

Abstract
Epithelioid glioblastoma (E‐GBM) was recently designated as a subtype of glioblastoma (GBM) by the World Health Organization (2016). E‐GBM is an aggressive and rare variant of GBM that primarily occurs in children and young adults. Although most characterized cases of E‐GBM harbor a mutation of the BRAF gene in which valine (V) is substituted by glutamic acid (E) at amino acid 600 (BRAF‐V600E), in addition to telomerase reverse transcriptase promoter mutations and homozygous CDKN2A/B deletions, the origins and cellular nature of E‐GBM remain uncertain. Here, we present a case of E‐GBM that exhibits antigenic and functional traits suggestive of microglia. Although no epithelial [e.g., CKAE1/3, epithelial membrane antigen (EMA)] or glial (e.g., GFAP, Olig2) markers were detected by immunohistochemical staining, the microglial markers CD68 and Iba1 were readily apparent. Furthermore, isolated E‐GBM‐derived tumor cells expressed microglial/macrophage‐related genes including cytokines, chemokines, MHC class II antigens, lysozyme and the critical functional receptor, CSF‐1R. Isolated E‐GBM‐derived tumor cells were also capable of phagocytosis and cytokine production. Treating E‐GBM‐derived tumor cells with the BRAF‐V600E inhibitor, PLX4032 (vemurafenib), resulted in a dose‐dependent reduction in cell viability that was amplified by addition of the CSF‐1R inhibitor, BLZ945. The present case provides insight into the cellular nature of E‐GBM and introduces several possibilities for effective targeted therapy for these patients.
Keywords: BRAF‐V600E, CSF‐1R, epithelioid glioblastoma, glioblastoma, microglia, vemurafenib
Introduction
Epithelioid glioblastoma (E‐GBM) is a clinical subtype of GBM that was included in the World Health Organization’s 2016 classification of central nervous system tumors (14). Histologically, E‐GBM is characterized by a dominant population of closely packed epithelioid cells and some rhabdoid cells with high mitotic activity, along with microvascular proliferation and necrosis (2). Immunohistochemically, E‐GBM cells generally express vimentin, S100 calcium binding protein P (S100P) and glial markers including glial fibrillary acidic protein (GFAP) and oligodendrocyte transcription factor (Olig2); however, some reports have described E‐GBMs that rarely express glial markers (2, 14). Clinically, E‐GBM is diagnosed primarily in children and young adults. The disease progresses rapidly and has a particularly poor prognosis with a median survival from the time of diagnosis of 5 months in children and 6 months in adults (14).
Over a decade ago, systematic genome‐wide screening analyses revealed that the point mutation BRAF‐V600E (i.e., a valine to glutamic acid substitution at position 600 of the serine/threonine‐specific protein kinase, B‐Raf) was detected in 50%–100% of E‐GBM cases that had been evaluated and in 75% of pleomorphic xanthoastrocytoma (PXA) cases, 47.4% of anaplastic PXAs (A‐PXAs) and 9% of pilocytic astrocytoma (PCA) cases (38). Recent studies have reported that E‐GBM cells commonly harbor telomerase reverse transcriptase (TERT) promoter mutations, homozygous cyclin‐dependent kinase inhibitor (CDKN2A/B) deletions and EGFR amplifications but lack both histone H3F3A K27M and IDH1/IDH2 mutations and the SWI/SNF‐related matrix‐associated actin‐dependent regulator of chromatin subfamily B member 1 (SMARCB1/INI1) and SMARC4/BRG1 (33, 36).
The BRAF‐V600E mutation is a common characteristic of many malignant tumors, including malignant melanoma (50%), papillary carcinoma of the thyroid (50%–90%), lung cancer (3%) and colorectal cancer (5%–10%). The BRAF‐V600E inhibitor PLX4032 (vemurafenib) has been approved for the treatment of malignant melanoma, thyroid papillary carcinoma and various types of lung cancer, and strong clinical responses have been reported (7, 10, 15, 22, 30). The clinical impact of PLX4032 on E‐GBM and related brain tumors is currently under investigation.
Despite substantial progress, the origins, lineage and genetic background of E‐GBM remain unclear. In this study, we characterized the cellular and tissue phenotypes of E‐GBM and assessed the inhibitory effect of PLX4032 both with and without the colony‐stimulating factor 1 receptor (CSF‐1R) antagonist, BLZ945, in assays targeting isolated E‐GBM cells.
Case presentation
A woman in her early twenties was admitted to our hospital with chief complaints of a headache and continuous vomiting for 1 month. She had no personal history of neoplastic disease, and her family history did not suggest a genetic predisposition to malignancy. Magnetic resonance imaging (MRI) revealed a massive lesion in her frontal lobe. The mass presented as a low‐intensity lesion on T1‐weighted images (Figure S1A) and a high‐intensity lesion on T2‐weighted images (Figure S1B); a ring‐enhancing lesion was detected on T1‐weighted postcontrast images (Figure S1C). Based on her clinical history and the MRI results, a diagnosis of glioblastoma multiforme (GBM) was considered. The tumor was surgically removed, and a diagnosis of E‐GBM was confirmed by microscopic evaluation of the tissue. Despite postoperative radiation therapy, she developed symptoms including seizures and diminishing consciousness 1 month later. At that time, MRI revealed hydrocephalus in association with tumor recurrence. Although a secondary procedure was performed to facilitate decompression, her condition deteriorated rapidly, and she died approximately 2 months after the initial diagnosis.
Materials and methods
Clinical study
All studies involving human participants were performed in accordance with the ethical standards of the institution and/or of the national research committee and comply with the 1964 Declaration of Helsinki and its later amendments and/or comparable ethical standards. The Ethics Committee of Hyogo College of Medicine reviewed and approved the study protocol (approval number: 0363, 3309). Informed written consent was obtained from the participant in this study.
Histological examination
The tumor tissue samples were fixed with formalin and embedded in paraffin. The paraffin sections (3‐μm thick) were stained with conventional hematoxylin and eosin staining. Immunohistochemistry was performed using the formalin‐fixed paraffin‐embedded (FFPE) tissues. After deparaffinization and epitope retrieval, the brain sections were incubated with primary antibodies against cytokeratin (#AE1/AE3‐601, Leica Biosystems, Newcastle, UK); epithelial membrane antigen (EMA, #M0613, DAKO, Tokyo, Japan); HMB‐45 (#M0634, DAKO); IDH1 R132H (#DIA‐H09, Dianova, Hamburg, Germany); INI1 (#612111, BD Biosciences, San Diego, CA, USA); histone H3.3 K27M mutation (#ABE413, Merck Millipore, Darmstadt, Germany); GFAP (#PA0032, Leica Biosystems, Newcastle, UK); Olig2 (#IG325, IBL International GmbH, Hamburg, Germany); S100P (#NCL‐L‐S100p, Leica Biosystems); epidermal growth factor wild‐type (#M7298, EGFR‐WT, DAKO); vimentin (#M0725, DAKO), BRAF‐V600E (#790‐5095, Roche Tissue Diagnostics, AZ, USA); p16INK4a (#705‐4793, Roche Tissue Diagnostics); CD45 (#M0754, DAKO), CD68 (#M0876, DAKO); Iba1 (#ab‐153696, Wako, Osaka, Japan), lysozyme (#N1515, DAKO); and CSF‐1R (#ab215441, Abcam, Cambridge, UK). Bound antibodies were reacted using secondary antibodies harboring universal immunoperoxidase (N‐Histofine Simple Stain Human MAX PO, Nichirei Corporation, Tokyo, Japan). Sections were stained with 3,3′‐diaminobenzidine tetrahydrochloride (Vector Laboratories Inc., Burlingame, CA, USA), followed by counterstaining with hematoxylin.
A double immunofluorescent study was performed using primary antibodies against Iba1 (ab5076, Abcam) and BRAF‐V600E (NBP2‐42702, Novus Bioscience, Centennial, CO, USA) or against GFAP (Leica Biosystems) and CSF‐1R (Abcam), followed by Alexa Fluor 488‐ or 555‐conjugated secondary antibodies (Molecular Probes, Eugene, OR), for 1 h at room temperature in the dark. Nuclear staining was performed with 4′,6‐diamidino‐2‐phenylindole (DAPI, Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD, USA). Images were acquired using a confocal laser microscope (LSM780; Carl Zeiss AG, Oberkochen, Germany).
Assessment of the immunoexpression of CSF‐1R
To evaluate CSF‐1R expression in gliomas, FFPE samples of E‐GBM and other gliomas were immunostained with CSF‐1R antibodies, and the expression was assessed by two blinded observers. The expression was scored according to the level of staining intensity and the percentage of positive cells as follows. In brief, the intensity of positive cells was evaluated on a 4‐point scale using the vessels as an internal positive control: 0 (no staining), 1 (weak; staining is weaker than that of the vessels), 2 (moderate; staining is equivalent to that of the vessels) and 3 (strong; staining is stronger than that of the vessels). The percentage of positive cells was also evaluated on a 4‐point scale: 1 (1%–10%), 2 (11%–50%), 3 (51%–90%) and 4 (91%–100%). Then, the scored values for intensity and percentage were multiplied, and the resultant values (0–12) were compared between E‐GBM and non‐E‐GBM samples.
Establishment of E‐GBM and other types of glioblastomas
Tumor cells from E‐GBM tumor tissues were obtained after passage through 18‐ to 22‐gauge needles under sterile conditions. The resulting cell suspensions were seeded into culture dishes and incubated in neurobasal medium (Thermo Fisher Scientific Inc., Waltham, MA, USA); epidermal growth factor (EGF, 20 ng/mL; PeproTech, Rocky Hill, NJ, USA); basic fibroblast growth factor (b‐FGF 20 ng/mL; PeproTech); GlutaMax (1: 100; Thermo Fisher Scientific); N2 supplement (Thermo Fisher Scientific); and 2.5% of fetal bovine serum. The culture medium was changed every 3 days. When the cells reached confluence, they were detached, reseeded and propagated under the same conditions. After three passages, the cells were plated at clonal density as previously described (31, 32, 41).
Conventional non‐E‐GBM types of glioma tissues obtained from two different patients were incubated in neurobasal medium (Thermo Fisher Scientific Inc.), EGF (20 ng/mL; PeproTech), b‐FGF (20 ng/mL; PeproTech), GlutaMax (1:100; Thermo Fisher Scientific) and N2 supplement (Thermo Fisher Scientific). Then, we established two types of glioma cell lines (glioma‐A and glioma‐B) by the same method as described above.
Cell lines and cell cultures
Two different commercially available phenotypes of human glioma cell lines [BRAF‐V600E mutant type (BRAF‐MT): AM38; and BRAF‐V600E wild‐type (BRAF‐WT): U251] were purchased from the Japanese Collection of Research Bioresources and maintained in Eagle’s minimum essential medium (Wako) according to the manufacturers’ instructions.
Human neural stem/progenitor cells derived from H9 embryonic stem cells (NSPCs; Part. No. SCC003, Millipore) and human brain‐derived astrocytes (ACs; #1800, ScienCell Research Laboratories, Carlsbad, CA, USA) were purchased and maintained according to the manufacturers’ instructions. In brief, the NSPCs were plated onto poly‐l‐ornithine/laminin coated dishes and maintained in ENStem‐A Expansion Medium (Millipore, SCM 004) with the addition of b‐FGF and l‐glutamine. ACs were plated on tissue culture dishes (Corning, NY) and maintained in Astrocyte Medium (ScienCell, #1801).
Immunofluorescent staining of cultured cells
The cells that were plated onto poly‐lysine‐coating culture chamber slides (Matsunami, Osaka, Japan) were fixed with ice‐cold methanol for 10 minutes, washed in phosphate‐buffered saline (PBS) and incubated for 30 minutes with 1% of bovine serum albumin (BSA) in PBS with Tween (PBST) for blocking. The cells were then incubated overnight with primary antibodies directed against Iba1 (ab5076, Abcam), BRAF‐V600E (NBP2‐42702, Novus Bioscience), CSF‐1R (Abcam) and IL‐6 (ab6672, Abcam) and diluted in PBST containing 0.3% of BSA. The cells were then washed twice with PBS and incubated with Alexa Fluor 488‐ or 555‐conjugated secondary antibodies (Molecular Probes) for 1 h at room temperature in the dark. Nuclear staining was performed using DAPI (Kirkegaard & Perry Laboratories, Inc.). Images were captured using a confocal laser microscope (LSM780; Carl Zeiss AG).
Detection of the BRAF‐V600E mutation
Genomic DNA was extracted from the tumor cells using DEXPAT (TAKARA, Kyoto, Japan). BRAF exon 15 was amplified by polymerase chain reaction (PCR) with AmpliTaq Gold (PerkinElmer, Norwalk, CT, USA) using the following oligonucleotide primer pairs: BRAF exon 15 forward, 5′‐AATATATTTCTTCATGAAG‐3′; and BRAF exon 15 reverse, 5′‐CATCCACAAAATGGATCCAG‐3′. After purifying the PCR products with the QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA, USA), direct sequencing was performed using ABI BigDye terminator ver. 3.1 (Applied Biosystems, Foster City, CA, USA) and the ABI Prism 3100‐Avant Genetic Analyzer (Applied Biosystems).
Phagocytosis assay
To determine whether the E‐GBM cells were capable of phagocytosis, the tumor cells were plated onto a standard culture dish and incubated for 2 days with FITC‐labeled beads (1‐μm diameter Fluoresbrite® YG carboxylate microspheres; Polysciences Inc., PA, USA) at a dilution of 1:5000, as described previously (37). Images were obtained using time‐lapse microscopy (Eclipse Ti; Nikon, Tokyo, Japan).
Microarray analysis
Total RNA was isolated from the E‐GBM tumor cells (three clonally cultured samples), the glioma‐A and glioma‐B cell lines, the AM38 and U251 cell lines, NSPCs and ACs using the RNeasy Micro Kit (QIAGEN). The microarray analysis, which evaluated RNA quality, labeling, hybridization, scanning and hierarchical clustering, was performed by Takara Bio Inc., Shiga, Japan. In brief, biotinylated cRNA was synthesized from isolated total RNA using the Gene Chip 3’IVT PLUS Reagent Kit (Affymetrix, Thermo Fisher Scientific Inc., Waltham, MA, USA). After validating production using a NanoDrop ND‐2000 Spectrophotometer, cRNA was hybridized for 16 h at 45°C to the GeneChip Human Genome U133 Plus 2.0 Array (Thermo Fisher Scientific, Inc.). The arrays were stained using the GeneChip Fluidics Station 450 (Affymetrix) and scanned using the GeneChip Scanner 3000 7G. A single array analysis was evaluated using Microarray Suite version 5.0 (MAS5.0). The trimmed mean target intensity of each array was randomly set to 500. Unsupervised hierarchical clustering was performed on each sample based on the normalized expression of all genes (total of 54 613 ID genes).
Cell viability assay
To measure the impact of the BRAFV600E inhibitor PLX4032 (Cayman Chemical, Ann Arbor, MI), the CSF‐1R inhibitor BLZ945 (APExBIO Technology, Houston, TX, USA) or their combination on cell viability, E‐GBM‐derived cells were seeded in flat bottom 96‐well plates and incubated under three different conditions: (i) PLX4032 alone (0, 1, 10, 20, 30 or 50 nM); (ii) BLZ945 alone (0, 10, 100, 200, 300 or 500 nM) or (iii) PLX4032 (0, 1, 10, 20, 30 or 50 nM) combined with BLZ945 (100 nM). A CellQuanti‐MTT cell viability assay kit (BioAssay Systems, Hayward, CA) was used to evaluate viability at 4 days according to the manufacturer's protocols. In brief, the MTT reagent (15 μL) was added to each well, followed by a 4 h incubation at 37°C. Solubilizer (100 μL) was then added to each well, followed by gentle shaking at room temperature for 1 h to dissolve insoluble formazan. The absorbance of the supernatants from each well was measured at 570 nm using a SpectraMax Plus 384 (Molecular Devices, San Jose, CA).
To evaluate the cell viability of the E‐GBM tumor cells on the same scale, we set the data obtained from the vehicle control and the cells that received 0 nM PLX4032 as the minimum (0) and maximum (100) points on the Y‐axis, respectively. Then, we converted the value obtained from PLX4032 treatment alone (0, 1, 10, 20, 30 and 50 nM) using the FORECAST function in Microsoft Excel accordingly. Similarly, we converted the value obtained from BLZ945 treatment alone (0, 10, 100, 200, 300 and 500 nM) or PLX4032 (0, 1, 10, 20, 30 and 50 nM) combine with BLZ945 (100 nM) using the FORECAST function in Microsoft Excel accordingly.
To investigate whether additional treatment with BLZ945 can inhibit the cell viability of E‐GBM tumor cells, the IC50 (50% inhibition concentration) value was calculated for the group treated with PLX4032 alone (0, 1, 10, 20, 30 and 50 nM) and for the group treated with PLX4032 (0, 1, 10, 20, 30 and 50 nM) combined with BLZ945 (100 nM) using XLfit5 software; the resultant values were compared between the two groups.
Effect of PLX4032 and BLZ945 on RAS‐MAPK phosphorylation
To investigate the effect of the drugs on molecular signaling, E‐GBM‐derived cells were subjected to serum and factor deprivation for 6 h, and then, cultured for 2 h under the following three conditions: (i) PLX4032 alone (0, 1, 5, 10, 20 or 30 nM); (ii) BLZ945 alone (0, 10, 50, 100, 500 or 1000 nM) or (iii) PLX4032 (0, 1, 5, 10, 20 or 30 nM) combined with BLZ945 (100 nM). Glioma cell lines (AM38 and U251) were used as a control and were subjected to PLX4032 treatment alone (0, 1, 5, 10, 20 or 30 nM) or PLX4032 treatment (0, 1, 5, 10, 20 or 30 nM) combined with BLZ945 (100 nM). After this treatment, the cells were lysed with CelLytic‐M lysis buffer (Sigma‐Aldrich, St. Louis, MO, USA) containing complete protease inhibitor cocktail tablets (Roche, Mannheim, Germany), a phosphatase inhibitor cocktail (Sigma‐Aldrich) and 1 μM sodium orthovanadate. The samples were loaded onto Bolt™ 4%–12% Bis‐Tris Plus gels (Invitrogen, CA, USA) and subjected to electrophoresis, followed by transfer to a membrane using the iBlot™ 2 Dry Blotting System (Life Technologies, CA, USA). To examine the inhibitory effect of PLX4032 and BLZ945 on the phosphorylation of signaling molecules downstream of B‐Raf, immunoblotting was performed using the iBind™ Western System (Life Technologies, CA, USA).
The membranes were incubated with the following primary antibodies: anti‐phospho‐p44/42 MAPK Abs (#9101, Cell Signaling Technology, Beverly, MA); anti‐p44/42 MAPK (#9102, Cell Signaling Technology); anti‐phospho‐STAT1 (#9167, Cell Signaling Technology); anti‐STAT1 (#9172, Cell Signaling Technology), anti‐phospho‐STAT3 (#9136, Cell Signaling Technology); anti‐STAT3 (#9139, Cell Signaling Technology); anti‐phospho‐STAT5 (#9351, Cell Signaling Technology), anti‐STAT5 (#94205, Cell Signaling Technology); and anti‐beta‐actin (A1978, Sigma‐Aldrich) as a loading control. The blots were developed using the ChemiDoc™Touch Imaging system (Bio‐RAD, CA).
Statistical analysis
The results are presented as the mean ± standard deviation. The data were analyzed by a one‐way analysis of variance followed by Bonferroni post hoc tests. Where indicated, comparisons between the two groups were performed using the Student’s t test. A P value of less than 0.05 was considered statistically significant.
Results
Histological findings of the E‐GBM case
As shown in Figure 1A, the tumor cells were loosely connected to one another, which typically occurs in epithelial‐derived cells, and they displayed rhabdoid features with eosinophilic cytoplasm. Necrosis was widely distributed. The epithelial cell markers CK AE1/AE3 and EMA were not detected by immunohistochemical staining; the tumor cells were also negative for isocitrate dehydrogenase 1 (IDH1) R132H, histone H3.3 K27M, Melan A and human melanoma black (HMB)‐45 (data not shown). By contrast, INI1 expression was retained throughout the cells, whereas p16 expression was completely absent (data not shown). Although most tumor cells rarely expressed the glial cell markers GFAP (Figure 1B) or Olig2 (Figure 1C), they expressed vimentin (Fiure 1D), S100P (Figure 1E), BRAF‐V600E (Figure 1F) and EGFR (Figureure 1G). These immunohistochemical findings were consistent with those reported previously for E‐GBM tissue (14). Interestingly, the tumor cells were also positive for CD45 (Figure S2A), which is a cell‐surface antigen common to all hematopoietic cell lineages that has also been detected in immature microglia (21). The microglial/macrophage markers CD68 (Figure 1H), Iba1 (Figure 1I) and lysozyme (Figure S2B) were also detected in the E‐GBM tumor cells.
Figure 1.
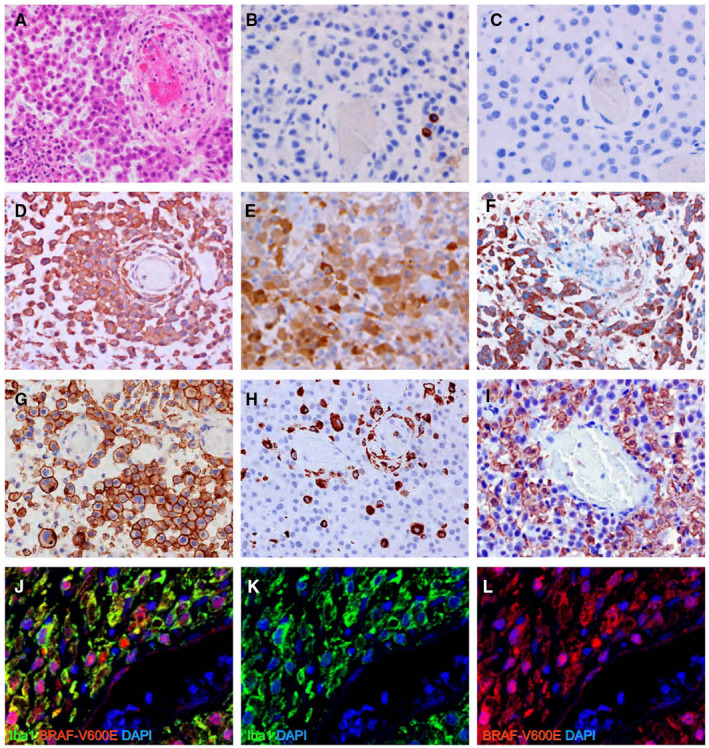
Histological examinations of E‐GBM. H&E staining displayed rhabdoid features with abundant eosinophilic cytoplasm in the tumor cells (A). Immunohistochemistry showed very few GFAP‐positive tumor cells (B) and no Olig2‐positive cells (C). Vimentin (D), S100P (E), BRAF‐V600E (F), EGFR‐WT (G), CD68 (H) and Iba1 (I) were all detected in these tissues. Double immunofluorescence for Iba1 and BRAF‐V600E documented the co‐expression of these markers in E‐GBM tumor cells; neither marker was detected in cells within blood vessels [Iba1 (J, K: green), BRAF‐V600E (J, L: red) and DAPI (J–L: blue)]. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; EGFR, epidermal growth factor wild‐type; E‐GBM, epithelioid glioblastoma; GFAP, glial fibrillary acidic protein; H&E, hematoxylin and eosin; Olig2, oligodendrocyte transcription factor; S100P, S100 calcium binding protein P.
The infiltration of macrophages (so‐called tumor‐associated macrophages; TAMs) has been well‐documented to occur around GBM (11). To eliminate the possibility that E‐GBM cells are derived from TAMs, we performed a double immunofluorescence study for Iba1 and BRAF‐V600E using FFPE samples. The Iba1+ cells were shown to co‐express BRAF‐V600E (Figure 1J–L), indicating that rather than TAMs, the tumor cells themselves expressed the microglia/macrophage marker Iba1.
Cell culture of E‐GBM tumor cells
To explore the functional properties of E‐GBM cells, we prepared single‐cell suspensions from the tissue samples. When cultured under conditions that prevent adherence to the substratum, the E‐GBM‐derived tumor cells exhibited rounded, immature shapes that resembled fetal macrophages (Figure 2A). When cultured under conditions that promote adherence, the isolated cells adopted ramified features with branching projections and evidence of phagocytosis and emperipolesis (Figure 2B). Direct sequencing of DNA extracted from adherent cells in the culture revealed the presence of the heterozygous BRAF‐V600E mutation (Figure 2C), and the adherent cells co‐expressed Iba1 and BRAF‐V600E (Figure 2D–F).
Figure 2.
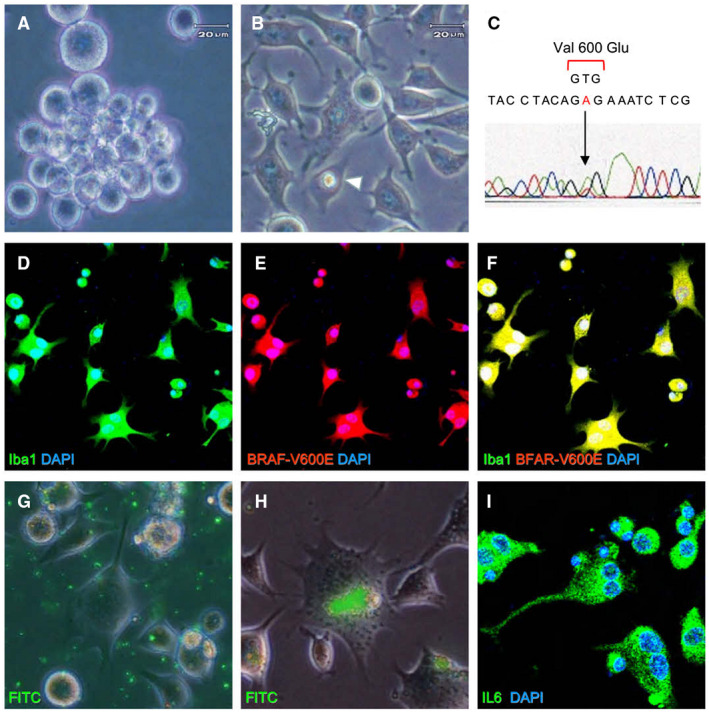
Characterization of E‐GBM‐derived tumor cells ex vivo. Isolated E‐GBM‐derived tumor cells were rounded and exhibited features suggesting immaturity when incubated in untreated culture plates (A). E‐GBM cells cultured in adherent conditions displayed ramifications and emperipolesis (arrowhead) (B). Direct sequencing of the BRAF amplicon from E‐GBM cells revealed a point mutation at codon 600 that resulted in the conversion of valine to glutamic acid (C). Isolated tumor cells co‐expressed Iba1 and BRAF‐V600E [Iba1 (D, F: green), BRAF‐V600E (E, F; red) and DAPI nuclear stain (D–F: blue)]. FITC‐labeled microbeads (G) were engulfed by E‐GBM‐derived tumor cells (H). Immunoreactive IL‐6 was detected as fine granular staining in the cytoplasm of tumor cells [IL‐6 (I: green) and DAPI (I: blue)]. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; E‐GBM, epithelioid glioblastoma.
Given these findings, we considered the possibility that the E‐GBM‐derived tumor cells may also have functional features consistent with those of microglia/macrophages. To explore this possibility, E‐GBM‐derived tumor cells from the single‐cell suspensions were incubated with FITC‐labeled microbeads (Figure 2G). When evaluated 2 days later, the microbeads could be clearly observed within the intracellular compartment. These results highlight the significant phagocytic potential of E‐GBM‐derived cells (Figure 2H).
Because microglia and related macrophages are capable of producing pro‐inflammatory cytokines in response to varied stimuli, we measured the IL‐6, TNFα and IL‐1β levels in the patient serum. We detected prominent elevation in the serum concentrations of IL‐6 (1550 pg/mL; normal range ≤4 pg/mL). Immunofluorescence staining of the E‐GBM‐derived tumor cells revealed the presence of IL‐6‐positive cytoplasmic fine granules (Figure 2I). Taken together, these results suggest that E‐GBM‐derived tumor cells are capable of generating a critical cytokine with complex pro‐inflammatory potential.
Microarray analysis of E‐GBM tumor cells and other types of cells
We then proceeded to examine the gene expression profiles of E‐GBM‐derived tumor cells from three clonally cultured samples via a comparison with those of our established gliomas (glioma‐A and glioma‐B), glioma cell lines (AM38 and U251), and non‐tumor neuroepithelial (NSPCs) and glial (ACs) lineage cell lines.
The principle component analysis (PCA) (Figure 3A) and heat mapping (Figure 3B) results indicated that the gene profiles of the E‐GBM‐derived tumor cells obtained from the three clonally cultured samples were very similar to all types of cells evaluated, indicating that they have similar phenotypes. The results also indicate that we could successfully establish E‐GBM tumor cells. The PCA analysis also showed that U251 cells were closer to ACs than to other types of cells, indicating that the U251 glioma cell line likely has features common to ACs. In support of this idea, the heat mapping results (Figure 3B) revealed strong GFAP expression in both U251 cells and ACs. Traditional lineage markers including nestin and sex‐determining region Y‐box 2 (Sox2) were readily detected in NSPCs. High levels of nestin were also expressed in E‐GBM‐derived tumor cells. Gliomas (glioma‐A and glioma‐B) and glioma cell lines (AM38 and U251) also expressed nestin and Sox2, consistent with previous findings (1, 3). Most interesting, the E‐GBM‐derived tumor cells displayed the marked expression of microglia and macrophage‐related transcripts, including cytokines IL‐1α and IL‐1β, chemokines CXCL1, CXCL3 and CXCL8, the chemokine receptor CCR1, MHC class II antigens (HLA‐DPA1 and HLA‐DRB4) and the microglia/macrophage cell‐surface cytokine receptor CSF‐1R. E‐GBM‐derived tumor cells also expressed lysozyme, the scavenger receptor CD163 molecular‐like 1 (CD163L1), which is a marker for tissue‐resident macrophages, and Mer tyrosine kinase (MERTK), which regulates phagocytosis.
Figure 3.

Microarray analysis. PCA (A) and heat mapping analysis (B) were performed for various types of cells, including E‐GBM tumor cells (three clonally cultured samples), glioma‐A and glioma‐B cells, AM38 and U251 cell lines, NSPCs and ACs. Abbreviations: ACs, astrocytes; E‐GBM, epithelioid glioblastoma; NSPCs, neural stem/progenitor cells; PCA, principle component analysis.
CSF‐1R expression of various types of cells, including E‐GBM tumor cells
CSF‐1R, also known as macrophage colony‐stimulating factor receptor (M‐CSFR), is a class III receptor tyrosine kinase. Human microglia express CSF‐1R, and their proliferation and survival depend directly on the CSF‐1/CSF‐1R signaling pathway (15). Double immunofluorescence staining of the cultured E‐GBM cells revealed that they co‐expressed CSF‐1R and BRAF‐V600E (Figure 4A–C). Furthermore, CSF‐1R expression was detected in the current case and also in four previously diagnosed E‐GBM cases (Figure 4D–M) (Table S1). These results indicate that the expression of this cell‐surface receptor is likely a common feature of E‐GBM.
Figure 4.

Histology and immunohistochemistry for CSF‐1R. E‐GBM‐derived tumor cells co‐expressed CSF‐1R (A, C: green) and BRAF‐V600E (B, C: red); DAPI nuclear staining (A–C: blue), H&E staining (D–H) and immunohistochemistry of CSF‐1R (I–M) in tissues from current (Case 1; D, I) and previously diagnosed cases of E‐GBM (Cases 2–5; E–H, J–M). Abbreviations: CSF‐1R, colony‐stimulating factor 1 receptor; E‐GBM, epithelioid glioblastoma.
To compare CSF‐1R expression among the gliomas, we performed an immunohistochemistry analysis using FFPE samples of these E‐GBM and additional glioma tissues, including PXA, A‐PXA, pilocytic astrocytoma (PCA), pilomyxoid astrocytoma (PMA), diffuse astrocytoma (DA), anaplastic astrocytoma (AA), oligodendroglioma (OD), anaplastic oligodendroglioma (AO) and non‐E‐GBM types of conventional GBM (GBM). The expression score was assessed according to the staining intensity and percentage of positive cells. The results showed that the scored values were significantly higher in E‐GBM (12 ± 0) than in non‐E‐GBM (2.5 ± 1.6) (P < 0.05) samples (Table S1).
Furthermore, we performed double immunofluorescence for CSF‐1R and GFAP using FFPE samples of current and previously diagnosed various types of gliomas, including E‐GBM, PXA, A‐PXA, conventional GBM and AA (Table S2). The results showed that CSF‐1R was highly and diffusely expressed in all E‐GBM samples (Figure 5A–C). No (Figure 5A) or only low levels of GFAP expression (Figure 5B,C) were observed in E‐GBM tumor cells. Compared with E‐GBM, GFAP expression was more strongly and diffusely observed in PXA (Figure 5D,E), A‐PXA (Figure 5F), GBM (Figure 5G–J,L) and AA (Figure 5K). In contrast, CSF‐1R was scattered and faintly expressed in PXA (Figure 5D,E) and A‐PXA (Figure 5F) and rarely and faintly expressed in GBM (Figure 5G–J,L) and AA (Figure 5K). These findings suggest that the strong and diffuse expression of CSF‐1R is likely more common in E‐GBM than in PXA and conventional GBM.
Figure 5.
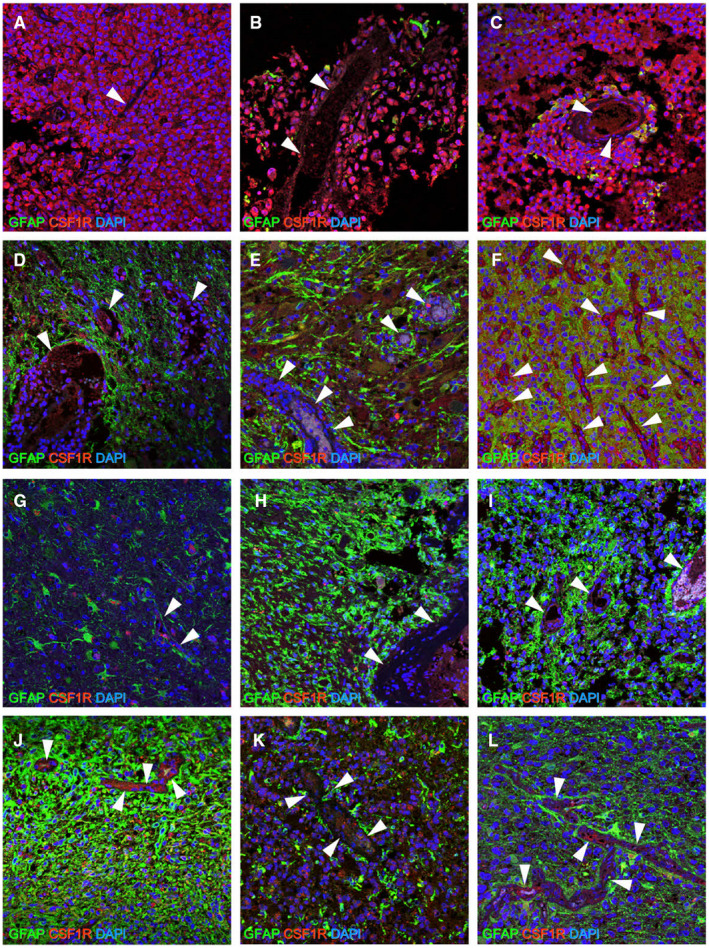
Double immunohistochemistry for CSF‐1R and GFAP. When CSF‐1R expression was compared to that of the vessels (arrow head), which was used as an internal positive control of CSF‐1R, strong and diffuse expression was detectable in all E‐GBM tumor cells [CSF‐1R (Cases 1–3; A–C: red) and DAPI (A–C: blue)]. By contrast, although CSF‐1R expression was observed in PXA [CSF‐1R (Cases 4 and 5; D, E: red) and DAPI (D, E: blue)] and A‐PXA [CSF‐1R (Case 6; F: red) and DAPI (F: blue)], its expression was not stronger than that of the vessels, and it was detectable only regionally. Similarly, in conventional GBM [CSF‐1R (Cases 7–10, 12; G–J, L: red) and DAPI (G–J, L: blue)] and AA [CSF‐1R (Case 11; K: red) and DAPI (K: blue)], CSF‐1R expression was not stronger than that in the vessels. No GFAP+ cells [GFAP (Case 1; A: green)] or only a few GFAP+ cells [GFAP (Cases 2 and 3; B, C: green)] were detectable in E‐GBM, whereas many GFAP+ cells were observed in PXA [Cases 4 and 5; GFAP (D, E: green)], A‐PXA [Case 6; GFAP (F: green)], conventional IDH‐WT GBM [Cases 7–10; GFAP (G–J: green)], IDH‐MT AA [GFAP (Case 11; K: green)] and IDH‐MT GBM [GFAP (Case 12; L: green)] .Abbreviations: AA, anaplastic astrocytoma; A‐PXA, atypical pleomorphic xanthoastrocytoma; CSF‐1R, colony‐stimulating factor 1 receptor; DAPI, 4′,6‐diamidino‐2‐phenylindole; E‐GBM, epithelioid glioblastoma; GBM, glioblastoma; GFAP, glial fibrillary acidic protein; MT, mutant; PXA, pleomorphic xanthoastrocytoma; WT, wild‐type.
Cell viability of E‐GBM tumor cells after treatment with PLX4032 and BLZ945
Our next goal was to examine the impact of the BRAF‐V600E inhibitor, PLX4032, and the CSF‐1R inhibitor, BLZ945, on E‐GBM‐derived tumor cells. First, E‐GBM‐derived tumor cells were treated for 2 h with various concentrations of PLX4032 [0 nM (Figure 6A), 10 nM (Figure 6B), or 30 nM (Figure 6C)]. Apoptosis and cell fragmentation were observed in the concentration of 30 nM of the reagent (Figure 6C).
Figure 6.

Effect of PLX4032 and BLZ945 on cell viability and intracellular signaling molecules. E‐GBM‐derived tumor cells were treated for 2 h with 0 nM (A), 10 nM (B) or 30 nM (C) PLX4032; at 30 nM, PLX4032‐induced apoptosis and cell fragmentation. Cell viability was reduced in E‐GBM‐derived tumor cells treated with 50 nM PLX4032 compared with the baseline (N = 4) (P < 0.05; vs. 0 nM PLX4032) (D). By contrast, the viability of E‐GBM‐derived tumor cells increased in response to more than 300 nM BLZ945 (N = 3) (P < 0.05; vs. 0 nM BLZ945) (E). Reduced viability of E‐GBM‐derived tumor cells was observed in response to more than 30 nM PLX4032 in the presence of 100 nM BLZ945 (P < 0.05; vs. 0 nM PLX4032) (N = 4) (F). Compared with the groups treated with PLX4032 alone, the IC50 value was significantly lower in the groups treated with PLX4032 in the presence of 100 nM BLZ945 (N = 4) (P < 0.05; vs. PLX4032 alone) (G). PLX4032 treatment (30 nM) completely inhibited the phosphorylation of MAPK (H). BLZ945 treatment alone (0–1000 nM) had no effect on any of the signaling molecules in this pathway (I). In the presence of 100 nM BLZ945, PLX4032 exerted complete inhibition of MAPK phosphorylation at lower (20 nM) concentrations (J). Abbreviations: E‐GBM, epithelioid glioblastoma; IC50, 50% inhibition concentration.
To examine the effect of PLX4032 on cell viability, E‐GBM‐derived tumor cells were cultured for 4 days with various concentrations of this inhibitor, followed by the MTT assay to assess cell viability. We found that the cell viability of E‐GBM‐derived tumor cells began to decrease with the addition of 20 nM PLX4032. Furthermore, we found that the cell viability of E‐GBM‐derived tumor cells was significantly decreased after 50 nM PLX4032 treatment (P < 0.05) (Figure 6D). Next, to examine the effect of BLZ945 treatment on cell viability, E‐GBM‐derived tumor cells were cultured for 4 days with various concentrations of this inhibitor. The cell viability of E‐GBM‐derived tumor cells was not significantly altered with lower doses (10, 100 or 200 nM) of BLZ945 treatment (Figure 6E). Interestingly, although 100 nM BLZ945 alone did not affect the cell viability of the E‐GBM‐derived tumor cells, the addition of 100 nM BLZ945 resulted in diminished viability at a much lower concentration of PLX4032 (concentrations greater than 30 nM; P < 0.05) (Figure 6F). These results suggest that the combination of the CSF‐1R inhibitor, BLZ945, with the BRAF‐V600E inhibitor, PLX4032, may be more effective than PLX4032 alone at reducing tumor cell viability. To confirm this finding, we compared the IC50 values between the two groups. The results showed that the IC50 value was significantly lower for combined treatment compared with PLX4032 treatment alone (P < 0.05) (Figure 6G).
Effect of PLX4032 and BLZ945 on the downstream signaling molecules
To investigate the inhibitory mechanism of these drugs, tumor cell cultures were challenged for 2 h with various concentrations of each inhibitor alone (0, 1, 5, 10, 20 or 30 nM of PLX4032 or 0, 10, 20, 50, 100 or 1000 nM of BLZ945) and with a combination of BLZ945 at 100 nM with the various concentrations of PLX4032 mentioned above. PLX4032 inhibited the phosphorylation of MAPK in a dose‐dependent manner; the complete inhibition of MAPK phosphorylation was detected in response to 30 nM PLX4032 (Figure 6H), which is consistent with the concentration at which the cell death of E‐GBM was observed (Figure 6C). CSF‐1R is a class III receptor tyrosine kinase and phosphorylate signaling molecule within the Ras‐RAF‐MAPK pathway. The CSF‐1R inhibitor, BLZ945, had no impact on the phosphorylation of MAPK (Figure 6I). Although seemingly counterintuitive, BRAF‐V600E protein expression results in the constitutive activation of the RAF‐MAPK signaling pathway, which may explain why CSF‐1R inhibition had no effect in this assay. However, when increasing concentrations of PLX4032 were introduced in combination with BLZ945, the phosphorylation of MAPK was completely inhibited at PLX4032 concentrations as low as 20 nM (Figure 6J). Because 20 nM PLX4032 is not highly effective on its own, this result suggests the existence of a specific relationship between PLX4032 and BLZ945. Of note, BLZ945 did not directly affect the phosphorylation of the JAK‐STAT signaling molecules STAT1, STAT3 and STAT5. However, we found that the administration of PLX4032 resulted in a gradual increase in STAT1 and STAT3 phosphorylation, inversely proportional to the observed levels of phosphor‐MAPK (Figure 6H–J). This finding may be associated with an alternative activation pathway for cytokine‐JAK‐STAT signaling.
Further, we investigated the effect of PLX4032 (0, 1, 5, 10, 20 and 30 nM) on non‐E‐GBM tumor cells using two different phenotypes of glioma cell lines (BRAF‐MT AM38 and BRAF‐WT U251). Although the phosphorylation of MAPK in BRAF‐WT U251 cell lines was not inhibited by various concentrations of PLX4032, it was inhibited in BRAF‐MT AM38 cell lines in a dose‐dependent manner (Figure S3). Next, we investigated the effect of PLX4032 (0, 1, 5, 10, 20 and 30 nM) combined with BLZ945 (100 nM). Similar to the results of PLX4032 treatment alone, the phosphorylation of MAPK was not inhibited in U251 cells (Figure S3). Although inhibitory effects were observed in the AM38 cells after the combined treatment, the phosphorylation of MAPK was inhibited at the same dose as PLX4032 alone. These results suggest that the additional treatment of BLZ945 did not enhance the inhibitory effect of PLX4032 on the phosphorylation of MAPK (Figure S3). Consequently, the combined therapy of PLX4032 and BLZ945 is specifically effective in E‐GBM.
Discussion
In this study, we established a primary cell culture from tumor tissue resected from a patient with E‐GBM. The definitive diagnosis of E‐GBM was made based on the following observations: histological features; the presence of immunoreactive for vimentin, S‐100P and INI1; and the absence of IDH1‐R132H, H3.3 K27M, Melan A and HMB‐45. The diagnosis of E‐GBM was confirmed by the detection of the BRAF‐V600E and TERT C228T promoter mutations and the absence of IDH1 and IDH2 mutations (data not shown). Likewise, resected tissues from the E‐GBM tumor displayed only faint expression of the glial marker, GFAP, consistent with previous reports (2, 14). In addition, although Olig2 was not detected in the current E‐GBM case, we cannot definitively conclude that no sections of this sample contain Olig2+ cells. Consistent with histological findings, the microarray analysis showed that the signal intensities for GFAP and/or Olig2 were very weak in the E‐GBM tumor cells obtained from the three clonally incubated samples, although the signal intensities for GFAP and/or Olig2 were highly detectable in other types of gliomas (glioma‐A and glioma‐B) and the glioma cell line U251.
The origins and cell lineages of E‐GBM tumors that rarely express or lack glial markers have not been clarified in the literature. Interestingly, the E‐GBM‐derived tumor cells in this study expressed various microglia/macrophage‐associated genes and were capable of phagocytosis and pro‐inflammatory cytokine production. These findings suggest that E‐GBM‐derived tumor cells may display microglia/macrophage traits.
Adult brains are composed of functional/anatomic structures defined as neurovascular units (NVUs) that consist of neural cells (neurons and astrocytes), endothelial cells and pericytes. During early brain development, neuroepithelial cells produce radial glia that can function as NSPCs; radial glia are ultimately converted into neurons, astrocytes and oligodendrocytes. Cells derived from conventional GBM tissue typically express glial markers, including GFAP (a marker for astrocytes) and Olig2 (a marker for oligodendrocyte progenitor cells (OPCs)). Therefore, conventional GBM is considered to derive from neural stem or glial progenitor cells (PCs), a group that also includes astrocytes and oligodendrocyte PCs (1). Furthermore, we evaluated the transcriptome E‐GBM‐derived tumor cells and noted profound distinctions between gene expression in these cells and the transcriptomes of both NSPCs and ACs (25). In contrast to what is known regarding conventional GBMs, our findings suggest that E‐GBMs might arise from a non‐neuroepithelial origin.
Although the precise origin of null glial marker expressing E‐GBM warrants further clarification, our established tumor cells exhibited major features of microglia/macrophage‐like cells; this result presents the possibility that E‐GBM‐derived tumor cells may originate, at least in part, from the microglia/macrophage lineage. In the brain, tissue‐resident macrophages include microglia and perivascular macrophages. Although the precise origin of brain‐resident macrophages remains uncertain, Kierdorf et al demonstrated that KIT+/CD45− erythromyeloid progenitor cells in the yolk sac generated KIT−/CD45+ myeloid precursors in the developing brain. These cells were identified as progenitors to Iba1+ pro‐microglia in the mid‐embryonal stage and ultimately emerged as mature, ramified microglia after birth (21). Among their findings, the myeloid precursors and embryonic microglia expressed the chemokine CXCL2 and the chemokine receptor CCR1, respectively. Interestingly, both CXCL2 and CCR1 were detected in our E‐GBM‐derived tumor cells. Clearly, E‐GBM‐derived tumor cells have traits in common with myeloid precursors and/or embryonic microglia.
It is also possible that E‐GBM‐derived tumor cells were originally neuroepithelial lineages that transformed to acquire the features of microglia. Cells from conventional GBM tumors can transform into other cell types, notably in response to hypoxia and in association with the epithelial–mesenchymal transition (17). Phillips et al reported that cells from a proneural glioma shifted to a mesenchymal signature upon recurrence and demonstrated the upregulation of CD44, STAT3 and vimentin and the downregulation of Olig2 (34). Similarly, Gouveia and colleagues reported that reprogramed brain pericytes transdifferentiated into microglia neuroepithelial glial lineages in response to hypoxia/ischemia (16). By contrast, other studies indicated that activated microglia were capable of transdifferentiating into neuroepithelial lineages, including astrocytes and oligodendrocytes, under similar conditions (42). These observations indicate that cell lineages within a given tumor type might vary in response to distinct pathological conditions. This hypothesis may also be explained by the evidence that nestin, which was observed in the current E‐GBM sample, was expressed in not only neuroepithelial lineages and NSPCs (28), but also in microglia lineages (26). In addition, E‐GBM is occasionally observed together with PXA, with cells undergoing a transition between the E‐GBM and PXA phenotypes (2). Further investigation will be necessary to determine whether the E‐GBM‐derived tumor cells isolated in this case are originally derived from microglia directly or from another cell lineage via transformation.
Korshunov and colleagues classified E‐GBM into three distinct categories based on specific gene expression patterns (24): type I, PXA‐like tumors with a favorable prognosis that predominantly occur in children and young adults; type II, IDHwt GBM‐like tumors with a poor prognosis that primarily occur in older adults, albeit with more frequent BRAF mutations; and type III, receptor tyrosine kinase (RTK1) pediatric GBM‐like neoplasms of intermediate prognosis in children and young adults that are associated with chromothripsis and platelet‐derived growth factor receptor A (PDGFRA) amplification. According to this classification, our case corresponds most closely to type II E‐GBM; however, we recognize that tumor cell phenotype and specific traits may vary among E‐GBM subtypes.
CSF‐1R, also known as M‐CSF, is a cell‐surface protein that is encoded by the CSF‐1R gene. It has been well‐documented that CSF1/CSF‐1R signaling plays an essential role in the regulation of survival, proliferation and differentiation of phagocytes such as microglia, macrophages and monocytes. In normal brains, low levels of CSF‐1R are expressed on vessel and neural cells, whereas high levels of CSF‐1R are observed in microglia and tissue‐resident macrophages (29). CSF‐1R is also known as an oncogene and is known to be expressed in various types of tumor cells (5), including gliomas (6, 23). Consistent with these findings, we found that CSF‐1R was faintly expressed in non‐E‐GBM types of conventional GBM, PXA, A‐PXA and AA. However, the scored value assessed by the intensity and percentage of cells positive for CSF‐1R demonstrated that the degree of its expression was significantly lower in non‐E‐GBM types of gliomas than in E‐GBM. Additionally, the microarray analysis showed that the signal intensity for CSF‐1R was highly detectable in E‐GBM tumor cells obtained from all three clonally incubated samples, whereas it was weakly detectable in other types of gliomas (glioma‐A and glioma‐B) and glioma cell lines (AM38 and U251). These findings indicate that strong and diffuse CSF‐1R expression is likely a common phenotype in E‐GBM, although further studies using more E‐GBM samples are necessary.
Inhibitors such as dabrafenib and vemurafenib that target BRAF‐V600E are recognized as important therapeutic breakthroughs for patients with malignant melanoma and papillary thyroid carcinoma (7, 10, 15, 22, 30). At this time, the efficacy of these drugs for GBM treatment is unclear; there are only a few reports from compassionate use trials that feature the use of BRAF‐V600E inhibitors in patients with E‐GBM (8, 9, 19, 39). Our study evaluates the efficacy of a BRAF‐V600E inhibitor and its interactions with E‐GBM cells in culture ex vivo. In this study, the BRAF‐V600E inhibitor, vemurafenib, was very effective in reducing tumor cell viability and inhibiting the phosphorylation of a critical intracellular signaling molecule. These results are consistent with those previously reported (19).
Drug resistance is a frequent confounding factor in BRAF‐V600E‐associated tumors. This is a result of the reactivation of the MAPK pathway by feedback activation of RTK and/or the increased expression of ARAF/CRAF, which results in the activation of alternative signaling pathways. To reduce drug resistance, the impact of BRAF inhibitors combined with other modalities including MEK and RTK inhibitors have been examined (12, 18, 19, 35). Given this important direction, we investigated the possibility of targeting CSF‐1R, which we detected on the E‐GBM‐derived tumor cells from our patient and other previously diagnosed patients. We determined that the CSF‐1R inhibitor, BLZ945, had no impact on signaling in E‐GBM‐derived tumor cells when acting alone. Additionally, we investigated whether BLZ945 alone could affect viability in E‐GBM‐derived tumor cells. Although the cell viability of E‐GBM‐derived tumor cells was not significantly altered with lower doses of BLZ945 treatment, it was significantly increased with higher doses of BLZ945 treatment. However, the combination of BLZ945 and PLX4032 effectively inhibited E‐GBM‐derived tumor cells, presumably through a cooperative mechanism. Although the precise mechanism remains unclear, this may relate in part to the effective blockade of feedback activation via CSF1R‐RAS‐RAF‐MAPK signaling. BRAF‐MEK inhibition has been reported to be effective in several recent clinical studies (4, 13, 19, 27); combined therapy with a MEK inhibitor has already been considered a therapeutic strategy for E‐GBM (19).
Interestingly, our evaluation revealed the alternative activation of STAT1 and STAT3 in association with the blockade of RAF‐RAS‐MAPK signaling. The JAK‐STAT signaling pathway is known to play an essential role in modulating the responses of immune and hematopoietic cells. We demonstrated that E‐GBM‐derived tumor cells generate IL‐6, which may promote alternative activation via this pathway. Interestingly, PLX4032 also induced alternative JAK‐STAT pathway activation in cases of malignant melanoma, a feature that has been related to drug resistance (20, 40). Taken together, these results suggest that STAT proteins may also be viable targets for combination therapy with PLX4032 for E‐GBM.
In conclusion, we established cultures of cells isolated from the tumor tissue of a patient with E‐GBM harboring the BRAF‐V600E mutation. These cells exhibited microglial features, including the expression of cell‐surface markers, phagocytosis and dependence on the CSF‐1R pathway for ongoing viability. The BRAF‐V600E inhibitor, PLX4032, was effective alone and in combination with BLZ945 at promoting cell death and diminishing phosphorylation in ex vivo studies. These results suggest that CSF‐1R and potentially the JAK‐STAT signaling pathway might be explored as targets for combined therapy for E‐GBM.
Conflict of interest
Department of therapeutic progress in brain diseases is financially supported by Daiichi Sankyo Co., Ltd., Nippon Zoki Pharmaceutical Co., Ltd. and CLEA Japan, Inc. The sponsors had no roles in this study, including those of study design, data collection, data analysis, data interpretation and manuscript writing.
Author contributions
NN: conception and design, collection and assembly of data, data analysis and interpretation, and manuscript writing; DS: data analysis and interpretation; TH: data analysis and interpretation, and manuscript editing; TT: data analysis and interpretation; MM: collection and assembly of data; TN: data analysis and interpretation, and financial support: SY: data analysis and interpretation; SH: data analysis and interpretation, and financial support. All authors read and approved the final manuscript.
Supporting information
Figure S1. Diagnosis and characterization of E‐GBM. Neuroimaging [T1‐weighted MRI (A), T2‐weighted MRI (B), and T1‐weighted postcontrast MRI (C)] on admission revealed a mass in the frontal lobes.
Figure S2. Immunohistochemistry for CD45 and lysozyme. Immunohistochemistry showed that CD45 (A) and lysozyme (B) were focally expressed in E‐GBM.
Figure S3. Western blot analysis using BRAF‐MT (AM38) and BRAF‐WT (U251) glioma cell lines. Western blotting showed that the phosphorylation of MAPK in U251 cells was not inhibited by PLX4032 alone or by PLX4032 combined with BLZ945. Although the phosphorylation of MAPK in AM38 cells was inhibited by PLX4032 alone or by PLX4032 combined with BLZ945, the inhibitory effect on the phosphorylation of MAPK by PLX4032 was not enhanced with the addition of BLZ945. Abbreviations: BRAF‐MT, BRAF‐V600E mutant type; BRAF‐WT, BRAF‐V600E wild type.
Table S1. Clinical information and CSF‐1R expression scores of samples of E‐GBM (Cases 1–5) and non‐E‐GBM types of gliomas (Cases 6–31).
Table S2. Clinical and genetic information of the gliomas assessed by double immunofluorescence.
Acknowledgments
We would like to thank A. Nakano‐Doi, T. Yokoyama, A. Narita, M. Okouchi and Y. Hashikura for their helpful assistance.
Data availability statement
The data set used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1. Alcantara Llaguno SR, Parada LF (2016) Cell of origin of glioma: biological and clinical implications. Br J Cancer 115:1445–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexandrescu S, Korshunov A, Lai SH, Dabiri S, Patil S, Li R et al (2016) Epithelioid glioblastomas and anaplastic epithelioid pleomorphic xanthoastrocytomas‐same entity or first cousins? Brain Pathol 26:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Annovazzi L, Mellai M, Caldera V, Valente G, Schiffer D (2011) SOX2 expression and amplification in gliomas and glioma cell lines. Cancer Genom Proteom 8:139–147. [PubMed] [Google Scholar]
- 4. Bahrami A, Hesari A, Khazaei M, Hassanian SM, Ferns GA, Avan A (2018) The therapeutic potential of targeting the BRAF mutation in patients with colorectal cancer. J Cell Physiol 233:2162–2169. [DOI] [PubMed] [Google Scholar]
- 5. Barbetti V, Morandi A, Tusa I, Digiacomo G, Riverso M, Marzi I et al (2014) Chromatin‐associated CSF‐1R binds to the promoter of proliferation‐related genes in breast cancer cells. Oncogene 33:4359–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bender AM, Collier LS, Rodriguez FJ, Tieu C, Larson JD, Halder C et al (2010) Sleeping beauty‐mediated somatic mutagenesis implicates CSF1 in the formation of high‐grade astrocytomas. Cancer Res 70:3557–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H et al (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF‐mutant melanoma. Nature 467:596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brown NF, Carter T, Kitchen N, Mulholland P (2017) Dabrafenib and trametinib in BRAFV600E mutated glioma. CNS Oncol 6:291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ceccon G, Werner JM, Dunkl V, Tscherpel C, Stoffels G, Brunn A et al (2018) Dabrafenib treatment in a patient with an epithelioid glioblastoma and BRAF V600E mutation. Int J Mol Sci 19:1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW et al (2017) Cellular and molecular identity of tumor‐associated macrophages in glioblastoma. Cancer Res 77:2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP et al (2012) EGFR‐mediated re‐activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2:227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dratkiewicz E, Simiczyjew A, Pietraszek‐Gremplewicz K, Mazurkiewicz J, Nowak D (2019) Characterization of melanoma cell lines resistant to vemurafenib and evaluation of their responsiveness to EGFR‐ and MET‐inhibitor treatment. Int J Mol Sci 21:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ellison DW, Kleinschmidt‐DeMasters BK, Park SH (2016) Epithelioid glioblastoma. In: WHO Classification of Tumors of the Central Nervous System, 4th edn. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 50–51. IARC Press: Lyon. [Google Scholar]
- 15. Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP et al (2012) Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose‐escalation trial. Lancet 379:1893–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gouveia A, Seegobin M, Kannangara TS, He L, Wondisford F, Comin CH et al (2017) The aPKC‐CBP pathway regulates post‐stroke neurovascular remodeling and functional recovery. Stem Cell Rep 9:1735–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iwadate Y (2016) Epithelial‐mesenchymal transition in glioblastoma progression. Oncol Lett 11:1615–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johanns TM, Ferguson CJ, Grierson PM, Dahiya S, Ansstas G (2018) Rapid clinical and radiographic response with combined dabrafenib and trametinib in adults with BRAF‐mutated high‐grade glioma. J Natl Compr Canc Netw 16:4–10. [DOI] [PubMed] [Google Scholar]
- 19. Kanemaru Y, Natsumeda M, Okada M, Saito R, Kobayashi D, Eda T et al (2019) Dramatic response of BRAF V600E‐mutant epithelioid glioblastoma to combination therapy with BRAF and MEK inhibitor: establishment and xenograft of a cell line to predict clinical efficacy. Acta Neuropathol Commun 7:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaoud TS, Mohassab AM, Hassan HA, Yan C, Van Ravenstein SX, Abdelhamid D et al (2019) NO‐releasing STAT3 inhibitors suppress BRAF‐mutant melanoma growth. Eur J Med Chem 186:111885. [DOI] [PubMed] [Google Scholar]
- 21. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG et al (2013) Microglia emerge from erythromyeloid precursors via Pu.1‐ and Irf8‐dependent pathways. Nat Neurosci 16:273–280. [DOI] [PubMed] [Google Scholar]
- 22. Kim KB, Cabanillas ME, Lazar AJ, Williams MD, Sanders DL, Ilagan JL et al (2013) Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF(V600E) mutation. Thyroid 23:1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Komohara Y, Horlad H, Ohnishi K, Fujiwara Y, Bai B, Nakagawa T et al (2012) Importance of direct macrophage‐tumor cell interaction on progression of human glioma. Cancer Sci 103:2165–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Korshunov A, Chavez L, Sharma T, Ryzhova M, Schrimpf D, Stichel D et al (2017) Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol 28:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kriegstein A, Alvarez‐Buylla A (2009) The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32:149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krishnasamy S, Weng YC, Thammisetty SS, Phaneuf D, Lalancette‐Hebert M, Kriz J (2017) Molecular imaging of nestin in neuroinflammatory conditions reveals marked signal induction in activated microglia. J Neuroinflammation 14:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M et al (2014) Combined vemurafenib and cobimetinib in BRAF‐mutated melanoma. N Engl J Med 371:1867–1876. [DOI] [PubMed] [Google Scholar]
- 28. Lendahl U, Zimmerman LB, McKay RD (1990) CNS stem cells express a new class of intermediate filament protein. Cell 60:585–595. [DOI] [PubMed] [Google Scholar]
- 29. Luo J, Elwood F, Britschgi M, Villeda S, Zhang H, Ding Z et al (2013) Colony‐stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J Exp Med 210:157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R et al (2014) Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation‐positive melanoma (BRIM‐3): extended follow‐up of a phase 3, randomised, open‐label study. Lancet Oncol 15:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakagomi N, Nakagomi T, Kubo S, Nakano‐Doi A, Saino O, Takata M et al (2009) Endothelial cells support survival, proliferation, and neuronal differentiation of transplanted adult ischemia‐induced neural stem/progenitor cells after cerebral infarction. Stem Cells 27:2185–2195. [DOI] [PubMed] [Google Scholar]
- 32. Nakagomi T, Taguchi A, Fujimori Y, Saino O, Nakano‐Doi A, Kubo S et al (2009) Isolation and characterization of neural stem/progenitor cells from post‐stroke cerebral cortex in mice. Eur J Neurosci 29:1842–1852. [DOI] [PubMed] [Google Scholar]
- 33. Nakajima N, Nobusawa S, Nakata S, Nakada M, Yamazaki T, Matsumura N et al (2018) BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions are frequent in epithelioid glioblastomas: a histological and molecular analysis focusing on intratumoral heterogeneity. Brain Pathol 28:663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD et al (2006) Molecular subclasses of high‐grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9:157–173. [DOI] [PubMed] [Google Scholar]
- 35. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D et al (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483:100–103. [DOI] [PubMed] [Google Scholar]
- 36. Rodriguez FJ, Scheithauer BW, Giannini C, Bryant SC, Jenkins RB (2008) Epithelial and pseudoepithelial differentiation in glioblastoma and gliosarcoma: a comparative morphologic and molecular genetic study. Cancer 113:2779–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rustenhoven J, Park TI, Schweder P, Scotter J, Correia J, Smith AM et al (2016) Isolation of highly enriched primary human microglia for functional studies. Sci Rep 6:19371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. [DOI] [PubMed] [Google Scholar]
- 39. Smith‐Cohn M, Davidson C, Colman H, Cohen AL (2019) Challenges of targeting BRAF V600E mutations in adult primary brain tumor patients: a report of two cases. CNS Oncol 8:CNS48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang K, Li Y, Song N, Che X, Hou K, Xu L et al (2019) Signal transducer and activator of transcription 3 inhibition enhances vemurafenib sensitivity in colon cancers harboring the BRAF(V600E) mutation. J Cell Biochem 120:5315–5325. [DOI] [PubMed] [Google Scholar]
- 41. Weiss S, Dunne C, Hewson J, Wohl C, Wheatley M, Peterson AC, Reynolds BA (1996) Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. J Neurosci 16:7599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yokoyama A, Sakamoto A, Kameda K, Imai Y, Tanaka J (2006) NG2 proteoglycan‐expressing microglia as multipotent neural progenitors in normal and pathologic brains. Glia 53:754–768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Diagnosis and characterization of E‐GBM. Neuroimaging [T1‐weighted MRI (A), T2‐weighted MRI (B), and T1‐weighted postcontrast MRI (C)] on admission revealed a mass in the frontal lobes.
Figure S2. Immunohistochemistry for CD45 and lysozyme. Immunohistochemistry showed that CD45 (A) and lysozyme (B) were focally expressed in E‐GBM.
Figure S3. Western blot analysis using BRAF‐MT (AM38) and BRAF‐WT (U251) glioma cell lines. Western blotting showed that the phosphorylation of MAPK in U251 cells was not inhibited by PLX4032 alone or by PLX4032 combined with BLZ945. Although the phosphorylation of MAPK in AM38 cells was inhibited by PLX4032 alone or by PLX4032 combined with BLZ945, the inhibitory effect on the phosphorylation of MAPK by PLX4032 was not enhanced with the addition of BLZ945. Abbreviations: BRAF‐MT, BRAF‐V600E mutant type; BRAF‐WT, BRAF‐V600E wild type.
Table S1. Clinical information and CSF‐1R expression scores of samples of E‐GBM (Cases 1–5) and non‐E‐GBM types of gliomas (Cases 6–31).
Table S2. Clinical and genetic information of the gliomas assessed by double immunofluorescence.
Data Availability Statement
The data set used and/or analyzed during the current study are available from the corresponding author on reasonable request.