasizing the great potential of parkin to be utilized as an E3 ligase for new approaches in targeted protein degradation. This finding is highly relevant as mechanisms of drug resistance have been recently reported for PROTAC approaches mediated by the most frequently used E3 ligases cereblon and VHL. [28]
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Craig M. Crews for providing the plasmids coding for the engineered HT7‐tagged E3 ligases HT7‐NEDD4L, MARCH5‐HT7, SIAH1‐HT7, HT7‐parkin, βTrCP‐HT7, and HT7‐CHIP that were previously reported by Ottis et al. [29] M.S. (SCHI 1408/2‐1), M.J. and N.W. (Ju295/14‐1 and 235777276/GRK1976), O.E. (Project‐ID 192904750 – SFB992) and W.S. (SI 868/15‐1) were supported by the Deutsche Forschungsgemeinschaft (DFG). J. Ovádi thanks the Hungarian National Research, Development and Innovation Office Grants OTKA [T‐112144] for support, and J. Oláh was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. Open access funding enabled and organized by Projekt DEAL.
M. Schiedel, A. Lehotzky, S. Szunyogh, J. Oláh, S. Hammelmann, N. Wössner, D. Robaa, O. Einsle, W. Sippl, J. Ovádi, M. Jung, ChemBioChem 2020, 21, 3371.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.12370469.v1)
References
- 1. De Ruijter A. J. M., Van Gennip A. H., Caron H. N., Kemp S., Van Kuilenburg A. B. P., Biochem. J. 2003, 370, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. North B. J., Marshall B. L., Borra M. T., Denu J. M., Verdin E., Mol. Cell 2003, 11, 437–444. [DOI] [PubMed] [Google Scholar]
- 3. Rothgiesser K. M., Erener S., Waibel S., Luscher B., Hottiger M. O., J. Cell Sci. 2010, 123, 4251–4258. [DOI] [PubMed] [Google Scholar]
- 4. North B. J., Rosenberg M. A., Jeganathan K. B., Hafner A. V., Michan S., Dai J., Baker D. J., Can Y., Wu L. E., Sauve A. A., van Deursen J. M., Rosenzweig A., Sinclair D. A., EMBO J. 2014, 33, 1438–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vaziri H., Dessain S. K., Eagon E. N., Imai S. I., Frye R. A., Pandita T. K., Guarente L., Weinberg R. A., Cell 2001, 107, 149–159. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Vaquero A., Sternglanz R., Reinberg D., Oncogene 2007, 26, 5505–5520; [DOI] [PubMed] [Google Scholar]
- 6b. Vaquero A., Scher M. B., Lee D. H., Sutton A., Cheng H. L., Alt F. W., Serrano L., Sternglanz R., Reinberg D., Genes Dev. 2006, 20, 1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Oliveira R. M., Sarkander J., Kazantsev A. G., Outeiro T. F., Front. Pharmacol. 2012, 3, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beirowski B., Gustin J., Armour S. M., Yamamoto H., Viader A., North B. J., Michán S., Baloh R. H., Golden J. P., Schmidt R. E., Sinclair D. A., Auwerx J., Milbrandt J., Proc. Natl. Acad. Sci. USA 2011, 108, E952–E961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Pais T. F., Szego E. M., Marques O., Miller-Fleming L., Antas P., Guerreiro P., de Oliveira R. M., Kasapoglu B., Outeiro T. F., EMBO J. 2013, 32, 2603–2616; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Eskandarian H. A., Impens F., Nahori M. A., Soubigou G., Coppee J. Y., Cossart P., Hamon M. A., Science 2013, 341, 1238858. [DOI] [PubMed] [Google Scholar]
- 10. Schiedel M., Robaa D., Rumpf T., Sippl W., Jung M., Med. Res. Rev. 2018, 38, 147–200. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Rumpf T., Schiedel M., Karaman B., Roessler C., North B. J., Lehotzky A., Oláh J., Ladwein K. I., Schmidtkunz K., Gajer M., Pannek M., Steegborn C., Sinclair D. A., Gerhardt S., Ovádi J., Schutkowski M., Sippl W., Einsle O., Jung M., Nat. Commun. 2015, 6, 6263; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Schiedel M., Rumpf T., Karaman B., Lehotzky A., Oláh J., Gerhardt S., Ovádi J., Sippl W., Einsle O., Jung M., J. Med. Chem. 2016, 59, 1599–1612. [DOI] [PubMed] [Google Scholar]
- 12. Schiedel M., Rumpf T., Karaman B., Lehotzky A., Gerhardt S., Ovadi J., Sippl W., Einsle O., Jung M., Angew. Chem. Int. Ed. 2016, 55, 2252–2256. [DOI] [PubMed] [Google Scholar]
- 13. Feldman J. L., Dittenhafer-Reed K. E., Kudo N., Thelen J. N., Ito A., Yoshida M., Denu J. M., Biochemistry 2015, 54, 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Sundriyal S., et al., J. Med. Chem. 2017, 60, 1928–1945; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Mellini P., et al., Chem. Sci. 2017, 8, 6400–6408; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Robaa D., Monaldi D., Wöinvsssner N., Kudo N., Rumpf T., Schiedel M., Yoshida M., Jung M., Chem. Rec. 2018, 18, 1701–1707. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Huisgen R., P Chem Soc London 1961, 357–396; [Google Scholar]
- 15b. Chan T. R., Hilgraf R., Sharpless K. B., Fokin V. V., Org. Lett. 2004, 6, 2853–2855. [DOI] [PubMed] [Google Scholar]
- 16. Swyter S., Schiedel M., Monaldi D., Szunyogh S., Lehotzky A., Rumpf T., Ovadi J., Sippl W., Jung M., Philos. Trans. R. Soc. London Ser. B 2018, 373, 20170083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schiedel M., Daub H., Itzen A., Jung M., ChemBioChem 2020, 21, 1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schiedel M., et al., J. Med. Chem. 2018, 61, 482–491. [DOI] [PubMed] [Google Scholar]
- 19. Toure M., Crews C. M., Angew. Chem. Int. Ed. 2016, 55, 1966–1973. [DOI] [PubMed] [Google Scholar]
- 20. Burslem G. M., Crews C. M., Cell 2020, 181, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Bondeson D. P., et al., Nat. Chem. Biol. 2015, 11, 611–617; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Lai A. C., Toure M., Hellerschmied D., Salami J., Jaime-Figueroa S., Ko E., Hines J., Crews C. M., Angew. Chem. Int. Ed. 2016, 55, 807–810; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Steinebach C., et al., Chem. Sci. 2020, 11, 3474–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Sakamoto K. M., Kim K. B., Verma R., Ransick A., Stein B., Crews C. M., Deshaies R. J., Mol. Cell. Proteomics 2003, 2, 1350–1358; [DOI] [PubMed] [Google Scholar]
- 22b. Okuhira K., Demizu Y., Hattori T., Ohoka N., Shibata N., Nishimaki-Mogami T., Okuda H., Kurihara M., Naito M., Cancer Sci. 2013, 104, 1492–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Lu J., et al., Chem. Biol. 2015, 22, 755–763; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Winter G. E., Buckley D. L., Paulk J., Roberts J. M., Souza A., Dhe-Paganon S., Bradner J. E., Science 2015, 348, 1376–1381; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23c. Zengerle M., Chan K. H., Ciulli A., ACS Chem. Biol. 2015, 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Remillard D., et al., Angew. Chem. Int. Ed. 2017, 56, 5738–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szabo A., et al., Sci. Rep. 2017, 7, 17070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Lu J., et al., Chem. Biol. 2015, 22, 755–763; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Burslem G. M., et al., Cell Chem. Biol. 2018, 25, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Vogelmann A., Robaa D., Sippl W., Jung M., Curr. Opin. Chem. Biol. 2020, 57, 8–16; [DOI] [PubMed] [Google Scholar]
- 27b. Mullard A., Nat. Rev. Drug Discovery 2019, 18, 237–239. [DOI] [PubMed] [Google Scholar]
- 28. Ottis P., et al., ACS Chem. Biol. 2019, 14, 2215–2223. [DOI] [PubMed] [Google Scholar]
- 29. Ottis P., Toure M., Cromm P. M., Ko E., Gustafson J. L., Crews C. M., ACS Chem. Biol. 2017, 12, 2570–2578. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Los G. V., et al., ACS Chem. Biol. 2008, 3, 373–382; [DOI] [PubMed] [Google Scholar]
- 30b. Encell L. P., et al., Curr. Chem. Genomics 2012, 6, 55–71; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30c. Hoelzel C. A., Zhang X., ChemBioChem 2020, 21, 1935–1946; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30d. Rivas-Pardo J. A., et al., Nat. Commun. 2020, 11, 2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schiedel M., Rumpf T., Karaman B., Lehotzky A., Gerhardt S., Ovadi J., Sippl W., Einsle O., Jung M., Angew. Chem. Int. Ed. 2016, 55, 2252–2256. [DOI] [PubMed] [Google Scholar]
- 32. Liu Y., Fares M., Dunham N. P., Gao Z., Miao K., Jiang X., Bollinger S. S., Boal A. K., Zhang X., Angew. Chem. Int. Ed. 2017, 56, 8672–8676. [DOI] [PubMed] [Google Scholar]
- 33. van Zundert G. C. P., Rodrigues J. P. G. L. M., Trellet M., Schmitz C., Kastritis P. L., Karaca E., Melquinod A. S. J., van Dijk M., de Vries S. J., Bonvin A. M. J. J., J. Mol. Biol. 2016, 428, 720–725. [DOI] [PubMed] [Google Scholar]
- 34. Heltweg B., Trapp J., Jung M., Methods 2005, 36, 332–337. [DOI] [PubMed] [Google Scholar]
- 35. Tsubuki S., Saito Y., Tomioka M., Ito H., Kawashima S., J. Biochem. 1996, 119, 572–576. [DOI] [PubMed] [Google Scholar]
- 36. Kolb H. C., Sharpless K. B., Drug Discovery Today 2003, 8, 1128–1137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary