

Abstract
β‐Thalassemia is an inherited blood disorder resulting from defects in hemoglobin production, leading to premature death of red blood cells (RBCs) or their precursors. Patients with transfusion‐dependent β‐thalassemia often need lifelong regular RBC transfusions to maintain adequate hemoglobin levels. Frequent transfusions may lead to iron overload and organ damage. Thus, there is a large unmet need for alternative therapies. Luspatercept, a first‐in‐class erythroid maturation agent, is the first approved therapy in the United States for the treatment of anemia in adult patients with β‐thalassemia who require regular RBC transfusions. The population pharmacokinetics and exposure‐response relationship of luspatercept were evaluated in 285 patients with β‐thalassemia. Luspatercept displayed linear and time‐invariant pharmacokinetics when administered subcutaneously once every 3 weeks. Body weight was the only clinically relevant covariate of luspatercept clearance, favoring weight‐based dosing. Magnitude and frequency of hemoglobin increase, if not influenced by RBC transfusions, was positively correlated with luspatercept area under the serum concentration‐time curve (AUC), 0.2‐1.25 mg/kg, whereas a significant reduction in RBC units transfused was observed in frequently transfused patients. The probability of achieving ≥33% or ≥50% reduction in RBC transfusion burden was similar across the time‐averaged AUC (0.6‐1.25 mg/kg), with the 1 mg/kg starting dose sufficient for most early responders (71%‐80%). Increasing luspatercept AUC (0.2‐1.25 mg/kg) did not increase incidence or severity of treatment‐emergent adverse events. These results provide a positive benefit‐risk profile for the recommended luspatercept doses (1‐1.25 mg/kg) in treating adult patients with β‐thalassemia who require regular RBC transfusions.
Keywords: anemia, beta‐thalassemia, biologics, exposure‐response, luspatercept, population pharmacokinetics
β‐Thalassemia is a red blood cell (RBC) disorder caused by mutations in the β‐globin gene. These mutations cause absent or reduced production of the β‐globin chains of hemoglobin (Hb), leading to maturation failure and apoptosis of erythroid precursors in the bone marrow and premature death of RBCs in the peripheral circulation. These processes, known as ineffective erythropoiesis and hemolysis, manifest with chronic anemia. 1 , 2 Before luspatercept, there was no approved therapy in the United States to treat anemia in adult patients with β‐thalassemia. In patients with more severe forms of β‐thalassemia, RBC transfusions have been the mainstay of therapy from childhood to adulthood. However, repeated transfusions may lead to iron overload and multiple organ failure. 1 , 2
Luspatercept is a recombinant fusion protein consisting of a modified form of the extracellular domain of human activin receptor type IIB (ActRIIB) linked to the human fragment crystallizable (Fc) domain of human immunoglobin G1. The ActRIIB receptor and its ligands are members of the transforming growth factor beta (TGF‐β) superfamily. 3 By binding several endogenous TGF‐β superfamily ligands, luspatercept leads to diminished Smad2/3 signaling, enhanced late‐stage erythroid maturation in the bone marrow, and improved hematology parameters in mouse models of β‐thalassemia. 4 , 5 In clinical trials of β‐thalassemia, luspatercept treatment led to sustained increases in Hb as well as reduced RBC transfusion frequency. 6 , 7 Luspatercept was well tolerated in these studies, with the maximum tolerated dose not reached at the highest clinical dose evaluated (1.25 mg/kg). 6 Luspatercept is the first drug approved in the United States to treat anemia from β‐thalassemia.
Here, we evaluate the population pharmacokinetics (PK) and exposure‐response relationship of luspatercept in patients with β‐thalassemia under a titration‐to‐response dosing regimen. These results were used to support the benefit‐risk assessments of the recommended dosage for the recently approved indication: treatment of anemia in adult patients with β‐thalassemia who require regular RBC transfusions.
Methods
Studies and Treatment
Institutional review boards or ethics committees at each site (Supplemental Table S1) approved the protocols before each study. Studies were conducted in compliance with Good Clinical Practice standards, as described in International Conference on Harmonization guidelines and in accordance with the ethical principles outlined in the Declaration of Helsinki. All patients provided written informed consent. This analysis was based on data from patients with βthalassemia in 3 studies: a phase 2 dose‐finding/expansion study (A536‐04; NCT01749540), a phase 2 extension study (A536‐06; NCT02268409) to which the patients from study A536‐04 were rolled over, and a pivotal phase 3 study (ACE‐536‐B‐THAL‐001 [BELIEVE]; NCT02604433). Luspatercept was administered subcutaneously once every 3 weeks. In the dose‐escalation cohorts of study A536‐04, the dose ranged from 0.2 to 1.25 mg/kg, and each patient received only 1 dose level. In the expansion cohort of study A536‐04, study A536‐06, and study ACE‐536‐B‐THAL‐001, the starting dose was 0.8 or 1 mg/kg; the dose could be increased in a stepwise manner (from 0.8 to 1 mg/kg and then to 1.25 mg/kg) if patients did not achieve clinical benefit (clinically meaningful reduction in RBC transfusion burden or increase in Hb) after at least 2 consecutive doses at the same dose level and did not meet the protocol‐specified criteria for dose decrease or delay. Patients in study A536‐04 received luspatercept for up to 5 doses, whereas patients in studies A536‐06 and ACE‐536‐B‐THAL‐001 could receive luspatercept for up to 5 years. Serial sampling of blood was performed in all patients to determine luspatercept concentration in serum. Hb level was assessed before each dose or more frequently during these studies. A brief summary of the study design, treatment, and PK/Hb sampling schedule is provided in Supplemental Table S2. Further details about the designs of these studies were described previously. 6 , 7
Bioanalytical Methodology
A fully validated enzyme‐linked immunosorbent assay, using goat polyclonal anti‐luspatercept antibody capture and sheep polyclonal anti‐human IgG1 horseradish peroxidase detection reagents, was performed to quantify luspatercept concentration in serum. The range of this assay was 50 to 600 ng/mL in 100% human serum with the standard curve fitted through 8 calibration standards using a 5‐parameter logistic fit. The lower limit of quantitation was 50 ng/mL. The interrun coefficient of variation was ≤12.0% and interrun accuracy was 97.7% to 107.6% of the nominal concentration.
Population PK Analysis
The population PK analysis was performed using NONMEM software (version 7.3; ICON Development Solutions, Ellicott City, Maryland) with the first‐order conditional estimation and the INTERACTION option. Perl Speak NONMEM (PsN version 4.6.0) was used to evaluate the PK model, and the results were further analyzed by R (version 3.5.1 or higher; The R Foundation for Statistical Computing, Vienna, Austria).
The population PK model was developed in 3 stages: structural model selection, covariate analysis, and model evaluation. A small number of postdosing concentrations (0.6%) were below the limit of quantitation and excluded from the analysis. Luspatercept concentrations were natural logarithm‐transformed before the analysis. One‐ and 2‐compartment models as well as nonlinear models were evaluated to describe the concentration‐time profiles of luspatercept. Residual variability was modeled using an additive error model. Interindividual variability was modeled using an exponential error model.
Structural model selection was based on statistical criteria, goodness‐of‐fit plots, and scientific plausibility. For hierarchical models, a reduction of >10 in the objective function value for 1 additional parameter was considered statistically significant (equivalent to P < .002 as assessed by the asymptotically χ2 distributed likelihood ratio test). For nonhierarchical models, a reduction in the Akaike information criterion value was considered an improvement.
The continuous and categorical candidate covariates tested are summarized in Table 1. In addition, subcutaneous injection locations (upper arm, thigh, and abdomen) were tested as a time‐varying covariate. The full‐model approach 8 was used in the covariate analysis, in which all covariate‐parameter relationships of interest were simultaneously incorporated into the model. The final model was derived from the full model by dropping statistically insignificant (95% confidence intervals [CIs] of the covariate effect parameter included the null value) or clinically unimportant (95%CIs of the covariate effect within 25% of the null value) covariates. Stability of the final model was evaluated using the nonparametric bootstrap approach (1000 replicates), and predictive performance of the final model was evaluated by visual predictive checks (VPC) (1000 simulations).
Table 1.
Summary of Patient Characteristics in Population Pharmacokinetic Analysis
| Patient Characteristics | Total (N = 285) |
|---|---|
| Sex, n (%) | |
| Female | 162 (56.8) |
| Male | 123 (43.2) |
| Race, n (%) | |
| White | 181 (63.5) |
| Asian | 82 (28.8) |
| Other (including uncollected or unreported) | 22 (7.7) |
| Renal impairment category, n (%) | |
| No (eGFR ≥90 mL/min/1.73 m2) | 245 (86.0) |
| Mild impairment (eGFR 60‐89 mL/min/1.73 m2) | 37 (13.0) |
| Moderate impairment (eGFR 30‐59 mL/min/1.73 m2) | 3 (1.1) |
| Splenectomy, n (%) | |
| Yes | 170 (59.6) |
| No | 115 (40.4) |
| Genotype, n (%) | |
| β0/β0 | 67 (23.5) |
| Non‐β0/β0 | 153 (53.7) |
| Missing (almost all from phase 2) | 65 (22.8) |
| Concurrent use of ICT, n (%) | |
| Yes | 242 (84.9) |
| No | 21 (7.4) |
| Missing | 22 (7.7) |
| Age (years), median (min‐max) | 32.0 (18.0‐66.0) |
| Weight (kg), median (min‐max) | 57.1 (34.1‐97.0) |
| Erythropoietin (U/L), median (min‐max) | 60.5 (2.4‐972.0) |
| RBCT burden (units/24 weeks), median (min‐max) | 14.1 (0‐34.0) |
| Total bilirubin (μmol/L), median (min‐max) | 32.8 (5.0‐246.0) |
| Albumin (g/L), median (min‐max) | 46.0 (30.0‐56.0) |
| Aspartate transaminase (U/L), median (min‐max) | 22.0 (10.0‐116.0) |
| eGFR (mL/min/1.73 m2), median (min‐max) | 120.0 (53.7‐314.0) |
eGFR, estimated glomerular filtration rate; ICT, iron chelation therapy; max, maximum; min, minimum; n, number of patients in each category; N, overall number of patients; RBCT, red blood cell transfusion.
Monte Carlo simulations were performed using the final model to evaluate the clinical relevance of statistically significant covariates. One hundred clinical trials, with each trial having the same number of patients and the same distribution of covariates as in the 3 clinical studies, were simulated for fixed (71 mg) and weight‐based (1.25 mg/kg) doses. Patients were grouped into 3 subpopulations according to distribution of their covariates: normal (10th‐90th percentiles), low (<10th percentile), and high (>90th percentile). Individual values of area under the concentration‐time curve at steady state (AUCss) and maximum concentration at steady state (Cmax.ss) were derived from the simulation. The percentage difference in the median exposure at low or high covariate values relative to median exposure at normal covariate values was computed using the following equation:
where EXP is steady‐state exposure (AUCss or Cmax.ss) and extreme is either the low or the high covariate values.
Exposure‐Response Analysis
Individual measures of luspatercept serum AUC used in exposure‐response analyses were generated based on empirical Bayes estimates of luspatercept apparent clearance (CL/F) from the final population PK model and actual dosing records.
Pharmacodynamic end points included change from baseline in Hb during weeks 1‐3 and change from baseline in RBC units transfused during weeks 1‐15. The efficacy end points were binary measures of achieving an erythroid response, defined as ≥33% or ≥50% reduction in RBC units transfused from baseline with a reduction of ≥2 RBC units for ≥12 consecutive weeks or ≥33% reduction in RBC units transfused from baseline for ≥24 consecutive weeks. 7 The exposure end point was AUCss of the starting dose for responses during weeks 1‐3 when the first dose was administered. The exposure end point was time‐averaged AUC (AUCavg) over a dosing interval (21 days) for responses determined during a given evaluation period when multiple doses were administered and dose modifications were allowed, calculated as (cumulative dose/[CL/F]/treatment days × 21 days). AUCavg was selected for exposure‐efficacy analyses based on several considerations. First, the effect of luspatercept on Hb lasted for approximately 8 weeks after dosing, 6 and an observed response could be because not only the current dose, but also to a cumulative effect of prior doses plus the current dose. Second, most responders experienced a reduction in RBC transfusion burden through the end of the evaluation period (week 24 or 48); thus, the AUCavg during the 24‐ or 48‐week window better reflected the overall exposure associated with efficacy. Finally, the time‐averaged AUC considered dose modifications during the evaluation period.
The records for treatment‐emergent adverse events (TEAEs) up to 60 days after the last dose as the cutoff dates were pooled from the 3 studies. The safety end points were binary measures of experiencing specified TEAEs, including serious TEAEs, ≥ grade 3 TEAEs, selected ≥ grade 1 TEAEs (asthenia, bone pain, bone pain‐like events, dizziness, hypertension, and myalgia), and TEAEs leading to dose reduction, interruption, or discontinuation. Selection of these TEAEs was based on the severity of TEAEs, imbalance in the incidence between luspatercept and placebo arms, and biologic consideration. The exposure end point was AUCss during the dosing interval when the first event of the specified TEAEs occurred (AUCTEAE), calculated as (actual dose/[CL/F]). The actual dose was the last luspatercept dose administered before or on the start day of the first event for patients who had the specified TEAEs, or the last dose during the evaluation period for patients who did not have any specified TEAEs. It was assumed that TEAEs were more likely to be associated with the most recent exposure level, as the frequency or severity of most TEAEs did not increase with administration of each higher dose.
Exposure‐response modeling for binary end points was conducted using logistic regression in R. Model fitting was performed by first fitting a univariate base model with the luspatercept exposure as the only covariate. For certain end points, the impact of risk factors was examined by adding the candidate covariates one by one to the base model and then in a full covariate model, including all potential factors. The final model was derived from the full model by dropping statistically insignificant factors. In addition, the exposure‐safety relationship over time was explored by Kaplan‐Meier curves stratified by luspatercept AUCTEAE groups, followed by Cox proportional hazards regression analysis. The efficacy and safety data from 109 patients receiving placebo were included in the graphs for visual comparison but were excluded from the exposure‐response modeling.
Results
Luspatercept Population PK Model
The PK population included 285 patients: 64 from study A536‐04 and 221 from study ACE‐536‐B‐THAL‐001. As summarized in Table 1, the patients were young (median age of 32 years), primarily white (63.5%) and Asian (28.8%), and 56.8% were female. The starting dose ranged from 0.2 to 1.25 mg/kg, but most patients (79.8%) received a starting dose of 1 mg/kg. During the first year of treatment, 34% of patients had their dose escalated to 1.25 mg/kg. There were 3680 quantifiable luspatercept serum concentration records collected on days 5 to 610 following the first dose.
A 1‐compartment model with first‐order absorption and elimination best described the concentration‐time profiles of luspatercept after subcutaneous injection. The model was parameterized in terms of the absorption rate constant (Ka), CL/F, and apparent volume of distribution (V1/F). The interindividual variability (IIV) was determined for CL/F and V1/F (Table 2). Inclusion of IIV for Ka led to large shrinkage, indicating insufficient data to inform the numerical estimation of this variable. The PK of luspatercept was linear over the studied dose range, as dose did not have a significant effect on CL/F, and a model in which luspatercept elimination described by a combination of linear and nonlinear (Michaelis‐Menten) terms did not converge. A time‐varying clearance model associated with disease dynamics 9 was ruled out because the observed luspatercept trough concentration was stable during the entire phase 3 study (Figure 1A). The mean elimination half‐life of luspatercept was approximately 11 days. The variability, as indicated by the coefficient of variation from descriptive statistics, for AUCss was 36%.
Table 2.
Parameter Estimates of Final Population Pharmacokinetic Model
| Bootstrap Estimates a | |||
|---|---|---|---|
| Parameter | NONMEM Estimate | Median | 95%CI |
| Fixed effect | |||
| CL/F, L/day | 0.532 | 0.532 | 0.500‐0.569 |
| V1/F, L | 8.39 | 8.39 | 7.97‐8.85 |
| Ka (1/day) | 0.409 | 0.410 | 0.354‐0.481 |
| Weight, kg on CL/F | 0.806 | 0.809 | 0.594‐1.04 |
| RBCT burden (units/24 weeks) on CL/F | −0.0118 | −0.0120 | −0.0186 to −0.00544 |
| Albumin, g/L on CL/F | −0.881 | −0.886 | −1.21 to −0.519 |
| Weight, kg on V1/F | 0.705 | 0.718 | 0.495‐0.916 |
| RBCT burden (units/24 weeks) on V1/F | −0.0141 | −0.0141 | −0.0189 to −0.00921 |
| Random effect | |||
| Interindividual variability of CL/F (%) | 34.7 | 34.1 | 28.2‐44.4 |
| Interindividual variability of V1/F (%) | 27.6 | 27.2 | 18.1‐36.2 |
| Residual variability (%) | 20.8 | 20.6 | 16.9‐25.2 |
CI, confidence interval; CL/F, apparent clearance; Ka, absorption rate constant; NONMEM, nonlinear mixed‐effects modeling software; RBCT, red blood cell transfusion; V1/F, apparent volume of distribution.
Estimated from nonparametric bootstrap procedure (1000 successful replicates).
Figure 1.
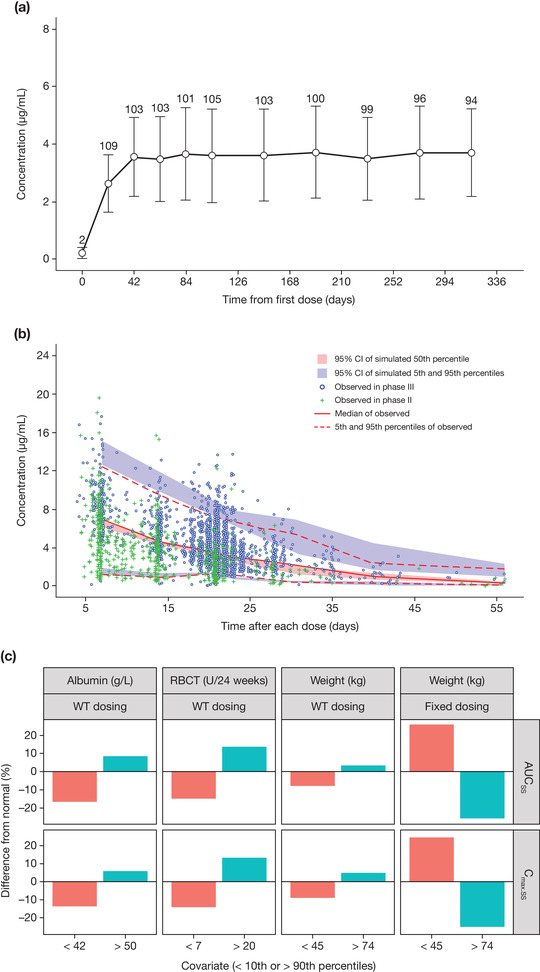
Pharmacokinetic profiles of luspatercept in adult patients with β‐thalassemia. (A) Observed mean (standard deviation) trough serum concentration of luspatercept in the pivotal phase 3 study. (B) Visual predictive check for the final population pharmacokinetic model of luspatercept. (C) Clinical relevance of statistically significant covariates. In (A), only patients who had a dose of 1 mg/kg without any dose modification during the entire pharmacokinetic evaluation period are included, and the number above each error bar shows the number of patients at each point. In (C), the numbers on the x axis represent values < the 10th percentile and > the 90th percentile for the corresponding covariate. % Difference from normal, % difference in median exposure at the low or high covariate values relative to the normal covariate values; AUCss, area under the concentration‐time curve at steady state; CI, confidence interval; Cmax.ss, maximum concentration at steady state; RBCT, baseline red blood cell transfusion; WT, body weight.
There was no obvious bias in the model‐predicted luspatercept concentrations at the population and individual levels or at any specific time (Supplemental Figure S1). Relative differences in parameters were <2% between the final model and bootstrap estimates (Table 2). The VPC plot (Figure 1B) showed that the observed concentration‐time course of luspatercept at the 5th, 50th, and 95th percentiles generally fell within the corresponding 95%CIs of simulated data, indicating that the model adequately characterized the main trend and associated variability of observed data.
Body weight, baseline RBC transfusion burden, and baseline albumin were statistically significant covariates of CL/F. Inclusion of these covariates reduced the IIV of CL/F from 41.2% in the structural model to 34.7% in the final model. The final covariate model for CL/F at the population level is described by the following equation (where RBCT is baseline RBC transfusion burden [RBC units/24 weeks]):
Body weight and baseline RBC transfusion burden were statistically significant covariates of V1/F. Inclusion of the 2 covariates reduced the IIV of V1/F from 33.2% in the structural model to 27.6% in the final model. The final covariate model for V1/F at the population level is described as follows:
The clinical relevance of these covariates was evaluated by PK simulation. The exposure difference between light or heavy patients and normal‐weight patients was predicted to be <10% for weight‐based dosing, but 25% for fixed dosing (Figure 1C). With weight‐based dosing, the exposure difference between patients with extreme values of RBC transfusion burden or albumin and patients with normal values of RBC transfusion burden or albumin was predicted to be <20% (Figure 1C).
Effects of other baseline characteristics of patients, such as age, sex, race (Asian versus white), mild to moderate renal impairment, liver enzymes (alanine transaminase and aspartate transaminase), total bilirubin, serum erythropoietin, β‐thalassemia genotype, and splenectomy status on CL/F or exposure were either insignificant or of low clinical relevance. Locations of subcutaneous injection and concurrent use of iron chelation therapy also had no effect on luspatercept PK.
Exposure‐Response for Hb
The exposure‐Hb relationship was evaluated during the first dosing interval (weeks 1‐3), as the greatest Hb increase during a dosing interval was observed after the first dose, 6 and the response was not confounded by dose modifications. To minimize the interference of RBC transfusions with Hb measures, only Hb values at >14 days after a transfusion in patients with lower transfusion burden (<12 RBC units/24 weeks at baseline) were included for this analysis. In 34 patients who had weekly Hb measures during weeks 1‐3 (all from study A536‐04), increasing luspatercept AUCss was associated with greater Hb increase (average of weekly measures; Figure 2A). This relationship suggested a mean increase of approximately 1 g/dL in Hb after the first dose at 1 mg/kg (mean AUCss = 129 µg·day/mL). In 70 patients who had trough Hb measures (on day 21) for the first dose, increasing luspatercept AUCss was associated with a greater probability of achieving a ≥1 g/dL increase in trough Hb (Figure 2B).
Figure 2.
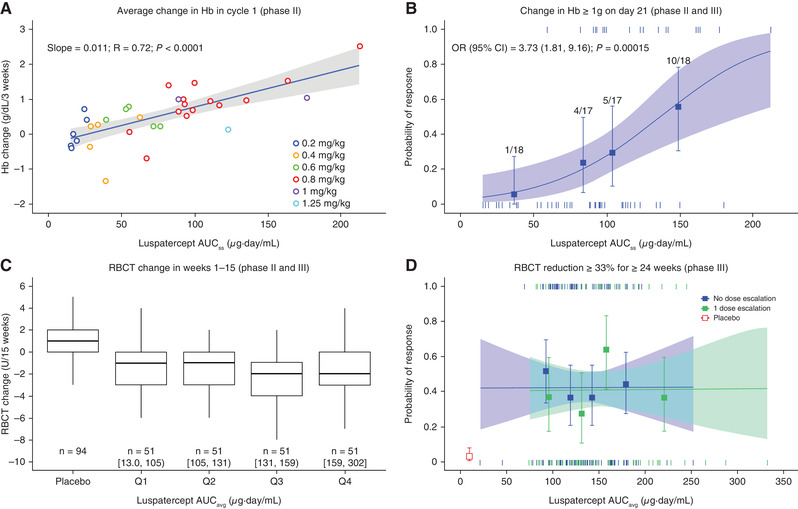
Association of luspatercept exposure with erythroid responses. (A) Linear regression analysis of average of Hb change from baseline as a function of luspatercept serum exposure in treatment cycle 1 in patients with a baseline RBC transfusion burden <12 RBC units/24 weeks (phase 2 study only). (B) Logistic regression analysis of the relationship between luspatercept serum exposure and the probability of achieving ≥1 g/dL increase in Hb on day 21 of treatment cycle 1 in patients with a baseline RBC transfusion burden <12 RBC units/24 weeks (data pooled from phase 2 and 3 studies). (C) Boxplot of change in RBC units transfused during weeks 1‐15 by luspatercept exposure in patients with a baseline RBC transfusion burden ≥12 RBC units/24 weeks (data pooled from phase 2 and 3 studies). (D) Logistic regression analysis of the relationship between luspatercept serum exposure and the probability of achieving ≥33% reduction in RBC transfusion burden for ≥24 weeks (phase 3 study only). In (A), (B), and (D), observed data (circles or squares) and 95%CIs (error bars) are presented along with the predicted regression fits (straight line or slanting lines) and 95%CIs (shaded area). In (B) or (D), vertical ticks at individual values of AUCss or AUCavg represent whether the patient achieved a response (at 1) or not (at 0). In (C), the range of AUCavg is shown for each quartile group. In (B), the numbers above the error bar show the number of patients with the event (numerator) and total number of patients (denominator) within each AUCss quartile. In (D), the red open square and error bar (bottom left) are data from placebo‐treated patients. AUCavg, average area under the concentration‐time curve during weeks 1‐48; AUCss, area under the concentration‐time curve at steady state for the starting dose; CI, confidence interval; Hb, hemoglobin; OR, odds ratio for 50 units of AUCss; Q, quartile; R, correlation coefficient; RBC, red blood cell; RBCT, RBC transfusion.
Exposure‐Response for RBC Units Transfused
The absolute change from baseline in RBC units transfused during weeks 1‐15 was compared across AUCavg quartiles in frequently transfused patients (≥12 RBC units/24 weeks at baseline). All patients in this analysis received an active luspatercept dose level (≥0.6 mg/kg); 97% received a maximum dose level (1‐1.25 mg/kg) during weeks 1‐15. The RBC units transfused were reduced in luspatercept‐treated patients but increased in placebo‐treated patients (Figure 2C). This difference was highly significant when comparing each AUCavg group with placebo group (P < .00001, Tukey test). There was no significant difference in RBC units transfused among luspatercept AUCavg quartile groups.
Exposure‐Response for Efficacy
Exposure‐response analysis was conducted for study ACE‐536‐B‐THAL‐001 (n = 221, patients with baseline RBC transfusion burden ≥6 RBC units/24 weeks) to assess the adequacy of the phase 3 titration dosage (1‐1.25 mg/kg) for efficacy. Stratification of the exposure‐efficacy curve by dose‐escalation status (Figure 2D) showed similar response rates between patients with and without dose escalation, suggesting negligible selection bias resulting from the titration‐to‐response dosing regimen. Therefore, all patients, regardless of their dose‐escalation status, were combined for the analysis. No significant exposure‐dependent trend was observed for any selected efficacy end point in multivariate analyses after accounting for effects of baseline risk factors (Table 3). However, the proportion of patients achieving a response in almost all luspatercept AUC quartile groups was significantly greater than that in placebo‐treated patients (P < .05 in logistic regression; example in Figure 2D).
Table 3.
Parameter Estimates of Final Multivariate Models for Selected Phase 3 Efficacy End Points in Luspatercept‐Treated Patients Who Required Regular Transfusions
| Parameter | Estimate | SE | OR | 95%CI of OR | P |
|---|---|---|---|---|---|
| ≥33% Reduction in weeks 13‐24 | |||||
| AUCavg24, μg·day/mL | 0.00620 | 0.00407 | 1.01 | 0.998‐1.01 | .1275 |
| Age, years | 0.0354 | 0.0154 | 1.04 | 1.01‐1.07 | .0210 |
| RBCT burden (RBC units/24 weeks) | −0.0985 | 0.0464 | 0.906 | 0.827‐0.993 | .0338 |
| ≥33% Reduction in weeks 37‐48 | |||||
| AUCavg48, μg·day/mL | 0.00571 | 0.00399 | 1.01 | 0.998‐1.01 | .1526 |
| Age, years | 0.0386 | 0.0161 | 1.04 | 1.01‐1.07 | .0168 |
| RBCT burden (RBC units/24 weeks) | −0.109 | 0.0498 | 0.896 | 0.813‐0.988 | .0281 |
| Genotype β0/β0 | −1.04 | 0.484 | 0.354 | 0.137‐0.913 | .0317 |
| ≥33% Reduction in any 12 weeks | |||||
| AUCavg48, μg·day/mL | 0.00172 | 0.00348 | 1.00 | 0.995‐1.01 | .621 |
| ≥33% Reduction in any 24 weeks | |||||
| AUCavg48, μg·day/mL | 0.00203 | 0.00376 | 1.00 | 0.995‐1.01 | .58901 |
| Age, years | 0.0461 | 0.0149 | 1.05 | 1.02‐1.08 | .00194 |
| RBCT burden (RBC units/24 weeks) | −0.111 | 0.0419 | 0.895 | 0.824‐0.971 | .00803 |
| Race: Asian (reference = non‐Asian) | −0.832 | 0.331 | 0.435 | 0.228‐0.833 | .01193 |
| Mild/moderate renal impairment | −1.11 | 0.479 | 0.330 | 0.129‐0.844 | .02070 |
| ≥50% Reduction in any 12 weeks | |||||
| AUCavg48, μg·day/mL | −0.00424 | 0.00361 | 0.996 | 0.989‐1.00 | .24111 |
| Age, years | 0.0423 | 0.0143 | 1.04 | 1.01‐1.07 | .00302 |
| RBCT burden (RBC units/24 weeks) | −0.146 | 0.0414 | 0.864 | 0.797‐0.938 | < .001 |
AUCavg24, average area under the concentration‐time curve from weeks 1 to 24; AUCavg48, average area under the concentration‐time curve from weeks 1 to 48; CI, confidence interval; OR, odds ratio; P, tail probability of the normal distribution (Wald test); RBCT, red blood cell transfusion; SE, standard error.
For the 12‐week evaluation period, a reduction of at least 2 units RBCs transfused was required for responders.
Age and RBC transfusion burden were identified as key baseline factors contributing to the variability in individual response to luspatercept (Table 3), with the probability of achieving efficacy decreased with higher baseline RBC transfusion burden (odds ratio [OR], 0.65‐0.74 per 3 RBC unit increase in RBC transfusion burden) and increased with older age (OR, 1.42‐1.59 per 10‐year increase in age). The presence of the β0/β0 genotype, mild/moderate renal impairment, and Asian race appeared to be associated with a reduced response for certain efficacy end points, but these effects were not consistently significant.
The dose level during the first response period was summarized for 2 efficacy end points. For ≥50% reduction in RBC transfusion burden in any 12‐week interval, 80.0% of responders (72 of 90 ) had no dose escalation, and 20.0% of responders (18 of 90) had a dose escalation before or during their first response event (Figure 3A). For a ≥33% reduction in RBC transfusion burden in any 24‐week interval, 70.7% of responders (65 of 92) had no dose escalation, and 29.3% of responders (27 of 92) had a dose escalation before or during their first response event (Figure 3B).
Figure 3.
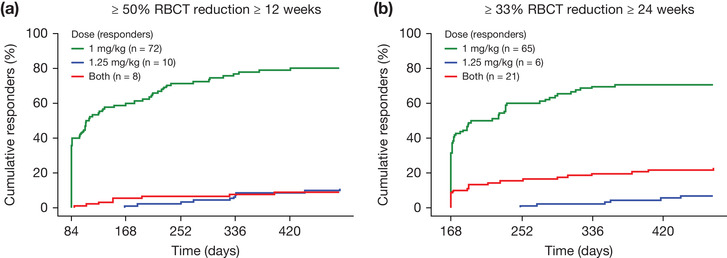
Cumulative response over time for the first event of transfusion reduction by dose level in responders (phase 3 study only). (A) ≥50% RBCT reduction with a reduction of ≥2 RBC units for ≥12 consecutive weeks. (B) ≥33% RBCT reduction for ≥24 consecutive weeks. RBC, red blood cell; RBCT, RBC transfusion.
Exposure‐Response for TEAEs
The safety population included all patients (N = 285) in the PK population. An exposure‐driven increase in an adverse event was not observed in univariate logistic regression analyses for any TEAEs tested (Supplemental Table S3). The effect of exposure was further assessed for TEAEs ≥ grade 3 and bone pain ≥ grade 1, which occurred more frequently in the luspatercept arm than in the placebo arm (P < .05). A flat exposure‐TEAE relationship (Figure 4A,B) was observed for both TEAEs during cycles 1‐2, when the patients were still on the starting dose. In the time‐to‐event analysis for both TEAEs, patients in the highest exposure group had lower incidence of the event at most times compared with patients in lower exposure groups (Figure 4C,D). Most bone pain events were low grade (< grade 3) and transient, often occurring in early treatment cycles.
Figure 4.
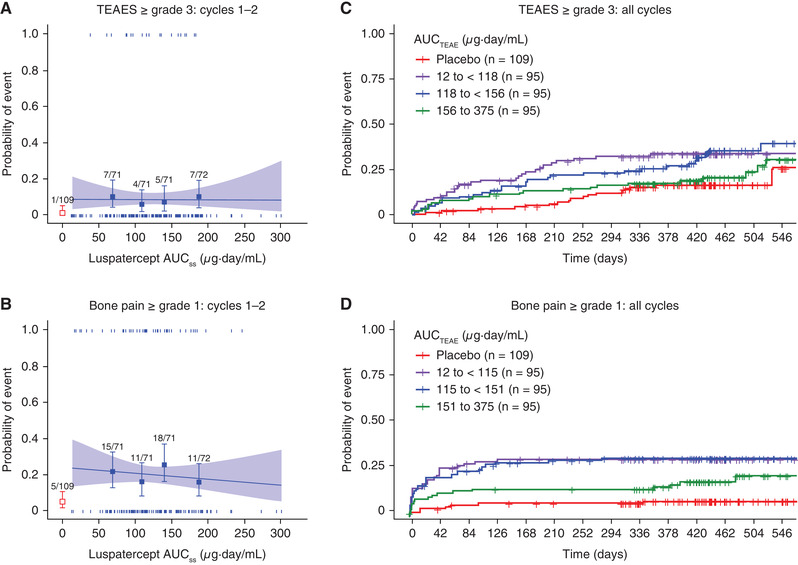
Association of luspatercept exposure with TEAEs ≥ grade 3 or bone pain ≥ grade 1. (A, B) Logistic regression analyses of the relationship between luspatercept serum exposure and the probability of experiencing the event during the first 2 treatment cycles. (C, D) Kaplan‐Meier curves of the time to the first event stratified by the exposure group during the entire study (all available treatment cycles). In (A) and (B), observed proportions (squares) and 95%CIs (error bars) are presented along with the predicted logistic regression fits (slanting lines) and 95%CIs (shaded area). Red open square and error bar (bottom left) are data from placebo‐treated patients. Vertical ticks at individual values of AUCTEAE represent whether the patient achieved an event (at 1) or not (at 0). The numbers above the error bars show the number of patients with the event (numerator) and total number of patients (denominator) within each AUCTEAE quartile. AUCss, area under the concentration‐time curve at steady state for the starting dose; AUCTEAE, area under the concentration‐time curve at steady state during the dosing period when the event occurred; CI, confidence interval; TEAE, treatment‐emergent adverse event.
In the Cox proportional hazards regression analysis for luspatercept‐treated patients (Table 4), the exposure‐associated reduction in TEAEs was marginally significant (P = .0538) for TEAEs ≥ grade 3 and statistically significant for bone pain ≥ grade 1 (P = .0153) after adjusting for the effects of baseline risk factors. Race (Asians vs non‐Asians) was the only baseline factor identified for TEAEs ≥ grade 3, with Asian patients experiencing increased events. Age, sex, weight, baseline RBC transfusion burden, baseline Hb, splenectomy status, β‐thalassemia genotype, and renal function had no statistically significant effect on the probability of experiencing ≥ grade 3 TEAEs. Race and splenectomy were significantly associated with bone pain, with decreased events in patients who were Asian or had splenectomy.
Table 4.
Parameter Estimates of Final Cox Models for Selected TEAEs
| Parameter | Estimate | SE | HR | 95%CI of HR | P |
|---|---|---|---|---|---|
| ≥ Grade 3 TEAEs | |||||
| Asian (vs non‐Asian) | 0.4736 | 0.2338 | 1.606 | 1.016‐2.539 | .0279 |
| AUCTEAE (μg·day/mL) × 50 | −0.2195 | 0.1169 | 0.803 | 0.639‐1.010 | .0538 |
| ≥ Grade 1 bone pain | |||||
| Asian (vs non‐Asian) | −1.3755 | 0.3507 | 0.253 | 0.127‐0.503 | .0002 |
| Splenectomy, yes | −0.6107 | 0.2366 | 0.543 | 0.342‐0.863 | .0216 |
| AUCTEAE (μg·day/mL) × 50 | −0.2718 | 0.1158 | 0.762 | 0.607‐0.956 | .0153 |
AUCTEAE, area under the concentration‐time curve at steady state during the dosing interval when the event occurred; CI, confidence interval; HR, hazard ratio; P, tail probability of the chi‐squared distribution (likelihood ratio test); SE, standard error; TEAE, treatment‐emergent adverse event.
Discussion
The PK of luspatercept in patients with β‐thalassemia was best described by a 1‐compartment model with first‐order absorption and elimination and time‐invariant CL/F. Model evaluation with goodness‐of‐fit plots, bootstrap procedures, and VPC demonstrated robust stability and predictive performance of the final PK model. Thus, the model was deemed appropriate for its intended purpose: evaluation of the need for dose individualization and generation of exposure metrics for use in exposure‐response analyses.
In this young patient population, age was not a significant covariate of luspatercept PK. Both CL/F and V1/F increased with heavier body weight. As suggested by PK simulation, the body weight‐based dosing would perform better than the fixed dosing by limiting over‐ or underexposing patients with extreme body weight. Thus, the effect of weight is considered clinically relevant. Luspatercept CL/F increased with decreasing albumin. This could be an indication of decreased efficiency of neonatal Fc receptor or elevated protein turnover. 9 , 10 Luspatercept CL/F and V1/F were also found to decrease with increasing baseline RBC transfusion burden, presumably reflecting poorer health status because of more severe disease. Despite the statistical significance of baseline albumin or RBC transfusion burden in the covariate analysis, their impact on luspatercept serum exposure appeared less clinically relevant, as <20% difference in luspatercept exposure was predicted for patients with extreme values of albumin or RBC transfusions when dosing was based on weight.
We demonstrated that the magnitude and frequency of Hb increase, if not influenced by RBC transfusions, was positively correlated with luspatercept AUC over the entire clinical dose range (0.2‐1.25 mg/kg). Because the ability of luspatercept to increase Hb is expected to reduce the need for RBC transfusions, the exposure‐Hb relationship provided the rationale for a starting dose of 1 mg/kg (leading to a 1 g/dL increase in Hb after the first dose) and for a further dose increase to 1.25 mg/kg (increasing proportion of patients achieving the effective exposure) in patients requiring regular transfusions for which the treatment goal was to reduce RBC transfusion burden. Consistent with the effectiveness of the selected dose levels, a significant reduction in RBC units transfused was observed in frequently transfused patients, 97% of whom received luspatercept doses of 1 to 1.25 mg/kg. The reduction was not statistically different among AUCavg quartile groups, suggesting that the exposure levels in most patients were within the response plateau region of the exposure‐response curve.
The titration‐to‐response regimen better mimicked the real‐world clinical practice and better reflected benefit‐risk considerations. The true exposure‐efficacy relationship for binary end points, however, could be obscured by selection bias because of dose escalation (eg, patients who received higher dose levels were more likely to be nonresponders compared with patients who remained at lower dose levels). Such selection bias was observed in the exposure‐efficacy analysis for patients with myelodysplastic syndromes. 11 However, this bias was considered minimum in the current exposure‐efficacy analysis for patients with β‐thalassemia, as the luspatercept AUC largely overlapped between 1 and 1.25 mg/kg given a 36% IIV. Moreover, the exposure‐efficacy trend was similar in patients with and without a dose escalation. The exposure‐response relationship was flat after adjusting for effects of baseline risk factors for all tested efficacy end points, indicating a near‐saturated drug effect at the phase 3 exposure range. To understand the contribution of each dose level to efficacy, luspatercept dose associated with the first response event was assessed for patients who achieved greater (≥50%) or more sustained (≥24 weeks) reduction in RBC transfusion burden. The 1 mg/kg starting dose was enough for most of the early responders (71%‐80%), and dose escalation increased the number of responders by at least 20%. These observations confirmed the appropriateness of the 1 mg/kg starting dose and suggested that dose escalation to 1.25 mg/kg might improve rate and durability of response. Starting at a lower effective dose followed by dose escalation depending on the patient's condition would also limit rapid Hb rise and unnecessary exposure to high drug levels. Overall, the exposure‐efficacy analysis demonstrated that the phase 3 dosages were sufficient for achieving meaningful and sustained reductions in RBC transfusion burden.
Under the phase 3 dosage, the variability in individual response to luspatercept was mainly decided by the individual difference in the severity of anemia. As such, the multivariate logistic models suggested that the response rate would be lower in patients with high baseline RBC transfusion burden or in younger patients. The apparent age effect was possibly attributed to multiple disease factors associated with more severe anemia, such as lack of splenectomy and the presence of the β0/β0 genotype, 1 both of which were more frequently seen in younger patients (data on file).
During the entire treatment period, a positive correlation was not observed between luspatercept exposure and any TEAEs tested. However, the incidence of TEAEs ≥ grade 3 was more frequent in the luspatercept arm than the placebo arm in the pivotal phase 3 study. In addition, bone pain was the most frequently reported treatment‐related TEAE that occurred in the luspatercept group. 7 Thus, further analyses were conducted for TEAE ≥ grade 3 and bone pain. During the first 2 cycles, when no dose modifications occurred and almost all patients had the same treatment duration, the exposure‐TEAE relationship was flat. Inspection of cumulative events for the 2 TEAEs over time during the entire study found that the increased TEAEs were mainly observed in the lower‐exposure groups; the incidence of the event was substantially lower in the highest AUCTEAE group compared with the lowest AUCTEAE group at most times. An inverse exposure‐TEAE relationship for luspatercept was also observed in other study populations. 11 The exposure‐TEAE relationship for the entire study could be confounded by dose increases over time or by individual variations in the treatment duration, during which certain adverse events manifest. Such an explanation is not considered likely for the current analysis, as the small dose increase (25%) fell within the individual exposure variability (36%) and the number of patients across AUCTEAE groups during the entire evaluation period was similar. It is possible that luspatercept could induce different biological responses between lower and higher exposures. Certain TEAEs may be mainly associated with biological responses to lower luspatercept exposure, which could be offset by the biological responses to higher luspatercept exposure. From a clinical perspective, the longer‐term exposure‐TEAE relationship is more informative on the safety of luspatercept dosage than that of the first 2 cycles. Overall, it can be concluded that increasing luspatercept exposure up to 1.25 mg/kg was not associated with increased incidence and severity of TEAEs.
A racial difference was observed in the exposure‐TEAE analysis, with Asian patients more likely to experience TEAEs ≥ grade 3, but less likely to experience bone pain. Such racial differences were not triggered by luspatercept treatment, as a similar trend was observed in placebo‐treated patients (data on file). Our analysis also suggested that the probability of experiencing bone pain would be smaller in patients who had a splenectomy. The cause underlying this observation remains unknown and is currently under investigation.
Conclusion
The results from population PK and exposure‐response analyses are informative on the benefit‐risk assessment for use of luspatercept in the treatment of adult patients with β‐thalassemia who require regular RBC transfusions. Body weight was the only clinically relevant covariate of luspatercept PK, supporting weight‐based dosing. Luspatercept exhibits a wide therapeutic margin in the target population. Although increasing luspatercept serum exposure increased Hb, and thus was expected to increase efficacy, it did not increase TEAEs. The favorable benefit‐risk profile in combination with moderate variability (approximately 36%) in serum exposure and individual variations in erythroid response support the titration‐to‐response regimen (1‐1.25 mg/kg once every 3 weeks) to maximize the efficacy potential of luspatercept. The adequacy of such dosage for efficacy was confirmed by the flat exposure‐efficacy relationship in the pivotal phase 3 study.
Conflicts of Interest
N.C., A.L., A.C.G., J.S., S.E.M., P.S., S.R., S.Z., and M.P. are employees of Bristol Myers Squibb. N.K. is an employee of Certara Strategic Consulting and a paid consultant. P.G.L., B.B., and J.G.R. are employees of Acceleron Pharma.
Funding
All clinical data were generated in studies sponsored by Bristol‐Myers Squibb Company or Acceleron Pharma.
Data Sharing
Data requests may be submitted to Celgene, a Bristol‐Myers Squibb Company, at https://vivli.org/ourmember/celgene/ and must include a description of the research proposal.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Acknowledgments
The authors thank all patients and their families and investigators and site staff who participated in these studies. This study was sponsored by Bristol‐Myers Squibb Company, Princeton, New Jersey, in collaboration with Acceleron Pharma, Cambridge, Massachusetts. The authors received editorial and writing support from Jacqueline Moy, PhD, from Excerpta Medica, funded by the sponsor. The authors are fully responsible for all content and editorial decisions for this article.
References
- 1. Rund D, Beta‐thalassemia Rachmilewitz E.. N Engl J Med. 2005;353(11):1135‐1146. [DOI] [PubMed] [Google Scholar]
- 2. Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155‐167. [DOI] [PubMed] [Google Scholar]
- 3. Schmierer B, Hill CS. TGFβ‐SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8(12):970‐982. [DOI] [PubMed] [Google Scholar]
- 4. Suragani RNVS, Cawley SM, Li R, et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine β‐thalassemia. Blood. 2014;123(25):3864‐3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suragani RNVS, Cadena SM, Cawley SM, et al. Transforming growth factor‐β superfamily ligand trap ACE‐536 corrects anemia by promoting late‐stage erythropoiesis. Nat Med. 2014;20(4):408‐414. [DOI] [PubMed] [Google Scholar]
- 6. Piga A, Perrotta S, Gamberini MR, et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with β‐thalassemia. Blood. 2019;133(12):1279‐1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cappellini MD, Viprakasit V, Taher AT, et al. A phase 3 trial of luspatercept in patients with transfusion‐dependent β‐thalassemia. N Engl J Med. 2020;382(13):1219‐1231. [DOI] [PubMed] [Google Scholar]
- 8. Byon W, Smith MK, Chan P, et al. Establishing best practices and guidance in population modeling: an experience with an internal population pharmacokinetic analysis guidance. CPT Pharmacometrics Syst Pharmacol. 2013;2(7):e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu C, Yu J, Li H, et al. Association of time‐varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin Pharmacol Ther. 2017;101:657‐666. PMID:28182273 [DOI] [PubMed] [Google Scholar]
- 10. Andersen JT, Sandlie I. The versatile MHC class I‐related FcRn protects IgG and albumin from degradation: implications for development of new diagnostics and therapeutics. Drug Metab Pharmacokinet. 2009;24(4):318‐332. [DOI] [PubMed] [Google Scholar]
- 11. Chen N, Kassir N, Laadem A, et al. Population pharmacokinetics and exposure–response of luspatercept, an erythroid maturation agent, in anemic patients with myelodysplastic syndromes. [published online ahead of print 2020] CPT Pharmacometrics Syst Pharmacol. 2020; 10.1002/psp4.12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.