

Abstract
Aims
Duchenne muscular dystrophy (DMD) is an X‐linked inherited disease due to dystrophin deficiency causing skeletal and cardiac muscle dysfunction. Affected patients lose ambulation by age 12 and usually die in the second to third decades of life from cardiac and respiratory failure. Symptomatic treatment includes the use of anti‐inflammatory corticosteroids, which are associated with side effects including weight gain, osteoporosis, and increased risk of cardiovascular disease. Novel treatment options include blockade of the renin–angiotensin–aldosterone system, because angiotensin as well as aldosterone contribute to persistent inflammation and fibrosis, and aldosterone blockade represents an efficacious anti‐fibrotic approach in cardiac failure. Recent preclinical findings enabled successful clinical testing of a combination of steroidal mineralocorticoid receptor antagonists (MRAs) and angiotensin converting enzyme inhibitors in DMD boys. The efficacy of MRAs alone on dystrophic skeletal muscle and heart has not been investigated. Here, we tested efficacy of the novel non‐steroidal MRA finerenone as a monotherapy in a preclinical DMD model.
Methods and results
The dystrophin‐deficient, utrophin haploinsufficient mouse model of DMD was treated with finerenone and compared with untreated dystrophic and wild‐type controls. Grip strength, electrocardiography, cardiac magnetic resonance imaging, muscle force measurements, histological quantification, and gene expression studies were performed. Finerenone treatment alone resulted in significant improvements in clinically relevant functional parameters in both skeletal muscle and heart. Normalized grip strength in rested dystrophic mice treated with finerenone (40.3 ± 1.0 mN/g) was significantly higher (P = 0.0182) compared with untreated dystrophic mice (35.2 ± 1.5 mN/g). Fatigued finerenone‐treated dystrophic mice showed an even greater relative improvement (P = 0.0003) in normalized grip strength (37.5 ± 1.1 mN/g) compared with untreated mice (29.7 ± 1.1 mN/g). Finerenone treatment also led to significantly lower (P = 0.0075) susceptibility to limb muscle damage characteristic of DMD measured during a contraction‐induced injury protocol. Normalized limb muscle force after five lengthening contractions resulted in retention of 71 ± 7% of baseline force in finerenone‐treated compared with only 51 ± 4% in untreated dystrophic mice. Finerenone treatment also prevented significant reductions in myocardial strain rate (P = 0.0409), the earliest sign of DMD cardiomyopathy. Moreover, treatment with finerenone led to very specific cardiac gene expression changes in clock genes that might modify cardiac pathophysiology in this DMD model.
Conclusions
Finerenone administered as a monotherapy is disease modifying for both skeletal muscle and heart in a preclinical DMD model. These findings support further evaluation of finerenone in DMD clinical trials.
Keywords: ACE/angiotensin receptors/renin–angiotensin system, Muscular dystrophy, Animal models of human disease, Cardiomyopathy, Mineralocorticoid receptors
Introduction
Duchenne muscular dystrophy (DMD) is a genetic, progressive, neuromuscular disease caused by a mutation in the dystrophin gene and characterized by striated muscle degeneration. 1 , 2 Subsequent conversion of muscle mass to fibrosis leads to loss of ambulation as well as cardiac and respiratory failure. About 1 in every 5000 boys is affected by this X‐linked disorder, 3 and there is still no long‐term treatment.
Current standard‐of‐care treatment for DMD consists of corticosteroids beginning in early childhood, with additional nocturnal ventilatory support and angiotensin converting enzyme inhibitors (ACEis), respectively, as respiratory and cardiac function declines. Corticosteroids are used to treat inflammation and have immunosuppressant properties that temporarily extend ambulation and control symptoms in DMD patients. 4 , 5 However, their use remains controversial, in part because the long‐term corticosteroid dosing is associated with many negative side effects including weight gain, osteoporosis, hormone deficiencies, increased risk of cardiovascular disease, and adverse behavioural changes. 6 , 7 Studies conducted in dystrophic mice also show continuous corticosteroid treatment leads to increased cardiac and muscle damage. 8 , 9 , 10
Negative consequences from long‐term use of corticosteroids and the progressive cardiomyopathy present in DMD even with ACEi treatment 11 , 12 justify the continued search for new therapies to improve the quality of life of DMD patients. Surprisingly, preclinical studies using DMD mouse models (utrn +/− ;mdx ‘Het’) demonstrated that treatment with mineralocorticoid receptor (MR) antagonists in combination with an ACEi not only improved cardiac function but also resulted in improved respiratory and limb muscle forces, reduction of ongoing muscle damage, and improved muscle membrane integrity. 13 , 14 , 15 These studies have also demonstrated that non‐specific MR antagonism (by spironolactone) and specific MR antagonism (by eplerenone) in respective combination with ACEi have comparable efficacy in muscular dystrophy in mice and that ACEi monotherapy improves muscle histopathology, but does not improve contractile function in DMD mice, strongly supporting an important role of MR in DMD pathophysiology. 14 , 16
MR are known to be present in many cell types including endothelial cells, myeloid cells and cardiomyocytes, and we showed that they are also present in all normal and dystrophic skeletal muscles. 17 , 18 Pathophysiological conditions like elevated aldosterone release, high dietary salt load, or increased generation of reactive oxygen species can cause an MR overactivation with subsequent expression of pro‐inflammatory and fibrotic proteins in the indicated cell types, which ultimately lead to cardiovascular damage and dysfunction. 19 Myeloid inflammatory cells are capable of synthesizing aldosterone and lead to increased aldosterone levels in dystrophic mouse muscles. 20 Blocking this signalling from chronic inflammation in dystrophic muscle likely explains the efficacy of MR antagonism.
Our team translated the preclinical cardiac benefits to a double‐blind placebo controlled clinical trial with a 2 year extension study demonstrating that MR antagonism added to ACEi further prevents cardiac dysfunction in DMD patients compared with ACEi alone. 21 , 22 We then demonstrated in a non‐inferiority clinical trial equivalency between spironolactone and eplerenone on cardiac parameters in DMD patients. 23
Our recent studies have demonstrated that a conditional knockout of MR in myofibers reproduces many parameters of efficacy of ACEi + MR antagonism in a DMD mouse model, but in vivo functions of MR antagonism alone, without ACEi, have never been investigated. 24 An ongoing clinical study with spironolactone alone in young DMD boys warrants further preclinical investigation of the effect of MR antagonism as a monotherapy on the later onset cardiac dysfunction. Moreover, skeletal muscle gene expression changes have been shown to result from in vivo treatment of dystrophic mice with steroidal MR antagonists (MRAs) plus ACEi, but cardiac gene expression in dystrophic mice treated with MRAs alone are missing. 13 , 17
The steroidal MRA spironolactone binds MR at high affinity but has off‐target effects on other hormone receptors including the androgen receptor, which causes the clinical side‐effect gynaecomastia in post‐pubescent males and influences treatment decisions in the male DMD population. While not shown in DMD trials to date, the steroidal MRAs spironolactone and eplerenone typically require careful monitoring for the potential adverse events of hyperkalaemia, particularly when given on top of inhibitors of the renin–angiotensin system such as ACEis or angiotensin receptor blockers to patients with concomitant kidney dysfunction.
Novel non‐steroidal MRAs such as finerenone have been identified recently. 25 , 26 These compounds have a different pharmacological profile in comparison with steroidal MRAs at least in preclinical studies. 27 , 28 Finerenone has greater MR selectivity than spironolactone and higher receptor affinity than eplerenone in vitro. 29 , 30 In a clinical trial of patients with chronic heart failure and chronic kidney disease, finerenone was at least as effective as spironolactone and was associated with significantly lower side effects. 31 Preclinical studies demonstrated that there is a greater reduction in cardiovascular end‐organ damage, fibrosis, and proteinuria with finerenone compared with eplerenone 25 , 32 , 33 and therefore generated the hypothesis that use of finerenone is associated with a more pronounced anti‐hypertrophic/anti‐fibrotic effect combined with a reduced risk of developing hyperkalaemia. 34 , 35
Therefore, it was the aim of the current study to investigate finerenone's disease‐modifying efficacy as a monotherapy in a preclinical DMD model and to explore for the first time the cardiac expression profile in MRA‐treated vs. untreated DMD animals.
Methods
Experimental animals and treatment
The Institutional Animal Care and Use Committee of The Ohio State University approved all protocols in compliance with the laws of the USA. All animal procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All personnel performing experiments and collecting data were blinded to treatment and genotypes of the animals throughout the study.
Dystrophin‐deficient, utrophin haploinsufficient (utrn +/−; mdx) Het mice on a C57BL/10 background were bred in‐house since their generation, genotyped as previously described, and used in this study. 15 , 36 C57BL/10 mice bred in‐house (originally obtained from Envigo) (n = 18; 11 male, 7 female) were used as wild‐type controls. Het mice were randomly assigned into treated and untreated groups (n = 18 per group) keeping male and female groups equal (9 male and 9 female mice per group). All mice were housed in the same room under standard temperature, humidity, and light/dark cycle conditions. Het treated mice were given a custom diet Teklad Rodent Chow #7912 containing 100 ppm (parts per million) finerenone (Bayer AG, Wuppertal, Germany) prepared by Research Diets, Inc. Untreated Het mice and C57BL/10 wild‐type control mice received Teklad Rodent Chow #7912 without medication. Medicated pellets were replaced every week. Body weights and food intake were monitored weekly to ensure an approximate drug dosage of approximately 3 mg/kg * day‐1. The finerenone chow dosage was calculated to reach a plasma concentration equivalent to once daily oral gavages between 1 and 10 mg/kg, which were previously shown to be efficacious in diverse chronic rat models. 25 A previous study in a mouse disease model revealed a free plasma drug concentration of 28 μg/L (equivalent to about 3xIC50 of finerenone at MR in vitro) after 4 weeks on a custom diet with 100 ppm finerenone. 37 This plasma exposure is similar to steady‐state exposures in male CD‐1 mice after a once daily gavage of 3 mg/kg (Bayer AG).
Mice were started on treatment at 4 weeks‐of‐age at weaning or left untreated and were all sacrificed at 20 weeks‐of‐age. Mice were euthanized by cervical dislocation without anaesthesia following the institutional animal care and use committee and American Veterinary Medical Association guidelines.
In vivo grip strength measurements
A grip strength meter (Columbus Instruments) was used to evaluate forelimb muscle strength according to the methods reported previously. 38 In brief, prior to the initiation of the experiments, mice were trained during two sessions occurring at least 2 days apart. At least 2 days after the second training period, five pulls were recorded by allowing the mice to grasp the bar on the meter and then pulling them gently by the tail. Each group of five pulls was followed by a 1 min rest and this procedure was repeated five times. The highest value in the first trial was used as the peak force produced from rested mice, and the highest value in the fifth trial was used as the peak force produced in fatigued mice. Exclusion criteria were set prior to commencing the study and included any pull in which the mouse grasps with only one forepaw or without resistance. The absolute grip strength in Newtons (N) recorded automatically on a digital force transducer (Chatillon, #DFE2‐002) and the weight of each mouse (g) was used to calculate grip strength force normalized to body weight. The same researcher, blinded to genotypes and treatment, performed all measurements in the vivarium where the animals were housed.
In vivo cardiac measurements
Magnetic resonance imaging (MRI) was performed on treated and untreated Het mice (n = 12 per group) 4 days before completing the treatment of the mice. During imaging, mice were anaesthetised with 1.5% isoflurane and electrodes for detection of the ECG signal were used, while animal temperature was maintained at 37°C in a 9.4 Tesla 30 mm bore system (Bruker Biospin) as previously described. 15 Heart rate (HR) was maintained at a minimum of 350 bpm during the imaging. Myocardial strain and strain rate were recorded using vector‐based tracking software (Vector Velocity Imaging, Siemens).
Electrocardiography (ECG) was performed in vivo in conscious and unrestrained mice just before euthanasia at 20 weeks‐of‐age. The body weight of each mouse was recorded, and ECG measurements were performed using the ECGenie system (Mouse Specifics, Inc.). Briefly, an ECG recording of approximately 8–10 s from each mouse was analysed during the time intervals when paws were in contact with the electrodes during a constant HR. 16 ECG measurements included QT interval, HR, as well as QT adjusted for HR variability (QTc).
In vitro extensor digitorum longus and diaphragm contraction force
Contractile functions of extensor digitorum longus (EDL) muscle were assessed according to methods described previously. 14 , 38 Briefly, isolated EDL muscle from each mouse was carefully dissected, and muscle was stretched to the optimal length while incubated at 30°C in Krebs–Henseleit solution continuously cycled. After 10 min, the muscles were subjected to tetanic contraction by stimulating for 250 ms at 150 Hz to measure baseline specific force. After a 5 min rest period, six eccentric contractions were performed by stimulating the muscle for 450 ms at 150 Hz with 3% stretch for the final 200 ms of the contraction. The first five stimulations were followed by a 5 min rest, and the fifth and sixth contraction were separated by a 15 min rest period. After the eccentric contraction, protocol was completed, muscles were weighed, and data were analysed using LabVIEW software (National Instruments, Austin, TX, USA). Force was normalized to cross‐sectional area to determine specific force.
Diaphragm force measurement was assessed as we have previously reported. 14 Diaphragm strips were carefully dissected (~3 mm wide). Platinum–iridium field electrodes were placed around strips at a controlled temperature of 37°C to stretch the muscle to the optimal length. After 10 min rest period, the muscle was stimulated for 250 ms during six tetanic contractions of 20, 50, 80, 120, 150, and 180 Hz with 2 min of rest between contractions. Output was recorded using LabVIEW (National Instruments), and specific force was expressed per unit cross sectional area (mN/mm2).
Histopathology analyses and quantification
The heart from each mouse in the study was divided transversely, and the top half was embedded in optimal‐cutting temperature freezing medium on isopentane cooled in a bath of liquid nitrogen and then stored at −80°C. Quadriceps muscles were removed, cut transversely, and processed using the same method. Sections of 8 μm thickness were cut from each frozen muscle using a cryostat model OTF 5000 (Bright). Muscle sections were stained with haematoxylin and eosin to assess quality before any fluorescent antibody staining. For immunofluorescence staining, heart and quadriceps sections were incubated with an antibody against mouse IgG (Alexa 488 goat anti‐mouse IgG, 1:200; Life Technologies) to quantify muscle damage. Heart muscle sections were also stained with rabbit anti‐mouse fibronectin (1:40, Abcam, ref# ab23750) and incubated with Alexa 555 goat anti‐rabbit IgG secondary antibody (1:200, Life Technologies, ref# A21429). Sections were mounted with VECTASHIELD mounting medium (H‐1000, Vector Laboratories) and counterstained with DAPI. Immunofluorescence stains were photographed using a Nikon Eclipse 800 microscope under a 10× objective with a SPOT RT slider digital camera and software, and images were processed with Adobe Photoshop CS6 software. Damage was quantified by the same individual who was blinded to genotype and treatment for all samples of the same tissue type and is reported as percentage of cross‐sectional area, as previously described. 14 , 16 , 38
RNA isolation and microarray analysis
Liquid nitrogen frozen heart ventricular tissues from three biological replicates from finerenone‐treated and untreated Het mice were pulverized with a mortar and pestle, and RNA was isolated with TRIzol reagent (Life Technologies #15596026), following the manufacturer's instructions. Only male mice were used for this experiment to limit variability. The RNA samples were DNase‐treated using RQ1 DNase (Promega #M610) and further purified using RNeasy mini kit (Qiagen #74104) as previously described. 13 , 39 RNA integrity was analysed with an Agilent 2100 Bioanalyzer (Agilent Technologies), and all samples had RNA integrity numbers above 7.3 and 260/280 ratios between 1.7 and 2.1.
One hundred nanograms of the RNA samples were lineally amplified, and 5.5 μg cDNA was labelled and fragmented using the GeneChip WT PLUS Reagent Kit (Affymetrix, Santa Clara, CA, USA), following the manufacturer's instructions. Labelled cDNA targets were hybridized to Affymetrix GeneChip® Clariom D array, mouse for 16 h at 45°C, rotating at 60 rpm. The arrays were washed and stained using the GeneChip Fluidics Station 450 and scanned using the GeneChip Scanner 3000 7G. Arrays were normalized using the gene‐level signal space transformation robust multi‐chip analysis algorithm in Expression Console software and comparisons were made in Transcriptome Analysis Console software (Affymetrix) using a cut‐off of two‐fold. Gene groups were assigned using the Functional Annotation clustering tool from the Database for Annotation, Visualization and Integrated Discovery (DAVID). Microarray data have been deposited in NCBI GEO under accession number GSE150302.
Data and statistical analysis
Summary values are presented as mean ± SEM and analysed using Minitab (Minitab, LLC, State College, PA, USA). Among groups, differences were evaluated using one‐way ANOVA. Dunnett's post‐hoc test was used to test for significant differences between each group compared with the untreated Het group, and a multiple comparison to find significant differences of EDL forces measured throughout the eccentric contraction was assessed using Bonferroni post‐hoc test. Student's t test was used to determine cardiac MRI significant differences between the finerenone‐treated Het and untreated Het group. Statistical significance was considered with P value ≤0.05.
Results
To determine whether finerenone improved the earliest sign of cardiomyopathy detectable in mouse models and DMD patients, cardiac strain rate was measured using MRI. The myocardial peak systolic strain rate at the base of the left ventricle showed a significant improvement (P = 0.0409) by a Student's t test comparing finerenone‐treated Het mice (0.39 ± 0.04 s‐1) with untreated Het mice (0.28 ± 0.03 s‐1) (Table 1 , Figure 1 ). Importantly, there was no statistically significant difference between the groups for the measured parameters of heart weight (P = 0.7587), heart weight normalized to body weight (P = 0.6568), or electrocardiographic parameters measured in conscious mice of heart rate (P = 0.3727), QT interval (P = 0.7693) or QTc adjusted for heart rate variability (P = 0.8570), which did not differ from previous published data 38 supporting the absence of negative consequences of drug treatment (Table 1 ). In addition, no evidence of arrhythmia was observed. Since Het mice model early stages of cardiomyopathy in humans with reduced strain rates, which occur years before reduced ejection fraction, both untreated and finerenone‐treated groups retained normal whole heart function at 20 weeks‐of‐age with left ventricular ejection fractions of 73 ± 2.7% and 72 ± 2.6%, respectively. Similarly, left ventricular end diastolic volumes were 0.05 ± 0.003 mL in finerenone‐treated compared with 0.05 ± 0.004 mL in untreated Het mice (P = 0.8320) and end systolic volumes were 0.01 ± 0.002mL in the finerenone‐treated group compared with 0.03 ± 0.014 mL (P = 0.3608) in untreated Het mice. No differences between male and female mice were observed in any of the measured cardiac or skeletal muscle parameters. Because reduced overall muscle strength and fatigue are clinically relevant features of DMD that contribute to reduced quality of life, we next measured forelimb grip strength in a blinded manner to determine effects of finerenone treatment on the whole animal. The assay consisted of five repeats of five pulls on a grip‐strength meter to determine the peak baseline grip strength during the first set and the peak grip strength after fatigue during the fifth set of pulls. Baseline total grip strength of finerenone‐treated Het mice (1.14 ± 0.04 N) was improved (P = 0.0265) compared with untreated Het mice (1.00 ± 0.05 N) (Table 1 ). Grip strength normalized to body weight was also significantly increased in rested (P = 0.0182) finerenone‐treated Het (40.3 ± 1.0 mN/g) compared with untreated Het (35.2 ± 1.5 mN/g) mice (Table 1 ; Figure 2 ). Furthermore, a greater relative improvement in normalized grip strength (P = 0.0003) was detected during the fifth trial in fatigued finerenone‐treated Het mice (37.5 ± 1.1 mN/g) compared with untreated Het mice (29.7 ± 1.1 mN/g) (Table 1 ; Figure 2 ).
Table 1.
Preclinical study outcome measurements after 16 weeks of treatment with finerenone
| Control C57 mean ± SEM (n) | Untreated Het mean ± SEM (n) | Finerenone treated mean ± SEM (n) | P value | ||
|---|---|---|---|---|---|
| ANOVA or Student's t test a | Dunnett untreated vs. finerenone | ||||
| Body weight (g) | 26.4 ± 0.7 (18) | 28.1 ± 0.9 (18) | 28.7 ± 0.8 (18) | 0.1152 | 0.8163 |
| Grip strength Trial 1 (N) | 1.50 ± 0.05 (15) | 1.00 ± 0.05 (15) | 1.14 ± 0.04 (16) | <0.0001 | 0.0265 |
| GS/BW Trial 1 (mN/g) | 58.2 ± 1.5 (15) | 35.2 ± 1.5 (15) | 40.3 ± 1.0 (16) | <0.0001 | 0.0182 |
| Grip strength Trial 5 (N) | 1.5 ± 0.05 (15) | 0.8 ± 0.04 (15) | 1.1 ± 0.04 (16) | <0.0001 | 0.0007 |
| GS/BW Trial 5 (mN/g) | 56.5 ± 1.7 (15) | 29.7 ± 1.1 (15) | 37.5 ± 1.1 (16) | <0.0001 | 0.0003 |
| Heart weight (g) | 0.120 ± 0.004 (18) | 0.12 ± 0.01 (18) | 0.12 ± 0.01 (18) | 0.7587 | 1.0000 |
| HW/BW (mg/g) | 4.5 ± 0.1 (18) | 4.4 ± 0.2 (18) | 4.3 ± 0.2 (18) | 0.6568 | 0.9321 |
| Heart rate (bpm) | 696 ± 14 (17) | 678 ± 15 (18) | 704 ± 11 (18) | 0.3727 | 0.2893 |
| QT (ms) | 46.0 ± 1.4 (17) | 46.9 ± 1.1 (18) | 45.8 ± 1.0 (18) | 0.7693 | 0.7277 |
| QTc (ms) | 48.8 ± 1.0 (17) | 49.3 ± 0.7 (18) | 49.3 ± 0.7 (18) | 0.8570 | 0.9997 |
| Base peak systolic strain rate lateral (endocardial circumferential S−1) | ND | 0.28 ± 0.03 (12) | 0.39 ± 0.04 (11) | 0.0409 a | |
| Diaphragm specific force (mN/mm2) | 98.0 ± 11.0 (15) | 151.8 ± 9.4 (18) | 139.0 ± 8.2 (17) | 0.0007 | 0.5228 |
| EDL specific force (mN/mm2) | 431 ± 19 (16) | 329 ± 13 (17) | 318 ± 21 (17) | <0.0001 | 0.8622 |
| Ecc1 (mN/mm2) | 400 ± 27 (17) | 310 ± 18 (18) | 298 ± 24 (18) | 0.0057 | 0.8965 |
| Ecc2 (% Ecc1) | 98 ± 1 (17) | 82 ± 2 (18) | 94 ± 5 (18) | 0.0026 | 0.0186 |
| Ecc5 (% Ecc1) | 86 ± 2 (17) | 51 ± 4 (18) | 71 ± 7 (18) | <0.0001 | 0.0075 |
| Post‐rest Ecc6 (% Ecc1) | 90 ± 2 (17) | 51 ± 4 (18) | 72 ± 6 (18) | <0.0001 | 0.0043 |
| Quad IgG (% CSA) | 0.30 ± 0.04 (18) | 5.6 ± 0.6 (18) | 5.8 ± 0.6 (18) | <0.0001 | 0.9644 |
| Heart IgG (% CSA) | 0.3 ± 0.1 (18) | 1.8 ± 0.3 (18) | 1.1 ± 0.2 (18) | 0.0002 | 0.0796 |
BW, body weight; CSA, percentage of cross‐sectional area; GS, grip strength; HW/BW, heart weight normalized by body weight; QT, QT interval; QTc, QT adjusted for heart rate variability; EDL, extensor digitorum longus; Ecc, EDL force after eccentric contraction.
Indicates Student's t‐test P values.
Measurements from treated (finerenone) and untreated dystrophic mice and wild type control mice (C57) are expressed as mean ± SEM. P values from ANOVA followed by Dunnett's post‐hoc test are shown for 21 parameters. Underlined P values indicate statistical significance (P ≤ 0.05).
Figure 1.
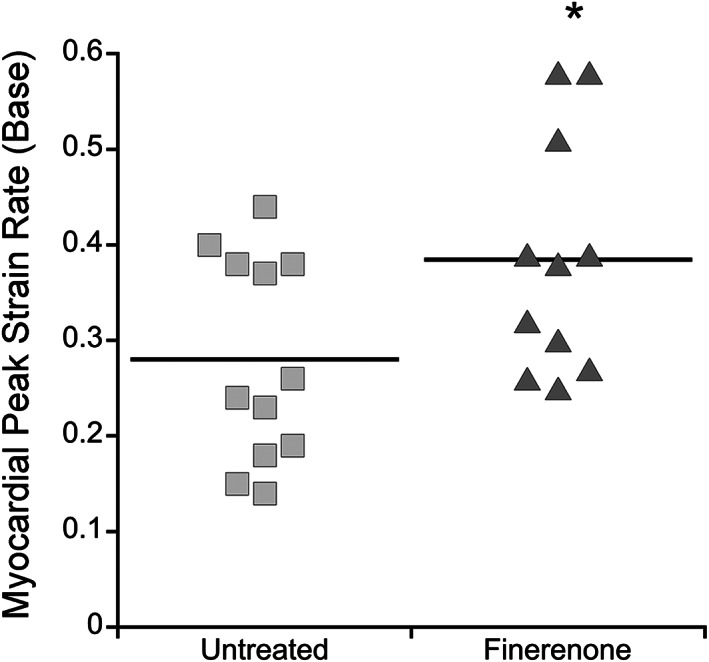
Finerenone treatment improves cardiac strain rate at the base of left ventricle. Dot plot showing in vivo myocardial peak strain rate measured by magnetic resonance imaging in untreated Het (n = 12) vs. finerenone‐treated Het mice (n = 11). Student's t test shows significant differences between groups (*P = 0.0409).
Figure 2.
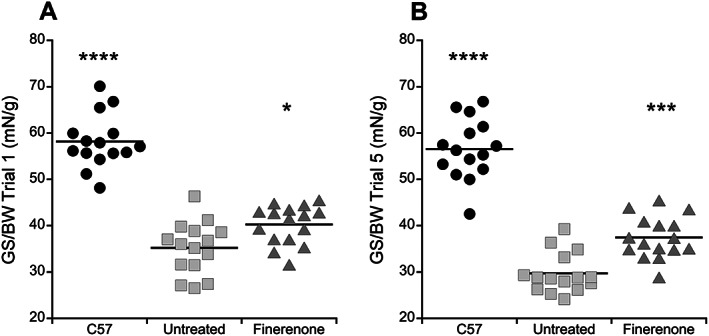
Finerenone improves normalized grip strength in Het mice at baseline and after fatigue. (A) Dot plot representing the highest peak force produced from rested mice during the first set of five pulls on a grip‐strength meter. Significant differences in normalized grip strength were detected between groups by ANOVA (P < 0.0001), and then a Dunnett's post‐hoc test was used to compare finerenone‐treated Het mice and untreated Het mice and showed a significant improvement in treated mice (P = 0.0182). (B) Dot plot representing the highest peak force produced from fatigued mice during the fifth set of five pulls on a grip‐strength meter. An even greater relative improvement was measured (P = 0.0003) in finerenone‐treated mice compared to untreated Het mice. The force measured in wild‐type (C57) mice is significantly larger compared with untreated het mice during Trial 1 (****P < 0.0001) and Trial 5 (P < 0.0001, Dunnett's post‐hoc test) validating the assay. Treatment groups: C57 (n = 15), untreated (n = 15), Finerenone (n = 16). Statistical significance: *P ≤ 0.05 and ***P ≤ 0.001 vs. untreated Hets. BW, body weight; GS, grip strength.
The absence of dystrophin in DMD patients and mouse models leads to an increased susceptibility of the muscle membrane to damage during activity, which can be experimentally measured as force reductions after lengthening or eccentric (Ecc) contractions of limb muscles. 40 Baseline EDL specific force was significantly different (P < 0.0001) between finerenone‐treated Het mice (318 ± 21 mN/mm2), untreated Het mice (329 ± 13 mN/mm2), and wild‐type control mice (431 ± 19 mN/mm2), but no significant differences were obtained when comparing finerenone‐treated dystrophic Het mice with untreated Het mice (P = 0.8622, by Dunnett's post‐hoc test) (Table 1 ). Similarly, there were no differences in specific force in diaphragm between finerenone‐treated Het mice (139.0 ± 8.2 mN/mm2) compared with untreated Het mice (151.8 ± 9.4 mN/mm2) (P = 0.5228, by Dunnett's post‐hoc test) (Table 1). However, contraction‐induced injury in EDL muscles from finerenone‐treated Het mice was reduced resulting in higher specific force generation throughout the eccentric contraction protocol compared with untreated Het mice (Table 1 ; Figure 3 A ). This protocol serves as a strong differentiator between wild‐type and dystrophic mice with damage accumulating in dystrophic muscles throughout the protocol (Figure 3 A ). During the second eccentric contraction, normalized tetanic force was significantly improved (P = 0.0186) in finerenone‐treated Het mice (94 ± 5% Ecc1) compared with untreated Het mice (82 ± 2% Ecc1) (Table 1 ; Figure 3 B ). During the fifth eccentric contraction, normalized specific force was further separated between (P = 0.0075) finerenone‐treated Het mice (71 ± 7% Ecc1) compared with untreated Het mice (51 ± 4% Ecc1) (Table 1 ; Figure 3 C ). To differentiate between contraction‐induced accumulated damage and fatigue, normalized tetanic force was measured again after a rest period (Ecc6). Again, the finerenone‐treated Het group (72 ± 6% Ecc1) generated significantly higher (P = 0.0043) specific force compared with untreated Het mice (51 ± 4% Ecc1), supporting that the prevention of force reduction was because of an effect of drug treatment on membrane stability and not fatigue (Table 1 ; Figure 3 ).
Figure 3.
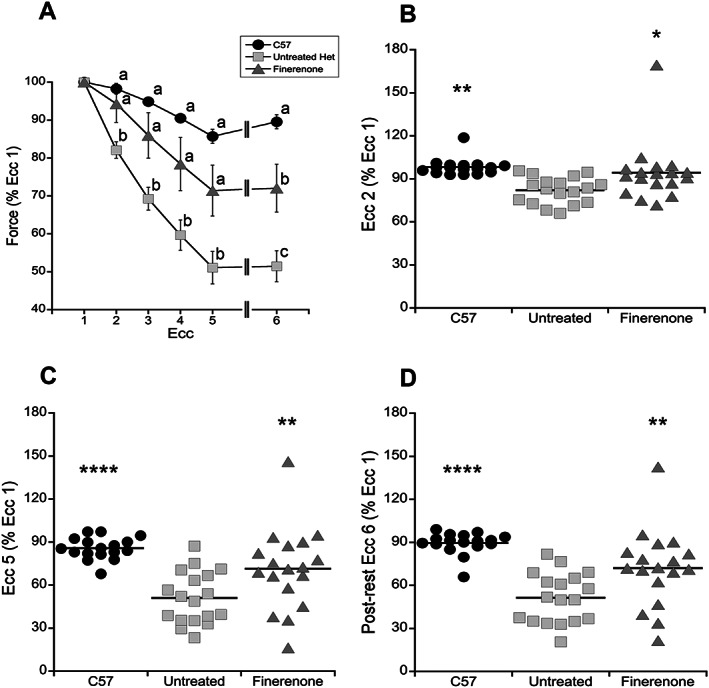
Finerenone treatment improves extensor digitorum longus (EDL) force generation during a contraction induced injury protocol. (A) EDL forces measured throughout the eccentric contraction (Ecc) protocol as a percentage of force generation during the first eccentric contraction (Ecc1). Bonferroni post‐hoc test was use to find significant differences throughout the eccentric contractions. Means that do not share a letter are significantly different from each other. (B–D) Measurements of EDL contractile force after Ecc2, Ecc5, and after a rest period (Ecc6) expressed as a percentage of force generated during Ecc1. The finerenone‐treated group has significantly higher relative EDL contractile force compared with untreated Het mice after Ecc2 (P = 0.0186), Ecc5 (P = 0.0075), and after the rest period (P = 0.0043, Dunnett's post‐hoc test). The same test was used to determine significant differences between wild‐type (C57) and untreated Het mice after Ecc2 (P = 0.0019), Ecc5 (P < 0.0001), and after the rest period (P < 0.0001). Treatment groups: C57 (n = 17), untreated (n = 18), Finerenone (n = 18). Statistical significance: *P ≤ 0.05, **P ≤ 0.01, and ****P ≤ 0.0001.
To evaluate the effect of finerenone on ongoing baseline muscle damage in dystrophic skeletal muscles and on accumulated and ongoing damage in heart, we also quantified the presence of serum IgG immunofluorescence within these tissues (Table 1 ). Quantification of IgG positive cross‐sectional area in quadriceps and heart muscle from wild‐type control (C57), untreated Het and finerenone‐treated Het mice revealed significant differences in the amount of damage (P < 0.0001 and P = 0.0002, respectively). Dunnett's post‐hoc test showed no differences between finerenone‐treated (5.8 ± 0.6%) and untreated Het (5.6 ± 0.6%) mice in the amount of IgG immunofluorescence in quadriceps, which were both quite low. However, hearts from finerenone‐treated Het mice (1.1 ± 0.2%) showed a trend towards less accumulated damage than untreated Het mice (1.8 ± 0.3%). Fibronectin immunofluorescence was used to show that most of this low percentage of IgG staining in heart was because of its accumulation in fibrotic tissue that had already replaced cardiac muscle (Figure 4 ).
Figure 4.
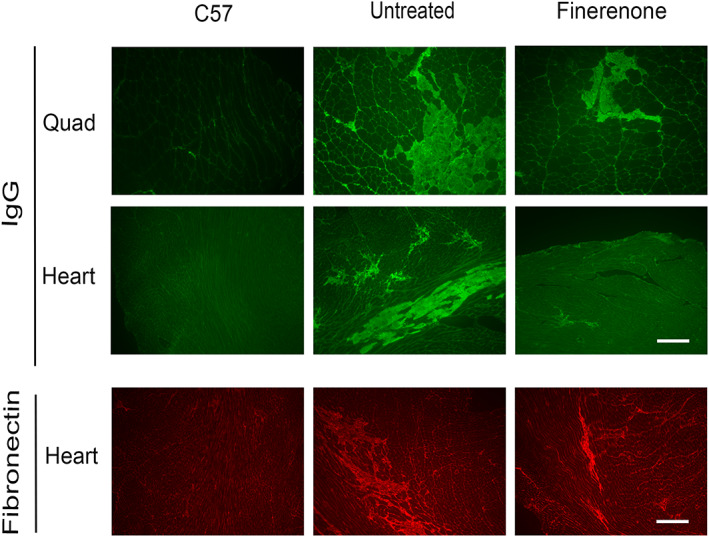
Representative images of immunofluorescence staining for serum IgG accumulation in quadriceps muscle and heart cross‐sections and fibronectin in heart from wild‐type (C57), untreated Het (untreated), and finerenone‐treated Het (finerenone) mice. Serum IgG is excluded from healthy quadriceps and heart tissue but accumulates in muscle fibres with membrane damage (quadriceps) and in both damaged cardiomyocytes as well as fibrotic areas that have replaced cardiomyocytes in heart. Fibronectin staining confirms the presence of small fibrotic areas present in Het hearts. Wild‐type control mice (C57) had no visible damage in either tissue. Data collected from 18 mice in each group: C57, untreated, and finerenone. Bar = 200 μm.
Previous studies have investigated the effect of MR‐regulated gene expression in the heart in models of advanced induced heart failure. 32 , 41 , 42 However, in this study, finerenone is able to prevent very early functional changes in cardiomyopathy progression detectable as cardiac strain rate abnormalities, which occur in DMD mouse models and patients far before reduced whole heart function. Therefore, we decided to assess gene expression changes in finerenone‐treated Het mice compared with untreated Het mice to identify potential very early cardiomyopathic changes that are targeted by non‐steroidal MR antagonism. We conducted a gene expression microarray analysis and observed that expression of 19 genes was increased and expression of eight genes was decreased (Table 2 ). Gene networks represented in the differentially expressed genes included circadian rhythm and ubiquitination (Table 2 ).
Table 2.
Gene expression microarray differences greater than two‐fold in ventricular heart tissue from finerenone‐treated Het vs. untreated Het mice
| Fold change (treated/untreated) | Gene symbol | Full gene name | Function |
|---|---|---|---|
| 3.01 | Wee1 | WEE 1 homologue 1 | Protein banding |
| 2.89 | Gm4899 | Predicted gene 4899 | Cellular respiration, ubiquinone binding |
| 2.87 | Per2 | Period circadian clock 2 | Circadian regulation of gene expression |
| 2.44 | Gm561 | Predicted gene 561 | Unknown function |
| 2.27 | Lrrc52 | Leucine rich repeat containing 52 | Ion transport |
| 2.17 | Alox5ap | Arachidonate 5‐lipoxygenase activating protein | Protein banding |
| 2.16 | Gm10260 | Predicted gene 10260 | Translation |
| 2.16 | Rps27a‐ps2 | Ribosomal protein S27A, pseudogene 2 | Ubiquinone binding |
| 2.14 | Gm11689 | Predicted gene 11689 | Unknown function |
| 2.13 | Rps27l | Ribosomal protein S27‐like | Ion binding, apoptosis, translation |
| 2.11 | Coq10b | Coenzyme Q10 homologue B | Cellular respiration, ubiquinone binding |
| 2.08 | Mrpl41 | Mitochondrial ribosomal protein L41 | Translation |
| 2.08 | Gm8430 | Predicted pseudogene 8430 | Ubiquinone binding |
| 2.06 | Bst2 | Bone marrow stromal cell antigen 2 | Immune system process, transmembrane |
| 2.05 | Wbp5 | WW domain binding protein 5 | Regulation of transcription |
| 2.03 | Ier3 | Immediate early response 3 | Protein banding, regulation of DNA repair, transmembrane, apoptosis |
| 2.02 | Per3 | Period circadian clock 3 | Circadian regulation of gene expression |
| 2.01 | Gm2000 | Predicted gene 2000 | Ribosomal protein |
| 2 | Il33 | Interleukin 33 | Transcription |
| −2.03 | Clock | Circadian locomotor output cycles kaput | Circadian regulation of gene expression |
| −2.04 | Adam19 | A disintegrin and metallopeptidase domain 19 (meltrin beta) | Protein banding |
| −2.2 | Mir467e | MicroRNA 467e | Micro‐ribonucleoprotein complex |
| −2.3 | Lbh | Limb‐bud and heart | Regulation of transcription, regulation of blood coagulation |
| −2.59 | Zfp36 | Zinc finger protein 36 | Regulation of cytokine production/immune response |
| −3.96 | Slc41a3 | Solute carrier family 41, member 3 | Protein banding |
| −4.85 | Cdkn1a | Cyclin‐dependent kinase inhibitor 1A (P21) | Apoptosis, protein banding |
| −5.71 | Arntl | Aryl hydrocarbon receptor nuclear translocator‐like | Circadian regulation of gene expression |
Discussion
This study is the first to demonstrate that treatment with the non‐steroidal MRA drug finerenone alone is able to improve the clinically relevant earliest diagnostic parameter of cardiomyopathy in DMD patients, cardiac strain rate. 21 , 43 , 44 In addition, finerenone is able to improve skeletal muscle strength and muscle membrane stability in this muscular dystrophy model. Finerenone in comparison with commonly used previous steroidal antagonists, spironolactone and eplerenone, combines high potency and selectivity. Previous preclinical and clinical studies have shown equivalent efficacy of spironolactone and eplerenone added to an ACEi or ARB in DMD mouse models and patients, but efficacy for MRA used alone has not been demonstrated for heart or skeletal muscles. 14 , 15 , 21 , 23 MRAs tend to cause less hypotension than ACEi or ARB, so MRAs alone, without these additional drugs, may be advantageous as a therapy for DMD patients who are typically normotensive. In cardiac clinical trials in DMD patients, spironolactone nor eplerenone added to ACEi or ARB reduced blood pressure nor changed salt balance from baseline over 3 years compared to standard therapy alone. 21 , 22 , 23 Typical measurements of renal function by urine or serum creatine are not used in DMD because of the large amount of muscle breakdown, but cystatin C has been used to demonstrate normal renal function in DMD patients. 45 , 46 Therefore, although finerenone results in less worsening of renal function in chronic kidney disease, 31 specific investigations of renal function were not included in the present study.
Finerenone led to overall increased baseline total and normalized grip strength, a measure of whole body muscle force that is reduced by dystrophin‐deficiency and a measure of muscle function that is relevant for patients with DMD. A major symptom experienced by DMD patients is fatigue due to their muscle weakness. Finerenone led to even greater significant improvements in normalized grip strength after five repeats of the grip strength protocol, which resulted in lower forces in untreated dystrophic mice compared with baseline measurements. This improvement in grip strength correlates with improved muscle forces observed in dystrophic mice with a conditional knockout allele of the MR specifically in skeletal muscle fibres. 24
Muscles lacking dystrophin are highly susceptible to damage during use. The typical state‐of‐the‐art preclinical measurement of this contraction‐induced injury is to record force throughout a repetitive lengthening contraction protocol in EDL. 47 In addition to improvements in muscle force and fatigue, finerenone also contributed to prevention of force drop during this protocol, supporting the ability of this treatment to partially prevent muscle injury. This observation was consistent with the direct membrane stabilizing effect of spironolactone we have previously demonstrated in a different assay that uses a laser to measure membrane stability, which is also greatly compromised in dystrophic muscles. 24 The ability of finerenone to prevent muscle damage is a key therapeutic effect for treating both the cardiac and skeletal muscle dysfunction in muscular dystrophies.Although this study demonstrates that finerenone prevents reduced cardiac strain rate, the earliest detectable functional sign of DMD cardiomyopathy in both mouse models and patients, it does not address the longevity of these effects, because of the lack of any DMD model that reproducibly progresses into heart failure. Mice lacking both copies of utrophin in addition to dystrophin (utrn −/− ;mdx) have a more severe cardiomyopathy in terms of fibrosis and reduced function than the utrn +/− ;mdx Het mice used here. However, half of utrophin/dystrophin‐deficient mice die from their skeletal muscle pathology by 10 weeks‐of‐age and the remainder by 20 weeks‐of‐age, 48 prohibiting comparison with untreated controls in chronic treatment studies such as that described here. Future studies using cell‐type specific knockout mouse models and improved models of DMD heart failure will be needed to separate the contribution from possible mechanisms to the cardiac benefits of MRAs in muscular dystrophy.
The presence of high levels of aldosterone synthase in myeloid cells from dystrophic skeletal muscles will need to be confirmed in cardiac pathology to determine whether blocking signalling from chronic inflammation contributes to finerenone's mechanism of action in heart. 20 Finerenone has previously been shown to reduce myocardial reactive oxygen species (ROS) in Zucker rats, a rat model of metabolic syndrome. 49 Because ROS also contributes to dystrophic cardiac pathology, this pathway may also contribute to finerenone's efficacy in this model. 50 , 51 The mechanisms underlying finerenone's improvement of skeletal muscle and cardiac muscle parameters are likely multi‐factorial, involving a combination of effects on membrane stabilization, mechanical force, and MR‐regulated gene expression changes in multiple cell types present in the dystrophic muscle microenvironment that have direct or indirect anti‐fibrotic effects.
Finerenone did not lead to a large number of persisting gene expression changes in ventricular heart tissue at the end of the treatment compared with untreated dystrophic hearts. A surprising finding was the presence of four genes involved in circadian rhythm. Finerenone treatment in dystrophic hearts increases Per2 and Per3, which compose the negative arm of the circadian clock, and decreases ArntI and Clock, which compose the positive arm of the clock. 52 These data suggest that finerenone has shifted or enhanced the amplitude of the clock. Disruption of the circadian clock in heart has been previously demonstrated to change the expression of numerous downstream inflammatory and fibrotic genes. 53 In our previous studies, of these genes, only Per3 was increased in quadriceps muscles of mice treated with lisinopril and spironolactone, but other genes associated with circadian rhythm, Dbp and Nr1d1, were also increased. 13 Per3 was increased by the addition of spironolactone to aldosterone treatment of normal human myotubes in culture. 39 Treatment of H9c2 cardiomyoblast‐like cells with aldosterone has previously been shown to regulate Per1 and initiate the cycling of Per2 and ArntI in these non‐endogenously cycling cells. 53 , 54 Myeloid MR conditional knockout mice demonstrated a reduction in the circadian rhythm of blood pressure. 53 , 55 These data suggest that MR antagonism may cause alterations in circadian rhythm that contribute to its beneficial effect on function, but further carefully timed studies will need to be carried out to determine the amplitude and frequency of expression of these oscillating genes. 52
A limitation of this gene expression data is that because of the extensive preclinical testing of each individual mouse, mice were sacrificed and heart tissue collected at two different times of the day: 10 a.m. and 1 p.m., because only two mice per day could be measured for muscle physiology. It is therefore possible that circadian rhythm gene expression differences with finerenone treatment are a side effect of dissection of tissues at different times of the day. All three of the untreated Het mice used for gene expression microarray were dissected at approximately 10 a.m., while one of the finerenone‐treated group was dissected at 10 a.m. and the other two were dissected at 1 p.m. Future mechanistic studies will need to address whether alterations in the heart clock are sufficient for improving dystrophic cardiomyopathy compared with other possible mechanisms of finerenone's function.
Conflict of Interest
This study was funded by Bayer AG, Wuppertal, Germany; PS and PK are full‐time employees of Bayer AG, all other authors have declared that no conflict of interest exists.
Funding
This work was funded by Bayer AG, Wuppertal, Germany.
Acknowledgements
We would like to thank Anna Bratasz and Michelle Williams for conducting the MRI scans in the Small Animal Imaging Core at the Davis Heart and Lung Research Institute. The authors also thank Paolo Fadda and Huabao Wang for microarray processing and their technical guidance at the Ohio State Genomic Shared Resource‐Comprehensive Cancer Center, microarray core. We also would like to thank Stefanie Breitenstein, Bayer AG clinical development, for discussion and support.
Lowe, J. , Kolkhof, P. , Haupt, M. J. , Peczkowski, K. K. , Rastogi, N. , Hauck, J. S. , Kadakia, F. K. , Zins, J. G. , Ciccone, P. C. , Smart, S. , Sandner, P. , Raman, S. V. , Janssen, P. M. L. , and Rafael‐Fortney, J. A. (2020) Mineralocorticoid receptor antagonism by finerenone is sufficient to improve function in preclinical muscular dystrophy. ESC Heart Failure, 7: 3983–3995. 10.1002/ehf2.12996.
References
- 1. Emery AEH. Duchenne Muscular Dystrophy, Second Edition ed. Oxford: Oxford University Press; 1993. [Google Scholar]
- 2. Guiraud S, Aartsma‐Rus A, Vieira NM, Davies KE, van Ommen GJ, Kunkel LM. The pathogenesis and therapy of muscular dystrophies. Annu Rev Genomics Hum Genet 2015; 16: 281–308. [DOI] [PubMed] [Google Scholar]
- 3. Mendell JR, Shilling C, Leslie ND, Flanigan KM, Al‐Dahhak R, Gastier‐Foster J, Kneile K, Dunn DM, Duval B, Aoyagi A, Hamil C, Mahmoud M, Roush K, Bird L, Rankin C, Lilly H, Street N, Chandrasekar R, Weiss RB. Evidence‐based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 2012; 71: 304–313. [DOI] [PubMed] [Google Scholar]
- 4. Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, Case LE, Clemens PR, Hadjiyannakis S, Pandya S, Street N, Tomezsko J, Wagner KR, Ward LM, Weber DR. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 2018; 17: 251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Griggs RC, Miller JP, Greenberg CR, Fehlings DL, Pestronk A, Mendell JR, Moxley RT 3rd, King W, Kissel JT, Cwik V, Vanasse M, Florence JM, Pandya S, Dubow JS, Meyer JM. Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology 2016; 87: 2123–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010; 9: 77–93. [DOI] [PubMed] [Google Scholar]
- 7. Guglieri M, Bushby K, McDermott MP, Hart KA, Tawil R, Martens WB, Herr BE, McColl E, Wilkinson J, Kirschner J, King WM, Eagle M, Brown MW, Willis T, Hirtz D, Shieh PB, Straub V, Childs AM, Ciafaloni E, Butterfield RJ, Horrocks I, Spinty S, Flanigan KM, Kuntz NL, Baranello G, Roper H, Morrison L, Mah JK, Manzur AY, McDonald CM, Schara U, von der Hagen M, Barohn RJ, Campbell C, Darras BT, Finkel RS, Vita G, Hughes I, Mongini T, Pegoraro E, Wicklund M, Wilichowski E, Bryan Burnette W, Howard JF, McMillan HJ, Thangarajh M, Griggs RC. Developing standardized corticosteroid treatment for Duchenne muscular dystrophy. Contemp Clin Trials 2017; 58: 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Janssen PM, Murray JD, Schill KE, Rastogi N, Schultz EJ, Tran T, Raman SV, Rafael‐Fortney JA. Prednisolone attenuates improvement of cardiac and skeletal contractile function and histopathology by lisinopril and spironolactone in the mdx mouse model of Duchenne muscular dystrophy. PLoS ONE 2014; 9: e88360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sali A, Guerron AD, Gordish‐Dressman H, Spurney CF, Iantorno M, Hoffman EP, Nagaraju K. Glucocorticoid‐treated mice are an inappropriate positive control for long‐term preclinical studies in the mdx mouse. PLoS ONE 2012; 7: e34204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bauer R, Straub V, Blain A, Bushby K, MacGowan GA. Contrasting effects of steroids and angiotensin‐converting‐enzyme inhibitors in a mouse of dystrophin‐deficient cardiomyopathy. Eur J Heart Fail 2009; 11: 463–471. [DOI] [PubMed] [Google Scholar]
- 11. Duboc D, Meune C, Pierre B, Wahbi K, Eymard B, Toutain A, Berard C, Vaksmann G, Weber S, Becane HM. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years' follow‐up. Am Heart J 2007; 154: 596–602. [DOI] [PubMed] [Google Scholar]
- 12. Hor KN, Mazur W, Taylor MD, Al‐Khalidi HR, Cripe LH, Jefferies JL, Raman SV, Chung ES, Kinnett KJ, Williams K, Gottliebson WM, Benson DW. Effects of steroids and angiotensin converting enzyme inhibition on circumferential strain in boys with Duchenne muscular dystrophy: a cross‐sectional and longitudinal study utilizing cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2011; 13: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chadwick JA, Bhattacharya S, Lowe J, Weisleder N, Rafael‐Fortney JA. Renin–angiotensin–aldosterone system inhibitors improve membrane stability and change gene‐expression profiles in dystrophic skeletal muscles. Am J Physiol Cell Physiol 2017; 312: C155–c168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lowe J, Floyd KT, Rastogi N, Schultz EJ, Chadwick JA, Swager SA, Zins JG, Kadakia FK, Smart S, Gomez‐Sanchez EP, Gomez‐Sanchez CE, Raman SV, Janssen PM, Rafael‐Fortney JA. Similar efficacy from specific and non‐specific mineralocorticoid receptor antagonist treatment of muscular dystrophy mice. J Neuromusc Diseases 2016; 3: 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rafael‐Fortney JA, Chimanji NS, Schill KE, Martin CD, Murray JD, Ganguly R, Stangland JE, Tran T, Xu Y, Canan BD, Mays TA, Delfin DA, Janssen PM, Raman SV. Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in Duchenne muscular dystrophy mice. Circulation 2011; 124: 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lowe J, Wodarcyk AJ, Floyd KT, Rastogi N, Schultz EJ, Swager SA, Chadwick JA, Tran T, Raman SV, Janssen PM, Rafael‐Fortney JA. The angiotensin converting enzyme inhibitor lisinopril improves muscle histopathology but not contractile function in a mouse model of Duchenne muscular dystrophy. J Neuromusc Diseases 2015; 2: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chadwick JA, Hauck JS, Lowe J, Shaw JJ, Guttridge DC, Gomez‐Sanchez CE, Gomez‐Sanchez EP, Rafael‐Fortney JA. Mineralocorticoid receptors are present in skeletal muscle and represent a potential therapeutic target. FASEB J 2015; 29: 4544–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang J, Young MJ. The mineralocorticoid receptor and its coregulators. J Mol Endocrinol 2009; 43: 53–64. [DOI] [PubMed] [Google Scholar]
- 19. Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol 2013; 9: 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chadwick JA, Swager SA, Lowe J, Welc SS, Tidball JG, Gomez‐Sanchez CE, Gomez‐Sanchez EP, Rafael‐Fortney JA. Myeloid cells are capable of synthesizing aldosterone to exacerbate damage in muscular dystrophy. Hum Mol Genet 2016; 25: 5167–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Raman SV, Hor KN, Mazur W, Halnon NJ, Kissel JT, He X, Tran T, Smart S, McCarthy B, Taylor MD, Jefferies JL, Rafael‐Fortney JA, Lowe J, Roble SL, Cripe LH. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol 2015; 14: 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raman SV, Hor KN, Mazur W, He X, Kissel JT, Smart S, McCarthy B, Roble SL, Cripe LH. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: results of a two‐year open‐label extension trial. Orphanet J Rare Dis 2017; 12: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raman SV, Hor KN, Mazur W, Cardona A, He X, Halnon N, Markham L, Soslow JH, Puchalski MD, Auerbach SR, Truong U, Smart S, McCarthy B, Saeed IM, Statland JM, Kissel JT, Cripe LH. Stabilization of early Duchenne cardiomyopathy with aldosterone inhibition: results of the multicenter AIDMD trial. J Am Heart Assoc 2019; 8: e013501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hauck JS, Lowe J, Rastogi N, McElhanon KE, Petrosino JM, Peczkowski KK, Chadwick AN, Zins JG, Accornero F, Janssen PML, Weisleder NL, Rafael‐Fortney JA. Mineralocorticoid receptor antagonists improve membrane integrity independent of muscle force in muscular dystrophy. Hum Mol Genet 2019; 28: 2030–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kolkhof P, Delbeck M, Kretschmer A, Steinke W, Hartmann E, Barfacker L, Eitner F, Albrecht‐Kupper B, Schafer S. Finerenone, a novel selective nonsteroidal mineralocorticoid receptor antagonist protects from rat cardiorenal injury. J Cardiovasc Pharmacol 2014; 64: 69–78. [DOI] [PubMed] [Google Scholar]
- 26. Kolkhof P, Nowack C, Eitner F. Nonsteroidal antagonists of the mineralocorticoid receptor. Curr Opin Nephrol Hypertens 2015; 24: 417–424. [DOI] [PubMed] [Google Scholar]
- 27. Bramlage P, Swift SL, Thoenes M, Minguet J, Ferrero C, Schmieder RE. Non‐steroidal mineralocorticoid receptor antagonism for the treatment of cardiovascular and renal disease. Eur J Heart Fail 2016; 18: 28–37. [DOI] [PubMed] [Google Scholar]
- 28. Dojki FK, Bakris G. Nonsteroidal mineralocorticoid antagonists in diabetic kidney disease. Curr Opin Nephrol Hypertens 2017; 26: 368–374. [DOI] [PubMed] [Google Scholar]
- 29. Pitt B, Filippatos G, Gheorghiade M, Kober L, Krum H, Ponikowski P, Nowack C, Kolkhof P, Kim SY, Zannad F. Rationale and design of ARTS: a randomized, double‐blind study of BAY 94‐8862 in patients with chronic heart failure and mild or moderate chronic kidney disease. Eur J Heart Fail 2012; 14: 668–675. [DOI] [PubMed] [Google Scholar]
- 30. Yang J, Young MJ. Mineralocorticoid receptor antagonists‐pharmacodynamics and pharmacokinetic differences. Curr Opin Pharmacol 2016; 27: 78–85. [DOI] [PubMed] [Google Scholar]
- 31. Pitt B, Kober L, Ponikowski P, Gheorghiade M, Filippatos G, Krum H, Nowack C, Kolkhof P, Kim SY, Zannad F. Safety and tolerability of the novel non‐steroidal mineralocorticoid receptor antagonist BAY 94‐8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: a randomized, double‐blind trial. Eur Heart J 2013; 34: 2453–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grune J, Benz V, Brix S, Salatzki J, Blumrich A, Hoft B, Klopfleisch R, Foryst‐Ludwig A, Kolkhof P, Kintscher U. Steroidal and nonsteroidal mineralocorticoid receptor antagonists cause differential cardiac gene expression in pressure overload‐induced cardiac hypertrophy. J Cardiovasc Pharmacol 2016; 67: 402–411. [DOI] [PubMed] [Google Scholar]
- 33. Grune J, Beyhoff N, Smeir E, Chudek R, Blumrich A, Ban Z, Brix S, Betz IR, Schupp M, Foryst‐Ludwig A, Klopfleisch R, Stawowy P, Houtman R, Kolkhof P, Kintscher U. Selective mineralocorticoid receptor cofactor modulation as molecular basis for finerenone's antifibrotic activity. Hypertension 2018; 71: 599–608. [DOI] [PubMed] [Google Scholar]
- 34. Bauersachs J. The ARTS of third‐generation mineralocorticoid receptor antagonists: achieving cardiovascular benefit with minimized renal side effects? Eur Heart J 2013; 34: 2426–2428. [DOI] [PubMed] [Google Scholar]
- 35. Naegele M, Hernandez AF, Ruschitzka F. Finerenone in heart failure: walking a fine line. Eur Heart J 2016; 37: 2115–2117. [DOI] [PubMed] [Google Scholar]
- 36. Deconinck N, Rafael JA, Beckers‐Bleukx G, Kahn D, Deconinck AE, Davies KE, Gillis JM. Consequences of the combined deficiency in dystrophin and utrophin on the mechanical properties and myosin composition of some limb and respiratory muscles of the mouse. Neuromuscul Disord 1998; 8: 362–370. [DOI] [PubMed] [Google Scholar]
- 37. Lavall D, Jacobs N, Mahfoud F, Kolkhof P, Bohm M, Laufs U. The non‐steroidal mineralocorticoid receptor antagonist finerenone prevents cardiac fibrotic remodeling. Biochem Pharmacol 2019; 168: 173–183. [DOI] [PubMed] [Google Scholar]
- 38. Lowe J, Kadakia FK, Zins JG, Haupt M, Peczkowski KK, Rastogi N, Floyd KT, Gomez‐Sanchez EP, Gomez‐Sanchez CE, Elnakish MT, Rafael‐Fortney JA, Janssen PML. Mineralocorticoid receptor antagonists in muscular dystrophy mice during aging and exercise. J Neuromuscul Dis 2018; 5: 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chadwick JA, Hauck JS, Gomez‐Sanchez CE, Gomez‐Sanchez EP, Rafael‐Fortney JA. Gene expression effects of glucocorticoid and mineralocorticoid receptor agonists and antagonists on normal human skeletal muscle. Physiol Genomics 2017; 49: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ichiyasu H, McCormack JM, McCarthy KM, Dombkowski D, Preffer FI, Schneeberger EE. Matrix metalloproteinase‐9‐deficient dendritic cells have impaired migration through tracheal epithelial tight junctions. Am J Respir Cell Mol Biol 2004; 30: 761–770. [DOI] [PubMed] [Google Scholar]
- 41. Lother A, Berger S, Gilsbach R, Rosner S, Ecke A, Barreto F, Bauersachs J, Schutz G, Hein L. Ablation of mineralocorticoid receptors in myocytes but not in fibroblasts preserves cardiac function. Hypertension 2011; 57: 746–754. [DOI] [PubMed] [Google Scholar]
- 42. Oakley RH, Cruz‐Topete D, He B, Foley JF, Myers PH, Xu X, Gomez‐Sanchez CE, Chambon P, Willis MS, Cidlowski JA. Cardiomyocyte glucocorticoid and mineralocorticoid receptors directly and antagonistically regulate heart disease in mice. Sci Signal 2019; 12: eaau9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hor KN, Kissoon N, Mazur W, Gupta R, Ittenbach RF, Al‐Khalidi HR, Cripe LH, Raman SV, Puchalski MD, Gottliebson WM, Benson DW. Regional circumferential strain is a biomarker for disease severity in duchenne muscular dystrophy heart disease: a cross‐sectional study. Pediatr Cardiol 2015; 36: 111–119. [DOI] [PubMed] [Google Scholar]
- 44. Hor KN, Wansapura J, Markham LW, Mazur W, Cripe LH, Fleck R, Benson DW, Gottliebson WM. Circumferential strain analysis identifies strata of cardiomyopathy in Duchenne muscular dystrophy: a cardiac magnetic resonance tagging study. J Am Coll Cardiol 2009; 53: 1204–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Braat E, Hoste L, De Waele L, Gheysens O, Vermeersch P, Goffin K, Pottel H, Goemans N, Levtchenko E. Renal function in children and adolescents with Duchenne muscular dystrophy. Neuromuscul Disord 2015; 25: 381–387. [DOI] [PubMed] [Google Scholar]
- 46. Viollet L, Gailey S, Thornton DJ, Friedman NR, Flanigan KM, Mahan JD, Mendell JR. Utility of cystatin C to monitor renal function in Duchenne muscular dystrophy. Muscle Nerve 2009; 40: 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moorwood C, Liu M, Tian Z, Barton ER. Isometric and eccentric force generation assessment of skeletal muscles isolated from murine models of muscular dystrophies. J Vis Exp 2013; 31: e50036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin‐dystrophin‐deficient mice as a model for Duchenne muscular dystrophy. Cell 1997; 90: 717–727. [DOI] [PubMed] [Google Scholar]
- 49. Lachaux M, Barrera‐Chimal J, Nicol L, Rémy‐Jouet I, Renet S, Dumesnil A, Wecker D, Richard V, Kolkhof P, Jaisser F, Ouvrard‐Pascaud A, Mulder P. Short‐ and long‐term administration of the non‐steroidal mineralocorticoid receptor antagonist finerenone opposes metabolic syndrome‐related cardio‐renal dysfunction. Diabetes Obes Metab 2018; 20: 2399–2407. [DOI] [PubMed] [Google Scholar]
- 50. Afzal MZ, Reiter M, Gastonguay C, McGivern JV, Guan X, Ge ZD, Mack DL, Childers MK, Ebert AD, Strande JL. Nicorandil, a nitric oxide donor and ATP‐sensitive potassium channel opener, protects against dystrophin‐deficient cardiomyopathy. J Cardiovasc Pharmacol Ther 2016; 21: 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Williams IA, Allen DG. The role of reactive oxygen species in the hearts of dystrophin‐deficient mdx mice. Am J Physiol Heart Circ Physiol 2007; 293: H1969–H1977. [DOI] [PubMed] [Google Scholar]
- 52. Young ME. The circadian clock within the heart: potential influence on myocardial gene expression, metabolism, and function. Am J Physiol Heart Circ Physiol 2006; 290: H1–H16. [DOI] [PubMed] [Google Scholar]
- 53. Fletcher EK, Morgan J, Kennaway DR, Bienvenu LA, Rickard AJ, Delbridge LMD, Fuller PJ, Clyne CD, Young MJ. Deoxycorticosterone/salt‐mediated cardiac inflammation and fibrosis are dependent on functional CLOCK signaling in male mice. Endocrinology 2017; 158: 2906–2917. [DOI] [PubMed] [Google Scholar]
- 54. Tanaka K, Ashizawa N, Kawano H, Sato O, Seto S, Nishihara E, Terazono H, Isomoto S, Shinohara K, Yano K. Aldosterone induces circadian gene expression of clock genes in H9c2 cardiomyoblasts. Heart Vessels 2007; 22: 254–260. [DOI] [PubMed] [Google Scholar]
- 55. Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schutz G, Lumeng CN, Mortensen RM. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest 2010; 120: 3350–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]