

Abstract
The recent emergence of oxidation state selective probes of cellular iron has produced a more nuanced understanding of how cells utilize this crucial nutrient to empower enzyme function, and also how ‘labile’ ferrous iron contributes to iron-dependent cell death (ferroptosis) and other disease pathologies including cancer, bacterial infections, and neurodegeneration. These findings, viewed in light of the Fenton chemistry promoted by ferrous iron, suggest a new category of therapeutics exhibiting ferrous iron–dependent pharmacology. While still in its infancy, this nascent field draws inspiration from the remarkable activity and tremendous clinical impact of the antimalarial artemisinin. Here we review recent insights into the role of labile ferrous iron in biology and disease, and describe new therapeutic approaches designed to exploit this divalent transition metal.
Keywords: Iron homeostasis, ferroptosis, ferritinophagy, targeted prodrugs, reactivity-based probes, activity-based probes
Iron Acquisition and Homeostasis
Around a third of all proteins are metalloproteins, with zinc, copper, and iron most prominent among the divalent metal ions that drive co-factor dependent enzyme function [1]. These activities include DNA & RNA synthesis and repair, epigenetic regulation, and cellular respiration, among many others. The ability to redox cycle between the ferrous (Fe2+) and ferric (Fe3+) states enables iron-dependent cofactors to fulfil their catalytic roles in a wide range of biochemical processes. Homeostatic control of cellular iron is carried out via dynamic mechanisms that regulate its absorption, transport, storage and mobilization [2]. Because the ferric ion is essentially water-insoluble, it must be bound to protein chaperones such as transferrin for transport and ferritin for storage. While ferric iron storage and transport has been well understood for decades, the much less well characterized cellular pool of ferrous iron has historically been referred to, somewhat unhelpfully, as the ‘chelateable’ or ‘labile’ iron pool (LIP) – the pool of iron subject to binding by exogenous reagents. Unlike enzyme-regulated iron co-factors or ferric iron stored in cellular ferritin, iron in the LIP undergoes spontaneous redox cycling, promoting Fenton-type reaction with oxygen and cellular hydroperoxides [2,3], producing oxidative stress and causing damage to cellular macromolecules and lipids [4]. Accordingly, the LIP and iron homeostasis generally must be carefully regulated, both at the level of the individual cell and the whole organism.
In mammals, total systemic iron is regulated through dietary uptake, with ferric iron absorbed at the apical membrane of the enterocyte through coordinated iron reduction by duodenal cytochrome B (DCYTB, encoded by the CYBRD1 gene) and imported via the divalent metal transporter (DMT1) [5]. At the basolateral membrane, an analogous process of export (via ferroportin) followed by oxidation (by hephaestin) enables loading of ferric iron ions onto transferrin for entry into systemic circulation. The distribution of iron between tissues and organs in mammals is primarily mediated by the cellular iron exporter ferroportin and its negative regulator hepcidin, a peptide hormone produced in the liver (or in autocrine fashion in some tissues) that binds to ferroportin and leads to its internalization and degradation [3]. Major sites of ferric iron storage include the liver and within plasma ferritin, while systemic iron is recycled from damaged and aged erythrocytes by splenic macrophages [6]. Thus, ferrous iron is produced transiently, as needed and “just in time”, while ferric iron in storage or transit comprises the major iron stores of the organism.
Iron handing at the cellular level involves analogous modes of iron uptake, recycling, storage, and export (Figure 1). Transferrin mediated iron uptake involves binding of holo-transferrin to the transferrin receptor, followed by endocytosis and endosome-lysosome acidification to release the otherwise insoluble ferric ion; reduction of the liberated Fe3+ to Fe2+ by STEAP3 is then followed by export via endolysosomal DMT1. The lysosome is also involved in ferritin iron recycling through the process of ferritinophagy [7]. A recent CRISPR functional screen found that maintenance of adequate cellular iron was an indispensable function of lysosome acidity [8], consistent with an essential role for the lysosome in iron uptake and ferritin recycling. As the Fe2+ ion exits the lysosome it enters the cytosolic labile iron pool (LIP), where it can be utilized directly by essential enzymes like ribonucleotide reductase that utilize a mononuclear iron cofactor, oxidized and incorporated into ferritin for storage, or incorporated into heme or Fe/S prosthetic groups in the mitochondria. Until very recently, it was unknown whether iron in the LIP was truly ‘labile’ (i.e., unbound) or whether cellular chaperones were involved in its trafficking. It has been proposed more recently that poly(rC)-binding protein (PCBP1) is an Fe2+ binding protein involved in the delivery of cytosolic iron to ferritin [9], and also to proteins involved in heme iron biosynthesis [10] and Fe/S cluster assembly [11]. Iron bound to PCBP1 is further ligated by a glutathione molecule, consistent with earlier work [12] suggesting glutathione-ligated Fe2+ as a component of the LIP. For the discussion that follows, it is important to appreciate that the LIP comprises a small percentage of total cellular iron, the vast majority of iron being safely stored in ferritin, or actively utilized in metalloproteins to perform specific tasks.
Figure 1.
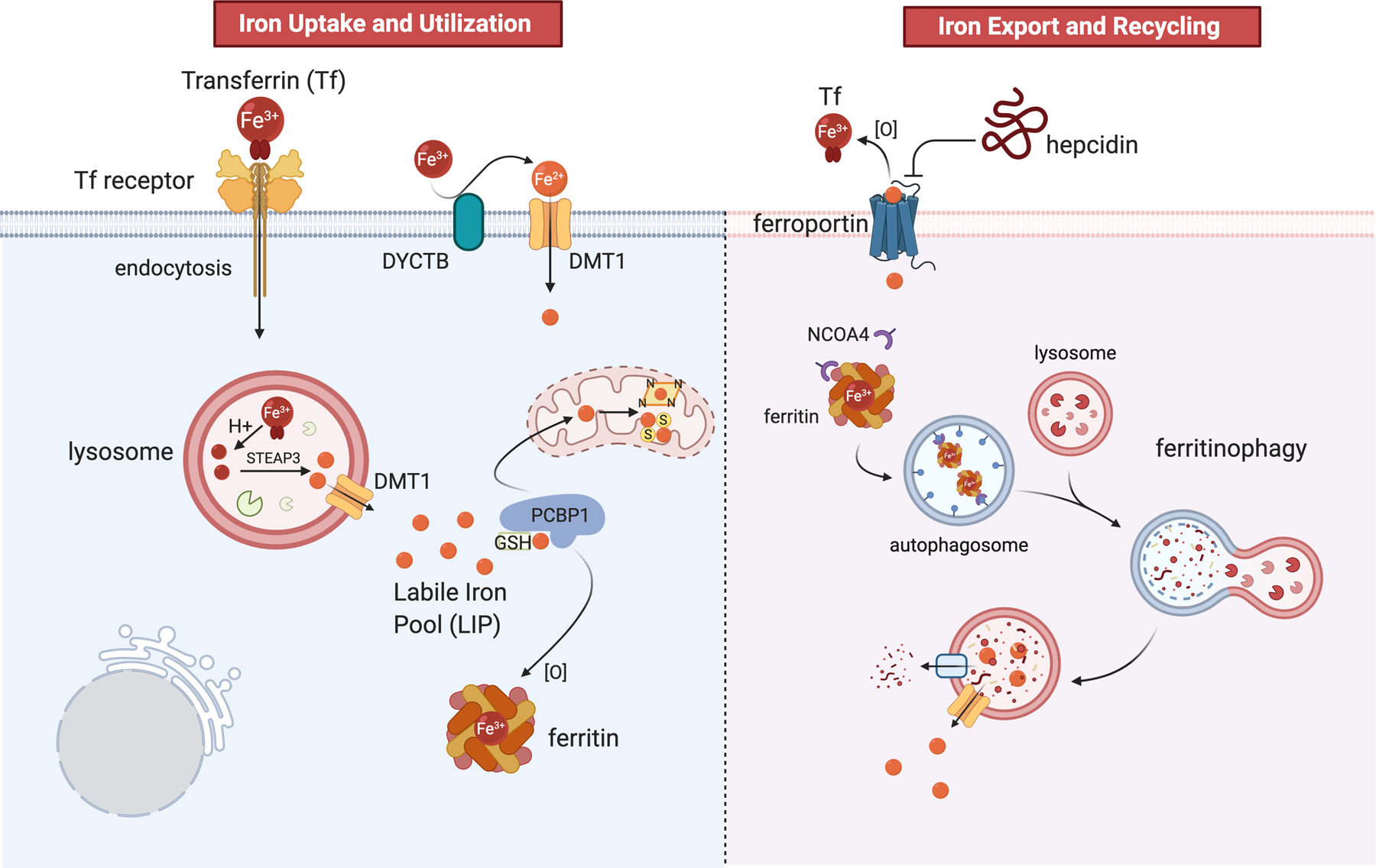
Summary of iron homeostasis in mammalian cells with a focus on ferrous iron (orange spheres). Figure created in BioRender (biorender.com)
With this understanding of iron metabolism, two principles for iron(II)-dependent pharmacology can be enunciated, 1) that redox-active ferrous iron is present at pharmacologically relevant concentrations within cells, but generally not in plasma or other extracellular compartments, and 2) that disease pathology is more likely to result from elevated ferrous iron levels (e.g., in the LIP, in mitochondria, in hypoxic cells) than from changes to total iron levels, which will be predominantly Fe3+ in storage. Unfortunately, traditional methods to measure total cellular iron, such as ICP-MS, are unable to distinguish ferrous from ferric states. Similarly, any approach that breaks the cell membrane prior to analysis will result in rapid oxidation of labile ferrous iron, unless special precautions are taken. PhenGreen SK, until recently the most widely utilized probe of ‘labile’ iron, is a chelation-based probe unable to reliably distinguish iron oxidation state and is furthermore susceptible to interference from other metal ions. Fortunately, the past five years have witnessed a revolution in the detection of the ferrous ion specifically, and this in turn has motivated new approaches and interest in iron(II)-dependent pharmacology.
First Selective Probes of Labile Ferrous Iron
The study of cellular ferrous iron has historically been hampered by a lack of chemical probes that could distinguish Fe2+ and Fe3+ ions [13]. Being relatively electron-poor, chelation of the ferric ion is characterized by sigma donation from electron-rich oxygen atoms of catechol or hydroxamate ligands. The more electron-rich ferrous iron can additionally engage in back-donation from metal d-orbitals to ligands capable of accepting such bonds – typically aromatic nitrogen heterocycles. This is why canonical (ferric) iron chelators like desferoxamine (DFO) employ hydroxamate ligands, while divalent metal ion probes like PhenGreen SK utilize a heteroaromatic 1,10-phenanthroline ligand. While some measure of selectivity can be achieved in this way, the key breakthrough in the detection of Fe2+ was a move from chelation-based to reactivity-based detection, as has been recently reviewed [14]. Thus, in 2014 Hirayama and Nagasawa described RhoNox-1 [15], the N-oxide form of fluorescein that is selectively reduced by Fe2+, producing a turn-on response (Figure 2). Further improvements to the RhoNox scaffold in terms of sensitivity and organelle-selective staining were subsequently achieved, yielding probes such as ER-SiRhoNox-1 [16], Mem-RhoNox [17], and Lyso-RhoNox [18], among others. Contemporaneously, our laboratory described 1,2,4-trioxolane (TRX) based scaffolds for Fe2+ detection [19,20], including a caged form of puromycin (TRX-PURO, Figure 2) able to detect Fe2+ in malaria parasites [21] and cancer cell lines [22] by immunofluorescence imaging with high specificity. Analogous to the reduction of the N–O bond in RhoNox probes, it is iron(II)-promoted reduction of a hindered O–O bond in TRX-bsed probes that promotes their activation. The TRX scaffold was subsequently utilized by us and others to produce FRET-based probes such as TRX-FRET [22] and FIP-1 [23] and the in vivo probes ICL-1 [24] and HNG [25], the later showing particular sensitivity to labile iron(II)heme. In 2019, Evans and Renslo described 18F-TRX [26], a Fe2+ probe for positron emission tomography (PET) that revealed an avidity for Fe2+ in several tumor types.
Figure 2.
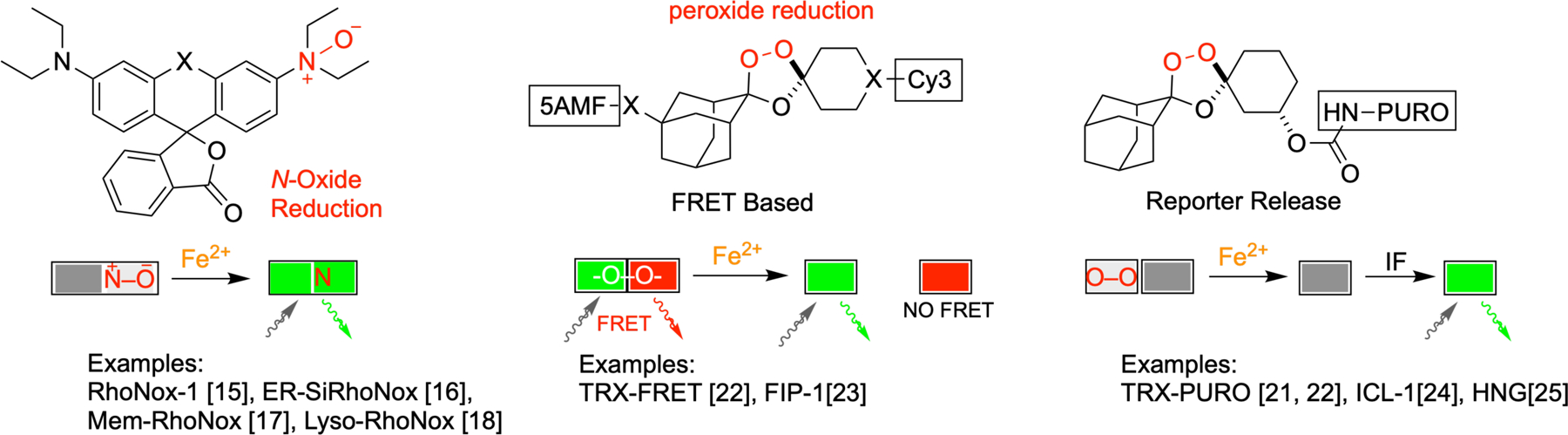
The first oxidation-state selective probes of ferrous iron rely on reactivity-based sensing. Three important classes of reactivity-based probes are shown, along with schematic representations of their modes of activation. RhoNox-based probes undergo iron(II)-promoted reduction of an N-oxide to produce a fluorescent signal. TRX-based probes include FRET-based turn-on and ratiometric probes, and caged reporter probes.
The availability of these new tools has enabled new insights into the role of ferrous iron in biology and disease pathology. Thus, RhoNox-class probes have confirmed iron reduction at the plasma and endolysosomal membranes [15,17] during cellular iron uptake, and revealed an elevation of lysosomal and ER-associated ferrous iron during ferroptotic cell death [18]. RhoNox probes have also linked hypoxia with elevated intracellular Fe2+ in tumor spheroids [16], and in cellular models of the blood-brain barrier [27] and the neural vascular barrier [28]. The trioxolane-based probes TRX-PURO and FIP-1 respectively revealed an elevation of the LIP in cancer cell lines [22] and in cells undergoing ferroptosis induced with the cystine/glutamate antiporter inhibitor erastin [23]. These probes have also revealed more unexpected roles for Fe2+ in the cell, such as a link between GPCR signaling and epigenetic regulation mediated by the LIP [29,30], and a possibly causative link between elevated Fe2+ and hepatic steatosis [31]. Among in vivo active TRX-type probes, ICL-1 has revealed elevated Fe2+ in mice infected with the bacterium Acinetobacter baumannii [24], while HNG detected heme(II)iron in the livers of mice following an acute hemolytic insult with phenylhydrazine [25]. Greater appreciation for the role of ferrous iron in disease will only increase as these new probes become more widely available and supplant the use of earlier, less selective probes.
Ferrous Iron-Dependent Pharmacology
The dysregulation of iron homeostasis has been associated with a wide range of disease states, from iron storage disorders [32], to cancer [33–35], neurodegenerative [36–38] and metabolic disease [39,40], and in infection by bacterial [41–43], viral [44], and eukaryotic [45–47] pathogens. Much as iron detection has historically relied on iron chelation, so have therapeutic strategies targeting iron been largely based on (ferric) iron chelation therapy [35,48,49] or on active drug uptake via conjugation to transferrin or bacterial iron siderophores [50–52]. While effective in certain disease contexts, chelation therapy can lead to anemia, and has been disappointing in the clinic as an approach to anti-infective or cancer therapy. However, the combination of natural chemical diversity and empiric traditional medicine has revealed an orthogonal approach to iron-based therapy exemplified today by standard-of-care antimalarial therapy with artemisinin-based agents. These endoperoxide-bearing sesquiterpenes are activated by ferrous iron heme liberated during parasite catabolism of hemoglobin in the erythrocyte [46]. Clinical success of this agent has driven a search for new chemotypes with analogous pharmacology, leading most notably to the 1,2,4-trioxolanes arterolane [53] and artefenomel [54,55] reported by Vennerstrom and co-workers. These wholly synthetic agents, like the artemisinins, exhibit highly iron(II)-selective reactivity (Box 1). Informed by careful mechanistic studies [56–59] detailing their iron(II)-dependent fragmentation, we and others have been inspired to expand the concept of ferrous iron-dependent pharmacology beyond its origins in antimalarial chemotherapy.
Box. 1. Antimalarial Endoperoxides.
The sesquiterpene artemisinin (qinghaosu) was first isolated in China in the 1970s as part of an effort to identify the active components in traditional Chinese herbal remedies [95]. While there was initial skepticism about the drug on account of the peroxide bond, clinical development of artemisinin-derived agents was ultimately undertaken and their clinical utility confirmed [96]. This success bred keen interest in the molecular pharmacology of these agents. Seminal mechanistic work by Posner in the 1990s involving synthetic artemisinin analogs, and later work by Vennerstrom involving synthetic 1,2,4-trioxolanes, established the crucial role of the peroxide bond in mediating the Fe2+ reactivity and the antimalarial effects of the artemisinin, arterolane, and artefenomel [46,59]. Space-filling models (Box 1, Figure I) of these agents reveal a hindered steric environment surround the peroxide bond. Fenton-type reaction with iron requires close inner-sphere coordination by Fe2+ prior to single-electron transfer that leads to O–O bond scission. Thus, the various C–H bonds surrounding the peroxide likely preclude interaction with co-factor Fe2+ bound to metalloproteins, while the major ferritin iron stores of the cell are both physically inaccessible and in a mineralized ferric and redox-stable state. Hence, it is the loosely chelated Fe2+ of the LIP and labile, unbound iron(II)heme that are in the correct redox state, and also able to physically approach the peroxide bond for reaction. These features are retained and exploited in the TRX-based probes and drug conjugates described herein.
Box 1, Figure I.
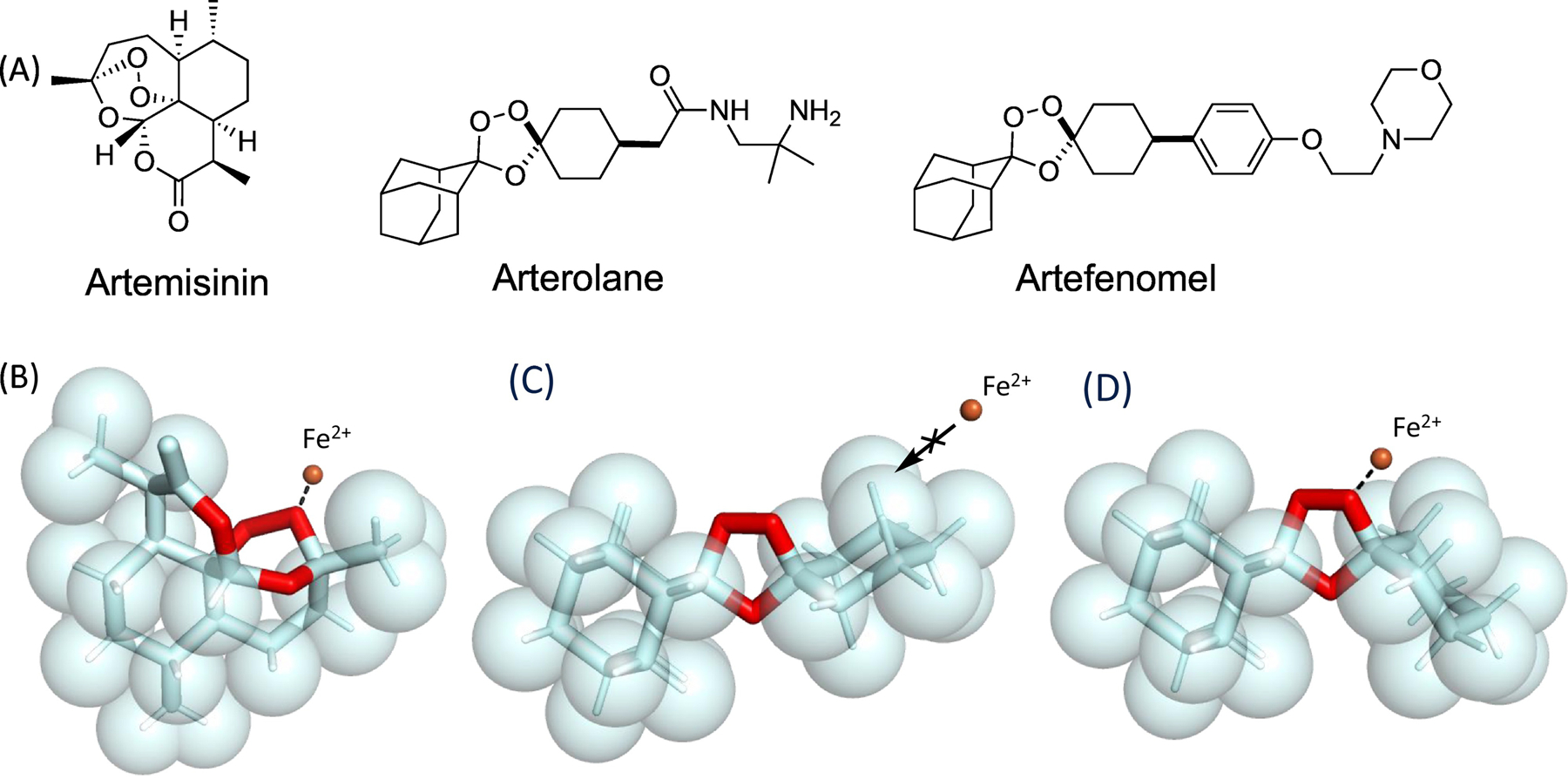
Structure-reactivity relationships of antimalarial peroxides. A. Structures of antimalarials artemisinin, arterolane, and artefenomel. B. Spacefilling representation of artemisinin showing approach (dashed line) of Fe2+ ion (orange sphere) to the more exposed of two peroxidic oxygen atoms. C. Spacefilling representation of arterolane pharmacophore in its major, unreactive conformer, with approach of the Fe2+ ion sterically blocked by proximal axial C–H bonds of the adamantane and cyclohexane rings. D. Spacefilling representation of the corresponding iron-reactive, peroxide equatorial conformer showing approach of the Fe2+ ion (orange sphere) to the exposed peroxidic oxygen atom. Artemisinin structure retrieved from the Cambridge Crystallographic Data Centrei and visualized using PyMol 2.4.0 (pymol.org). Arterolane pharmacophore structure minimized using MarvinSketch 6.3.0 (chemaxon.com) and visualized using PyMol 2.4.0 (pymol.org).
Ferrous Iron-Dependent Pharmacology in Bacterial Infection
The innate immune response to bacterial infection in mammals, known as nutritional immunity, comprises a diverse array of iron limiting and withholding strategies carried out via the ferroportin/hepcidin axis [43,60], the iron-binding protein lactoferrin [41], and the iron siderophore-scavenging protein lipocalin-2 [61]. In response, pathogenic bacteria deploy diverse adaptive responses to overcome iron withholding, including uptake and degradation of host heme iron, and assimilation of iron from transferrin, ferritin and lactoferrin [41]. The latter is achieved through the expression of small molecule siderophores with exceptionally high binding affinity for the Fe3+ ion [42]. The reliance of bacteria on siderophore mediated iron uptake has motivated an impressive volume of work on antibiotic-siderophore conjugates as notional ‘Trojan Horse’ therapies [52,62–64], the agent cefiderocol [65] being the first to receive marketing approval from the FDA, in 2019.
While siderophore-antibiotic conjugates necessarily exploit binding and uptake of Fe3+, several recent studies provide evidence for Fe2+ utilization by bacterial pathogens, which in turn suggests the potential to develop reactivity-based, ferrous iron-targeted therapy. Thus, Nolan and co-workers recently showed that the mammalian protein calprotectin sequesters the Fe2+ ion in response to bacterial infection and induces an iron starvation response in the pathogen Pseudomonas aeruginosa [66,67]. Evolution of a host response around Fe2+ (analogous to Fe3+ binding by lactoferrin) suggests that bacterial pathogens are able to exploit Fe2+ during infection. Indeed, organisms that evolved on the anoxic earth (prior to the ‘great oxidation’ that accompanied early photosynthesis) would require the means to exploit forms iron that predominate under such conditions. The most widely distributed of these bacterial Fe2+ transporters are members of the Feo system [68,69] and are known to be exploited by pathogens such as P. aeruginosa that often colonize hypoxic niches, paradoxically including the lungs of cystic fibrosis (CF) patients [70]. Most provocatively, a recent study [71] of live Escherichia coli using Mössbauer and EPR spectroscopy paints a dramatically different picture of bacterial iron homeostasis in which labile Fe2+ is in fact the predominant iron species present during exponential stages of bacterial growth.
The in vivo probe ICL-1 was recently applied [24] to study mice infected with the Gram-negative pathogen Acinetobacter baumannii (Figure 3A). Comprising the TRX-caged form of D-aminoluciferin, ICL-1 was shown in a series of cell free and cell-based experiments to luminesce (with luciferin and ATP), strictly contingent on its initial uncaging via reaction with Fe2+. Other metal ions (including Fe3+) and cellular reductants or oxidants were unable to uncage ICL-1, thereby confirming its utility as a selective probe of Fe2+. In luciferase-expressing mice infected with A. baumannii, or mock-infected as a control, ICL-1 revealed a substantial increases in bioluminescent signal across a range of tissues that were subsequently shown by ex vivo analysis to also be major sites of infection. By contrast, mock infected controls showed only minimal bioluminescent signal that was focused around the site of ICL-1 injection in the peritoneum (Figure 3A). Further ex vivo analysis of organs/tissues from both infected and mock-infected mice revealed that total iron in these tissues was not significantly elevated as a result of infection (the exception being in the liver, where ferritin iron accumulation in response to infection is to be expected). These findings with ICL-1 suggest that ferrous iron concentrations are elevated at sites of infection in a live animal, a perhaps surprising result given that nutritional immunity predicts iron limitation during infection. Further studies will be required to determine whether the observed effects derive from changes in host iron disposition, bacterial utilization or acquisition of iron, an emergent property of the infection microenvironment, or some combination of these factors. Whatever the case, the iron avidity of infected tissues revealed by ICL-1 clearly suggests the possibility of targeting antibiotics to infected tissues with analogous TRX-based drug conjugates.
Figure 3.
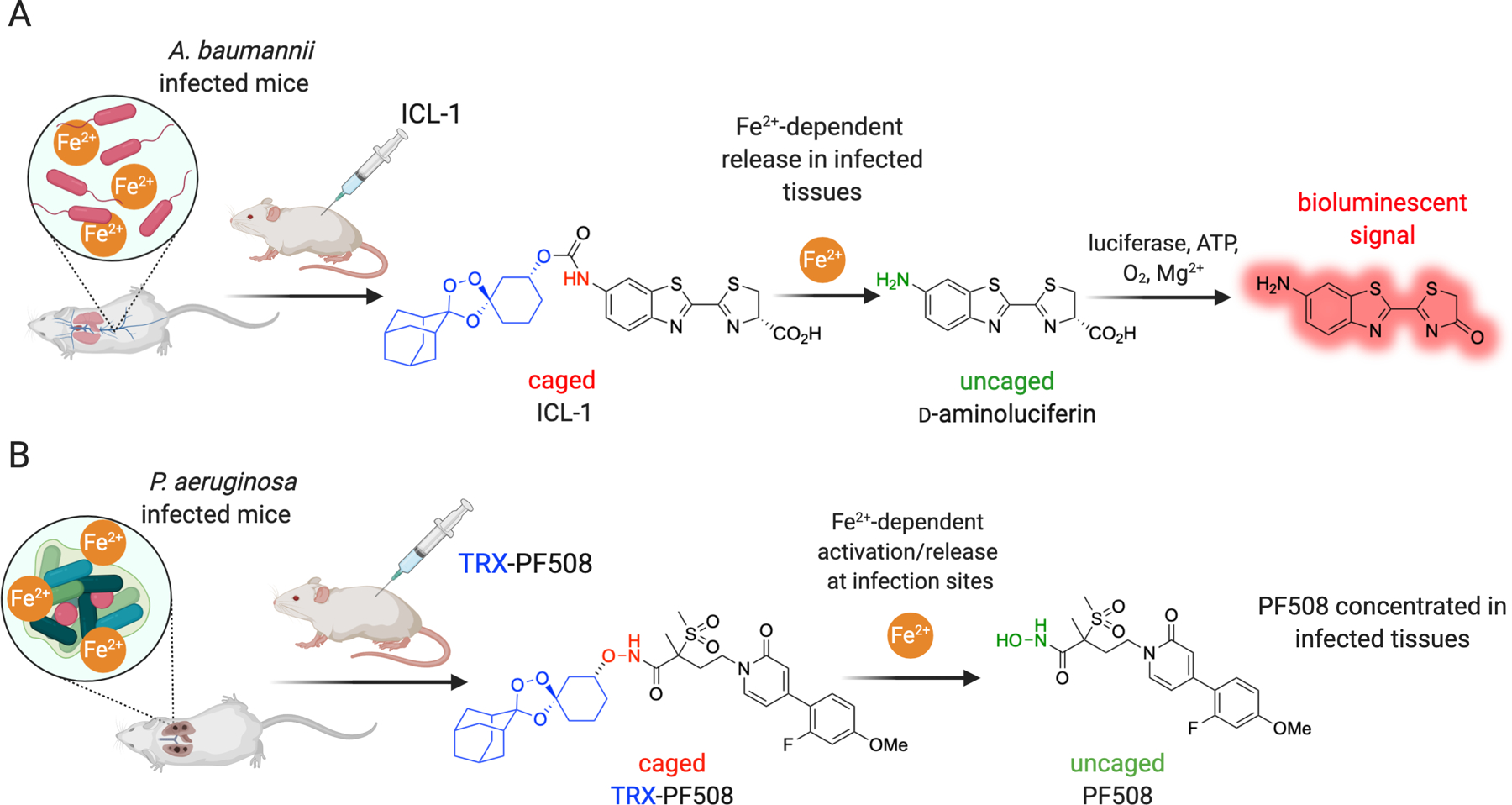
Iron(II)-based detection and treatment of mice infected with Gram-negative pathogens. A. Scheme illustrating bioluminescence imaging of Fe2+ at sites of infection using ICL-1 in FVB-Luc+ mice infected with A. baumannii. B. Scheme illustrating Fe2+ targeted therapy using the caged LpxC inhibitor TRX-PF508 in BALB/c mice bearing an acute lung infection with the Gram-negative pathogen P. aeruginosa.
The bacterial deacetylase LpxC performs the first committed step in lipopolysaccharide biosynthesis and this, combined with an absence of the pathway in mammals, has made this metalloenzyme a promising antibacterial drug target. Inhibition of LpxC has most often been approached with hydroxamate-based ligation of the enzyme’s zinc center. While hydroxamates can be highly potent inhibitors, the susceptibility of this functional group to proteolytic or metabolic degradation, including the formation of genotoxic intermediates [72], has hampered their clinical development. Our laboratory explored the use of a known LpxC inhibitor (PF-5081090) as the hydroxamate-linked conjugate TRX-PF508, which we surmised would be inactive toward LpxC and other metalloenzyme off-targets in the absence of Fe2+. Activation by Fe2+ at infection sites would liberate the LpxC inhibitor in its parent form (Figure 3B), and the desired iron(II)-dependent reactivity was confirmed in control experiments [73]. The antimicrobial activity of the conjugate in Minimum Inhibitory Concentration (MIC) assays was modest however, which suggested inefficient activation and/or limited access to Fe2+ in the bacterial cytoplasm in this in vitro setting. In contrast, when administered to mice in an acute P. aeruginosa lung infection model, TRX-PF508 produced significant reductions in bacterial load in the lung, and did so more robustly than in mice treated with the parent LpxC inhibitor at an equimolar dose. This implied a drug concentrating effect at sites of infection and was consistent with the compound’s predicted iron(II)-dependent pharmacology. Moreover, the observation with this compound of robust in vivo efficacy despite poor MICs, suggested that P. aeruginosa can utilize pools of Fe2+ in vivo that are not present in in vitro MIC assays. In fact, a microenvironment rich in Fe2+ has been demonstrated clinically in the case of chronic lung infection by P. aeruginosa in cystic fibrosis patients [74].
Ferrous Iron-Dependent Pharmacology in Cancer
It has been long been recognized that cancers exhibit an avidity for iron that seems to drive proliferation and metastasis [33,34]. Upregulation of the transferrin receptor is the canonical mechanism associated with this archetype but recent years have seen a striking array of novel mechanisms come to light. The appetite of the cancer cell for iron has been dubbed ‘iron addiction’ [75] and indeed, appears to follow in lockstep with dependence upon ongoing signaling from commonly mutated oncogenes. Thus, the seminal finding [76] that c-Myc regulates iron storage (ferritin) and regulatory (IRP2) genes was further corroborated when the c-Myc driven cell line PC3 was found, using TRX-PURO, to be among the most avid for Fe2+ across a panel of lines from different tumor types [77]. In two important studies from 2008, the lipid hydroperoxidase glutathione peroxidase 4 (GPX4) was found to promote survival of neurons [78] and mutant Ras driven cancer cells [79], seminal work that would help position GPX4 at the center of a pro-survival pathway that protects cells from an iron-dependent, nonapoptotic form of cell death subsequently dubbed ferroptosis [80]. Drug-tolerant ‘persister’ cells were similarly found to be highly dependent on GPX4 function for survival and therefore highly susceptible to GPX4 inhibition [81,82]. The hepcidin/ferroportin axis (Figure 1) can also contribute to ‘ferroaddiction’ via systemic liver hepcidin, or via hepcidin produced by the tumor itself in autocrine fashion, retaining tumoral iron and leading to more aggressive disease and poorer prognosis in breast [83], prostate [84], and pancreatic [85] cancers. In the case of gliomas, high-grade tumors of a mesenchymal subtype have been shown to possess significantly higher expression of transferrin receptor and lysosomal ferrireductase (STEAP3) as compared to low grade tumors [86]. How avidity for iron, and ferrous iron specifically, supports and promotes these aggressive and resistant cancer phenotypes remains largely unknown. One can speculate however, that ready access to soluble and metabolically active Fe2+ ion would be of significant benefit to rapidly proliferating cells, insofar as this would support the various iron-dependent enzyme functions required for cell growth and division. It appears a Faustian bargain is struck in which the benefits of elevated Fe2+ are exploited by managing (e.g., through greater reliance on GPX4) the dangerous levels of oxidative stress that result.
Given the avidity of cancer cells for iron, it is unsurprising that artemisinins and other antimalarial peroxides have been evaluated for their anticancer effects. In the most comprehensive of these studies, artesunate, dihydroartemisinin, arterolane, and artefenomel were profiled across 91 human cancer cell lines [87]. While these compounds showed measurable cytotoxicity in nearly all cell lines examined, their potencies landed squarely in the micromolar range, which compares unfavorably with the low-nanomolar effect of these same compounds in malaria parasites. Likely the difference stems from the presence in parasites of pathogenic free iron(II)heme, and a more robust cellular response to oxidative stress in cancer cells, many of which are likely pre-adapted to survive in a ferroaddicted state. Although these antimalarial compounds would appear to have limited clinical potential as single agents, they might yet find utility as adjuvant therapies, for example in combination with a GPX4 inhibitor as has been recently proposed [88]. Testing such hypotheses will however require the development of GPX4 inhibitors with suitable in vivo properties. It is also worth noting that the ferroptosis-inducing small molecule FINO2 [89,90] which bears an endoperoxide function, can be regarded as a starting point for the development of anticancer endoperoxides that exploit the ferroptosis pathway for therapeutic ends.
While arterolane itself has modest effects on cancer cells, we judged that TRX-based ferrous iron-targeting could be employed to direct much more potent therapeutics selectively to ferroaddicted cancer cells. To explore this notion, we employed a highly potent cyclobenzindoline (CBI) class DNA-alkylator and studied the resulting conjugate TRX-CBI in xenograft models of predicted high iron avidity [77]. Consistent with expectations, TRX-CBI conferred a potent low-nM insult on cell lines like PC3 that are ferroaddicted as judged with TRX-PURO. Importantly, TRX controls lacking the CBI payload, or non-peroxidic conjugates unable to release the CBI payload were at least 1000-fold less potent than TRX-CBI, confirming that the potent cell-killing effects of TRX-CBI derive from release of the free CBI payload. The effective caging of CBI toxicity by the TRX moiety allowed us to safely achieve >20-fold higher dose and in vivo exposure levels for TRX-CBI in mouse xenografts, which in turn produced dramatically superior cytocidal effect response when compared to CBI administered at its MTD. Moreover, the tumor-shrinking effects of TRX-CBI were achieved while largely avoiding CBI-associated hepatotoxicity in the same mice. These encouraging findings suggest that selective targeting of cancer cells through ferrous iron-dependent pharmacology represents a promising new way to deliver cancer chemotherapeutics.
In a recent preprint [bioRxiv 2020.05.12.088971] the Collisson and Renslo laboratories explored ferrous iron-dependent drug delivery in mutant KRAS-driven cancer cells and xenograft models. Several FDA-approved inhibitors of the KRAS effector MEK are currently available, but these agents uniformly exhibit dose-limiting toxicity resulting from chronic MEK1/2 inhibition in healthy tissues [91] at and below MTDs established in phase I trials. After establishing with both TRX-PURO and SiRhoNox [16] an elevated pool of Fe2+ in KRAS-driven pancreatic ductal adenocarcinoma (PDA) cells, we synthesized TRX-COBI, the ferrous iron-activated form of the approved MEK inhibitor cobimetinib. As hoped, TRX-COBI behaved as a bona fide MEK inhibitor in PDA cells, and ablated MAPK signaling in tumors of patient derived xenograft (PDX) and syngeneic PDA mouse models, as judged by pERK staining. Most importantly, TRX-COBI achieved effective MAPK blockade in PDA models as well as in autochothanous KrasLSL_G12D-driven lung adenocarcinoma, while sparing MAPK signaling in normal, tumor-free tissues from the same animals. Moreover, thinning of the epidermal layer in mouse tail skin (a model of clinically relevant MEK-related toxicity) was wholly avoided in TRX-COBI treated animals, while clearly evident in cobimetinib and binimetinib treated animals. Finally, we observed superior efficacy (Figure 4) with reduced weigh loss when employing TRX-COBI (in place of cobimetinib) in SHP-2 inhibitor (SHP2i) combination therapy of a KRASG12C-driven lung adenocarcinoma PDX model, consistent with improved systemic tolerability of TRX-COBI as compared to cobimetinib. Thus the significance of ferrous-iron dependent pharmacology in the context of mutant KRAS-driven malignancies could lie in the ability to achieve potent yet tolerable parallel and/or vertical pathway blockade, thus avoiding the inter- and intra-pathway signaling loops that enable tumor escape from single-agent blockade [92].
Figure 4.
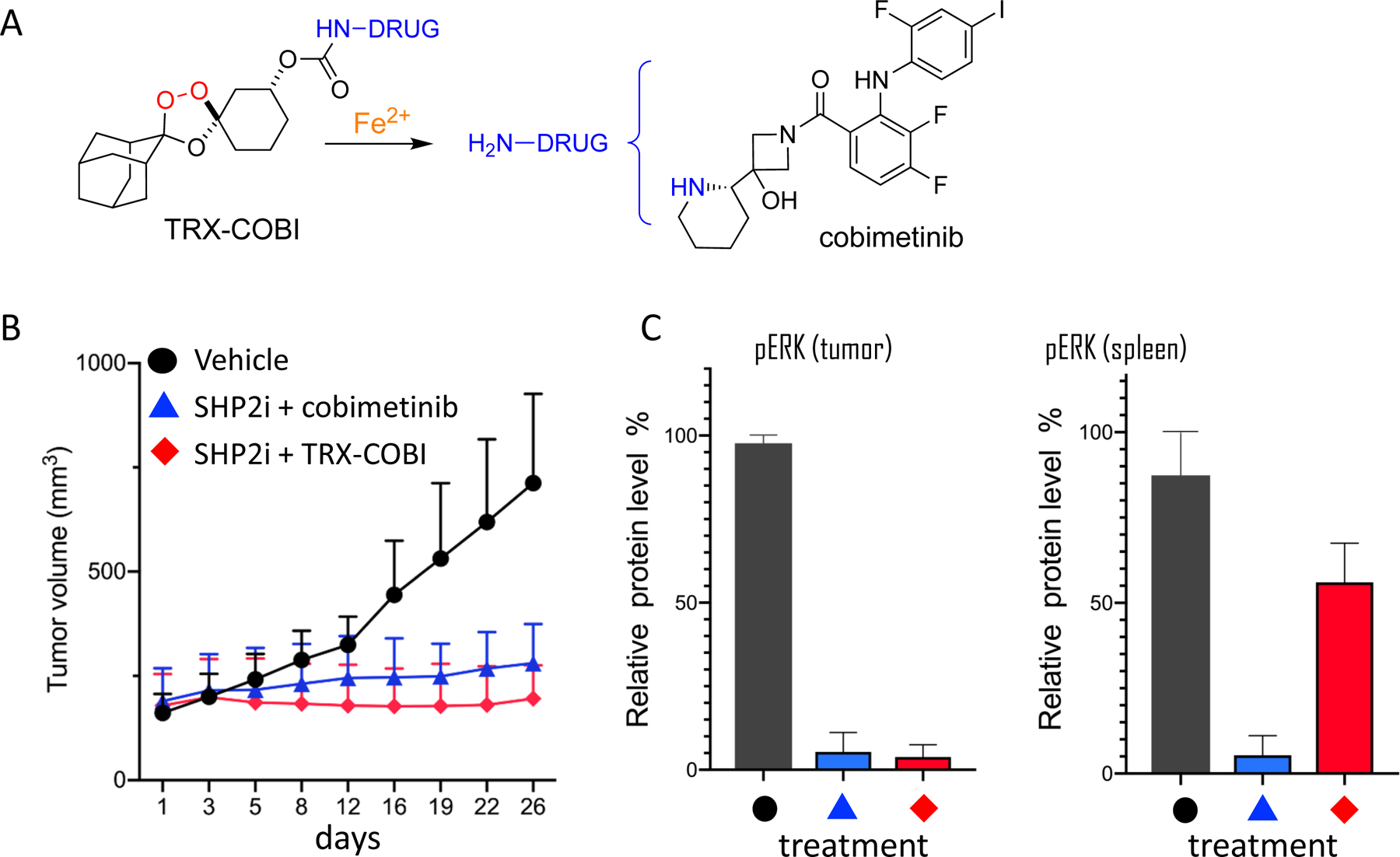
Efficacy of iron(II)-targeted forms of FDA-approved MEK inhibitor cobimetinib. A. Structure of TRX-DRUG conjugates such as TRX-COBI, which releases free cobimetinib upon reaction with Fe2+. B. Tumor volume over time in mice bearing KRAS lung adenocarcinoma and treated with vehicle, SHP2 inhibitor + cobimetinib, or SHP2i + TRX-COBI at equivalent, equimolar doses. C. Phospho-ERK staining of tumor in spleen from animals studied in panel B, indicating selective activation of TRX-COBI in tumor. Unpublished data reproduced from bioRxiv 2020.05.12.088971.
Concluding Remarks and Future Perspectives
Here we have detailed recent progress in the detection of iron with oxidation-state specificity and described some of the new biological findings and therapeutic approaches that have resulted from these advances. In science and in drug discovery, inquiry is often biased toward what is ‘easy’ to study (or to drug), rather than what is most important or relevant to study (or drug). The study of iron homeostasis has thus relied on (ferric) iron sequestration or supplementation experiments, and quantified iron load using experimental approaches that are blind to what is almost certainly the more relevant analyte – labile ferrous iron. Mirroring this state of affairs, iron-focused drug therapy is overwhelmingly focused on ferric iron chelation (e.g., in iron overload diseases), when it is labile ferrous iron that is associated with diseases of much greater burden to humankind – malaria most notably, but apparently also many of the most aggressive and untreatable cancers and, more tenuously, also in neurogenerative disease.
Much remains to be learned and understood about the role of ferrous iron under conditions of normal and aberrant homeostasis, and its relative distribution in normal and diseased tissues (see Outstanding Questions). Will these conditions be best treated with drugs designed to restore a normal homeostatic state, or through reactivity-based approaches that exploit elevated ferrous iron levels to achieve tissue selectivity, as detailed in some recent work highlighted herein. The unprecedented clinical impact of artemisinin therapy in malaria would seem to argue for the latter approach, and yet decades after its introduction, artemisinin resides in a lonely niche of the pharmacopeia. This fact can be attributed in part to a reticence of drug discovery scientists to pursue reactivity-based pharmacology. However, a significant proportion of currently used drugs have been found – often well after their discovery and approval – to act unexpectedly via covalent, reactive mechanisms [93]. Increasing appreciation for this fact has seen a resurgence of work on drugs designed from the outset to exhibit reactivity-based pharmacology, with cysteine-reactive drugs and drug candidates becoming commonplace. Next-generation trioxolane antimalarials like artefenomel combine exquisitely selective iron(II)-dependent reactivity with excellent pharmacokinetic and safety profiles in human subjects [94]. The utility of this chemotype as applied to reactivity-based probes and in ferrous-iron dependent drug delivery, as reviewed here, suggests a bright future for the study of iron homeostasis and metabolism, and in the treatment of any disease state in which ferrous iron levels are dysregulated.
Outstanding Questions.
How precisely do elevated labile ferrous iron concentrations promote cell growth and proliferation in cancer?
Is the labile iron pool necessarily elevated in cells that are sensitive to, or actively undergoing ferroptosis?
What is the source and nature of the free ferrous iron that appears to be mobilized during bacterial infection?
Which normal tissues and cells harbor elevated ferrous iron concentrations under conditions of normal iron homeostasis?
Is there a link between ferroptotic cell death, aberrant accumulation of ferrous iron, and neurodegenerative disease?
Which iron regulatory proteins might be targeted to limit the labile iron pool and what effect would such intervention have in proliferative or neurodegenerative disease?
Highlights.
Iron cycles between the ferric and ferrous oxidation states to empower enzyme function but also during its uptake, export, and storage, with the ferrous state being generated transiently and in limited concentrations to prevent oxidative damage to the cell.
Until very recently, it has not been possible to study the ferrous ion specifically, largely due to its much poorer ligation by small molecule or protein siderophores, as compared to the ferric ion.
The emerging role of ferrous iron in a form of iron-dependent cell death (ferroptosis), and in supporting the proliferation of some of the most aggressive cancers has given new urgency to the study of cellular ferrous iron in biology and disease.
The clinical success of the antimalarial artemisinin provides inspiration as well as a chemical and conceptual framework for the development of a new class of therapeutic agents exhibiting ferrous iron dependent pharmacology.
Acknowledgements
This work was supported by US National Institutes of Health R01 Grant AI105106 to AR Renslo
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclaimer Statement
EA Collisson and AR Renslo are co-founders of Tatara Therapeutics, Inc.
Resources
References
- 1.Rosenzweig AC (2002) Metallochaperones: bind and deliver. Chem. Biol 9, 673–677 [DOI] [PubMed] [Google Scholar]
- 2.Aisen P et al. (2001) Chemistry and biology of eukaryotic iron metabolism. Int. J. Biochem. Cell Biol 33, 940–959 [DOI] [PubMed] [Google Scholar]
- 3.Ganz T (2013) Systemic iron homeostasis. Physiol. Rev 93, 1721–1741 [DOI] [PubMed] [Google Scholar]
- 4.Sies H and Jones DP (2020) Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol 21, 363–383 [DOI] [PubMed] [Google Scholar]
- 5.Hentze MW et al. (2010) Two to tango: regulation of Mammalian iron metabolism. Cell 142, 24–38 [DOI] [PubMed] [Google Scholar]
- 6.Nairz M et al. (2015) ‘Ride on the ferrous wheel’--the cycle of iron in macrophages in health and disease. Immunobiology 220, 280–294 [DOI] [PubMed] [Google Scholar]
- 7.Asano T et al. (2011) Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol. Cell. Biol 31, 2040–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber RA et al. (2020) Maintaining iron homeostasis is the key role of lysosomal acidity for cell proliferation. Mol. Cell 77, 645–655.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi H et al. (2008) A cytosolic iron chaperone that delivers iron to ferritin. Science 320, 1207–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryu M-S et al. (2017) PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J. Clin. Invest 127, 1786–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel SJ et al. (2019) A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat. Chem. Biol 15, 872–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hider RC and Kong XL (2011) Glutathione: a key component of the cytoplasmic labile iron pool. Biometals 24, 1179–1187 [DOI] [PubMed] [Google Scholar]
- 13.Carter KP et al. (2014) Fluorescent sensors for measuring metal ions in living systems. Chem. Rev 114, 4564–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aron AT et al. (2018) Activity-based sensing fluorescent probes for iron in biological systems. Curr. Opin. Chem. Biol 43, 113–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niwa M et al. (2014) A new class of high-contrast Fe(II) selective fluorescent probes based on spirocyclized scaffolds for visualization of intracellular labile iron delivered by transferrin. Org. Biomol. Chem 12, 6590–6597 [DOI] [PubMed] [Google Scholar]
- 16.Hirayama T et al. (2017) A universal fluorogenic switch for Fe(ii) ion based on N-oxide chemistry permits the visualization of intracellular redox equilibrium shift towards labile iron in hypoxic tumor cells. Chem. Sci 8, 4858–4866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niwa M et al. (2018) Fe(II) Ion Release during Endocytotic Uptake of Iron Visualized by a Membrane-Anchoring Fe(II) Fluorescent Probe. ACS Chem. Biol 13, 1853–1861 [DOI] [PubMed] [Google Scholar]
- 18.Hirayama T et al. (2019) Organelle-specific analysis of labile Fe(ii) during ferroptosis by using a cocktail of various colour organelle-targeted fluorescent probes. Metallomics 11, 111–117 [DOI] [PubMed] [Google Scholar]
- 19.Fontaine SD et al. (2014) Efficient and stereocontrolled synthesis of 1,2,4-trioxolanes useful for ferrous iron-dependent drug delivery. Org. Lett 16, 5776–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahajan SS et al. (2011) A fragmenting hybrid approach for targeted delivery of multiple therapeutic agents to the malaria parasite. ChemMedChem 6, 415–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fontaine SD et al. (2015) Drug delivery to the malaria parasite using an arterolane-like scaffold. ChemMedChem 10, 47–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spangler B et al. (2016) A reactivity-based probe of the intracellular labile ferrous iron pool. Nat. Chem. Biol 12, 680–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aron AT et al. (2016) An Endoperoxide Reactivity-Based FRET Probe for Ratiometric Fluorescence Imaging of Labile Iron Pools in Living Cells. J. Am. Chem. Soc 138, 14338–14346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aron AT et al. (2017) In vivo bioluminescence imaging of labile iron accumulation in a murine model of Acinetobacter baumannii infection. Proc. Natl. Acad. Sci. USA 114, 12669–12674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu S et al. (2020) Learning from Artemisinin: Bioinspired Design of a Reaction-Based Fluorescent Probe for the Selective Sensing of Labile Heme in Complex Biosystems. J. Am. Chem. Soc 142, 2129–2133 [DOI] [PubMed] [Google Scholar]
- 26.Muir RK et al. (2019) Measuring Dynamic Changes in the Labile Iron Pool in Vivo with a Reactivity-Based Probe for Positron Emission Tomography. ACS Cent. Sci 5, 727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imai T et al. (2019) Intracellular Fe2+ accumulation in endothelial cells and pericytes induces blood-brain barrier dysfunction in secondary brain injury after brain hemorrhage. Sci. Rep 9, 6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui D et al. (2019) Hypoxia-induced disruption of neural vascular barrier is mediated by the intracellular induction of Fe(II) ion. Exp. Cell Res 379, 166–171 [DOI] [PubMed] [Google Scholar]
- 29.Camarena V et al. (2017) cAMP signaling regulates DNA hydroxymethylation by augmenting the intracellular labile ferrous iron pool. Elife 6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huff TC et al. (2020) Oscillatory cAMP signaling rapidly alters H3K4 methylation. Life Sci. Alliance 3, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Protchenko O et al. (2020) Iron chaperone PCBP1 protects murine liver from lipid peroxidation and steatosis. Hepatology DOI: 10.1002/hep.31328 [DOI] [PMC free article] [PubMed]
- 32.De Domenico I et al. (2008) Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol 9, 72–81 [DOI] [PubMed] [Google Scholar]
- 33.Kwok JC and Richardson DR (2002) The iron metabolism of neoplastic cells: alterations that facilitate proliferation? Crit. Rev. Oncol. Hematol 42, 65–78 [DOI] [PubMed] [Google Scholar]
- 34.Torti SV and Torti FM (2013) Iron and cancer: more ore to be mined. Nat. Rev. Cancer 13, 342–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown RAM et al. (2020) Altered iron metabolism and impact in cancer biology, metastasis, and immunology. Front. Oncol 10, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belaidi AA and Bush AI (2016) Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J. Neurochem 139 Suppl 1, 179–197 [DOI] [PubMed] [Google Scholar]
- 37.Masaldan S et al. (2019) Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med 133, 221–233 [DOI] [PubMed] [Google Scholar]
- 38.Ndayisaba A et al. (2019) Iron in Neurodegeneration - Cause or Consequence? Front. Neurosci 13, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wessling-Resnick M (2010) Iron homeostasis and the inflammatory response. Annu. Rev. Nutr 30, 105–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Britton LJ et al. (2016) Iron and non-alcoholic fatty liver disease. World J. Gastroenterol 22, 8112–8122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carver PL (2018) The Battle for Iron between Humans and Microbes. Curr. Med. Chem 25, 85–96 [DOI] [PubMed] [Google Scholar]
- 42.Holden VI and Bachman MA (2015) Diverging roles of bacterial siderophores during infection. Metallomics 7, 986–995 [DOI] [PubMed] [Google Scholar]
- 43.Palmer LD and Skaar EP (2016) Transition metals and virulence in bacteria. Annu. Rev. Genet 50, 67–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wessling-Resnick M (2018) Crossing the iron gate: why and how transferrin receptors mediate viral entry. Annu. Rev. Nutr 38, 431–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark MA et al. (2014) Influence of host iron status on Plasmodium falciparum infection. Front. Pharmacol 5, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Neill PM and Posner GH (2004) A medicinal chemistry perspective on artemisinin and related endoperoxides. J. Med. Chem 47, 2945–2964 [DOI] [PubMed] [Google Scholar]
- 47.Rosenthal MR and Ng CL (2020) Plasmodium falciparum Artemisinin Resistance: The Effect of Heme, Protein Damage, and Parasite Cell Stress Response. ACS Infect. Dis 6, 1599–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalinowski DS and Richardson DR (2005) The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev 57, 547–583 [DOI] [PubMed] [Google Scholar]
- 49.Crielaard BJ et al. (2017) Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov 16, 400–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mislin GLA and Schalk IJ (2014) Siderophore-dependent iron uptake systems as gates for antibiotic Trojan horse strategies against Pseudomonas aeruginosa. Metallomics 6, 408–420 [DOI] [PubMed] [Google Scholar]
- 51.Schalk IJ and Mislin GLA (2017) Bacterial iron uptake pathways: gates for the import of bactericide compounds. J. Med. Chem 60, 4573–4576 [DOI] [PubMed] [Google Scholar]
- 52.Lin Y-M et al. (2019) Synthetic sideromycins (skepticism and optimism): selective generation of either broad or narrow spectrum Gram-negative antibiotics. Biometals 32, 425–451 [DOI] [PubMed] [Google Scholar]
- 53.Vennerstrom JL et al. (2004) Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 430, 900–904 [DOI] [PubMed] [Google Scholar]
- 54.Charman SA et al. (2011) Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc. Natl. Acad. Sci. USA 108, 4400–4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong Y et al. (2017) Structure-Activity Relationship of the Antimalarial Ozonide Artefenomel (OZ439). J. Med. Chem 60, 2654–2668 [DOI] [PubMed] [Google Scholar]
- 56.Tang Y et al. (2005) Dispiro-1,2,4-trioxane analogues of a prototype dispiro-1,2,4-trioxolane: mechanistic comparators for artemisinin in the context of reaction pathways with iron(II). J. Org. Chem 70, 5103–5110 [DOI] [PubMed] [Google Scholar]
- 57.Wang X et al. (2007) Spiro- and dispiro-1,2-dioxolanes: contribution of iron(II)-mediated one-electron vs two-electron reduction to the activity of antimalarial peroxides. J. Med. Chem 50, 5840–5847 [DOI] [PubMed] [Google Scholar]
- 58.Creek DJ et al. (2008) Relationship between antimalarial activity and heme alkylation for spiro- and dispiro-1,2,4-trioxolane antimalarials. Antimicrob. Agents Chemother 52, 1291–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Creek DJ et al. (2007) Iron-mediated degradation kinetics of substituted dispiro-1,2,4-trioxolane antimalarials. J. Pharm. Sci 96, 2945–2956 [DOI] [PubMed] [Google Scholar]
- 60.Cassat JE and Skaar EP (2013) Iron in infection and immunity. Cell Host Microbe 13, 509–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flo TH et al. (2004) Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432, 917–921 [DOI] [PubMed] [Google Scholar]
- 62.Neumann W et al. (2018) Esterase-Catalyzed Siderophore Hydrolysis Activates an Enterobactin-Ciprofloxacin Conjugate and Confers Targeted Antibacterial Activity. J. Am. Chem. Soc 140, 5193–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu R et al. (2018) A Synthetic Dual Drug Sideromycin Induces Gram-Negative Bacteria To Commit Suicide with a Gram-Positive Antibiotic. J. Med. Chem 61, 3845–3854 [DOI] [PubMed] [Google Scholar]
- 64.Schalk IJ (2018) Siderophore-antibiotic conjugates: exploiting iron uptake to deliver drugs into bacteria. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases 24, 801. [DOI] [PubMed] [Google Scholar]
- 65.Lee YR and Yeo S (2020) Cefiderocol, a New Siderophore Cephalosporin for the Treatment of Complicated Urinary Tract Infections Caused by Multidrug-Resistant Pathogens: Preclinical and Clinical Pharmacokinetics, Pharmacodynamics, Efficacy and Safety. Clin Drug Investig DOI: 10.1007/s40261-020-00955-x [DOI] [PMC free article] [PubMed]
- 66.Nakashige TG et al. (2015) Human calprotectin is an iron-sequestering host-defense protein. Nat. Chem. Biol 11, 765–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zygiel EM et al. (2019) The human innate immune protein calprotectin induces iron starvation responses in Pseudomonas aeruginosa. J. Biol. Chem 294, 3549–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Minandri F et al. (2016) Role of Iron Uptake Systems in Pseudomonas aeruginosa Virulence and Airway Infection. Infect. Immun 84, 2324–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lau CKY et al. (2016) Bacterial ferrous iron transport: the Feo system. FEMS Microbiol. Rev 40, 273–298 [DOI] [PubMed] [Google Scholar]
- 70.Tyrrell J and Callaghan M (2016) Iron acquisition in the cystic fibrosis lung and potential for novel therapeutic strategies. Microbiology (Reading, Engl.) 162, 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wofford JD et al. (2019) Evidence that a respiratory shield in Escherichia coli protects a low-molecular-mass FeII pool from O2-dependent oxidation. J. Biol. Chem 294, 50–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shen S and Kozikowski AP (2016) Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors--What Some May Have Forgotten or Would Rather Forget? ChemMedChem 11, 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blank BR et al. (2019) Targeting Mobilization of Ferrous Iron in Pseudomonas aeruginosa Infection with an Iron(II)-Caged LpxC Inhibitor. ACS Infect. Dis 5, 1366–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hunter RC et al. (2013) Ferrous iron is a significant component of bioavailable iron in cystic fibrosis airways. MBio 4, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basuli D et al. (2017) Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene 36, 4089–4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu KJ et al. (1999) Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 283, 676–679 [DOI] [PubMed] [Google Scholar]
- 77.Spangler B et al. (2016) A Novel Tumor-Activated Prodrug Strategy Targeting Ferrous Iron Is Effective in Multiple Preclinical Cancer Models. J. Med. Chem 59, 11161–11170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seiler A et al. (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8, 237–248 [DOI] [PubMed] [Google Scholar]
- 79.Yang WS and Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol 15, 234–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dixon SJ et al. (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Viswanathan VS et al. (2017) Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hangauer MJ et al. (2017) Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pinnix ZK et al. (2010) Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med 2, 43ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tesfay L et al. (2015) Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res 75, 2254–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Toshiyama R et al. (2018) Association of iron metabolic enzyme hepcidin expression levels with the prognosis of patients with pancreatic cancer. Oncol. Lett 15, 8125–8133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han M et al. (2018) Six-Transmembrane Epithelial Antigen of Prostate 3 Predicts Poor Prognosis and Promotes Glioblastoma Growth and Invasion. Neoplasia 20, 543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hooft van Huijsduijnen R et al. (2013) Anticancer properties of distinct antimalarial drug classes. PLoS One 8, e82962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen G-Q et al. (2020) Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ 27, 242–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abrams RP et al. (2016) Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. ACS Chem. Biol 11, 1305–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gaschler MM et al. (2018) FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol 14, 507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao Y and Adjei AA (2014) The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol 11, 385–400 [DOI] [PubMed] [Google Scholar]
- 92.Caunt CJ et al. (2015) MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat. Rev. Cancer 15, 577–592 [DOI] [PubMed] [Google Scholar]
- 93.Bauer RA (2015) Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 20, 1061–1073 [DOI] [PubMed] [Google Scholar]
- 94.Phyo AP et al. (2016) Antimalarial activity of artefenomel (OZ439), a novel synthetic antimalarial endoperoxide, in patients with Plasmodium falciparum and Plasmodium vivax malaria: an open-label phase 2 trial. Lancet Infect. Dis 16, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Klayman DL (1985) Qinghaosu (artemisinin): an antimalarial drug from China. Science 228, 1049–1055 [DOI] [PubMed] [Google Scholar]
- 96.Faurant C (2011) From bark to weed: the history of artemisinin. Parasite 18, 215–218 [DOI] [PMC free article] [PubMed] [Google Scholar]