

Building on the status quo of metastasis research, this review highlights key bottlenecks in the spatiotemporal progression of metastasis and provides a framework for propelling bench-to-bedside translation of fundamental preclinical discoveries.
Abstract
Historically, therapy of metastatic disease has essentially been limited to using strategies that were identified and established to shrink primary tumors. The limited efficacy of such treatments on overall patient survival stems from diverging intrinsic and extrinsic characteristics of a primary tumor and metastases originating therefrom. To develop better therapeutic strategies to treat metastatic disease, there is an urgent need to shift the paradigm in preclinical metastasis research by conceptualizing metastatic dissemination, colonization, and growth as spatiotemporally dynamic processes and identifying rate-limiting vulnerabilities of the metastatic cascade. Clinically, while metastatic colonization remains the most attractive therapeutic avenue, comprehensive understanding of earlier steps may unravel novel metastasis-restricting therapies for presurgical neoadjuvant application. Moving beyond a primary tumor-centric view, this review adopts a holistic approach to understanding the spatial and temporal progression of metastasis. After reviewing recent developments in metastasis research, we highlight some of the grand challenges and propose a framework to expedite mechanism-based discovery research feeding the translational pipeline.
Introduction
Cancer is an evolutionary disease in which, most often, a sporadic genetic alteration bestows a growth advantage to an otherwise physiologically normal cell (Hanahan and Weinberg, 2011). Over time, mutations accumulate, resulting in a neoplastic lesion that subsequently, although rarely, turns malignant. The early 1900s witnessed advances in surgical techniques and development of aggressive chemo- and radiotherapies for treating primary tumors. The declaration of the “war on cancer” in 1971 led researchers to discover cancer-causing genes, including oncogenes and tumor suppressors, and to develop strategies to therapeutically target them in the hope of restricting cancer growth (Wheeler and Wang, 2013). With the turn of the century and successful realization of the Human Genome Project, global oncology research entered the new era of genomic medicine.
The availability of a reference genome and affordable technological platforms to undertake next-generation sequencing fueled large-scale intercontinental consortia such as The Cancer Genome Atlas, the Catalogue of Somatic Mutations in Cancer, the Encyclopedia of DNA Elements, the International Cancer Genome Consortium, and the recently concluded Pan-Cancer Analysis of Whole Genomes Consortium (ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020; Garraway and Lander, 2013; Shaw and Maitra, 2019). These large-scale concerted efforts have revolutionized our understanding of the genetic makeup of cancer and expanded our knowledge from a few hundred genes to instead mapping genetic interactions at the whole-genome level. The explosion of genomic data also led to a comprehensive characterization of different subtypes and grades of cancers relying on the underlying mutational landscape and maladapted signal transduction pathways (Garraway and Lander, 2013; Suhail et al., 2019). These research efforts have enormously expanded the mechanistic understanding of interpatient and intratumor heterogeneity and have diversified clinicians’ armamentarium to curtail cancer growth. Alongside these developments, changes in lifestyle and medical practices, including large-scale screening approaches to identify asymptomatic patients, resulted in a decline of cancer incidence from the peak observed in 1991 (Siegel et al., 2020). Today, despite achieving multiple clinical milestones in curbing primary disease, metastasis remains the primary cause of cancer-related mortality.
Distant metastasis may be considered the pinnacle of tumor cell evolution. Individual cells or a subset of cancer cells break the physical boundaries of a primary tumor and manage to execute a series of rate-limiting steps to eventually colonize secondary sites (Welch and Hurst, 2019). Metastasis-initiating cells (MICs; reviewed in Celià-Terrassa and Kang, 2016; Oskarsson et al., 2014) potentially colonize distant sites in cancer patients already at the time of initial diagnosis. MICs often exhibit very distinct transcriptomic and phenotypic characteristics when compared with the originating primary tumor (Celià-Terrassa and Kang, 2016). Hence, it comes without surprise that metastatic tumors are much more resistant to therapies effective in debulking the originating primary tumor (Brabletz et al., 2013). Indeed, the ever-growing repertoire of oncology drugs has only modestly prolonged overall survival (OS) of metastatic patients during the past two decades, and the 5-yr survival of stage IV cancer patients remains <20% for the majority of solid tumors to this day (Siegel et al., 2020). Hence, there is an urgent need to better understand the pathogenesis of metastasis to identify bottlenecks in the metastatic process, thereby developing mechanism-based therapies for restricting metastatic progression. Moreover, it is imperative to perceive a metastatic tumor as an independent entity rather than treat it as an extension of the primary tumor. Along with reviewing the recent developments in the field of metastasis, this review aims at recognizing grand challenges and providing a framework to stimulate discussion on possible avenues to overcome these bottlenecks in order to identify targets for antimetastatic therapy.
Status quo and challenges
Successful metastatic growth reflects the survival of MICs through a series of rate-limiting steps over the duration of months to years (Welch and Hurst, 2019). While the majority of tumors remain focally confined and benign, a minority turn malignant when tumor cells break through local boundaries to invade adjacent normal tissue. Subsequently, malignant cells may penetrate into blood and lymphatic vessels to spread throughout the body. Rarely, these disseminated tumor cells manage to evade the biophysical stress in the circulation and the local immune attack at a secondary site to progress to macrometastatic lesions. This multistep sequential process of malignant cells disseminating from a primary tumor to colonize a noncontiguous site is referred to as the “metastatic cascade.” The limited mechanistic knowledge and the overwhelming complexity of the process make metastasis an unresolved enigma to this day. In the following section, we highlight recent seminal discoveries that contributed to the limited understanding of the metastatic cascade.
Go versus grow state of malignant cells
The epithelium consists of differentiated epithelial cells tightly woven in continuous sheets that rely on the underlying stroma for nourishment and growth cues. Cancerous transformation of an epithelial cell makes it independent of external growth factors and offers unrestricted proliferation to yield a tumor (Hanahan and Weinberg, 2011). Successively, a subset of tumor cells may undergo so-called epithelial-to-mesenchymal transition (EMT), a developmental process that fosters cellular mobility (Fig. 1 A; Kalluri and Weinberg, 2009). The induction of the EMT signaling program results in (1) repressing epithelial adhesion genes, allowing tumor cells to detach from the epithelial sheet, and (2) activating mesenchymal migratory genes to assist the invasion of tumor cells into the basement membrane and surrounding tissues (Aiello and Kang, 2019). Moreover, EMT bestows stem cell–like properties on the tumor cells, thereby promoting their ability to rapidly adapt and survive in a hostile environment (Mani et al., 2008). While the EMT program aids the initial steps of the metastatic cascade—namely invasion, intravasation, survival in the circulation and, potentially, extravasation—the reverse program, mesenchymal-to-epithelial transition (MET), has been implicated in facilitating the outgrowth of seeded malignant cells (Chaffer et al., 2007; Tsai et al., 2012). Concurrently, EMT features are prominently visible in micrometastases, whereas macrometastases largely display epithelial characteristics (Aiello et al., 2016), suggesting that the reversal of EMT is crucial for successful metastatic colonization (Fig. 1 B). The transient nature of the EMT and MET processes are an inherent major contributor to tracing these processes in archived pathology specimens.
Figure 1.

EMT. (A) During development, epithelial cells undergo a set of reversible, yet sequential, morphological changes to acquire mesenchymal characteristics. (B) Tumor cells activate similar developmental programs to attain a mesenchymal state that not only bolsters their migratory ability but also assists in the survival of disseminated cells in the circulation and upon extravasation at a noncontiguous site. Seeded MICs revert to an epithelial state to actively proliferate and subsequently colonize the metastatic site. The figure was generated with BioRender.com.
The fate of tumor cells undergoing EMT/MET is regulated by several key EMT-inducing transcription factors (EMT-TFs), such as SNAIL, TWIST, and ZEB (Brabletz, 2012). Diverging phenotypes have been observed in the genetic knockouts of individual EMT-TFs. For example, while the genetic deletion of either SNAIL or TWIST did not influence tumor progression and metastasis (Fischer et al., 2015; Zheng et al., 2015), depletion of the EMT activator ZEB1 resulted in a reduced fraction of tumors progressing to high-grade cancers and consequently impeded the formation of distant metastases (Krebs et al., 2017). These controversies stem not only from technical issues such as incomplete deletion efficacy but also compensatory mechanisms and overall difficulties in proving that an epithelial metastasis actually underwent EMT (Williams et al., 2019). In an attempt to induce a mesenchymal state, scientists genetically deleted E-cadherin in tumor cells. E-cadherin–depleted cancer cells showed a multifold reduction in the number of metastatic foci as compared with control cells (Padmanaban et al., 2019). Mechanistically, loss of epithelial identity rendered tumor cells vulnerable to environmental stress and induced apoptotic signals, suggesting a crucial dependence of seeded metastatic cells on MET for successful colonization. Together, these observations have instigated intense research effort to decipher the underlying molecular mechanisms that control the temporal regulation of EMT/MET during metastasis.
To explain these findings, EMT/MET, rather than being a binary event (comprising the epithelial and mesenchymal phenotypes), was proposed to reflect a spectrum of hybrid phenotypes where cancer cells may activate the EMT/MET program to various degrees in a contextual manner (Brabletz et al., 2018; Nieto et al., 2016; Williams et al., 2019). Indeed, screening a large panel of cell surface markers identified subpopulations of cells displaying different extents (partial to full) of EMT coexisting in a primary tumor (Pastushenko and Blanpain, 2019; Pastushenko et al., 2018). Overall, the induction of the EMT program endows malignant cells to enter a migratory “go” state to enable their body-wide spread at a compromise of their proliferative capacity, whereas the MET program allows a seeded cell to “grow” and colonize a distant organ (Nieto, 2017; Suhail et al., 2019). Recently, the EMT International Association published a white paper to guide research on EMT/MET with the goals to reduce misinterpretation of published scientific data and to provide a framework for propelling international collaboration (Yang et al., 2020a). With an increasing understanding of tumor cell plasticity and its vital importance during metastasis and response to therapy, it is critical to answer questions such as what factors control the extent of EMT induction in tumor cells, whether different tissue microenvironments instigate different EMT-TFs in metastasizing cancer cells, and if disseminated tumor cells can be kept dormant by restricting them in their mesenchymal state.
Hematogenous versus lymphogenous route of metastatic dissemination
Rapid expansion of the vascular network is essential to support tumor growth. In turn, tumor-associated blood and lymphatic vessels serve as escape routes from the primary site (Fig. 2), because both routes give malignant cells access to the systemic circulation and thereby to distant organs (Paduch, 2016). Lack of interendothelial tight junctions and low-shear fluid flow conditions present the lymphatic drainage as a favorable route for dissemination. Indeed, clinical observations have long reported a much higher frequency of melanoma- and carcinoma-associated regional LNs (for diagnostic reasons, also called sentinel LNs) to be positive for cancer cells, such as in patients with sarcomas (Wong and Hynes, 2006). Furthermore, LNs have been proposed to assist in selecting cancer cells with high metastatic potential, which may subsequently drain into the blood either via the thoracic duct or by infiltrating LN blood vessels (Krishnan et al., 2003). Yet, multicenter phase III clinical trials reported no long-term survival advantage for melanoma patients undergoing complete dissection of draining LNs as compared with observation only (Faries et al., 2017; Leiter et al., 2016). Similarly, lack of additional benefit was reported for women with invasive breast cancer undergoing additional axillary LN dissection compared with sentinel lymphadenectomy only (Giuliano et al., 2017). Concurrent with lymphadenectomy trials, genomic comparison of matched primary and metastatic melanoma specimens unveiled that parallel seeding of genetically distinct tumor clones gives rise to regional and distant metastatic tumors (Sanborn et al., 2015). Likewise, phylogenetic mapping of hypermutable genomic regions revealed that lymphatic and distant organ metastases originated from different primary tumor clones in nearly two-thirds of colorectal cancer patients (Naxerova et al., 2017). However, in the remaining one-third of patients, liver and LN metastases shared a common tumor cell progeny, suggesting either simultaneous dissemination of an individual tumor clone via lymphatic and hematogenous routes or sequential seeding, wherein LNs acted as a transient reservoir for distant liver metastasis.
Figure 2.

Individual and collective dissemination of tumor cells. Neoplastic cells can enter the circulation via either a hematogenous or lymphogenous route. Tumor cells tend to cluster together or may aggregate with immune cells to survive in the circulation. While the sentinel LNs often present a wide interlesion tumor cell heterogeneity, the clonal composition of metastases at the distant organ site displays substantially lower complexity. The figure was generated with BioRender.com.
While clinical studies so far suggest no additional survival advantage for patients undergoing sentinel LN removal, it remains experimentally inconclusive whether distant metastases originate solely from tumor cells disseminated via the hematogenous route with no contribution from the lymphogenous route. In preclinical mouse models, focal photoconversion of fluorescent protein Dendra2 in LN-seeded tumor cells allowed researchers to trace the origin of distant metastases (Pereira et al., 2018). Indeed, nearly 70% of lung-metastasized tumor cells were mapped to the LN origin, indicating that LNs act as an intermediary reservoir for distant metastases. Likewise, intralymphatic inoculation of tumor cells led to rapid colonization of LN and direct intravasation into LN blood vessels without involving the thoracic duct (Brown et al., 2018). Concomitantly, active lymphangiogenesis was observed at distant metastatic sites, including the lungs. This was positively correlated with metastatic burden, indicating that lymphatic vessels seed malignant cells to secondary sites (Ma et al., 2018). Furthermore, genetic ablation of lymphatic vessels in tumor-bearing mice strongly suppressed metastatic dissemination without influencing the growth of primary tumors (Chen et al., 2018). Indeed, performing lymphadenectomy at the time of primary tumor resection resulted in long-term survival of mice in a spontaneously metastasizing melanoma model that relies on the lymphogenous route for distant metastases (Gengenbacher et al., 2020). Moreover, exposure to the lymphatic environment was described to protect disseminated melanoma cells from undergoing ferroptosis and facilitate distant metastasis (Ubellacker et al., 2020).
While these preclinical studies solidly establish the importance of the lymphatic route for distant metastases, the contribution of lymphogenous spread to fatal distant metastases remains debated in clinical practice. Future research will need to focus on investigating varying reliance of different cancer entities on the lymphatic route for dissemination and whether dissimilar levels of immunogenicity of tumor cells prime the host immune cells and influence their cytotoxic response toward tumor cells at distant organs. Another major question is the biological relevance of peri- and intratumoral lymphatics and whether the presence of either can predict the preferential route of metastatic spread. The better mechanistic understanding of hematogenous versus lymphogenous spread may have immediate translational implications because it may lead to the development of stratifying procedures to identify subpopulations of patients likely to benefit from lymphadenectomy during primary tumor surgery.
Single versus collective dissemination of metastatic cancer cells
A large number of circulating tumor cells (CTCs) may enter the circulation. Yet, only a select few complete the journey to finally give rise to metastatic colonies (estimated to be <0.02% based on the experimental preclinical models; Luzzi et al., 1998; Micalizzi et al., 2017; Strilic and Offermanns, 2017). While the primary tumor offers an immunosuppressive environment for cancer cells, CTCs face the host immune attack in the circulation. Likewise, a combination of other stresses challenge the survival of CTCs, including anoikis as well as high shear and oxidative stress (Strilic and Offermanns, 2017). Circulating emboli of multiple tumor cells and the association of CTCs with immune cells may provide a survival advantage and enhance the metastatic potential as compared with individual CTCs (Cheung and Ewald, 2016; Micalizzi et al., 2017; Strilic and Offermanns, 2017). Indeed, intravenous injection of tumor cell clumps resulted in significantly higher numbers of pulmonary metastases compared with single tumor cells (Liotta et al., 1976). Likewise, breast cancer cells showed collective dissemination with aggregated cells, forming a multifold higher number of lung metastatic colonies compared with the injection of dissociated cells (Cheung et al., 2013, 2016). Indeed, CTC clusters were described to exhibit an altered epigenetic landscape when compared with matched single CTCs, including enrichment of gene pathways related to stemness and proliferation (Gkountela et al., 2019). Likewise, association with neutrophils was found to be highly advantageous for the survival of CTCs (Szczerba et al., 2019). Mechanistically, neutrophil-associated CTCs expressed higher levels of genes related to cell cycle progression and benefited from various neutrophil-derived cytokines and growth signals. Concomitantly, the presence of circulating neutrophil–CTC clusters was positively correlated with disease progression in patients with advanced breast cancer (Egeblad and de Visser, 2019). Similarly, platelet aggregation not only allowed CTCs to evade the innate immune response but also facilitated transendothelial migration (Schumacher et al., 2013; Stegner et al., 2014; Strilic and Offermanns, 2017). Platelet–tumor cell contact promoted EMT-like transition and facilitated metastatic progression (Labelle et al., 2011). Overall, collective dissemination provides metastatic tumor cells a better chance to survive in the circulation (Fig. 2).
Several lineage-tracing studies have shown that metastasis arising from clustered CTCs is often oligoclonal in nature (Cheung and Ewald, 2016). CTC clusters composed of genetically distinct clones displayed a 50–100-fold higher probability than individual clones of forming distant metastasis (Aceto et al., 2014; Cheung et al., 2016). Intriguingly, while a large fraction of peritoneal and diaphragm metastases were found to be polyclonal, liver and lung metastases were largely monoclonal in a genetically engineered mouse model of pancreatic cancer (Maddipati and Stanger, 2015). Such organ-specific variations in the clonal composition of metastatic tumors may arise from differences in niche-specific tropic factors.
In line with preclinical studies, nearly 30% of patients with melanoma, breast, and prostate cancer had detectable circulating CTC clusters (Sarioglu et al., 2015). Clinically, the presence of circulating tumor emboli correlated with poor progression-free survival and OS of patients with small-cell lung cancer (Hou et al., 2012). Further phylogenetic assessment of paired primary and metastatic tumor specimens revealed complex polyclonal origins of metastatic tumors (Turajlic and Swanton, 2016). Polyclonal seeding contributed to 59% of LN and 29% of distant metastatic tumors (Hu et al., 2020). Correspondingly, a wide interlesion tumor cell heterogeneity in draining LNs was hypothesized to select the fittest tumor clones that will eventually seed distant organ sites (Reiter et al., 2020). Metastatic cross-seeding, where clones from a metastatic lesion seed secondary sites, adds another layer of complexity in defining the origin and clonality of resulting metastatic cancers in patients with advanced prostate cancer (Gundem et al., 2015).
The polyclonal nature of metastatic tumors poses a major hurdle to treating them, because therapy-resistant clones emerge and result in disease relapse (Massagué and Obenauf, 2016). Future research will need to deconvolute the alterations in the clonality of metastatic tumors during therapy as well as the mechanisms underlying site-specific bias in the clonality of metastasis-initiating clusters. While preclinical studies have unambiguously associated CTC clusters with poor prognosis, future clinical studies will have to investigate if the variable presence of tumor microemboli can be exploited as an independent prognostic determinant to assess an individual patient’s response to therapy, especially considering that metastatic seeding may have occurred before initial diagnosis. Last, the comprehensive multiomic analysis of CTCs may allow clinicians to tailor personalized therapy aimed at eradicating seeded metastatic cells.
Microenvironmental control of metastatic colonization
Disseminated tumor cells may stay undetected in a dormant state for long periods (ranging from months to even decades) at noncontiguous sites before beginning to actively grow (Massagué and Obenauf, 2016). This two-step process, involving a forced dormancy followed by a reawakening of malignant cells, remains the least well-understood step of the metastatic cascade. The unprecedented complexity of metastatic colonization arises from a wide array of synergistically acting tumor cell–intrinsic and tumor cell–extrinsic factors. While the genetic evolution of tumor cells and intrinsic drivers have been studied in much detail (as recently reviewed in Phan and Croucher, 2020; Summers et al., 2020), the notion that malignant cells rely on the tissue microenvironment for colonization has gained momentum only in the last decade (Liu and Cao, 2016). Host cells, including vascular, mesenchymal, resident, and infiltrating immune cells, together constitute a metastatic niche to host disseminated cells and impose various molecular checkpoints that an MIC must overcome for successful colonization (Fig. 3; Peinado et al., 2017).
Figure 3.

Metastatic niche–imposed molecular checkpoints. Seeded tumor cells closely interact with different stromal cells within the metastatic niche. MICs escape dormancy by inducing the angiogenic switch (activation of otherwise quiescent ECs to induce sprouting angiogenesis), remodel the ECM, and orchestrate an immunosuppressive environment to collectively favor the colonization process. The figure was generated with BioRender.com.
Blood vessel–lining endothelial cells (ECs) serve as a docking interface for CTCs, which eventually transmigrate to abluminal vascular niches. Malignant cells rely on ECs not only for nourishment but also for different growth cues, called “angiocrine factors” (Butler et al., 2010; Pasquier et al., 2020; Singhal and Augustin, 2020). In adults, resting ECs express the decoy chemokine receptor DARC, which may, by physically interacting with KAI1/CD82 on CTCs, induce tumor cell dormancy (Bandyopadhyay et al., 2006). Gene expression of DARC and KAI1 was found negatively correlated with metastatic progression for different cancer entities. Likewise, vascular niche–expressed TSP1 promoted senescence of seeded tumor cells (Ghajar et al., 2013). Similarly, EC-derived IGFBP7 acted as a tumor-suppressive checkpoint. Consequently, the loss of IGFBP7 enhanced aggressiveness and facilitated chemoresistance by activating stemness-related gene signatures in disseminated cells (Cao et al., 2017). In contrast to the resting vasculature, activated vascular niches support the growth of metastatic tumors. For example, the maladapted bone vasculature was recently shown to promote metastatic colonization by instigating MET in breast cancer CTCs in a SOX2/9-dependent manner (Esposito et al., 2019). Overall, ECs impart instructive cues to dictate the fate of a seeded malignant cell.
Tumor cells traveling to distant sites are trapped in the next capillary bed that they encounter, simply because the diameter of a capillary is smaller than the diameter of tumor cells. The next capillary bed is therefore oftentimes the preferred site of metastatic colonization (i.e., peripheral tumors giving rise to lung metastases or gastrointestinal tumors metastasizing to the liver). Yet, tumor cells may also pass through the first capillary bed to colonize a distant site independent of anatomical trapping (e.g., prostate tumors preferentially metastasizing bones or melanomas to the brain). Such secondary colonization may be driven largely by physiological compatibility of CTCs to an environment more conducive in some organs than in others. The nonrandom metastatic pattern of many tumors was observed in pathology specimens >130 yr ago and led to the seed-and-soil hypothesis of metastasis; i.e., the seed (the tumor cell) must fall on fertile soil (the target organ) in order to successfully form a metastasis (Fidler, 2003; Peinado et al., 2017). The understanding of the mechanisms driving site-specific metastatic patterns of different cancer indications remains limited to this day (reviewed in Celià-Terrassa and Kang, 2018; Obenauf and Massagué, 2015).
Organotypic blood vessels have long been known to mitigate selective infiltration of hematopoietic cells and orchestrate cytokine amplification in different organs during pathological conditions such as cancer (De Palma et al., 2017). Lung ECs strongly up-regulate ANG2 during metastatic progression. In turn, autocrine-acting ANG2, in a STAT3-dependent manner, resulted in the up-regulation of chemoattractant molecules such as IL-6, CCL2, and the adhesion molecule ICAM1 that facilitated the recruitment of protumorigenic myeloid cells to the metastatic niche (Srivastava et al., 2014). In a similar experiment, sustained activation of endothelial NOTCH signaling resulted in the overexpression of a plethora of inflammation-inducing chemokines (Wieland et al., 2017). The enhanced secretion of chemoattractant signals led to the infiltration of myeloid-derived suppressor cells into the metastatic niche, thereby generating a protumorigenic environment. Correcting the vascular niche by either genetic deletion or pharmacological targeting of EC-derived angiocrine factors suppressed metastatic growth and prolonged OS of mice (Kim et al., 2017; Srivastava et al., 2014; Wieland et al., 2017).
Mesenchymal cells provide a scaffold for more specialized epithelial cells to reside and perform their physiological functions (Nelson and Bissell, 2006). Additionally, mesenchymal cells secrete balanced amounts of soluble factors, including mitogens and morphogens, for maintaining tissue architecture. These developmental programs are often hijacked by cancer-associated fibroblasts to aid metastatic progression (Sahai et al., 2020). In the presence of a primary tumor, lung mesenchymal/perivascular cells were shown to activate the pluripotency gene Klf4, thereby switching to a more proliferative synthetic state (Murgai et al., 2017). Subsequently, an increased number of perivascular cells results in the enhanced deposition of extracellular matrix (ECM) and facilitates metastatic colonization of intravenously injected cancer cells. Furthermore, integrin-β3low perivascular cells regulate metastatic growth via paracrine release of various cytokines (Wong et al., 2020). In a very compelling experiment, researchers implanted tumors in young and aged mice and discovered that fibroblasts secrete the WNT antagonist sFRP2 to drive melanoma metastasis and foster resistance to targeted therapy such as vemurafenib (Kaur et al., 2016). Likewise, the loss of fibroblast-secreted HAPLN1 was associated with an increased risk of distant metastasis (Ecker et al., 2019; Kaur et al., 2019).
An integrated multiomic analysis of human metastatic biopsies of high-grade serous ovarian cancer revealed a matrix index, an ECM-associated molecular signature, that profoundly correlated with poor prognosis (Pearce et al., 2018). Furthermore, the matrix index was inversely correlated with the infiltration of CD8+ T cells, suggesting that a stiffened matrix may limit the host immune response to invading malignant cells. Indeed, the reversal of ECM stiffening in the liver improved the efficacy of antiangiogenic therapies for metastatic colorectal cancers (Shen et al., 2020). Looking forward, modifications in the matrix composition at the metastatic site could prove beneficial to enhance the efficacy of immune checkpoint (IC) inhibitors toward metastatic cancers. While the crucial role of cancer-associated fibroblasts has been well recognized, future research will also need to investigate the influence of dysfunctional mesenchymal cells on ECM remodeling within the metastatic niche and its influence on metastatic colonization.
The primary tumor offers an immunosuppressive environment, largely composed of myeloid cells, for transformed epithelial cells to evade the host immune response and to proliferate unrestrictedly (Altorki et al., 2019; Lim et al., 2018; Ritter and Greten, 2019). Metastasis can occur during early tumorigenesis, and similar modulation of the metastatic site can be linked to the outgrowth of metastatic colonies (El-Kenawi et al., 2020; Garner and de Visser, 2020; Swierczak and Pollard, 2020). For example, malignant cells evoked a systemic inflammatory response by granulocyte colony-stimulating factor–mediated expansion of neutrophils, which, in turn, suppressed cytotoxic CD8+ T cells and permitted disseminated tumor cells to establish ectopic colonies (Coffelt et al., 2015). Postsurgical adjuvant administration of low-dose epigenetic therapy could interfere with the trafficking of myeloid cells into the metastatic site by down-regulating key chemokine receptors, thereby resulting in long-term disease-free survival in preclinical cancer models (Lu et al., 2020). Similarly, natural killer cells actively pruned metastatic colonies by eliminating single-seeded CTCs while sparing CTC clusters (Lo et al., 2020). Malignant cells instigated regenerative transcriptomic programs to escape natural killer cell–mediated immunoediting in a SOX9-dependent manner (Laughney et al., 2020). Likewise, multiplexed interrogation of clinical metastatic specimens revealed that the evolution of tumor clones was closely associated with changes in the immune microenvironment at the metastatic site (Angelova et al., 2018). Moreover, ECM remodeling induced by neutrophil-extruded proteases was sufficient to awaken otherwise dormant metastatic clones (Albrengues et al., 2018). Neutrophil extracellular traps could additionally facilitate the proliferation of seeded tumor cells in an integrin-dependent manner (Yang et al., 2020b). Overall, the past decade has witnessed major scientific discoveries expanding the hitherto minimalistic understanding of the metastatic niche and its contribution to the most deadly stages of metastasis. Yet, recent knowledge has stimulated additional questions and opened novel avenues for future research. Envisioning an enormous potential of metastatic niche–targeted therapies across the spectrum of cancer entities, it is crucial to foster global research efforts to deconvolute the spatiotemporal evolution of the metastatic niche and to explore potential ways to correct it.
Perspectives
As cancer outpaces cardiovascular diseases to become the most common cause of death in high-income and middle-income nations (Dagenais et al., 2020), there is an urgent need for a paradigm shift in the pursuit of metastasis-focused oncology research from being largely fundamental discovery to rather translational therapy oriented. The recent seminal discoveries outlined in this review have identified molecular mechanisms underlying different aspects of metastatic progression that offer avenues for clinical translation. With a clear vision of translation, it is crucial to intensify preclinical efforts to expand the existing portfolio of therapy targets to prevent not only the formation of metastases but also the growth of established metastatic lesions. In this section, we discuss focus areas that we believe have the potential not only to expedite the pace of metastatic research but also to bridge the gap between the bench and the bedside.
Adopting latest technologies to advance metastasis research
As in the past, technology will continue to advance oncology research. Next-generation sequencing has enormously expanded the knowledge of tumor-intrinsic drivers as well as interpatient heterogeneity and their differential response to therapy (Shaw and Maitra, 2019). The increasing availability of single-cell analysis platforms allows researchers to investigate tumor heterogeneity and alterations in the metastatic microenvironment at the highest possible resolution (Lim et al., 2020). Recent benchmarking efforts have resulted in white papers to standardize single-cell transcriptomics, thereby guiding individual laboratories as well as global consortia to generate high-quality datasets that can be unified for large-scale meta-analysis (Ding et al., 2020; Mereu et al., 2020; Rood et al., 2019; Stuart et al., 2019). In addition to providing quantitative gene expression information, spatial transcriptomics adds another layer of information regarding the physical location of the analyzed cell in the tissue microenvironment (Eng et al., 2019; Rodriques et al., 2019; Ståhl et al., 2016).
The field of imaging has similarly witnessed groundbreaking discoveries in recent years. The growing repertoire of tissue clearance techniques allows three-dimensional (3D) histopathological analysis of both preclinical and clinical specimens (Ueda et al., 2020). Whole-tissue 3D high-resolution imaging enabled investigators to study intratumoral plasticity and precisely stage clinical biopsies (Rios et al., 2019). Likewise, noninvasive whole-body imaging relying on the differential refractive index of malignant cells could trace micrometastases at single-cell resolution (Kubota et al., 2017). The combination of tissue clearing with a deep learning–based automated detection platform allowed the mapping of dormant malignant cells that had escaped therapy (Pan et al., 2019). More recently, with use of a lipid-permeable fluorescent labeling system, metastatic niche cells in the vicinity of a seeded tumor cell could be isolated to dissect contact-based reprogramming of the metastatic microenvironment (Ombrato et al., 2019).
Recapitulating every aspect of the multistep nature of human tumor progression in preclinical research models is probably the most challenging bottleneck in the development of metastasis-targeted therapies. 3D organoid cultures and zebrafish avatars preserve tumor heterogeneity and have therefore rapidly emerged as drug-screening platforms. Yet, the inherent lack or mismatch of stroma limits their applicability for metastasis research (Drost and Clevers, 2018; Fazio et al., 2020). While the toolbox of cancer researchers has been expanding at a rapid pace, mouse tumor models remain the most powerful and indispensable tool for metastasis research (Gengenbacher et al., 2017). Different strategies have been employed to capture different aspects of metastatic disease in the mouse (see text box). Correspondingly, machine learning–based executable computational models can be established by integrating acquired knowledge from experimental preclinical and clinical studies (Clarke and Fisher, 2020). Such computational models are anticipated to simulate already in the near future the dynamic multicellular cross-talk in the metastatic niche and will not only accelerate preclinical research but also potentially help clinicians to choose a therapy based on genomic data of a patient’s tumor.
Modeling metastasis in mice
Mouse tumor models are indispensable experimental tools for oncology research because they truthfully capture the intimate cross-talk between cancer cells and their local microenvironment. Autochthonous tumors in genetically engineered and environmentally induced tumor models have enormously expanded our understanding of the early steps of tumor progression. Yet, the heavy multifocal primary tumor burden often marks the ethical endpoint of the experiment, thereby limiting the evaluation of metastasis in such models. Contrastingly, grafted cell line–based or patient-derived tumors grow focally and can therefore be surgically removed to allow sufficient time for spontaneous metastasis formation to occur at distant organs. For multifocal autochthonous models, a potential solution might be tumor-derived allografts, which metastasize from single orthotopically transplanted tumor fragments or organoids that can subsequently be surgically resected. In line with clinical practice, only surgical removal of a primary tumor can truthfully make metastasis rate limiting for tumor progression and mortality of mice.

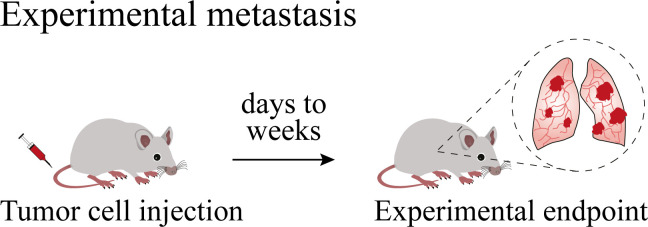
In experimental metastasis models, cancer cell lines are systemically (mostly intracardially, intraperitoneally, intrasplenically, or intravenously) injected for developing organ-specific metastasis. Experimental metastasis models allow investigation of the colonization process at a predetermined organ site, but in the absence of preconditioning of the metastatic niche by primary tumor-derived factors.
Elucidating the host response to metastatic progression
With the approval of stroma-targeting therapies including antiangiogenic drugs and IC inhibitors as part of first-line therapy for multiple metastatic cancers, the past decade has witnessed a fundamental shift in the conception of the stroma from being merely supportive of metastatic growth to actively guarding the colonization of seeded cancer cells. The path forward will require the adoption of a holistic approach with a temporal analysis of the metastatic process rather than focusing on end-stage metastatic disease. Indeed, a comprehensive systems map of the metastatic microenvironment, integrating dynamic evolution of different cellular components, will identify disease stage–specific critical nodes and facilitate the process of target identification. Such an extensive database will serve as a reference resource for studying the multicellular molecular cross-talk and will highlight signaling redundancies that may mediate therapy resistance and disease relapse. Beyond the metastatic site, tumor cell–derived factors perturb physiological homeostasis of the hematopoietic system and profoundly affect distant organs such as skeletal muscle and adipose tissue (Alečković et al., 2019; Biswas and Acharyya, 2020). These systemic alterations are frequently associated with poor quality of a patient’s life and reduced tolerance of antimetastatic therapies. Understanding factors that contribute to these systemic alterations will allow designing innovative solutions to improve the healthspan of a patient, not merely the lifespan.
Exploiting novel strategies to target metastasis
Most of the current clinically approved therapies have limited to no efficacy in patients with late-stage cancers (Anderson et al., 2019). Hence, there is an immediate necessity to identify biological mechanism–based therapies that can effectively restrict metastatic disease. Keeping the clinical therapeutic window in mind, ideal antimetastatic therapies should target invasiveness and migratory ability and/or limit the survival of disseminated tumor cells at the secondary site (Yoo et al., 2018). The term “antimigrastatics” refers to a class of drugs that do not influence the proliferation of tumor cells, but rather interfere with the invasiveness and motility of malignant cells (Gandalovičová et al., 2017). For example, a Tie1 function–blocking antibody was recently demonstrated to impede the extravasation of CTCs at the metastatic site, thereby curtailing metastatic progression in an antimigrastatic mode of action (Singhal et al., 2020).
Exploiting the plasticity of cancer cells is another innovative approach that has been exploited by combining the antidiabetic drug rosiglitazone and trametinib (mitogen-activated protein kinase kinase 1/2 inhibitor) to target metastasis in various breast cancer models (Ishay-Ronen et al., 2019). Mechanistically, intervention with rosiglitazone forced disseminated tumor cells to transdifferentiate into postmitotic adipocytes, resulting in the repression of metastases. Likewise, dysregulation of intracellular levels of reactive oxygen species via either timed inhibition of MCT1 or destabilization of BACH1 had detrimental effects on the survival of disseminated tumor cells and led to decreased distant metastasis (Lignitto et al., 2019; Tasdogan et al., 2020; Wiel et al., 2019).
Despite the potentially unprecedented curative nature of IC therapy, only small subsets of patients with advanced solid tumors benefit from it. Preclinical studies suggest that antiangiogenic therapies can improve the efficacy of IC therapies by promoting selective infiltration of cytotoxic CD8+ T cells (Allen et al., 2017; Fukumura et al., 2018; Munn and Jain, 2019; Schmittnaegel et al., 2017). In a recently concluded phase III trial, the combination of atezolizumab (anti–PD-L1) and bevacizumab (anti–vascular endothelial growth factor) was found superior to the current first-line therapy sorafenib for patients with unresectable (including metastatic) hepatocellular carcinoma (Finn et al., 2020). In addition, several angiogenic factors were found up-regulated in pretreatment tumor biopsies derived from metastatic melanoma patients who failed to respond as compared with patients who responded to anti–PD-1 treatment (Hugo et al., 2016), suggesting that analysis of a panel of vascular markers may help to preselect patients with a higher chance of responding to IC therapies. Overall, these preclinical and clinical data not only highlight the antimetastatic potential of combining stroma-targeted drugs but also emphasize the underappreciated role of stromal biomarkers in the stratification of patients receiving IC therapies.
Concluding remarks
Driven by the inherent genomic instability, malignant cells overcome multiple host-imposed checkpoints to establish distant colonies that frequently evade administered therapies and present a nonpredictable disease progression trajectory. As highlighted in this review, despite seminal discoveries of the last decade, numerous key unanswered questions continue to make metastasis largely a black box for researchers and clinicians alike. It is becoming increasingly evident that tackling metastasis requires a holistic approach that not only considers the tumor ecosystem but also takes into consideration a plethora of environmental confounding factors such as a person’s lifestyle. Looking forward, the metastasis research community needs to move beyond the candidate gene approach and invest in developing a multifactorial model that integrates existing experimentally and clinically acquired data. In concert with artificial intelligence, a comprehensive systems map of the metastatic ecosystem will potentially allow clinicians to predict a patient’s response to therapy in silico, thereby empowering them to make informed decisions.
Acknowledgments
The authors regret that, owing to space limitations, they could not cite all original research articles and related references on this topic.
Work in the authors’ laboratory is supported by funds from the Deutsche Forschungsgemeinschaft, European Research Council, the Helmholtz Association, and Heidelberg University. Figures were generated with BioRender.com.
Author contributions: All authors researched data for the article; substantially contributed to discussion of the content; and wrote, reviewed, and edited the manuscript before submission.
References
- Aceto N., Bardia A., Miyamoto D.T., Donaldson M.C., Wittner B.S., Spencer J.A., Yu M., Pely A., Engstrom A., Zhu H., et al. 2014. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 158:1110–1122. 10.1016/j.cell.2014.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello N.M., and Kang Y.. 2019. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 216:1016–1026. 10.1084/jem.20181827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello N.M., Bajor D.L., Norgard R.J., Sahmoud A., Bhagwat N., Pham M.N., Cornish T.C., Iacobuzio-Donahue C.A., Vonderheide R.H., and Stanger B.Z.. 2016. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat. Commun. 7:12819 10.1038/ncomms12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrengues J., Shields M.A., Ng D., Park C.G., Ambrico A., Poindexter M.E., Upadhyay P., Uyeminami D.L., Pommier A., Küttner V., et al. 2018. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 361:eaao4227 10.1126/science.aao4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alečković M., McAllister S.S., and Polyak K.. 2019. Metastasis as a systemic disease: molecular insights and clinical implications. Biochim. Biophys. Acta Rev. Cancer. 1872:89–102. 10.1016/j.bbcan.2019.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen E., Jabouille A., Rivera L.B., Lodewijckx I., Missiaen R., Steri V., Feyen K., Tawney J., Hanahan D., Michael I.P., and Bergers G.. 2017. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 9:eaak9679 10.1126/scitranslmed.aak9679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altorki N.K., Markowitz G.J., Gao D., Port J.L., Saxena A., Stiles B., McGraw T., and Mittal V.. 2019. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat. Rev. Cancer. 19:9–31. 10.1038/s41568-018-0081-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R.L.T., Balasas T., Callaghan J., Coombes R.C., Evans J., Hall J.A., Kinrade S., Jones D., Jones P.S., Jones R., et al. Cancer Research UK and Cancer Therapeutics CRC Australia Metastasis Working Group . 2019. A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 16:185–204. 10.1038/s41571-018-0134-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova M., Mlecnik B., Vasaturo A., Bindea G., Fredriksen T., Lafontaine L., Buttard B., Morgand E., Bruni D., Jouret-Mourin A., et al. 2018. Evolution of metastases in space and time under immune selection. Cell. 175:751–765.e16. 10.1016/j.cell.2018.09.018 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S., Zhan R., Chaudhuri A., Watabe M., Pai S.K., Hirota S., Hosobe S., Tsukada T., Miura K., Takano Y., et al. 2006. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat. Med. 12:933–938. 10.1038/nm1444 [DOI] [PubMed] [Google Scholar]
- Biswas A.K., and Acharyya S.. 2020. Understanding cachexia in the context of metastatic progression. Nat. Rev. Cancer. 20:274–284. 10.1038/s41568-020-0251-4 [DOI] [PubMed] [Google Scholar]
- Brabletz T. 2012. To differentiate or not--routes towards metastasis. Nat. Rev. Cancer. 12:425–436. 10.1038/nrc3265 [DOI] [PubMed] [Google Scholar]
- Brabletz T., Lyden D., Steeg P.S., and Werb Z.. 2013. Roadblocks to translational advances on metastasis research. Nat. Med. 19:1104–1109. 10.1038/nm.3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T., Kalluri R., Nieto M.A., and Weinberg R.A.. 2018. EMT in cancer. Nat. Rev. Cancer. 18:128–134. 10.1038/nrc.2017.118 [DOI] [PubMed] [Google Scholar]
- Brown M., Assen F.P., Leithner A., Abe J., Schachner H., Asfour G., Bago-Horvath Z., Stein J.V., Uhrin P., Sixt M., and Kerjaschki D.. 2018. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science. 359:1408–1411. 10.1126/science.aal3662 [DOI] [PubMed] [Google Scholar]
- Butler J.M., Kobayashi H., and Rafii S.. 2010. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer. 10:138–146. 10.1038/nrc2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z., Scandura J.M., Inghirami G.G., Shido K., Ding B.S., and Rafii S.. 2017. Molecular checkpoint decisions made by subverted vascular niche transform indolent tumor cells into chemoresistant cancer stem cells. Cancer Cell. 31:110–126. 10.1016/j.ccell.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celià-Terrassa T., and Kang Y.. 2016. Distinctive properties of metastasis-initiating cells. Genes Dev. 30:892–908. 10.1101/gad.277681.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celià-Terrassa T., and Kang Y.. 2018. Metastatic niche functions and therapeutic opportunities. Nat. Cell Biol. 20:868–877. 10.1038/s41556-018-0145-9 [DOI] [PubMed] [Google Scholar]
- Chaffer C.L., Thompson E.W., and Williams E.D.. 2007. Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs. 185:7–19. 10.1159/000101298 [DOI] [PubMed] [Google Scholar]
- Chen Y., Keskin D., Sugimoto H., Kanasaki K., Phillips P.E., Bizarro L., Sharpe A., LeBleu V.S., and Kalluri R.. 2018. Podoplanin+ tumor lymphatics are rate limiting for breast cancer metastasis. PLoS Biol. 16:e2005907 10.1371/journal.pbio.2005907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K.J., and Ewald A.J.. 2016. A collective route to metastasis: Seeding by tumor cell clusters. Science. 352:167–169. 10.1126/science.aaf6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K.J., Gabrielson E., Werb Z., and Ewald A.J.. 2013. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 155:1639–1651. 10.1016/j.cell.2013.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K.J., Padmanaban V., Silvestri V., Schipper K., Cohen J.D., Fairchild A.N., Gorin M.A., Verdone J.E., Pienta K.J., Bader J.S., and Ewald A.J.. 2016. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA. 113:E854–E863. 10.1073/pnas.1508541113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke M.A., and Fisher J.. 2020. Executable cancer models: successes and challenges. Nat. Rev. Cancer. 20:343–354. 10.1038/s41568-020-0258-x [DOI] [PubMed] [Google Scholar]
- Coffelt S.B., Kersten K., Doornebal C.W., Weiden J., Vrijland K., Hau C.S., Verstegen N.J.M., Ciampricotti M., Hawinkels L.J.A.C., Jonkers J., and de Visser K.E.. 2015. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 522:345–348. 10.1038/nature14282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagenais G.R., Leong D.P., Rangarajan S., Lanas F., Lopez-Jaramillo P., Gupta R., Diaz R., Avezum A., Oliveira G.B.F., Wielgosz A., et al. 2020. Variations in common diseases, hospital admissions, and deaths in middle-aged adults in 21 countries from five continents (PURE): a prospective cohort study. Lancet. 395:785–794. 10.1016/S0140-6736(19)32007-0 [DOI] [PubMed] [Google Scholar]
- De Palma M., Biziato D., and Petrova T.V.. 2017. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer. 17:457–474. 10.1038/nrc.2017.51 [DOI] [PubMed] [Google Scholar]
- Ding J., Adiconis X., Simmons S.K., Kowalczyk M.S., Hession C.C., Marjanovic N.D., Hughes T.K., Wadsworth M.H., Burks T., Nguyen L.T., et al. 2020. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 38:737–746. 10.1038/s41587-020-0465-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost J., and Clevers H.. 2018. Organoids in cancer research. Nat. Rev. Cancer. 18:407–418. 10.1038/s41568-018-0007-6 [DOI] [PubMed] [Google Scholar]
- Ecker B.L., Kaur A., Douglass S.M., Webster M.R., Almeida F.V., Marino G.E., Sinnamon A.J., Neuwirth M.G., Alicea G.M., Ndoye A., et al. 2019. Age-related changes in HAPLN1 increase lymphatic permeability and affect routes of melanoma metastasis. Cancer Discov. 9:82–95. 10.1158/2159-8290.CD-18-0168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M., and de Visser K.E.. 2019. Sticking together helps cancer to spread. Nature. 566:459–460. 10.1038/d41586-019-00341-4 [DOI] [PubMed] [Google Scholar]
- El-Kenawi A., Hänggi K., and Ruffell B.. 2020. The immune microenvironment and cancer metastasis. Cold Spring Harb. Perspect. Med. 10:a037424 10.1101/cshperspect.a037424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng C.L., Lawson M., Zhu Q., Dries R., Koulena N., Takei Y., Yun J., Cronin C., Karp C., Yuan G.C., and Cai L.. 2019. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 568:235–239. 10.1038/s41586-019-1049-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito M., Mondal N., Greco T.M., Wei Y., Spadazzi C., Lin S.C., Zheng H., Cheung C., Magnani J.L., Lin S.H., et al. 2019. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol. 21:627–639. 10.1038/s41556-019-0309-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faries M.B., Thompson J.F., Cochran A.J., Andtbacka R.H., Mozzillo N., Zager J.S., Jahkola T., Bowles T.L., Testori A., Beitsch P.D., et al. 2017. Completion dissection or observation for sentinel-node metastasis in melanoma. N. Engl. J. Med. 376:2211–2222. 10.1056/NEJMoa1613210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio M., Ablain J., Chuan Y., Langenau D.M., and Zon L.I.. 2020. Zebrafish patient avatars in cancer biology and precision cancer therapy. Nat. Rev. Cancer. 20:263–273. 10.1038/s41568-020-0252-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler I.J. 2003. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer. 3:453–458. 10.1038/nrc1098 [DOI] [PubMed] [Google Scholar]
- Finn R.S., Qin S., Ikeda M., Galle P.R., Ducreux M., Kim T.Y., Kudo M., Breder V., Merle P., Kaseb A.O., et al. IMbrave150 Investigators . 2020. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 382:1894–1905. 10.1056/NEJMoa1915745 [DOI] [PubMed] [Google Scholar]
- Fischer K.R., Durrans A., Lee S., Sheng J., Li F., Wong S.T., Choi H., El Rayes T., Ryu S., Troeger J., et al. 2015. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 527:472–476. 10.1038/nature15748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumura D., Kloepper J., Amoozgar Z., Duda D.G., and Jain R.K.. 2018. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat. Rev. Clin. Oncol. 15:325–340. 10.1038/nrclinonc.2018.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandalovičová A., Rosel D., Fernandes M., Veselý P., Heneberg P., Čermák V., Petruželka L., Kumar S., Sanz-Moreno V., and Brábek J.. 2017. Migrastatics—anti-metastatic and anti-invasion drugs: promises and challenges. Trends Cancer. 3:391–406. 10.1016/j.trecan.2017.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner H., and de Visser K.E.. 2020. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat. Rev. Immunol. 20:483–497. 10.1038/s41577-019-0271-z [DOI] [PubMed] [Google Scholar]
- Garraway L.A., and Lander E.S.. 2013. Lessons from the cancer genome. Cell. 153:17–37. 10.1016/j.cell.2013.03.002 [DOI] [PubMed] [Google Scholar]
- Gengenbacher N., Singhal M., and Augustin H.G.. 2017. Preclinical mouse solid tumour models: status quo, challenges and perspectives. Nat. Rev. Cancer. 17:751–765. 10.1038/nrc.2017.92 [DOI] [PubMed] [Google Scholar]
- Gengenbacher N., Singhal M., Mogler C., Hai L., Milde L., Abdul Pari A.A., Besemfelder E., Fricke C., Baumann D., Gehrs S., et al. 2020. Timed Ang2-targeted therapy identifies the Angiopoietin-Tie pathway as key regulator of fatal lymphogenous metastasis. Cancer Discov.:CD-20-0122 10.1158/2159-8290.CD-20-0122 [DOI] [PubMed] [Google Scholar]
- Ghajar C.M., Peinado H., Mori H., Matei I.R., Evason K.J., Brazier H., Almeida D., Koller A., Hajjar K.A., Stainier D.Y., et al. 2013. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 15:807–817. 10.1038/ncb2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliano A.E., Ballman K.V., McCall L., Beitsch P.D., Brennan M.B., Kelemen P.R., Ollila D.W., Hansen N.M., Whitworth P.W., Blumencranz P.W., et al. 2017. Effect of axillary dissection vs no axillary dissection on 10-year overall survival among women with invasive breast cancer and sentinel node metastasis: the ACOSOG Z0011 (alliance) randomized clinical trial. JAMA. 318:918–926. 10.1001/jama.2017.11470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkountela S., Castro-Giner F., Szczerba B.M., Vetter M., Landin J., Scherrer R., Krol I., Scheidmann M.C., Beisel C., Stirnimann C.U., et al. 2019. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell. 176:98–112.e14. 10.1016/j.cell.2018.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundem G., Van Loo P., Kremeyer B., Alexandrov L.B., Tubio J.M.C., Papaemmanuil E., Brewer D.S., Kallio H.M.L., Högnäs G., Annala M., et al. ICGC Prostate Group . 2015. The evolutionary history of lethal metastatic prostate cancer. Nature. 520:353–357. 10.1038/nature14347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., and Weinberg R.A.. 2011. Hallmarks of cancer: the next generation. Cell. 144:646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hou J.M., Krebs M.G., Lancashire L., Sloane R., Backen A., Swain R.K., Priest L.J., Greystoke A., Zhou C., Morris K., et al. 2012. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. 30:525–532. 10.1200/JCO.2010.33.3716 [DOI] [PubMed] [Google Scholar]
- Hu Z., Li Z., Ma Z., and Curtis C.. 2020. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 52:701–708. 10.1038/s41588-020-0628-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. 2016. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 165:35–44. 10.1016/j.cell.2016.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium 2020. Pan-cancer analysis of whole genomes. Nature. 578:82–93. 10.1038/s41586-020-1969-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishay-Ronen D., Diepenbruck M., Kalathur R.K.R., Sugiyama N., Tiede S., Ivanek R., Bantug G., Morini M.F., Wang J., Hess C., and Christofori G.. 2019. Gain fat—lose metastasis: converting invasive breast cancer cells into adipocytes inhibits cancer metastasis. Cancer Cell. 35:17–32.e6. 10.1016/j.ccell.2018.12.002 [DOI] [PubMed] [Google Scholar]
- Kalluri R., and Weinberg R.A.. 2009. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119:1420–1428. 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur A., Webster M.R., Marchbank K., Behera R., Ndoye A., Kugel C.H. III, Dang V.M., Appleton J., O’Connell M.P., Cheng P., et al. 2016. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature. 532:250–254. 10.1038/nature17392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur A., Ecker B.L., Douglass S.M., Kugel C.H. III, Webster M.R., Almeida F.V., Somasundaram R., Hayden J., Ban E., Ahmadzadeh H., et al. 2019. Remodeling of the collagen matrix in aging skin promotes melanoma metastasis and affects immune cell motility. Cancer Discov. 9:64–81. 10.1158/2159-8290.CD-18-0193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.J., Kwon S.H., Yun J.H., Jeong H.S., Kim H.R., Lee E.H., Ye S.K., and Cho C.H.. 2017. STAT3 activation in endothelial cells is important for tumor metastasis via increased cell adhesion molecule expression. Oncogene. 36:5445–5459. 10.1038/onc.2017.148 [DOI] [PubMed] [Google Scholar]
- Krebs A.M., Mitschke J., Lasierra Losada M., Schmalhofer O., Boerries M., Busch H., Boettcher M., Mougiakakos D., Reichardt W., Bronsert P., et al. 2017. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 19:518–529. 10.1038/ncb3513 [DOI] [PubMed] [Google Scholar]
- Krishnan J., Kirkin V., Steffen A., Hegen M., Weih D., Tomarev S., Wilting J., and Sleeman J.P.. 2003. Differential in vivo and in vitro expression of vascular endothelial growth factor (VEGF)-C and VEGF-D in tumors and its relationship to lymphatic metastasis in immunocompetent rats. Cancer Res. 63:713–722. [PubMed] [Google Scholar]
- Kubota S.I., Takahashi K., Nishida J., Morishita Y., Ehata S., Tainaka K., Miyazono K., and Ueda H.R.. 2017. Whole-body profiling of cancer metastasis with single-cell resolution. Cell Rep. 20:236–250. 10.1016/j.celrep.2017.06.010 [DOI] [PubMed] [Google Scholar]
- Labelle M., Begum S., and Hynes R.O.. 2011. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 20:576–590. 10.1016/j.ccr.2011.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughney A.M., Hu J., Campbell N.R., Bakhoum S.F., Setty M., Lavallée V.P., Xie Y., Masilionis I., Carr A.J., Kottapalli S., et al. 2020. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med. 26:259–269. 10.1038/s41591-019-0750-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiter U., Stadler R., Mauch C., Hohenberger W., Brockmeyer N., Berking C., Sunderkötter C., Kaatz M., Schulte K.W., Lehmann P., et al. German Dermatologic Cooperative Oncology Group (DeCOG) . 2016. Complete lymph node dissection versus no dissection in patients with sentinel lymph node biopsy positive melanoma (DeCOG-SLT): a multicentre, randomised, phase 3 trial. Lancet Oncol. 17:757–767. 10.1016/S1470-2045(16)00141-8 [DOI] [PubMed] [Google Scholar]
- Lignitto L., LeBoeuf S.E., Homer H., Jiang S., Askenazi M., Karakousi T.R., Pass H.I., Bhutkar A.J., Tsirigos A., Ueberheide B., et al. 2019. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell. 178:316–329.e18. 10.1016/j.cell.2019.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim B., Woodward W.A., Wang X., Reuben J.M., and Ueno N.T.. 2018. Inflammatory breast cancer biology: the tumour microenvironment is key. Nat. Rev. Cancer. 18:485–499. 10.1038/s41568-018-0010-y [DOI] [PubMed] [Google Scholar]
- Lim B., Lin Y., and Navin N.. 2020. Advancing cancer research and medicine with single-cell genomics. Cancer Cell. 37:456–470. 10.1016/j.ccell.2020.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta L.A., Saidel M.G., and Kleinerman J.. 1976. The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res. 36:889–894. [PubMed] [Google Scholar]
- Liu Y., and Cao X.. 2016. Characteristics and significance of the pre-metastatic niche. Cancer Cell. 30:668–681. 10.1016/j.ccell.2016.09.011 [DOI] [PubMed] [Google Scholar]
- Lo H.C., Xu Z., Kim I.S., Pingel B., Aguirre S., Kodali S., Liu J., Zhang W., Muscarella A.M., Hein S.M., et al. 2020. Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat. Can. 1:709–722. 10.1038/s43018-020-0068-9 [DOI] [PubMed] [Google Scholar]
- Lu Z., Zou J., Li S., Topper M.J., Tao Y., Zhang H., Jiao X., Xie W., Kong X., Vaz M., et al. 2020. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature. 579:284–290. 10.1038/s41586-020-2054-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzi K.J., MacDonald I.C., Schmidt E.E., Kerkvliet N., Morris V.L., Chambers A.F., and Groom A.C.. 1998. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 153:865–873. 10.1016/S0002-9440(10)65628-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q., Dieterich L.C., Ikenberg K., Bachmann S.B., Mangana J., Proulx S.T., Amann V.C., Levesque M.P., Dummer R., Baluk P., et al. 2018. Unexpected contribution of lymphatic vessels to promotion of distant metastatic tumor spread. Sci. Adv. 4:t4758 10.1126/sciadv.aat4758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddipati R., and Stanger B.Z.. 2015. Pancreatic cancer metastases harbor evidence of polyclonality. Cancer Discov. 5:1086–1097. 10.1158/2159-8290.CD-15-0120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani S.A., Guo W., Liao M.J., Eaton E.N., Ayyanan A., Zhou A.Y., Brooks M., Reinhard F., Zhang C.C., Shipitsin M., et al. 2008. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 133:704–715. 10.1016/j.cell.2008.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J., and Obenauf A.C.. 2016. Metastatic colonization by circulating tumour cells. Nature. 529:298–306. 10.1038/nature17038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu E., Lafzi A., Moutinho C., Ziegenhain C., McCarthy D.J., Álvarez-Varela A., Batlle E., Sagar D., Grün D., Lau J.K., et al. 2020. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat. Biotechnol. 38:747–755. 10.1038/s41587-020-0469-4 [DOI] [PubMed] [Google Scholar]
- Micalizzi D.S., Maheswaran S., and Haber D.A.. 2017. A conduit to metastasis: circulating tumor cell biology. Genes Dev. 31:1827–1840. 10.1101/gad.305805.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn L.L., and Jain R.K.. 2019. Vascular regulation of antitumor immunity. Science. 365:544–545. 10.1126/science.aaw7875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgai M., Ju W., Eason M., Kline J., Beury D.W., Kaczanowska S., Miettinen M.M., Kruhlak M., Lei H., Shern J.F., et al. 2017. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat. Med. 23:1176–1190. 10.1038/nm.4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naxerova K., Reiter J.G., Brachtel E., Lennerz J.K., van de Wetering M., Rowan A., Cai T., Clevers H., Swanton C., Nowak M.A., et al. 2017. Origins of lymphatic and distant metastases in human colorectal cancer. Science. 357:55–60. 10.1126/science.aai8515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C.M., and Bissell M.J.. 2006. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 22:287–309. 10.1146/annurev.cellbio.22.010305.104315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M.A. 2017. Context-specific roles of EMT programmes in cancer cell dissemination. Nat. Cell Biol. 19:416–418. 10.1038/ncb3520 [DOI] [PubMed] [Google Scholar]
- Nieto M.A., Huang R.Y., Jackson R.A., and Thiery J.P.. 2016. EMT: 2016. Cell. 166:21–45. 10.1016/j.cell.2016.06.028 [DOI] [PubMed] [Google Scholar]
- Obenauf A.C., and Massagué J.. 2015. Surviving at a distance: organ-specific metastasis. Trends Cancer. 1:76–91. 10.1016/j.trecan.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ombrato L., Nolan E., Kurelac I., Mavousian A., Bridgeman V.L., Heinze I., Chakravarty P., Horswell S., Gonzalez-Gualda E., Matacchione G., et al. 2019. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature. 572:603–608. 10.1038/s41586-019-1487-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T., Batlle E., and Massagué J.. 2014. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. 14:306–321. 10.1016/j.stem.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanaban V., Krol I., Suhail Y., Szczerba B.M., Aceto N., Bader J.S., and Ewald A.J.. 2019. E-cadherin is required for metastasis in multiple models of breast cancer. Nature. 573:439–444. 10.1038/s41586-019-1526-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paduch R. 2016. The role of lymphangiogenesis and angiogenesis in tumor metastasis. Cell Oncol. (Dordr.). 39:397–410. 10.1007/s13402-016-0281-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C., Schoppe O., Parra-Damas A., Cai R., Todorov M.I., Gondi G., von Neubeck B., Böğürcü-Seidel N., Seidel S., Sleiman K., et al. 2019. Deep learning reveals cancer metastasis and therapeutic antibody targeting in the entire body. Cell. 179:1661–1676.e19. 10.1016/j.cell.2019.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier J., Ghiabi P., Chouchane L., Razzouk K., Rafii S., and Rafii A.. 2020. Angiocrine endothelium: from physiology to cancer. J. Transl. Med. 18:52 10.1186/s12967-020-02244-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastushenko I., and Blanpain C.. 2019. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 29:212–226. 10.1016/j.tcb.2018.12.001 [DOI] [PubMed] [Google Scholar]
- Pastushenko I., Brisebarre A., Sifrim A., Fioramonti M., Revenco T., Boumahdi S., Van Keymeulen A., Brown D., Moers V., Lemaire S., et al. 2018. Identification of the tumour transition states occurring during EMT. Nature. 556:463–468. 10.1038/s41586-018-0040-3 [DOI] [PubMed] [Google Scholar]
- Pearce O.M.T., Delaine-Smith R.M., Maniati E., Nichols S., Wang J., Böhm S., Rajeeve V., Ullah D., Chakravarty P., Jones R.R., et al. 2018. Deconstruction of a metastatic tumor microenvironment reveals a common matrix response in human cancers. Cancer Discov. 8:304–319. 10.1158/2159-8290.CD-17-0284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H., Zhang H., Matei I.R., Costa-Silva B., Hoshino A., Rodrigues G., Psaila B., Kaplan R.N., Bromberg J.F., Kang Y., et al. 2017. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer. 17:302–317. 10.1038/nrc.2017.6 [DOI] [PubMed] [Google Scholar]
- Pereira E.R., Kedrin D., Seano G., Gautier O., Meijer E.F.J., Jones D., Chin S.M., Kitahara S., Bouta E.M., Chang J., et al. 2018. Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science. 359:1403–1407. 10.1126/science.aal3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan T.G., and Croucher P.I.. 2020. The dormant cancer cell life cycle. Nat. Rev. Cancer. 20:398–411. 10.1038/s41568-020-0263-0 [DOI] [PubMed] [Google Scholar]
- Reiter J.G., Hung W.T., Lee I.H., Nagpal S., Giunta P., Degner S., Liu G., Wassenaar E.C.E., Jeck W.R., Taylor M.S., et al. 2020. Lymph node metastases develop through a wider evolutionary bottleneck than distant metastases. Nat. Genet. 52:692–700. 10.1038/s41588-020-0633-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios A.C., Capaldo B.D., Vaillant F., Pal B., van Ineveld R., Dawson C.A., Chen Y., Nolan E., Fu N.Y., Jackling F.C., et al. 3DTCLSM Group . 2019. Intraclonal plasticity in mammary tumors revealed through large-scale single-cell resolution 3D imaging. Cancer Cell. 35:618–632.e6. 10.1016/j.ccell.2019.02.010 [DOI] [PubMed] [Google Scholar]
- Ritter B., and Greten F.R.. 2019. Modulating inflammation for cancer therapy. J. Exp. Med. 216:1234–1243. 10.1084/jem.20181739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriques S.G., Stickels R.R., Goeva A., Martin C.A., Murray E., Vanderburg C.R., Welch J., Chen L.M., Chen F., and Macosko E.Z.. 2019. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 363:1463–1467. 10.1126/science.aaw1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rood J.E., Stuart T., Ghazanfar S., Biancalani T., Fisher E., Butler A., Hupalowska A., Gaffney L., Mauck W., Eraslan G., et al. 2019. Toward a common coordinate framework for the human body. Cell. 179:1455–1467. 10.1016/j.cell.2019.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E., Astsaturov I., Cukierman E., DeNardo D.G., Egeblad M., Evans R.M., Fearon D., Greten F.R., Hingorani S.R., Hunter T., et al. 2020. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer. 20:174–186. 10.1038/s41568-019-0238-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn J.Z., Chung J., Purdom E., Wang N.J., Kakavand H., Wilmott J.S., Butler T., Thompson J.F., Mann G.J., Haydu L.E., et al. 2015. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc. Natl. Acad. Sci. USA. 112:10995–11000. 10.1073/pnas.1508074112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarioglu A.F., Aceto N., Kojic N., Donaldson M.C., Zeinali M., Hamza B., Engstrom A., Zhu H., Sundaresan T.K., Miyamoto D.T., et al. 2015. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods. 12:685–691. 10.1038/nmeth.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittnaegel M., Rigamonti N., Kadioglu E., Cassará A., Wyser Rmili C., Kiialainen A., Kienast Y., Mueller H.J., Ooi C.H., Laoui D., and De Palma M.. 2017. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 9:eaak9670 10.1126/scitranslmed.aak9670 [DOI] [PubMed] [Google Scholar]
- Schumacher D., Strilic B., Sivaraj K.K., Wettschureck N., and Offermanns S.. 2013. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell. 24:130–137. 10.1016/j.ccr.2013.05.008 [DOI] [PubMed] [Google Scholar]
- Shaw K.R.M., and Maitra A.. 2019. The status and impact of clinical tumor genome sequencing. Annu. Rev. Genomics Hum. Genet. 20:413–432. 10.1146/annurev-genom-083118-015034 [DOI] [PubMed] [Google Scholar]
- Shen Y., Wang X., Lu J., Salfenmoser M., Wirsik N.M., Schleussner N., Imle A., Freire Valls A., Radhakrishnan P., Liang J., et al. 2020. Reduction of liver metastasis stiffness improves response to bevacizumab in metastatic colorectal cancer. Cancer Cell. 37:800–817.e7. 10.1016/j.ccell.2020.05.005 [DOI] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., and Jemal A.. 2020. Cancer statistics, 2020. CA Cancer J. Clin. 70:7–30. 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- Singhal M., and Augustin H.G.. 2020. Beyond angiogenesis: Exploiting angiocrine factors to restrict tumor progression and metastasis. Cancer Res. 80:659–662. 10.1158/0008-5472.CAN-19-3351 [DOI] [PubMed] [Google Scholar]
- Singhal M., Gengenbacher N., La Porta S., Gehrs S., Shi J., Kamiyama M., Bodenmiller D.M., Fischl A., Schieb B., Besemfelder E., et al. 2020. Preclinical validation of a novel metastasis-inhibiting Tie1 function-blocking antibody. EMBO Mol. Med. 12:e11164 10.15252/emmm.201911164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava K., Hu J., Korn C., Savant S., Teichert M., Kapel S.S., Jugold M., Besemfelder E., Thomas M., Pasparakis M., and Augustin H.G.. 2014. Postsurgical adjuvant tumor therapy by combining anti-angiopoietin-2 and metronomic chemotherapy limits metastatic growth. Cancer Cell. 26:880–895. 10.1016/j.ccell.2014.11.005 [DOI] [PubMed] [Google Scholar]
- Ståhl P.L., Salmén F., Vickovic S., Lundmark A., Navarro J.F., Magnusson J., Giacomello S., Asp M., Westholm J.O., Huss M., et al. 2016. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 353:78–82. 10.1126/science.aaf2403 [DOI] [PubMed] [Google Scholar]
- Stegner D., Dütting S., and Nieswandt B.. 2014. Mechanistic explanation for platelet contribution to cancer metastasis. Thromb. Res. 133(Suppl 2):S149–S157. 10.1016/S0049-3848(14)50025-4 [DOI] [PubMed] [Google Scholar]
- Strilic B., and Offermanns S.. 2017. Intravascular survival and extravasation of tumor cells. Cancer Cell. 32:282–293. 10.1016/j.ccell.2017.07.001 [DOI] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M. III, Hao Y., Stoeckius M., Smibert P., and Satija R.. 2019. Comprehensive integration of single-cell data. Cell. 177:1888–1902.e21. 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhail Y., Cain M.P., Vanaja K., Kurywchak P.A., Levchenko A., Kalluri R., and Kshitiz. 2019. Systems biology of cancer metastasis. Cell Syst. 9:109–127. 10.1016/j.cels.2019.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers M.A., McDonald M.M., and Croucher P.I.. 2020. Cancer cell dormancy in metastasis. Cold Spring Harb. Perspect. Med. 10:a037556 10.1101/cshperspect.a037556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swierczak A., and Pollard J.W.. 2020. Myeloid cells in metastasis. Cold Spring Harb. Perspect. Med. 10:a038026 10.1101/cshperspect.a038026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczerba B.M., Castro-Giner F., Vetter M., Krol I., Gkountela S., Landin J., Scheidmann M.C., Donato C., Scherrer R., Singer J., et al. 2019. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. 566:553–557. 10.1038/s41586-019-0915-y [DOI] [PubMed] [Google Scholar]
- Tasdogan A., Faubert B., Ramesh V., Ubellacker J.M., Shen B., Solmonson A., Murphy M.M., Gu Z., Gu W., Martin M., et al. 2020. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature. 577:115–120. 10.1038/s41586-019-1847-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J.H., Donaher J.L., Murphy D.A., Chau S., and Yang J.. 2012. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 22:725–736. 10.1016/j.ccr.2012.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S., and Swanton C.. 2016. Metastasis as an evolutionary process. Science. 352:169–175. 10.1126/science.aaf2784 [DOI] [PubMed] [Google Scholar]
- Ubellacker J.M., Tasdogan A., Ramesh V., Shen B., Mitchell E.C., Martin-Sandoval M.S., Gu Z., McCormick M.L., Durham A.B., Spitz D.R., et al. 2020. Lymph protects metastasizing melanoma cells from ferroptosis. Nature. 585:113–118. 10.1038/s41586-020-2623-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H.R., Ertürk A., Chung K., Gradinaru V., Chédotal A., Tomancak P., and Keller P.J.. 2020. Tissue clearing and its applications in neuroscience. Nat. Rev. Neurosci. 21:61–79. 10.1038/s41583-019-0250-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch D.R., and Hurst D.R.. 2019. Defining the hallmarks of metastasis. Cancer Res. 79:3011–3027. 10.1158/0008-5472.CAN-19-0458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler D.A., and Wang L.. 2013. From human genome to cancer genome: the first decade. Genome Res. 23:1054–1062. 10.1101/gr.157602.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiel C., Le Gal K., Ibrahim M.X., Jahangir C.A., Kashif M., Yao H., Ziegler D.V., Xu X., Ghosh T., Mondal T., et al. 2019. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell. 178:330–345.e22. 10.1016/j.cell.2019.06.005 [DOI] [PubMed] [Google Scholar]
- Wieland E., Rodriguez-Vita J., Liebler S.S., Mogler C., Moll I., Herberich S.E., Espinet E., Herpel E., Menuchin A., Chang-Claude J., et al. 2017. Endothelial notch1 activity facilitates metastasis. Cancer Cell. 31:355–367. 10.1016/j.ccell.2017.01.007 [DOI] [PubMed] [Google Scholar]
- Williams E.D., Gao D., Redfern A., and Thompson E.W.. 2019. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer. 19:716–732. 10.1038/s41568-019-0213-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S.Y., and Hynes R.O.. 2006. Lymphatic or hematogenous dissemination: how does a metastatic tumor cell decide? Cell Cycle. 5:812–817. 10.4161/cc.5.8.2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P.P., Muñoz-Félix J.M., Hijazi M., Kim H., Robinson S.D., De Luxán-Delgado B., Rodríguez-Hernández I., Maiques O., Meng Y.M., Meng Q., et al. 2020. Cancer burden is controlled by mural cell-β3-integrin regulated crosstalk with tumor cells. Cell. 181:1346–1363.e21. 10.1016/j.cell.2020.02.003 [DOI] [PubMed] [Google Scholar]
- Yang J., Antin P., Berx G., Blanpain C., Brabletz T., Bronner M., Campbell K., Cano A., Casanova J., Christofori G., et al. EMT International Association (TEMTIA) . 2020a Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 21:341–352. 10.1038/s41580-020-0237-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Liu Q., Zhang X., Liu X., Zhou B., Chen J., Huang D., Li J., Li H., Chen F., et al. 2020b DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 583:133–138. 10.1038/s41586-020-2394-6 [DOI] [PubMed] [Google Scholar]
- Yoo B., Fuchs B.C., and Medarova Z.. 2018. New directions in the study and treatment of metastatic cancer. Front. Oncol. 8:258 10.3389/fonc.2018.00258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Carstens J.L., Kim J., Scheible M., Kaye J., Sugimoto H., Wu C.C., LeBleu V.S., and Kalluri R.. 2015. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 527:525–530. 10.1038/nature16064 [DOI] [PMC free article] [PubMed] [Google Scholar]