

Abstract
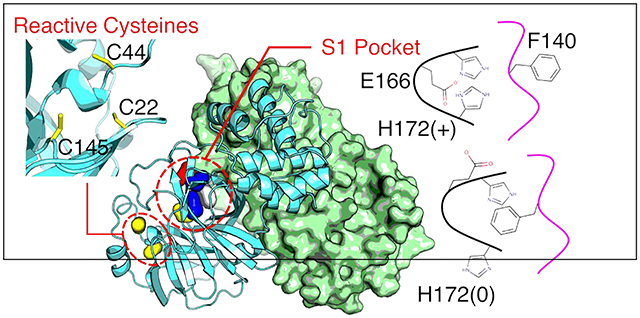
The SARS coronavirus 2 (SARS-CoV-2) main protease (Mpro) is an attractive broad-spectrum antiviral drug target. Despite the enormous progress in structure elucidation, the Mpro’s structure-function relationship remains poorly understood. Recently, a peptidomimetic inhibitor has entered the clinical trial; however, small-molecule orally available antiviral drugs have yet to be developed. Intrigued by a long-standing controversy regarding the existence of an inactive state, we explored the proton-coupled dynamics of the Mpros of SARS-CoV-2 and the closely related SARS-CoV using a newly developed continuous constant pH molecular dynamics (MD) method and microsecond fixed-charge all-atom MD simulations. Our data supports a general base mechanism for Mpro’s proteolytic function. The simulations revealed that protonation of His172 alters a conserved interaction network that upholds the oxyanion loop, leading to a partial collapse of the conserved S1 pocket, consistent with the first and controversial crystal structure of SARS-CoV Mpro determined at pH 6. Interestingly, a natural flavonoid binds SARS-CoV-2 Mpro in the close proximity to a conserved cysteine (Cys44), which is hyper-reactive according to the CpHMD titration. This finding offers an exciting new opportunity for small-molecule targeted covalent inhibitor design. Our work represents a first step towards the mechanistic understanding of the proton-coupled structure-dynamics-function relationship of CoV Mpros; the proposed strategy of designing small-molecule covalent inhibitors may help accelerate the development of orally available broad-spectrum antiviral drugs to stop the current pandemic and prevent future outbreaks.
Graphical abstract
INTRODUCTION
The ongoing COVID-19 pandemic has claimed nearly 1.5 million human lives worldwide, but effective therapeutic intervention has yet to be developed. COVID-19 is caused by a new coronavirus called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, GenBank accession code: MN908947.31). Belonging to the genus Betacoronavirus, SARS-CoV-2 is closely related to several bat SARS-like coronaviruses, and SARS-CoV which caused an outbreak in 2002–2003 as well as MERS-CoV (Middle East Respiratory Syndrome coronavirus) which caused an outbreak in 2012.1 The related genus Alphacoronavirus includes human coronaviruses that cause the common cold.2 The replicase gene of coronaviruses encodes two polyproteins (pp1a and pp1ab), which are processed into functional nonstructural proteins to form the viral replication/transcription complex by two cysteine proteases:3 the main protease (Mpro, also known as 3CLpro due to the similarity to picornavirus 3C protease) and the papain-like protease (PLpro).3 The Mpro cleaves the larger polyprotein pp1ab at 11 sites, with the sequence of (small)-X-(Leu/Phe/Met)-Gln↓(Gly/Ala/Ser), where X denotes any amino acid and ↓ indicates the cleavage site. Note, Gln in the P1 position is absolutely required by the Mpro and the substrate specificity pattern is not shared by any human protease.3 The S1′, S1, and S2 substrate binding pockets are conserved among the Mpros of SARS-, MERS-, and SARS-like bat-CoVs.3 Thus, the Mpro is an attractive broad-spectrum antiviral drug target for combating the COVID-19 pandemic and preventing future outbreaks through the zoonotic transmission of SARS-like bat coronaviruses to human.3,4
SARS-CoV-2/CoV Mpros have been well characterized by crystallography. To date, Protein Data Bank (PDB) contains 54 and 188 entries of X-ray crystal structures for SARS-CoV and SARS-CoV-2 Mpros, respectively, most of which are inhibitor-bound complexes.5–10 The rapid structure determination led to the discovery of the first broad-spectrum lead inhibitor against SARS, MERS, and the related coronavirus Mpros.2,5 Most recently, Pfizer initiated a clinical trial of PF-07304814, a prodrug which metabolizes into a ketone-based peptidomimetic inhibitor PF-00835231 against the Mpros of SARS-CoV-2/CoV and other coronaviruses.11 Despite the progress, the structure-function relationship and conformational plasticity of the Mpros remain poorly understood. Such information is urgently needed to advance the broad-spectrum antiviral drug design.
SARS-CoV-2/CoV Mpros function as a dimer.2,3 With 96% overall sequence identity and 100% identity in the active site, their structures are nearly superimposable.5 The Mpro protomer is comprised of three domains (Fig. 1a). The chymotrypsin-like β-barrel domains I (residues 10–99) and II (residues 100–182) embrace the Cys-His catalytic dyad at the interface, while the α-helical domain III (residues 198–303) is connected to domain II via a long loop called L3 (residues 183–197). Dimerization of Mpro is mainly supported by the domain III interactions with domain III and the N-terminal loop (residues 1–10) of the opposite protomer.12 In particular, residues 1–7 (called N-finger13) form hydrogen bonds and electrostatic interactions with the S1 pocket (Fig. 1c), and have been considered critical for both dimerization and active site integrity of SARS-CoV Mpro.13 Deletion or mutation of Arg414 and Gly1115 was shown to completely inactivate SARS-CoV Mpro, while deletion of residues 1–3 maintained 76% enzymatic activity.14 Deletion of residues 1–5 in the related transmissible gastroenteritis virus Mpro almost completely abolished the activity.16
Figure 1. Crystal structure and pH-dependent activity for SARS-CoV-2/CoV Mpros.
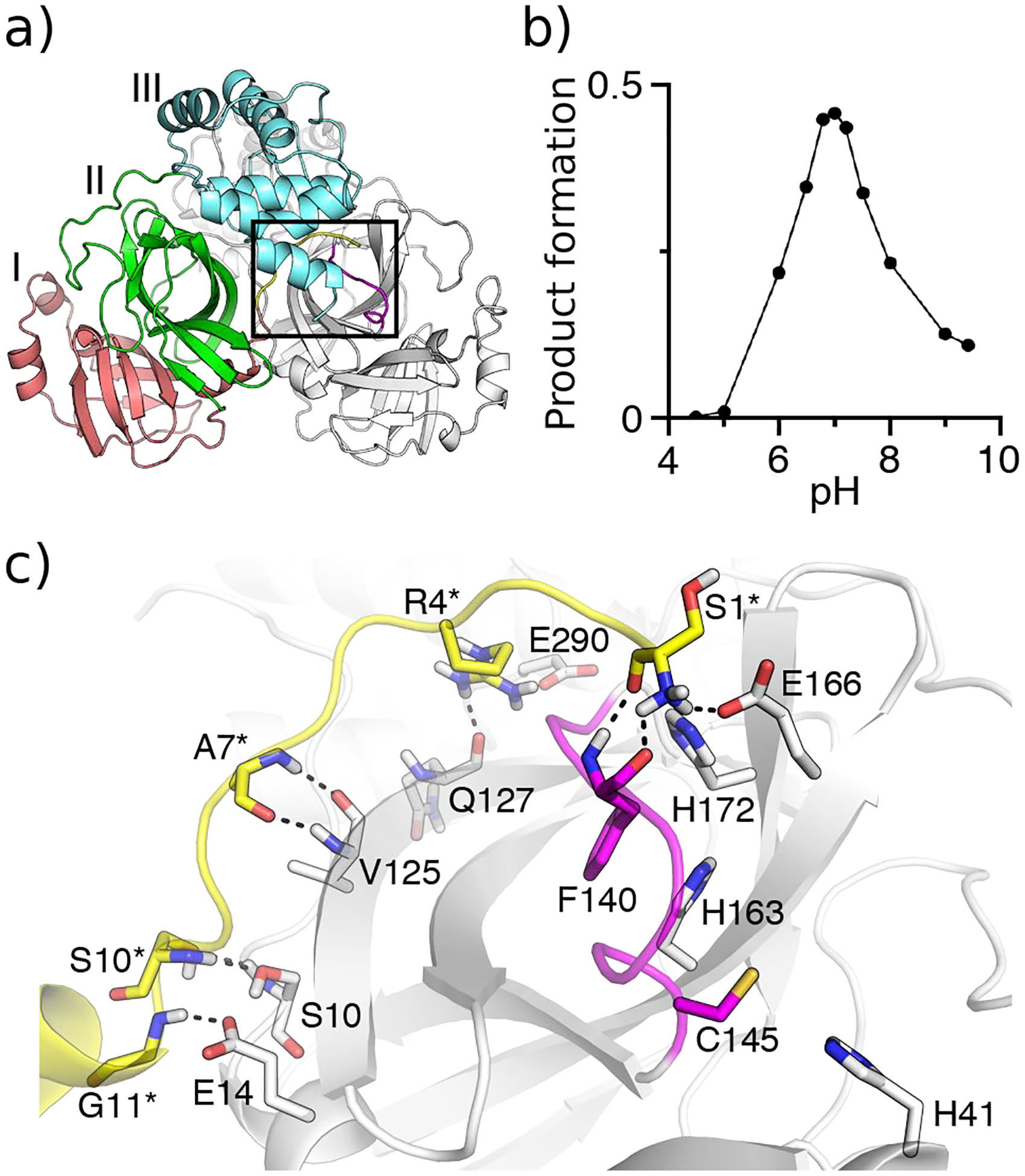
a) X-ray crystal structure of SARS-Cov-2 Mpro dimer (PDB: 6y2g5). Protomer A is colored gray, while domains I, II, and III in protomer B are colored red, green, and cyan, respectively. The oxyanion loop L1 (residues 138–145) is colored magenta and the N-finger loop (residues 1–10) as well as G11 are colored yellow. b) pH-activity profile of SARS-CoV Mpro determined by the HPLC-based peptide cleavage assay. c) A zoomed-in view of the S1 specificity pocket in protomer A and the N-finger loop in protomer B. Residues involved in the dimer interface interactions are explicitly shown (those in protomer B denoted with an asterisk). The conserved S1 pocket residues Phe140, His163, Glu166, His172, and the catalytic dyad Cys145 and His41 are explicitly shown.
Mpro’s substrate binding site is comprised of the canonical S1–S4 pockets, whereby the S1 pocket is formed by the sidechains of His163, Phe140 and the backbones of Met165, Glu166, and His172, all of which are conserved (Fig. 1c). In the majority of crystal structures of SARS-CoV-2 Mpro, Glu166 forms a hydrogen bond with His172, while His163 forms an aromatic stacking with Phe140 (Fig. 1c and Fig. S1). Additionally, the sidechain of Glu166 and backbone of Phe140 form hydrogen bonds with the amino group of Ser1 (amino terminus) of the opposite protomer (Fig. 1c and Fig. S1). The oxyanion hole of the cysteine protease is formed by the backbone amides of Gly143, Ser 144, and Cys145,5,13 and the oxyanion loop (residues 138–145, also called L1) forms a wall of the S1 pocket (Fig. 1c).
The proteolytic activity of SARS-CoV Mpro displays a bell-shaped pH profile, with the peak at pH 7.017 (or 7.418) and sharp decreases at lower or higher pH (Fig. 1b); however, the molecular mechanism of the pH dependence is not understood. Interestingly, a pH 6 crystal structure of SARS-CoV Mpro (PDB: 1uj113) which captured two independent protomers in the asymmetric unit revealed an inactive conformation for protomer B, in which the oxyanion loop is moved towards the S1 pocket and the hydrogen bond between Glu166 and His172 is broken. Hilgenfeld, Rao, and coworkers hypothesized that the pH-dependent activity change arises from the protonation state switches of His163 and His172.13,17 Based on the crystal structures,13,17 His163 and His172 were suggested as neutral and charged at physiological pH, respectively, and consequently, the protonation of His163 was hypothesized to induce the aforementioned conformational changes in the S1 pocket.17 Shortly after, the authenticity of the inactive conformation was questioned, by arguing that a fusion tag added to the termini from the recombinant protein expression system may have destabilized the Ser1 interactions with the S1 pocket of the opposite protomer, causing it to collapse.19
Recently, a X-ray structure of SARS-CoV-2 Mpro dimer (PDB: 6y2g5) revived the debate regarding the existence of an inactive Mpro protomer and its pH dependence. In protomer B of this crystal structure, the Glu166–His172 hydrogen bond is broken, similar to the inactive protomer of the pH 6 structure of SARS-CoV Mpro (PDB: 1UJ113), although the large movement of the oxyanion loop is absent.5 Testing the hypothesis of an inactive state by crystallography proves challenging, as most crystal structures of Mpros were determined in space group C2 which captures only one protomer in the asymmetric unit which favors a more ordered structure.
To shed light on the above controversy, we conducted a computational study of SARS-CoV-2/CoV Mpros, employing the newly developed GPU-accelerated implicit-solvent continuous constant pH MD (CpHMD)20,21 to determine the protonation states and μs-long conventional fixed-charge MD simulations to characterize the detailed conformational changes. Our data confirms the general base mechanism for Mpro’s proteolytic function and suggests that protonation of His172 (and not His163) induces a partial collapse of the S1 pocket, consistent with the aforementioned pH 6 crystal structure of SARS-CoV Mpro. The simulations also predict an alternative Cys residue for targeted covalent inhibitor design. Taken together, our work represents a first step towards a mechanistic understanding of the pH-dependent structure-dynamics-function relationship of SARS-CoV-2/Cov Mpros and contributes urgently needed knowledge for broad-spectrum antiviral drug design.
RESULTS AND DISCUSSION
Protonation states of SARS-CoV-2 Mpro.
The Mpro protomer has 12 Cys and 7 His residues, including the catalytic dyad Cys145 and His41 as well as the absolutely conserved His163, His164, and His172 in the S1 pocket. Because the solution pKa’s of Cys and His (8.5 and 6.5, respectively) are only 1 pH unit away from the physiological pH, it is important to determine their protonation states in the protein before conducting a fixed-charged MD study. We performed the pH replica-exchange GBNeck2-CpHMD simulations,20,21 which have been shown to give accurate pKa estimates, particularly for Cys22 and His21 The simulations were initiated from the aforementioned crystal structure of SARS-CoV-2 Mpro (PDB: 6y2g5), whereby all sidechains of Asp/Glu/His/Cys/Lys were allowed to titrate. 9 pH replicas were used over a pH range of 5–9, with a total sampling time of 495 ns. The calculated pKa’s were well converged (Fig. S2).
CpHMD showed that the catalytic dyad residues Cys145 and His41 are both neutral at physiological pH, suggesting that the cleavage reaction of the Mpro follows a general base and not an ion-pair mechanism like SARS-CoV-2/CoV PLpros,23 consistent with the pH-dependent activity data of SARS-CoV Mpro.18 The latter gave the pKa’s of 8.34 and 6.38 for Cys145 and His41, respectively.18 The calculated pKa of His41 (6.6/6.7 for the two protomers, Table 1) is in excellent agreement with experiment, while the calculated pKa of Cys145 is overestimated by about 2 units. Trajectory snapshots showed that the deprotonated form of Cys145 requires stabilization by several hydrogen bonds that are formed after extensive conformational sampling, consistent with our previous findings regarding the deprotonation of cysteines.22,24 We defer the detailed investigation of this topic to a future study.
Table 1.
Calculated pKa’s of relevant Cys and His residues in the two protomers of SARS-CoV-2/CoV Mprosa
| Residue | SARS-CoV-2 | SARS-CoV | ||
|---|---|---|---|---|
| A | B | A | B | |
| C22 | 7.5 | 6.8 | 6.8 | 7.0 |
| C44 | 7.0 | 9.2 | 4.2 | 5.8 |
| C145 | neutral | ~9.4b | neutral | neutral |
| H41 | 6.6 | 6.7 | 6.2 | 6.5 |
| H163 | neutral | neutral | neutral | neutral |
| H164 | neutral | neutral | neutral | neutral |
| H172 | 6.6 | 6.6 | 6.6 | 7.7c |
Calculations based on the crystal structures of SARS-CoV-2 (PDB: 6y2g5) and SARS-CoV (PDB: 1uk213) Mpros. A complete list of pKa’s is given in SI Table 1. For residues that did not titrate in the simulation pH range, their protonation states are given.
Estimated based on partial deprotonation at pH 7.0–9.0.
See discussion in the main text.
Importantly, CpHMD titration gave the pKa’s of His163 and His164 below 5, whereas the pKa of His172 is 6.6/6.6 (Table 1). Thus, our data supports the hypothesis that His163 is neutral, but it contradicts the hypothesis that His172 is charged at physiological pH.5,13,17 Consequently, our data suggests that the protonation state switch of His172 (and not the proposed His1635,13,17) is responsible for the conformational changes of the S1 pocket at pH 6.
Conformational changes in the S1 pocket of SARS-CoV-2 Mpro.
To test the hypothesis that the protonation of His172 leads to a conformational deactivation of SARS-CoV-2 Mpro, we carried out two independent runs of 2-μs fixed-charged MD starting from the crystal structure (PDB: 6y2g5). In run 1, His172 was neutral in protomer A and charged in protomer B, while in run 2, His172 was charged in protomer A and neutral in protomer B. All other residues were fixed in the standard protonation states, as determined by the CpHMD titration for physiological pH (see Table S1). During the simulations, the dimer and each protomer remained stable with a Cα root-mean-square deviation (RMSD) below 3 Å (Fig. S3 and Fig. S4). However, the RMSD of the oxyanion loop in the monomer with the charged His172 increased to about 2 Å within 500 ns, whereas that in the monomer with the neutral His172 stayed below 1 Å throughout the 2-μs simulations (Fig. S5).
To understand the impact of the His172 protonation state, we first examine its interaction with Glu166 and the latter interaction with His163 by calculating the probability distributions of the sidechain minimum distances. In both simulation runs, the charged His172 forms a salt bridge (weak hydrogen bond) with Glu166 (distance of ~3.5 Å) however, the interaction between the neutral His172 and Glu166 is flexible, as evident from the distance distribution showing two peaks at 4.5/6 Å in the first run (Fig. 2a) and 3.5/6 Å in the second run (Fig. S6). Interestingly, in the presence of the charged His172, Glu166 also forms a weak hydrogen bond with His163 (distance of ~3.5 Å), whereas with the neutral His172, the Glu166–His163 interaction is largely absent, with the most probable distance of 7 Å in the first run (Fig. 2b) and 6.2 Å in the second run (Fig. S6). Trajectory snapshots and the χ1 angle distributions revealed that the sidechain of Glu166 adopts different rotamer states dependent on the protonation state of His172. In the presence of the charged His172, Glu166 prefers the g− state (χ1 of 300°), whereas with the neutral His172, it mainly samples the trans state (χ1 of 180°, similar to the crystal structure value) and occasionally adopts the g+ state (χ1 of 9°, Fig. 2d and Fig. S6). These data suggest that when His172 is charged, Glu166 is locked in the g− state, interacting with both His172 and His163, whereas when His172 is neutral, Glu166 sidechain is more flexible, sampling both trans and g+ states and loosely interacting with His172 and His163.
Figure 2. Conformational changes of the S1 pocket in SARS-CoV-2 Mpro is coupled to the switch of His172 protonation state.
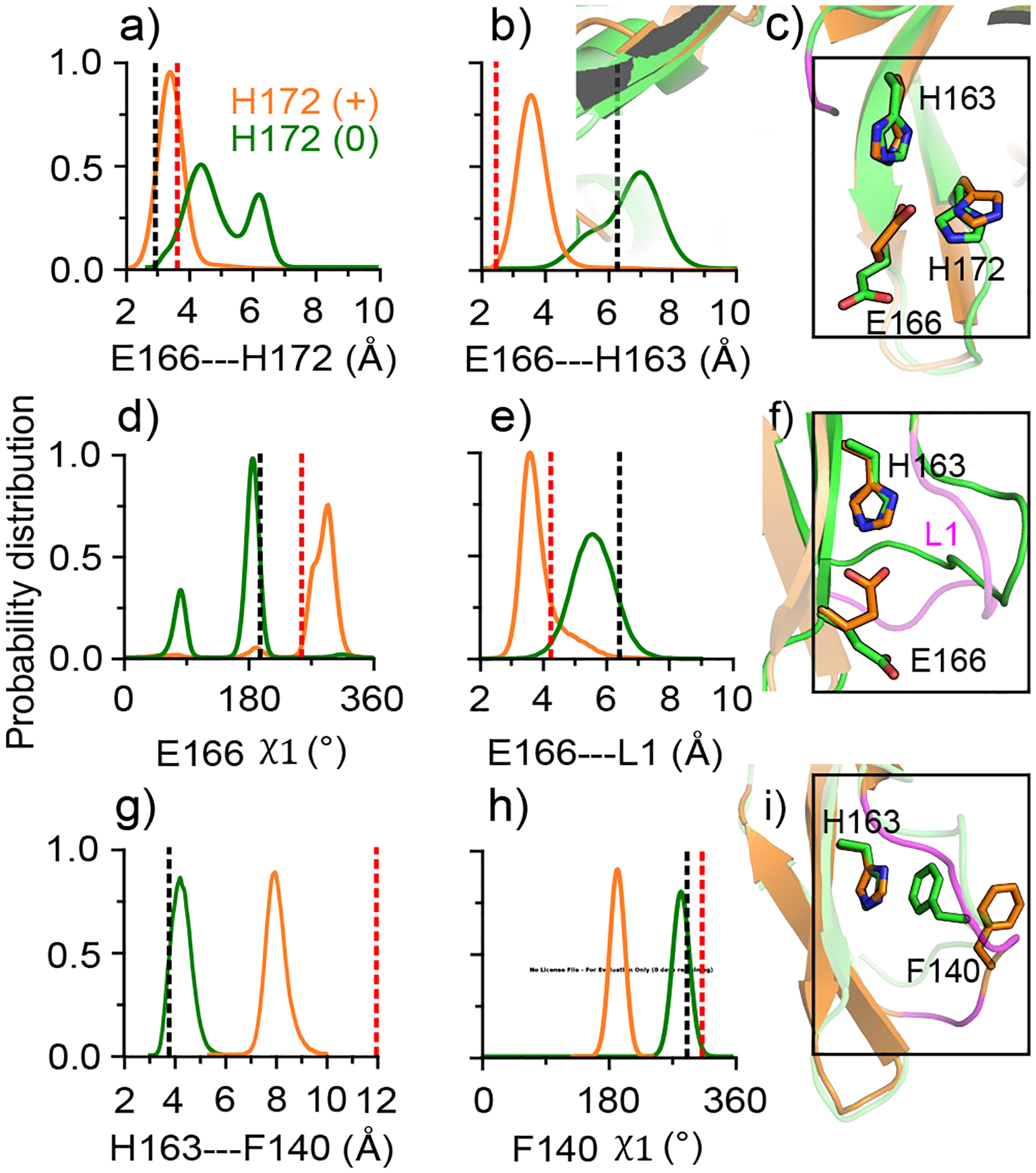
a), b) Probability distribution of the minimum distance between the carboxylate oxygens of Glu166 and the imidazole nitrogens of His172 (a) and His163 (b). d) Distribution of the χ1 angle of Glu166. e) Distribution of the distance between the center of mass of Glu166 (carboxylate oxygens) and the oxyanion loop (Cα atoms of residues 138–145). g) Distribution of the distance between the center of mass of the aromatic rings of His163 and Phe140. h) Distribution of the χ1 angle of Phe140. Data for the protomer with neutral and charged His172 are colored green and orange, respectively. All plots were based on simulation run 1 (run 2 data given in Fig. S6). The black and red dashed lines indicate the corresponding values from the protomer A of the SARS-CoV-2 Mpro structure (PDB: 6y2g5), and the inactive protomer B of the SARS-CoV Mpro structure (PDB: 1uj113). c), f) and i) Snapshots showing the conformational differences between the the protomers with neutral (green) and charged (orange) His172. The oxyanion loop is colored magenta.
Movement of the oxyanion loop in SARS-CoV-2 Mpro.
The protonation state of His172 is also coupled to the conformation of the oxyanion loop L1. The Cα RMSD of L1 increased to 1.5–2 Å in the protomer with charged His172, but it remained below 1 Å in the protomer with neutral His172 (Fig. S5). Trajectory snapshots revealed that L1 is moved closer to the S1 pocket when His172 is charged. In the presence of the charged His172, the distance between the center of mass of Glu166 and that of L1 is moved to ~4 Å, whereas with neutral His172, the distance is ~6 Å (Fig. 2e and Fig. S6). The L1 movement is likely due to the change in the interaction between Phe140 on L1 and His163. Crystal structures of SARS-Cov-2/CoV Mpros show an aromatic ring stacking interaction between them, which is thought to stabilize L1 in the active form.5,13 Simulations showed that the stacking interaction is maintained when His172 is neutral (distance of ~4 Å) and disrupted when His172 is charged (distance of ~8 Å, Fig. 2g, Fig. S6). Interestingly, the loss in the aromatic stacking is correlated with a change in the sidechain conformation of Phe140, which adopts a trans rotamer state (χ1 angle ~180°) in the protomers with charged His172 and a g− state (χ1 angle ~300°) in the protomers with neutral His172 (Fig. 2h, i and Fig. S6).
Comparison to crystal structures of active and inactive Mpro protomers.
The conformational changes of the S1 pocket induced by the protonation of His172 are consistent with the differences between the crystal structures of the active protomer of SARS-CoV-2 Mpro (PDB 6y2g5) and the inactive protomer of SARS-CoV Mpro (PDB: 1uj1, see Fig. 2 dashed lines). Two seeming discrepancies are worth noting. While the Glu166–His172 interaction in the presence of neutral His172 is flexible in our simulations, the aforementioned crystal structure shows a hydrogen bond. This discrepancy may be due to the crystal vs. solution condition. In fact, about 10 crystal structures of SARS-CoV-2 Mpro display a corresponding distance of ~5 Å (Fig. S1), in line with the simulation data (Fig. 2a and Fig. S6). Another seeming discrepancy is in the rotamer state of Phe140. In the simulations, Phe140 switches to a trans rotamer (χ1 of 180°) with the charged His172, whereas in the crystal structure of the inactive protomer B of SARS-CoV Mpro (PDB: 1uj113), the χ1 angle remains similar to that in the active protomer. This may be explained by the extremely low electron density of the Phe140 sidechain atoms in the X-ray structure (B-factor nearly 100).
N-finger interactions and dimer stability in SARS-CoV-2 Mpro.
Having tested our hypothesis that protonation of His172 leads to a partial collapse of the S1 pocket, we turn to its interactions with the N-finger, which were thought to be disrupted at low pH.5,13 Note, the positions of the first two N-terminal residues were missing in the pH 6 crystal structure of SARS-CoV Mpro (PDB: 1uj113). In most of the crystal structures of SARS-CoV-2 Mpro, the interactions between Phe140/His172A/B and Ser1B/A are stable, while the Glu166A/B–Ser1B/A distance varies between 2.2 and 5.3 Å (Fig. S1).
In the simulation run 1, the interactions between Glu166A/His172A (neutral His172A) and Ser1B are largely maintained, and the interaction between Phe140A and Ser1B is flexible (Fig. 3). In contrast, the interactions between Phe140B/Glu166B/His172B (charged His172B) and Ser1A are completely abolished (Fig. 3). In the simulation run 2, the differences are more pronounced; all three interactions sample the crystal structure values when His172 is neutral but become disrupted when His172 is charged (Fig. S7).
Figure 3. Interactions between the S1 pocket residues and Ser1 of the opposite protomer in SARS-CoV-2 Mpro are disrupted by the protonation of His172.
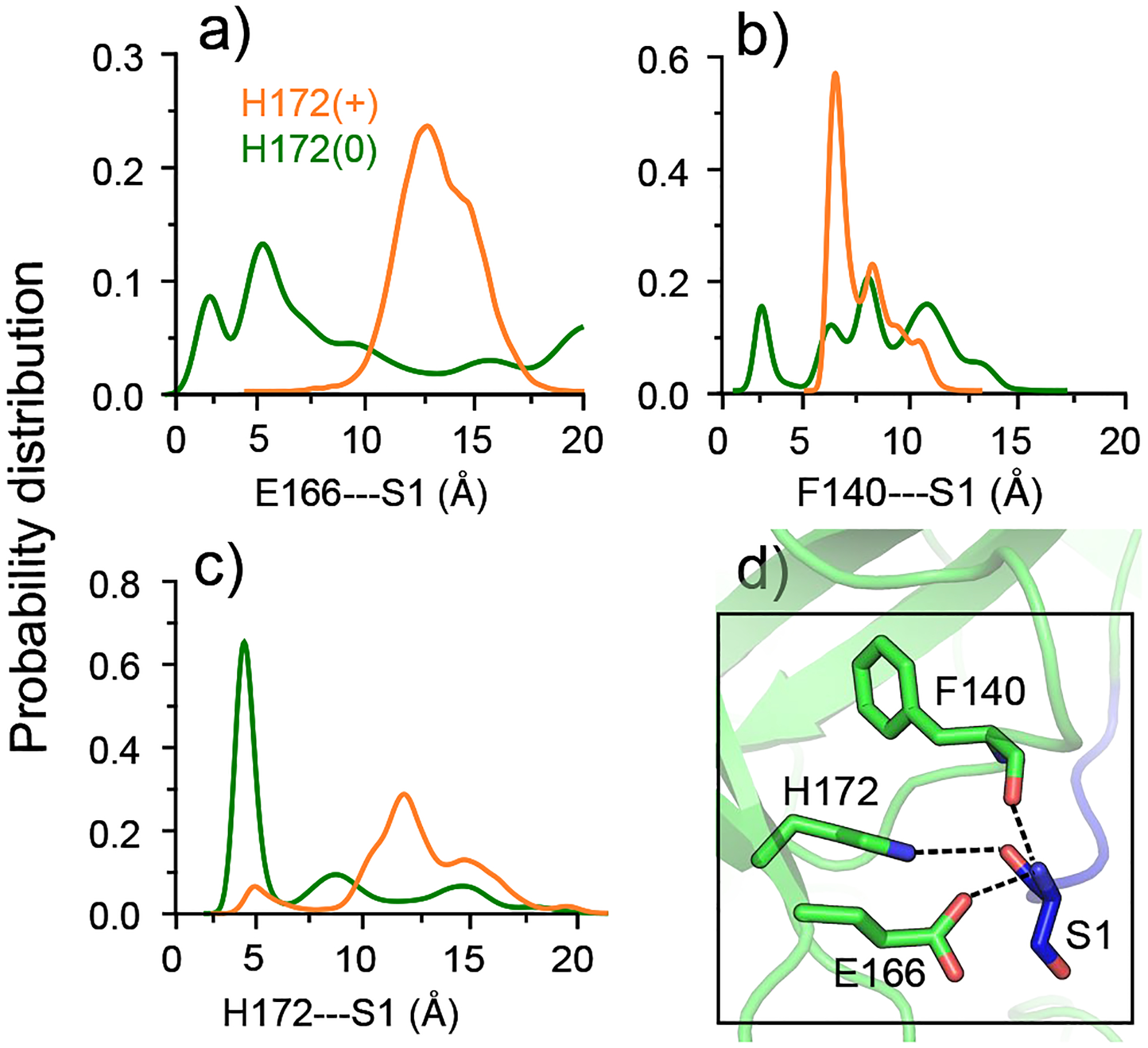
a), b), c) Probability distributions of the Glu166A/Phe140A/His172A–Ser1B distances are colored green; distributions of the Glu166B/Phe140B/His172B–Ser1A distances are colored orange. His172 is neutral in protomer A and charged in protomer B. The Glu166/Phe140–Ser1 distance refers to that between the N-terminal amino nitrogen and the nearest carboxyl oxygen of Glu166 or the carbonyl oxygen of Phe140. The His172–Ser1 distance is refers that between the backbone carbonyl oxygen of Ser1 and the nearest imidazole nitrogen of His172. d) Zoomed-in view of the interactions between the S1 pocket (protomer A) and Ser1 (protomer B) in the X-ray structure of SARS-CoV-2 Mpro (PDB: 6y2g5). Hydrogen bonds are shown in dashed lines.
To further investigate the effect of His172 protonation on the dimerization stability, we examined several hydrogen bond contacts formed between the two protomers in the crystal structure (PDB: 6y2g5). Distance distributions show that all interactions, Gln127A/B/ Glu290A/B–Arg4B/A, Ser10A/B–Ser10B/A, Glu14A/B–Gly11B/A, and Val125A/B–Ala7B/A, remain stable regardless of the His172 protonation state (Fig. S8), which is consistent with the dimer stability in both simulation runs (Fig. S3).
Protonation states and proton-coupled dynamics of SARS-CoV Mpro.
To further corroborate our findings, we conducted CpHMD titration and fixed-charged MD simulations of SARS-CoV Mpro starting from the crystal structure (PDB: 1uk2,13 convergence plots are given in Fig. 9). The CpHMD titration results of SARS-CoV Mpro are consistent with those from SARS-CoV-2 Mpro (Table 1 and Table S1). The pKa’s of the catalytic His41 and Cys145 remain neutral at physiological pH (Table 1), supporting a general base mechanism for Mpro’s proteolytic function.18 Consistently, His163 and His164 remain neutral above pH 4.5. However, while the pKa of His172A is identical to that in SARS-CoV-2 Mpro, the pKa of His172B is one unit higher. The difference can be attributed to the loss of the interaction with Ser1A in the crystal structure (PDB: 1uk213). Due to the charged amino group, the His172B–Ser1A interaction would destabilize the charged form of His172B and thereby lowering its pKa.
To test the hypothesis that protonation of His172 leads to the conformational deactivation of the S1 pocket in SARS-CoV Mpro, we performed a 2-μs fixed-charged MD simulation starting from the aforementioned crystal structure (PDB: 1uk213), in which His172 was fixed in neutral and charged states in protomers A and B, respectively. The differences in the S1 pocket interactions between protomer A and B are in complete agreement with those of SARS-CoV-2 Mpro (Fig. S10). We did not test the S1 pocket interactions with Ser1, as the hydrogen bonds between the S1 pocket residues of protomer B and Ser1 of protomer A in the the X-ray structure of SARS-CoV Mpro are absent (PDB: 1uk213).
New opportunity for small-molecule broad-spectrum covalent inhibitor design.
Currently, the majority of potent broad-spectrum antiviral inhibitors are large substrate mimetics carrying an electrophilic warhead that covalently binds with the catalytic Cys145 in SARS-CoV-2/CoV Mpros, e.g., the α-ketoamide based inhibitors from the Hilgenfeld lab5 and Pfizer’s ketone-based inhibitor PF-00835231 that has entered clinical trial.11 The latter is promising, as it also showed additive/synergistic effect in combination with remdesivir which targets the RNA polymerase. However, delivery of large peptidomimetic compounds such as Pfizer’s inhibitor11 often requires intravenous infusion, which necessitates hospital visit and lowers patient compliance. Thus, orally available small-molecule antiviral drugs are more desirable.
Interestingly, the CpHMD titration revealed that Cys22 and Cys44, which are conserved among the Mpros of SARS-CoV-2/CoV, MERS-CoV, and SARS-like bat-CoV, are significantly more nucleophilic than Cys145, as they are readily deprotonated at physiological pH (Table 1). The hyper-reactivity of Cys44 and its promixity to the substrate binding pocket make it a particularly attractive site of covalent linkage. A recent crystal structure showed that galangin (3,5,7-trihydroxyflavone), a naturally occurring flavonoid with anti-tumor activities,25,26 is bound to SARS-CoV-2 Mpro in a position with a minimum heavy-atom distance of 3.9 Å to either Cys44 or Cys145 (Fig. 4). Thus, we envision the design of targeted covalent inhibitors of Mpros by installing a weakly electrophilic warhead on ganlangin to enable the covalent bond formation with the Cys44 thiolate. It is also conceivable that two warheads can be installed on both ends of ganlangin to bond with both Cys44 and Cys145, allowing superior potency. These strategies may lead to novel small-molecule broad-spectrum covalent inhibitors that have not been attempted so far.
Figure 4. A natural flavonoid compound offers a starting point for designing small-molecule broad-spectrum targeted covalent inhibitors.
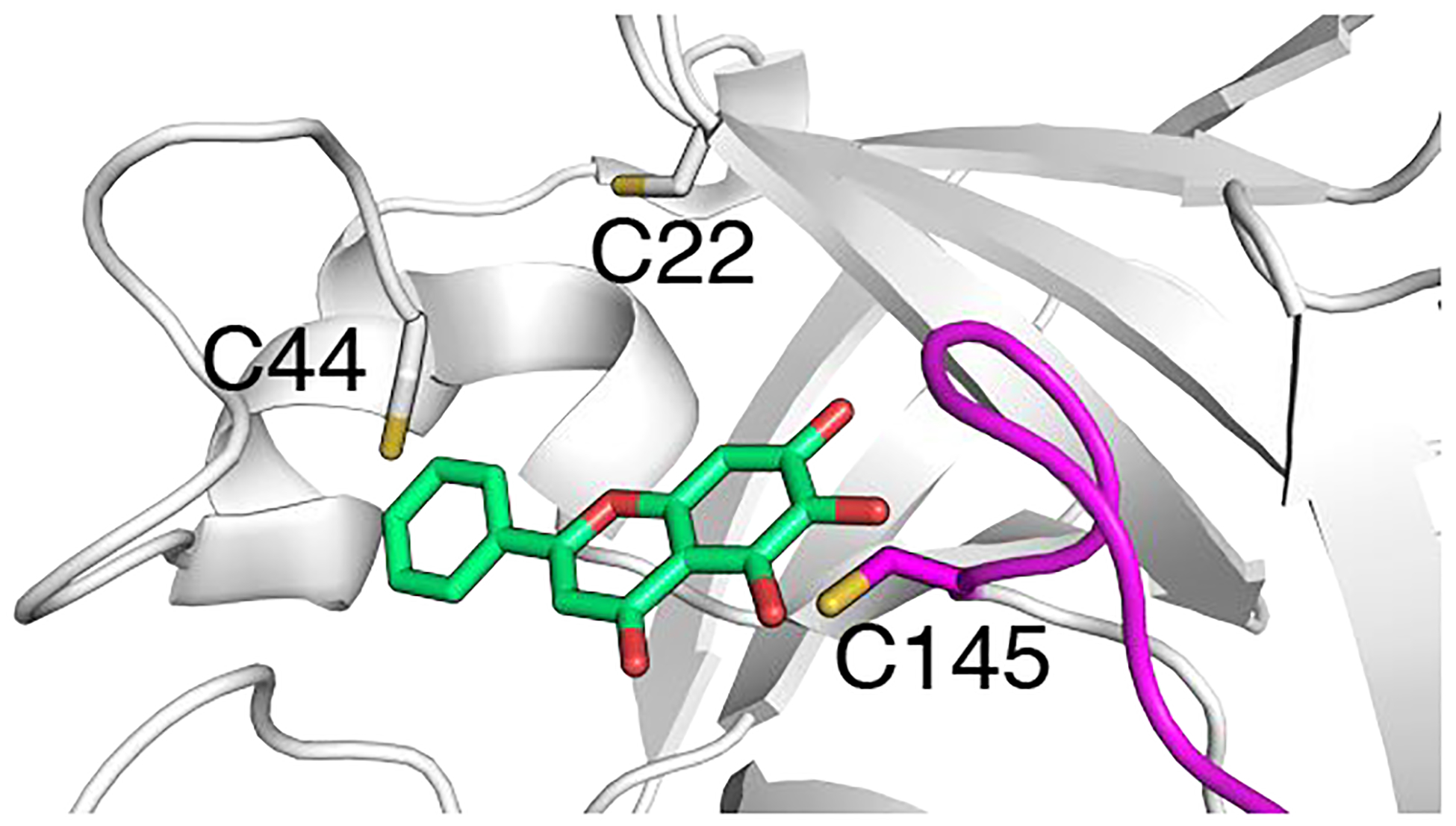
X-ray structure of SARS-CoV-2 Mpro in complex with galangin (PDB: 6m2n) shows that it is in the proximity of both Cys44 and Cys145.
CONCLUSION
Our simulations showed that the protonation state of His172 profoundly impacts the conformation of the S1 pocket through an interaction network involving His172, Glu166, His163, and Phe140. When His172 is neutral, Glu166 is flexible and does not form a hydrogen bond with His163, which allows His163 to maintain the aromatic stacking with Phe140 and thereby keeping the oxyanion loop in the active open conformation. However, when His172 is charged, it forms a salt bridge with Glu166, which locks Glu166 in the g− rotameric state, stabilizing the hydrogen bond interaction with His163. The latter results in a disruption of the stacking interaction between His163 and Phe140 and consequently a collapse (closing) of the oxyanion loop towards the S1 pocket. Consistent with the pH 6 crystal structure of SARS-CoV Mpro, our data provides a detailed molecular mechanism of the conformational deactivation and loss of catalytic activity of SARS-CoV-2/CoV Mpros with decreased pH. Our future work will address the mechanism of the Mpro’s activity decrease at high pH. We speculate that deprotonation of the amino terminus may play a significant role, as the solution pKa of the α-amino group is 8.027 and in the proximity of the negatively charged Glu166, the pKa may shift somewhat higher.
The present study has several caveats worth pointing out. In the CpHMD simulations, the N-terminus was acetylated which may have reduced the stability of the dimer interface. In the fixed-charge simulations, only the protonation state of His172 was varied as it is one of the two residues suggested by experiment to possibly make an impact on the S1 pocket; however, His41 may also become protonated at similar pH (e.g., pH 6) and this effect was neglected in the present study. With regards to the dimerization stability, the fixed-charge simulations of SARS-CoV-2 Mpro demonstrated that His172 protonation disrupted the S1 pocket interactions with Ser1 but not other dimer interface contacts. The latter could be a result of the limited sampling time.
While a complete elucidation of the pH-dependent conformational mechanisms awaits future investigation, the present work offers a first step in understanding the proton-coupled structure-dynamics-function relationships of coronavirus Mpros. Our finding of the hyper-reactive cysteine and the proposed strategies of designing small molecule targeted covalent inhibitors may help accelerate the development of orally available broad-spectrum antiviral drugs to stop the current pandemic and prevent future outbreaks.
METHODS AND PROTOCOLS
System Preparation for CpHMD simulations.
The coordinates of the X-ray crystal structures of SARS-CoV-2 (PDB: 6y2g,5 determined at pH 8.5) and SARS-CoV (PDB 1uk2,13 determined at pH 8) Mpros were retrieved from the PDB. Both structures contain two independent protomers in the asymmetric unit. The N- and C-termini of each protomer were acetylated and amidated, respectively (free in the fixed-charge simulations). Missing hydrogens were added using the HBUILD facility in CHARMM (C38b2);28 dummy hydrogens for titration of Asp/Glu were added using a custom CHARMM script.29 A short energy minimization was performed for the hydrogen positions using the steepest decent and Newton-Raphson methods for 20 steps with the heavy atoms constrained. The force field parameters and coordinate files were constructed using the LEAP utility in AMBER.30 The protein was represented by the ff14sb force field31 and solvent was represented by the GBNeck2 (igb=8) implicit-solvent model.32 The default mbondi3 intrinsic Born radii, except for His and Cys, for which the following radii optimized for CpHMD were used: 1.17 Å for imidazole nitrogen atoms of His20 and 2.0 Å for sulfur atom of Cys.33 Following energy minimization using the steepest decent and conjugate-gradient algorithms for 5000 and 1000 steps, respectively, the system underwent restrained equilibration at pH 7 in four stages (2000 MD steps each). The force constant in the heavy-atom restraint was 5, 2, 1, and 0 kcal/mol/Å2 in the four stages. The final structure was used for the CpHMD titration simulations.
pH replica-exchange CpHMD simulations.
The CpHMD titration simulations were performed with the GPU-accelerated GBNeck2-CpHMD method21 in Amber18.30 The pH replica-exchange protocol34 was used to accelerate convergence. To allow for the use of one or two GPU cards, we applied the newly implemented asynchronous replica-exchange scheme.23 For SARS-CoV-2 Mpro, 9 replicas over the pH range 5–9 with an interval of 0.5 pH unit were used, with an aggregate sampling time of 495 ns. For SARS-CoV Mpro, 9 replicas over the pH range 4.5–8.5 were used, with an aggregate sampling time of 360 ns. All Asp, Glu, His, Cys, and Lys sidechains were allowed to titrate, with the CpHMD default model pKa’s of 3.8, 4.2, 6.5, 8.5, and 10.4, respectively.21 The simulations were run at 300 K with an ionic strength of 0.15 M and an effectively infinite cutoff (999 Å) for nonbonded interactions. The SHAKE algorithm was used to constrain the bonds involving the hydrogens to allow for a 2-fs time step. For pKa calculations and analysis, data from the first 20 ns per replica were discarded.
Conventional fixed-charge all-atom MD simulations.
The initial structures for SARS-CoV-2/CoV Mpros (PDB: 6y2g5 and 1uk213) prepared using the LEAP utility in Amber.30 The termini of each protomer were left in the free form. Hydrogens were added according to the appropriate protonation states (see below). Sodium and chloride ions were added to represent a neutral system with a 150-mM physiological salt concentration. Two 2-μs all-atom fixed-charged MD simulations were performed for SARS-CoV-2/CoV Mpros (PDB: 6y2g5), whereby the protonation states (except for His172) were fixed to those as determined by CpHMD for physiological pH, i.e., Asp(−)/Glu(−)/His(0)/Cys(0). In the simulation run 1, His172 was neutral in protomer A and charged in protomer B, whereas in the simulation run 2, His172 was charged in monomer A and neutral in monomer B. As a comparison, one 2-μs simulation for SARS-CoV Mpro was performed using the same protocol as the run 1 of SARS-CoV-2 Mpro.
All simulations were performed with Amber18.30 The Mpro dimer was placed in a truncated octahedral water box, with a minimum of 11 Å between the protein heavy atoms and the edges of the water box. The protein was represented by the ff14SB force field31 and water represented by the TIP3P35 model. Long-range electrostatic interactions were treated with the particle-Mesh Ewald method.36 A non-bonded cutoff of 8 Å was used. SHAKE was turned on to allow a 2-fs time step. Initially, the system was energy minimized by applying 5000 steps of steepest descent followed by 5000 steps of conjugate gradient minimization with a force constant of 25 kcal/mol/Å2 to the solute heavy atoms. The force constant was then reduced to 5 kcal/mol/Å2 and system was heated from 100 K to 300 K in the canonical ensemble for 50 ps followed by 250-ps restrained and 100 ps unrestrained equilibration in the isothermal-isobaric ensemble with the isotropic Berendsen barostat.37 Finally, the production runs of 2 μs each were performed, starting from a different random initial velocity seed for each system. All analysis was performed with the Amber module CPPTRAJ38 using the last 1 μs of each trajectory.
Supplementary Material
Acknowledgement
Financial support from the National Institutes of Health (GM09888) is acknowledged. JAH acknowledges a fellowship from American Foundation for Pharmaceutical Education.
Footnotes
Supporting Information Available
Supporting Information contains supplemental tables and figures.
References
- (1).Wu F; Zhao S; Yu B; Chen Y-M; Wang W; Song Z-G; Hu Y; Tao Z-W; Tian J-H; Pei Y-Y; Yuan M-L; Zhang Y-L; Dai F-H; Liu Y; Wang Q-M; Zheng J-J; Xu L; Holmes EC; Zhang Y-Z A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zhang L; Lin D; Kusov Y; Nian Y; Ma Q; Wang J; von Brunn A; Leyssen P; Lanko K; Neyts J; de Wilde A; Snijder EJ; Liu H; Hilgenfeld R α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem 2020, 63, 4562–4578. [DOI] [PubMed] [Google Scholar]
- (3).Hilgenfeld R From SARS to MERS: Crystallographic Studies on Coronaviral Proteases Enable Antiviral Drug Design. FEBS J. 2014, 281, 4085–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Needle D; Lountos GT; Waugh DS Structures of the Middle East Respiratory Syndrome Coronavirus 3C-like Protease Reveal Insights into Substrate Specificity. Acta Crystallogr. D 2015, 71, 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; Duan Y; Yu J; Wang L; Yang K; Liu F; Jiang R; Yang X; You T; Liu X; Yang X; Bai F; Liu H; Liu X; Guddat LW; Xu W; Xiao G; Qin C; Shi Z; Jiang H; Rao Z; Yang H Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [DOI] [PubMed] [Google Scholar]
- (7).Rathnayake AD; Zheng J; Kim Y; Perera KD; Mackin S; Meyerholz DK; Kashipathy MM; Battaile KP; Lovell S; Perlman S; Groutas WC; Chang K-O 3C-like Protease Inhibitors Block Coronavirus Replication in Vitro and Improve Survival in MERS-CoV–Infected Mice. Sci. Transl. Med 2020, 12, eabc5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y; Wang J Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jin Z; Zhao Y; Sun Y; Zhang B; Wang H; Wu Y; Zhu Y; Zhu C; Hu T; Du X; Duan Y; Yu J; Yang X; Yang X; Yang K; Liu X; Guddat LW; Xiao G; Zhang L; Yang H; Rao Z Structural Basis for the Inhibition of SARS-CoV-2 Main Protease by Antineoplastic Drug Carmofur. Nat. Struct. Mol. Biol 2020, 27, 529–532. [DOI] [PubMed] [Google Scholar]
- (10).Kneller DW; Phillips G; O’Neill HM; Jedrzejczak R; Stols L; Langan P; Joachimiak A; Coates L; Kovalevsky A Structural Plasticity of SARS-CoV-2 3CL Mpro Active Site Cavity Revealed by Room Temperature X-Ray Crystallography. Nat. Commun 2020, 11, 3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hoffman RL; Kania RS; Brothers MA; Davies JF; Ferre RA; Gajiwala KS; He M; Hogan RJ; Kozminski K; Li LY; Lockner W; Lou J; Marra MT; Jr LJM; Murray BW; Nieman A; Noell S; Planken SP; Rowe T; Ryan K; Smith GJ; Solowiej JE; Steppan CM; Taggart B The Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem 2020, 63, 12725–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shi J; Sivaraman J; Song J Mechanism for Controlling the Dimer-Monomer Switch and Coupling Dimerization to Catalysis of the Severe Acute Respiratory Syndrome Coronavirus 3C-Like Pro-tease. J. Virol 2008, 82, 4620–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Yang H; Yang M; Ding Y; Liu Y; Lou Z; Zhou Z; Sun L; Mo L; Ye S; Pang H; Gao GF; Anand K; Bartlam M; Hilgenfeld R; Rao Z The Crystal Structures of Severe Acute Respiratory Syndrome Virus Main Protease and Its Complex with an Inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hsu W-C; Chang H-C; Chou C-Y; Tsai P-J; Lin P-I; Chang G-G Critical Assessment of Important Regions in the Subunit Association and Catalytic Action of the Severe Acute Respiratory Syndrome Coronavirus Main Protease. J. Biol. Chem 2005, 280, 22741–22748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chen S; Hu T; Zhang J; Chen J; Chen K; Ding J; Jiang H; Shen X Mutation of Gly-11 on the Dimer Interface Results in the Complete Crystallographic Dimer Dissociation of Severe Acute Respiratory Syndrome Coronavirus 3C-like Protease: Crystal Structure with Molecular Dynamics Simulations. J. Biol. Chem 2008, 283, 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Anand K; Palm GJ; Mesters JR; Siddell SG; Ziebuhr J; Hilgenfeld R Structure of Coronavirus Main Proteinase Reveals Combination of a Chymotrypsin Fold with an Extra α-Helical Domain. EMBO J. 2002, 21, 3213–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tan J; Verschueren KH; Anand K; Shen J; Yang M; Xu Y; Rao Z; Bigalke J; Heisen B; Mesters JR; Chen K; Shen X; Jiang H; Hilgenfeld R pH-Dependent Conformational Flexibility of the SARS-CoV Main Proteinase (Mpro) Dimer: Molecular Dynamics Simulations and Multiple X-Ray Structure Analyses. J. Mol. Biol 2005, 354, 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huang C; Wei P; Fan K; Liu Y; Lai L 3C-like Proteinase from SARS Coronavirus Catalyzes Substrate Hydrolysis by a General Base Mechanism. Biochemistry 2004, 43, 4568–4574. [DOI] [PubMed] [Google Scholar]
- (19).Xue X; Yang H; Shen W; Zhao Q; Li J; Yang K; Chen C; Jin Y; Bartlam M; Rao Z Production of Authentic SARS-CoV Mpro with Enhanced Activity: Application as a Novel Tag-Cleavage Endopeptidase for Protein Overproduction. J. Mol. Biol 2007, 366, 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Huang Y; Harris RC; Shen J Generalized Born Based Continuous Constant pH Molecular Dynamics in Amber: Implementation, Benchmarking and Analysis. J. Chem. Inf. Model 2018, 58, 1372–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Harris RC; Shen J GPU-Accelerated Implementation of Continuous Constant pH Molecular Dynamics in Amber: pKa Predictions with Single-pH Simulations. J. Chem. Inf. Model 2019, 59, 4821–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Harris RC; Liu R; Shen J Predicting Reactive Cysteines with Implicit-Solvent-Based Continuous Constant pH Molecular Dynamics in Amber. J. Chem. Theory Comput 2020, 16, 3689–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Henderson JA; Verma N; Harris RC; Liu R; Shen J Assessment of Proton-Coupled Conformational Dynamics of SARS and MERS Coronavirus Papain-like Proteases: Implication for Designing Broad-Spectrum Antiviral Inhibitors. J. Chem. Phys 2020, 153, 115101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Liu R; Yue Z; Tsai C-C; Shen J Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J. Am. Chem. Soc 2019, 141, 6553–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chien S-T; Shi M-D; Lee Y-C; Te C-C; Shih Y-W Galangin, a Novel Dietary Flavonoid, Attenuates Metastatic Feature via PKC/ERK Signaling Pathway in TPA-Treated Liver Cancer HepG2 Cells. Cancer Cell Int 2015, 15, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kong Y; Feng Z; Chen A; Qi Q; Han M; Wang S; Zhang Y; Zhang X; Yang N; Wang J; Huang B; Zhang Q; Xiang G; Li W; Zhang D; Wang J; Li X The Natural Flavonoid Galangin Elicits Apoptosis, Pyroptosis, and Autophagy in Glioblastoma. Front. Oncol 2019, 9, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Thurlkill RL; Grimsley GR; Scholtz JM; Pace CN {{pK}} Values of the Ionizable Groups of Proteins. Protein Sci. 2006, 15, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Brooks B; Brooks C; MacKerell A; Nilsson L; Petrella R; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner A; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor R; Post C; Pu J; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York D; Karplus M CHARMM: The Biomolecular Simulation Program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Khandogin J; Brooks CL Constant pH Molecular Dynamics with Proton Tautomerism. Biophys. J 2005, 89, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham T III; Cruzeiro VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK; Gohlke H; Goetz AW; Greene D; Harris R; Homeyer N; Huang Y; Izadi S; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein DJ; Merz KM; Miao Y; Monard G; Nguyen C; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling CL; Smith J; Salomon-Ferrer R; Swails J; Walker RC; Wang J; Wei H; Wolf RM; Wu X; Xiao L; York DM; Kollman PA AMBER 2018. 2018. [Google Scholar]
- (31).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nguyen H; Roe DR; Simmerling C Improved Generalized Born Solvent Model Parameters for Protein Simulations. J. Chem. Theory Comput 2013, 9, 2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liu R; Yue Z; Tsai C-C; Shen J Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J. Am. Chem. Soc 2019, 141, 6553–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wallace JA; Shen JK Continuous Constant pH Molecular Dynamics in Explicit Solvent with pH-Based Replica Exchange. J. Chem. Theory Comput 2011, 7, 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jorgensen WL Efficient Drug Lead Discovery and Optimization. Acc. Chem. Res 2009, 42, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Salomon-Ferrer R; Götz AW; Poole D; Le Grand S; Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput 2013, 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- (37).Berendsen HJC; Postma JPM; van Gunsteren WF; Di-Nola A; Haak JR Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar]
- (38).Roe DR; Cheatham TE PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.