

Abstract
The recent pandemic caused by SARS-CoV-2 has led the world to a standstill, causing a medical and economic crisis worldwide. This crisis has triggered an urgent need to discover a possible treatment strategy against this novel virus using already-approved drugs. The main protease (Mpro) of this virus plays a critical role in cleaving the translated polypeptides that makes it a potential drug target against COVID-19. Taking advantage of the recently discovered three-dimensional structure of Mpro, we screened approved drugs from the Drug Bank to find a possible inhibitor against Mpro using computational methods and further validating them with biochemical studies. The docking and molecular dynamics study revealed that DB04983 (denufosol) showed the best glide docking score, −11.884 kcal/mol, and MM-PBSA binding free energy, −10.96 kcal/mol. Cobicistat, cangrelor (previous computational studies in our lab), and denufosol (current study) were tested for the in vitro inhibitory effects on Mpro. The IC50 values of these drugs were ∼6.7 μM, 0.9 mM, and 1.3 mM, respectively, while the values of dissociation constants calculated using surface plasmon resonance were ∼2.1 μM, 0.7 mM, and 1.4 mM, respectively. We found that cobicistat is the most efficient inhibitor of Mpro both in silico and in vitro. In conclusion, cobicistat, which is already an FDA-approved drug being used against HIV, may serve as a good inhibitor against the main protease of SARS-CoV-2 that, in turn, can help in combating COVID-19, and these results can also form the basis for the rational structure-based drug design against COVID-19.
Introduction
Coronaviruses (CoVs) are known to cause mild to severe illness in humans.1−3 The current pandemic COVID-19 is also caused by another CoV known as severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). This was first reported from Wuhan City of China in November 2019. Since then, it has caused 49.6 million confirmed infections and over 1,245,717 deaths worldwide (https://www.who.int/emergencies/diseases/novel-coronavirus-2019). SARS-CoV-2 is highly contagious that spreads through respiratory droplets expelled by infected people while coughing and sneezing. These viruses have the ability to get transferred and replicated from animals to humans.4 The global lockdown imposed to control the spread of the virus has led to a grave economic crisis all over the world. To date, there is no drug or vaccine available in the market to fight this deadly virus. Hence, we need to revise and reinvent our strategy to fight this pandemic. Developing a drug from scratch would be a time-consuming process and in the present chaos of pandemic, there is an urgent need for a treatment strategy. Drug repurposing could be the better option to seek effective treatment in a short period of time. Recently, several drug repurposing studies on SARS-CoV-2 have been published.5,6 The clinical trial of a lopinavir–ritonavir drug for COVID-19 has been indicated on the top of the drug repurposing study.7 The main druggable targets of SARS-CoV-2 that have taken the attention of the scientists worldwide are the spike protein (S), NSP-13, NSP-16 RNA-dependent RNA polymerase, helicase, envelope, nucleocapsid phosphoprotein, and papain-like protease (PLpro).8−10 Out of these drug targets, the main focus has been on Mpro because it is essential for the life cycle of SARS-CoV-2. It proteolytically cleaves the overlapping pp1a and pp1ab polyproteins encoded from the genome of this virus. The functional polypeptides are needed for SARS-CoV-2 replication and transcription.11 This enzyme functions like a cysteine protease that exists as a homodimer composed of two monomers of identical conformations. Each monomer consists of three domains, I (residues 8–101) and II (residues 102–184) that also include an antiparallel β-barrel, which is similar to the trypsin-like serine proteases. Domain III is formed by a cluster of five α-helices.12,13 MPro has a specific cleavage site, which allows it to cleave the polypeptide after the glutamine residue. On the other hand, PLpro can recognize ubiquitin and hence, its inhibitors can interfere with the deubiquitinase system.14 It is also evident from the literature that the cleavage site of Mpro is very different from that of any human proteases. These features make Mpro an attractive drug target.15
Several drug repurposing studies against Mpro have been published,16−20 but very few of them have validated their results in vitro.21 In this study, Mpro has been virtually screened against the compound library of the Drug Bank, and then best hits were subjected to molecular dynamic simulations. The best compound identified from the Drug Bank and two previously identified FDA-approved compounds cobicistat and cnagrelor22 were further subjected to validation through biochemical studies.
Results and Discussion
Molecular Docking Analysis
The main protease of SARS-CoV-2 is a homodimeric protein where each molecule is a protomer. Each molecule of this protease comprises three domains, domain I (8–101), domain II (102–184), and domain III (201–303). The active site is constituted by dyad amino acid residues His41 and Cys145 that are present between domain I and domain II. The active site is surrounded by many hydrophobic and hydrophilic residues. The active site residues are conserved among all the known CoVs. This kind of protease is not present in humans, which makes it an attractive drug target against COVID-19. The docking studies revealed that out of all the drugs screened against this protein, four drugs, DB02338, DB01753, DB04158, and DB04983, showed a high glide docking score and glide energy. The chemical structures of the shortlisted compounds are presented in Table 1.
Table 1. 2D Chemical Structures of the Shortlisted Drugs.

Binding Analysis of DB02338
NADPH fits in the active site and interacts with the catalytic dyad amino acid residues His41 through a hydrogen bond and Cys145 through a hydrophobic interaction. This complex is stable with a docking score of −10.303 kcal/mol and glide energy of −100.345 kcal/mol (Table 2). The other amino acid residues found forming a hydrogen bond with NADPH are Thr25, His41, Cys44, Asn142, Glu166, Arg188, and Gln189.
Table 2. Docking Score and Glide Energy (in kcal/mol) of the Co-Crystal (O6K) Inhibitor and Shortlisted Drugs from the Drug Bank against Mpro-COVID-19 (PDB ID: 6Y2F).
| compounds | hydrogen binding interactions | distance (Å) | hydrophobic interactions | docking score (kcal/mol) | glide energy (kcal/mol) |
|---|---|---|---|---|---|
| co-crystal (O6K) α-ketoamide | Asn142(N—H···O) | 3.23 | Thr25, Thr26, Leu27, His41, Val42, Met49, Tyr54, Phe140, Leu141, Ser144, Cys145, His163, His164, Met165, Leu167, Pro168, Asp187, Arg188, Thr190, and Ala191 | –10.983 | –78.884 |
| Gly143(N—H···O) | 2.77 | ||||
| Glu166(N—H···O) | 2.72 | ||||
| Glu166(N—H···O) | 2.95 | ||||
| Gln189(N—H···O) | 2.87 | ||||
| Gln192(N—H···O) | 3.18 | ||||
| DB04158 (6-(adenosine tetraphosphate-methyl)-7,8-dihydropterin) | (N—H···O)Glu47 | 2.80 | Thr24, Thr25, Thr26, His41, Cys44, Thr45, Met49, Cys145, Met165, Leu167, Pro168, Asp187, Arg188, Gln189, and Ala191. | –9.958 | –110.383 |
| Asn142(N–H···O) | 3.21 | ||||
| Gly143(N—H···O) | 2.97 | ||||
| (O—H···O)His164 | 2.51 | ||||
| (N—H···O)Glu166 | 3.01 | ||||
| (O—H···O)Glu166 | 2.55 | ||||
| (O—H···O)Glu166 | 2.99 | ||||
| (O—H···O)Thr190 | 2.71 | ||||
| Thr190(N—H···O) | 3.08 | ||||
| Gln192(N—H···O) | 2.65 | ||||
| DB02338(NADPH) | Thr25(O—H···O) | 3.12 | Leu27, Val42, Thr45, Ser46, Glu47, Met49, Leu141, Ser144, Cys145, His163, His164, Met165, Leu167, Pro168, and Gln192. | –10.303 | –100.345 |
| His41(N—H···O) | 2.80 | ||||
| (O—H···O)His41 | 3.36 | ||||
| (O—H···O)Cys44 | 2.57 | ||||
| (O—H···O) Asn142 | 2.63 | ||||
| (O—H···O) Asn142 | 2.40 | ||||
| Gly143(N—H···O) | 2.90 | ||||
| (O—H···O)Glu166 | 3.01 | ||||
| (O—H···O)Glu166 | 2.79 | ||||
| (O—H···O)Glu166 | 2.57 | ||||
| (N—H···O)Arg188 | 2.97 | ||||
| Gln189(N—H···N) | 3.05 | ||||
| (N—H···O)Thr190 | 2.98 | ||||
| DB04983 (Denufosol) | (O—H···O) Thr25 | 2.88 | His41, Met49, Gly138, Ser139, Leu141, Asn142, Ser144, Cys145, His163, His164, Met165, Leu167, Pro168, Gly170, His172, Asp187, Arg188, and Gln189 | –11.884 | –89.995 |
| (N—H···O) Thr26 | 3.08 | ||||
| Thr26(N—H···O) | 2.93 | ||||
| (O—H···O)Glu47 | 2.52 | ||||
| (O—H···O) Asn142 | 3.08 | ||||
| Asn142(N—H···O) | 2.95 | ||||
| Gly143(N—H···O) | 2.89 | ||||
| Glu166(N—H···O) | 2.99 | ||||
| Gln189(N—H···O) | 3.17 | ||||
| (N—H···O)Thr190 | 3.29 | ||||
| DB01753 (−oxo-nicotinamide-adenine dinucleotide phosphate) | (N—H···O)Cys44 | 2.79 | His41, Glu47, Met49, Phe140, Leu141, Gly143, Ser144, Cys145, His163, Met165, Leu167, Pro168, His172, Asp187, and Ala191 | –11.154 | –89.690 |
| (N—H···O)Phe140 | 3.06 | ||||
| Asn142(O—H···N) | 2.79 | ||||
| Glu166(N—H···N) | 3.26 | ||||
| (O—H···O)Glu166 | 2.72 | ||||
| (O—H···O)Gln189 | 2.87 | ||||
| (O—H···O)Gln189 | 2.56 |
The amino acid residues Leu27, Val42, Thr45, Ser46, Met49, Leu141, Cys145, His163, His164, Pro168, and Gln192 contribute toward hydrophobic interactions with NADPH (Figure 1A).
Figure 1.
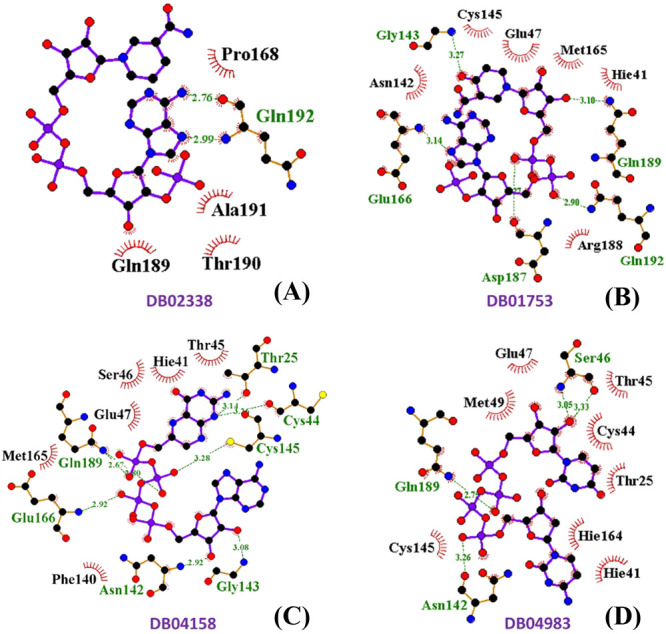
2D ligand interaction diagram of SARS-CoV-2 Mpro with (A) NADPH (DB02338), (B) oxo-nicotinamide adenine dinucleotide phosphate (DB01753), (C) 6-(adenosine tetraphosphate methyl)-7,8-dihydropterin (DB04158), and (D) denufosol (DB04983).
Binding Analysis of DB01753
The drug DB01753 also sits in the active site of this protease with a docking score of −11.154 kcal/mol and glide energy of −89.690 kcal/mol (Table 2). In this case also, both the catalytic residues interact with this compound through hydrophobic interactions. Cys44, Phe140, Asn142, Glu166, and Gln189 formed hydrogen bond interactions, and His41, Glu47, Met49, Phe140, Leu141, Ser144, Cys145, His163, Pro168, His172, and Ala191 are the major contributors toward the hydrophobic forces in stabilizing DB01753 in the active site (Figure 1B).
Binding Analysis of DB04158
The drug DB04158 is found interacting with catalytic dyad residues His41 and Cys145 through hydrophobic interactions with a docking score of −9.958 kcal/mol and glide energy of −110.38 kcal/mole (Table 2). It further forms hydrogen bonds mainly with amino acid residues Glu47, Asn142, His164, Glu166, and Thr190. The main amino acid residues that interact with this compound through hydrophobic interactions are Thr24, Thr25, Thr26, His41, Cys145, Pro168, Arg188, and Gln189 (Figure 1C).
Binding Analysis of DB04983
The drug DB04983 (denufosol) is an FDA-approved drug, which also interacts with both the catalytic dyad amino acids His41 and Cys145 through hydrophobic interactions with a docking score of −11.884 kcal/mol and glide energy score of −89.995 kcal/mol (Table 2). It forms strong hydrogen bonds with amino acid residues Thr25, Thr26, Glu47, Asn142, Glu166, Gln189, and Thr190. The amino acid residues His41, Met49, Ser139, Leu141, Asn142, Ser144, Cys145, His163, Leu167, Asp187, Arg188, and Gln189 contribute toward stabilization of denufosol through hydrophobic interactions (Figure 1D).
Molecular Dynamics Simulations
The MD trajectories of all protein–ligand complexes were first analyzed via monitoring the RMSD fluctuations as a function of run length and are shown in Figure 2.
Figure 2.
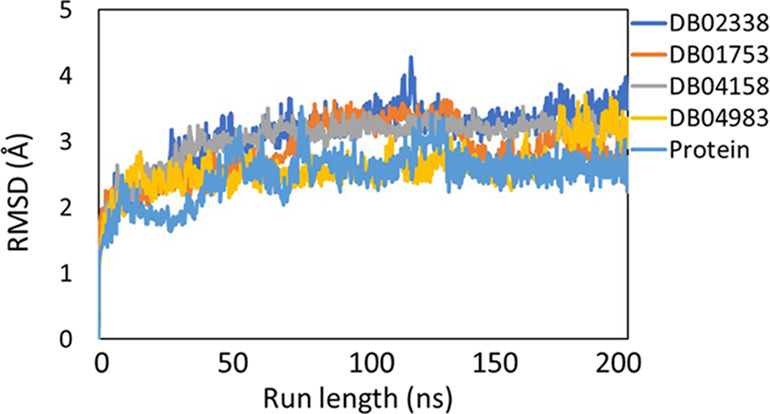
RMSD fluctuations of the protein backbone (light blue), DB02338 (dark blue), DB01753 (orange), DB04158 (gray), and DB04983 (yellow) during 200 ns molecular dynamics simulation runs on Mpro. This plot indicates that there were no significant fluctuations during the entire course of simulations.
As seen from Figure 2, all the systems fluctuations (<3 Å) were stable throughout the trajectory, indicating that the systems displayed no huge dynamical/conformational changes with time. The radius of gyration (Rg) as a function of run length (Figure 3) showed that all the systems have a similar Rg profile and there is no significant compression/expansion of the system. The total number of hydrogen bonds between the ligand and the protein was computed as a function of time and is shown in Figure 4.
Figure 3.
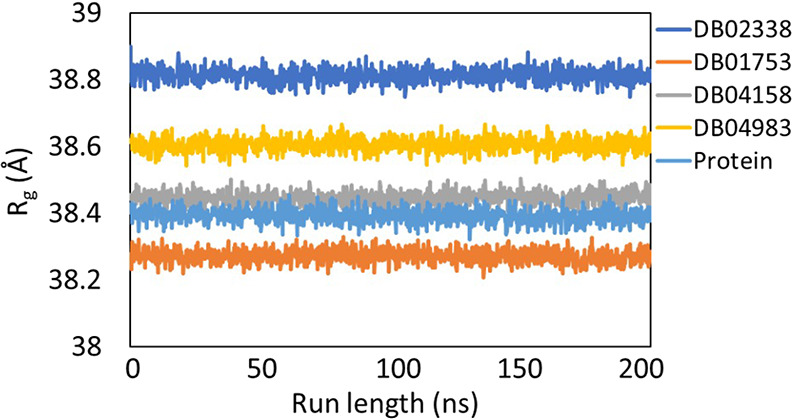
Plot showing the change in radius of gyration (Rg) of the protein backbone (light blue), DB02338 (dark blue), DB01753 (orange), DB04158 (gray), and DB04983 (yellow) during 200 ns molecular dynamics simulation runs on Mpro.
Figure 4.
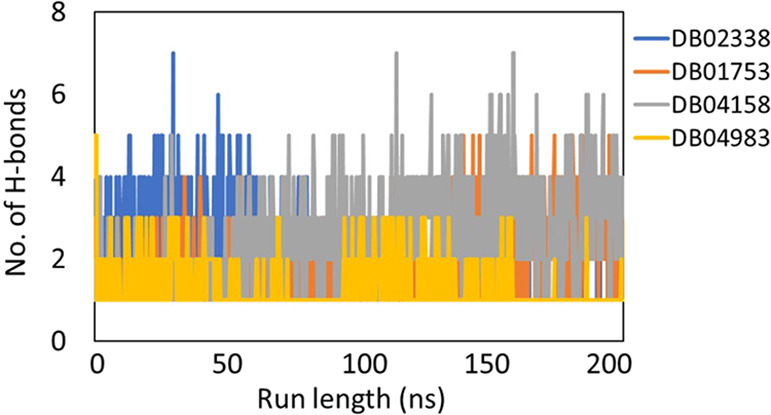
Plot showing the number of stable hydrogen bonds as a function of run length for all the ligands: DB02338 (dark blue), DB01753 (orange), DB04158 (gray), and DB04983 (yellow) during 200 ns molecular dynamics simulation runs on Mpro.
The average number of hydrogen bonds, calculated from the last 50 ns of the simulation trajectories, were 1 (for DB02338), 2 (for DB01753), 3 (for DB04158), and 2 (for DB04983), respectively. The interactions of the ligand in the active site of the protein are displayed in the form of 2D plots in Figure 4. It can be easily seen in Figure 4 that the molecule DB04158 showed an intense hydrogen bond network as well as a snug fit conformation, leading to several van der Waals contacts. Using the hydrogen bond network displayed by the representative structures in the ligand-bound complexes (Figure 1), we assigned each interaction a number as in the Figure1 (#1, #2, etc.). We then used the Cpptraj tool in AMBER to calculate the time-evolved distances between these interactions, and the same is displayed in Figure 5. In the case of DB01753, only 2 or 3 h bonds are clearly persistent all along the simulation, while one #5 is only established at the end of the trajectory, and #2 is also forming a metastable state. The same is true for H bond #3 and #4 of DB04983.
Figure 5.

Variations in the bond distances between donors and acceptors as a function of time.
Finally, the energetics of the system was estimated using the MM-PBSA approach using 100 frames from the last 50 ns of the simulations trajectories. The binding free energies (with respective standard errors) are −8.73 kcal/mol (0.25) (for DB02338), −9.91 kcal/mol (0.33) (for DB01753), −13.36 kcal/mol (0.39) (for DB04158), and −10.96 kcal/mol (0.29) (for DB04983). These values suggested that the ligands have shown strong binding with protein.
Molecular Interactions Using Surface Plasmon Resonance (SPR)
The sensograms of compounds cobicistat, cangrelor, and denufosol were recorded for five different concentrations (Figure 6). The association rate constants (kon) and dissociation rate constants (koff) for the binding of the inhibitor to Mpro were determined using BIA evaluation 3.0 software provided by the manufacturer (GE Healthcare, Sweden, UK). The values of equilibrium dissociation constants, KD (M), were then calculated by the relation KD = koff/kon using the same software.
Figure 6.
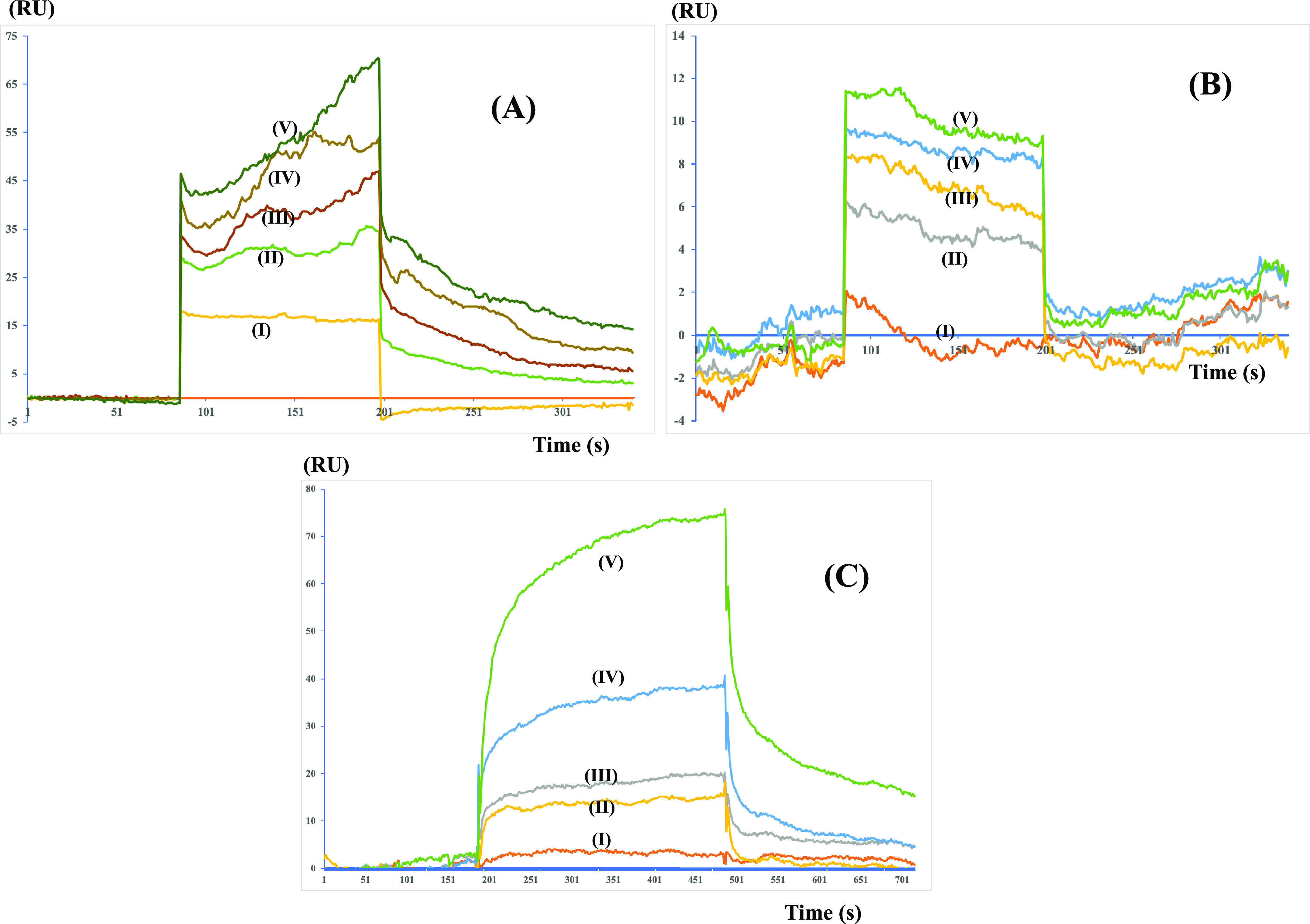
SPR sensograms showing the mode of binding of (A) cobicistat, (B) cangrelor, and (C) denufosol to the immobilized Mpro on the CM-5 chip. Cobicistat was found to show the best KD value of 2.1 μM followed by 0.7 mM for cangrelor and 1.44 mM for denufosol.
The values of dissociation constants (KD) for the compounds cobicistat, cangrelor, and denufosol were determined to be 2.1 × 10–6, 6.4 × 10–4, and 1.4 × 10–3 M, respectively. These values are in agreement with the values of the binding free energy estimated using the MM-PBSA method after MD simulations.
Enzyme Activity Assay
The activity of Mpro was measured using the universal protease activity assay, which uses casein as a substrate by Sigma Aldrich. The assay is based on the measurement of free tyrosine liberated on digestion of casein by an active Mpro enzyme. The protocol used to perform it and the calculation of enzyme activity are as described in the Experimental Section. The IC50 values for the compounds cobicistat, cangrelor, and denufosol determined using the plots (Figure 7) are 6.7 ± 0.6 × 10–6, 9.0 ± 0.5 × 10–4, and 1.3 ± 0.6 × 10–3 M, respectively.
Figure 7.
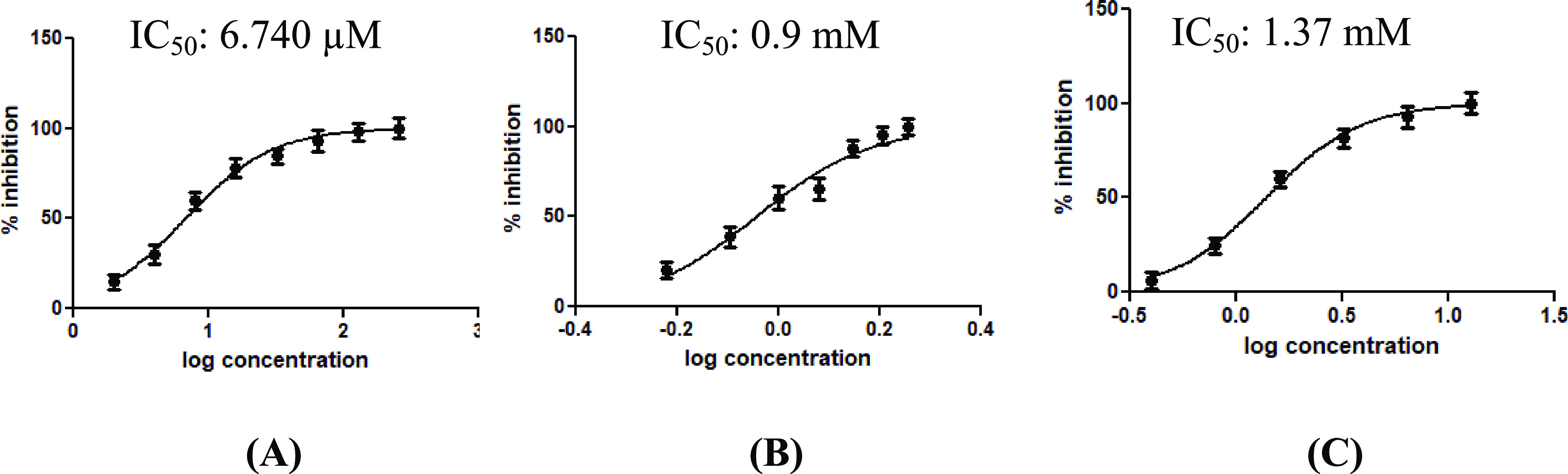
Inhibitory activity profile of the tested compounds (A) cobicistat, (B) cangrelor, and (C) denufosol. The corresponding IC50 values are also indicated.
Conclusions
The current pandemic is caused by a highly infectious novel coronavirus that demands the development of multiple treatment plans as soon as possible. The lack of targeted treatments has caused crisis and posed a serious challenge to the government and health care professionals worldwide. Drug discovery is a lengthy process of developing and validating drugs from scratch. A computational drug repurposing strategy can offer better options in a short period of time. The cleavage site of Mpro is unique, and it is not present in any human proteases, which makes it an attractive drug target against this disease. The catalytic mechanism of Mpro has been explained recently using QM/MM methods,23,24 and the high-resolution structures of Mpro with its inhibitors are available in the Protein Data Bank. These coordinates were used to screen against drug libraries, which includes the Zinc Database22 and Drug Bank.
Various drug repurposing studies against Mpro of SARS-CoV-2 have been done,22 but very few have validated these identified compounds in vitro. In this study, we have biochemically validated our lead drugs that were the best in terms of docking score and binding free energy values post molecular dynamics simulations. Cobicistat and cangrelor had docking scores of −12.31 and −12.28. kcal/mol, respectively. The MM-PBSA binding free energy values are −11.41 and – 6.93 kcal/mol, respectively.16 Denufosol was identified as the best drug out of the screened drugs from the Drug Bank with a docking score of −11.88 kcal/mol and MM-PBSA binding free energy value of −10.96 kcal/mol.
The active site of Mpro has been defined into four subsites named as S1, S1′, S2, and, S4.25 The information about the key residues constituting these subsites are given in Table 3.
Table 3. Key Amino Acid Residues Forming the Specific Subsites of the Active Site of MPro.
| subsite | amino acid residues |
|---|---|
| S1 | Phe140, Asn142, His163, Glu166, and His173 |
| S1′ | Thr26, Glu143, and Cys145 |
| S2 | His41, Cys44, Met49, His164, Met165, Val186, and Arg188 |
| S4 | Pro168 and Gln189 |
Cobicistat interacts with the amino acid residues of all the four subsites, namely, S1, S1′, S2, and S4, whereas cangrelor and denufosol interact with the amino acid residues of S1, S1′, and S4 only (Figure 8).
Figure 8.
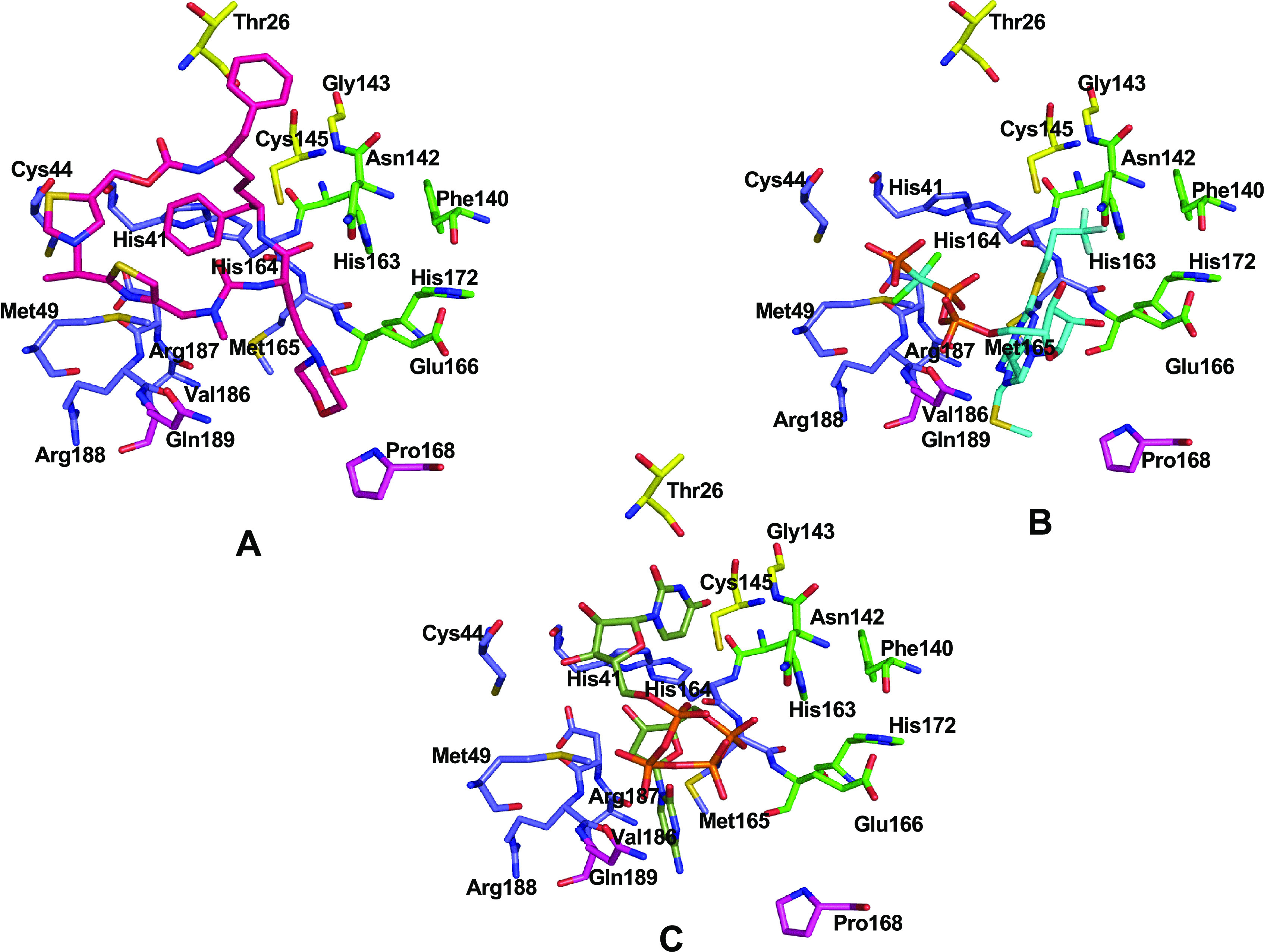
Three-dimensional individual snapshots of (A) cobicistat (pink), (B) cangrelor (cyan), and (C) denufosol (black) in the active site of Mpro. Subsite residues S1 (green), S1′, (yellow), S2 (blue), and S4 (magenta) are also indicated.
All these three drugs were superimposed on each other, and they were found to interact with various parts of the binding site (Figure 9).
Figure 9.
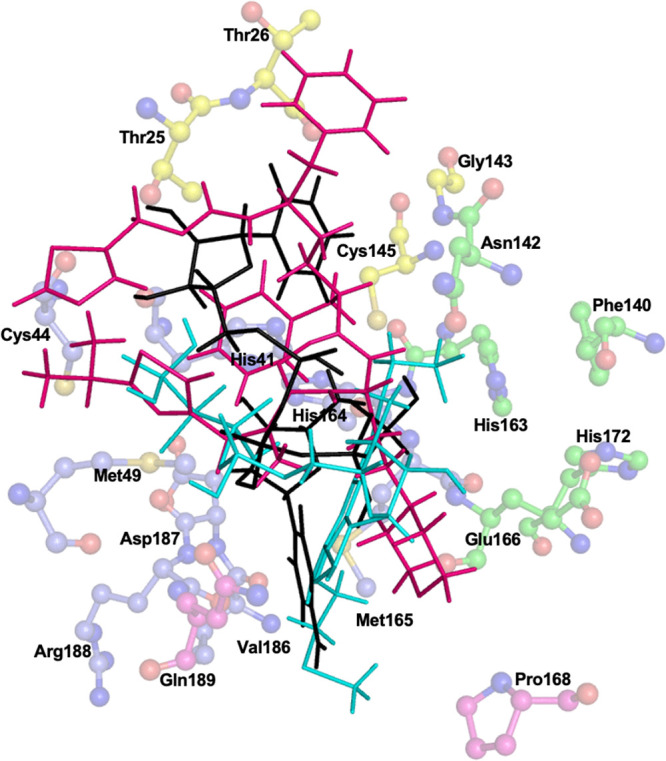
Three-dimensional representation of all the three FDA-approved drugs cobicistat (pink), cangrelor (cyan), and denufosol (black) in the active site of Mpro. The key amino acid residues forming S1 (green), S1′ (yellow), S2 (blue), and S4 (magenta) are also indicated.
So, the three best compounds, cobicistat, cangrelor, and denufosol, that we got from our computational analysis approach were subjected to validation through biochemical studies with Mpro. The binding affinity of these compounds was calculated in real time using SPR technology. The molecular interaction curves after evaluation gave the highest KD values of 2.1 μM in the case of cobicistat. All these drugs were also checked for their inhibitory effects on protease activity of Mpro using the universal protease activity assay, which uses casein as a substrate by Sigma Aldrich. IC50 values were calculated using the different concentrations of these drugs in the enzyme activity assay. We found that cobicistat showed the best value of IC50 at 6.7 μM.
Cobicistat is the FDA-approved drug molecules, which is currently being used for the treatment of HIV infections, and it can also be used as a potential inhibitor of the main protease of SARS-CoV-2 as indicated by our recent computational and biochemical studies. A recent clinical trial of cobicistat is underway against COVID-19 in the USA (NCT04252274), although there are limitations in this study including small sample size and short follow up. However, this is the first clinical trial of cobicistat against COVID-19. More detailed studies are required to establish cobicistat a potential therapeutic option against COVID-19.
Experimental Section
Molecular Docking
Protein and Ligand Preparation
The docking and analysis of the binding modes of different drugs, which were taken from the Drug Bank into the binding site of the main protease of SARS-CoV-2, were performed by the glide module of Schrodinger Maestro. Taking advantage of the recently solved structure of the main protease of SARS-CoV-2, it was selected as a target for docking studies. The coordinates of this protein were obtained from the Protein Data Bank (PDB ID: 6Y2F). The crystal structure was processed, refined, optimized, and energy minimized using the Protein Preparation Wizard of Schrödinger (2017).26,27 The OPLS-3 force field was used to minimize and optimize the protein atoms before docking.28 The missing loops and the side chains were built with a prime module. The grid box was generated using the receptor grid generation module around the center of the co-crystal ligand of the protein to ensure that all the extended conformations of the ligand could fit within the grid box.29 The co-crystal ligand was redocked in the active site to validate the docking protocol. The ligands were retrieved from the Drug Bank in SDF file formats. All the ligands were also energy minimized and optimized using OPLS-3 and addition of hydrogen atoms. Ligands were also converted to their correct chiral forms, tautomeric forms, and ionization states to further minimize the energy.30 We have also used various other programs for docking like SwissDock31 and PatchDock,32 and results were further validated using FireDock. Overall, we observed that the Glide Dock and FireDock results are similar. The results are discussed in the Supporting Information.
Screening the Compounds
The glide docking module was used for screening the compounds. A total of 2100 FDA-approved drugs from the Drug Bank database were used for HTV.29 Compound screening was done against the active site grid box using HTV’s docking module, and 50 drugs were selected on the basis of their best values of docking score, glide energy, and number of hydrogen bond interactions. In the next step, these selected compounds were further docked with extra precision by keeping the grid box rigid using extra precision (XP) glide docking. A total of four drugs having the best values of docking score were selected from XP docking.
Induced-Fit Docking
These four selected drugs were further subjected to induced-fit docking (IFD) using the molecular modeling software GLIDE of Maestro v11.29 The energy-minimized main protease active site residues were chosen to generate the grid box, which was subjected to the IFD studies. The energetically favorable docked poses were obtained for all four drugs, and the best poses were chosen on the basis of glide energy, docking score, hydrogen bond, and hydrophobic interactions.
Molecular Dynamics Simulations
Molecular dynamics simulation techniques have been used to explore bio-macromolecular interactions and evolved as a state-of-the-art technique in recent years.33 MD simulations have been used successfully to explore the complex protein folding pathways,34 protein–ligand binding,35−37 nucleic acid structure and interactions,38−41 and macromolecular mechanisms.40,41 In the present study, we performed MD simulations on the docked structures to explore the binding of the ligand with protein as a function of time in explicit solvent conditions (to mimic the biological environment).42,43 The MD simulations were performed using AMBER software.44 The ligand was optimized using the HF/6-31G* theory and base set in Gaussian0945−47 followed by the generation of ligand parameters for MD simulations (RESP charges, bond, angle, and dihedral parameters). We used the modified ff99SB48 AMBER library for the protein counterpart of the complex. The protein–ligand systems were solvated in a cubic water box with TIP3P water molecules, and sufficient counterions were added to maintain the electroneutrality of the systems considered for the study. The ions were modeled using the parameters in the literature.49 The periodic boundary conditions particle mesh Ewald treatment50 (for long-range electrostatics) and SHAKE algorithm (to constrain hydrogens) were implemented in all the systems. The initial setup underwent minimization to remove any close contacts in the systems. The minimization was performed by using 10,000 conjugate gradients and the 10,000 steepest decent cycles. These systems were then heated to room temperature for 50 ps in NVT conditions and equilibrated for 5 ns. Finally, all the systems underwent 200 ns-long MD simulations. For comparisons, we simulated the native protein (without any bound ligand) using the protocol discussed above. The simulations trajectories were analyzed using the Cpptraj tool51 in AMBER for computing the RMSD and Rg fluctuations as a function of time. The Cpptraj code was also utilized to calculate the number of hydrogen bonds between the ligand and protein throughout the simulations using the default hydrogen bond distance (3.5 Å) and angle (120°) values. The ligand-binding sites were analyzed with the LigPlot+ tool.52 The binding free energies of the protein–ligand complexes were estimated using the MM-PBSA methodology.53,54
Cloning, Expression, and Purification
A gene encoding main protease (Mpro) from SARS-CoV-2 with codons optimized for expression in E. coli was synthesized by MWG Eurofins and cloned into the PGEX-6p-1 (GE Healthcare), which was a gift from Prof. Rolf Hilgenfeld (Germany). This construct had Mpro’s own cleavage site (SAVLQ↓SGFRK; the arrow indicates the cleavage site) at the N-terminus in order to generate an authentic N-terminus during its expression by auto cleavage at this site. An authentic N-terminus plays an important role in forming the functionally active conformation of the S1 pocket of the substrate binding site.55 Similarly, in order to generate an authentic C-terminus, the site for a modified PreScission (SGVTFQ↓GP; the six-residue sequence at the C-terminus of the SARS-CoV-2 Mpro was used as P6–P1 for PreScission cleavage) protease was added just before the His6-tag to the construct.56 The clone obtained was transformed into E. coli BL21 (DE3) expression host cells. After confirming the expression of protein in these cells, the purification was carried out. A single colony was picked and inoculated in 10 mL of Luria–Bertani (LB) containing ampicillin at a concentration of 100 μg/mL overnight at 37 °C with shaking at 250 rpm. Secondary inoculation was done by adding this culture to 1000 mL of the LB medium, which contained 100 μg/mL ampicillin. It was then placed in an orbital shaker at 37 °C with shaking at 250 rpm until the optical density (OD) of the culture at 600 nm reached the level of 0.8 followed by addition of 0.5 mM isopropyl-d-thiogalactoside (IPTG) for induction of the Mpro gene and culture was kept shaking for 5 h at 37 °C. After 5 h, cells were harvested by centrifugation at 6000 rpm at 4 °C for 15 min. The supernatant was discarded, and the cell pellet was resuspended in lysis buffer (50 mM potassium phosphate buffer containing 150 mM NaCl, pH 7.8). The sonicator (model UP50H, Hielscher, Brandenburg, Germany) was used for the disruption of cells followed by high-speed centrifugation at 15,000 rpm for 40 min. The clear lysate obtained was loaded onto a Ni-NTA (Ni2+–nitrilotriacetate) super flow column (Qiagen, Hilden, Germany) pre-equilibrated with lysis buffer. The column was washed with 50 mL of lysis buffer containing 40 mM imidazole to remove nonspecific binding proteins followed by elution using lysis buffer containing 300 mM imidazole. The eluted target protein fraction was mixed with PreScission protease at a molar ratio of 5:1 and dialyzed against buffer (50 mM potassium phosphate, 150 mM NaCl, 1 mM DTT, pH 7.8) at 4 °C overnight.55 The mixture was then loaded onto the GSTtrap FF (GE Healthcare) column, which resulted in a free His-tag, His-tag-free protein, and protein with uncleaved His-tag in the collected flow through. The flow through was then loaded onto a Ni-NTA column equilibrated with lysis buffer. The flow through collected from this column contained a His-tag-free Mpro with authentic N- and C-termini. The collected protein in buffer (50 mM potassium phosphate, 150 mM NaCl, 1 mM DTT, pH 7.8) was concentrated using an Amicon ultrafiltration device (Millipore) with a molecular mass cutoff of 3 kDa. The purity of the protein was finally checked by SDS-PAGE, which clearly showed a single band of an approximate monomer weight of 33 kDa.
Molecular Interactions Using Surface Plasmon Resonance (SPR)
Binding studies of Mpro from SARS CoV-2 with compounds cobicistat (DB09065), cangrelor (DB06441), and denufosol (DB04983) were carried out using the technique surface plasmon resonance. The binding studies using a BIAcore-3000 (Biacore Inc., Uppsala, Sweden) were carried out in our lab. Mpro was immobilized on a Biacore chip CM5 using the amine coupling by the coupling agents EDC and NHS. After immobilization, a total of 1000 RU (response unit) was achieved for the immobilized protein. Five different concentrations of ligands cangrelor, cobicistat, and denufosol were run on the immobilized Mpro at a flow rate of 10 μL/min. The concentrations for ligands cobicistat, cangrelor, and denufosol used are 4 × 10–6, 8 × 10–6, 1.6 × 10–5, 3.2 × 10–5, and 6.4 × 10–5 M; 2 × 10–4, 4 × 10–4, 8 × 10–4, 1.6 × 10–3, and 3.2 × 10–3 M; and 2 × 10–3, 4 × 10–3, 8 × 10–3, 1.6 × 10–2, and 3.2 × 10–2 M, respectively. The regeneration of bound analytes was achieved by injecting 50 mM NaOH for 60 s at a flow rate of 30 μL/min.
Enzyme Activity Assay
For enzyme inhibition studies on Mpro, universal, non-specific protease activity assay by Sigma Aldrich, which uses casein as a substrate, was used. When casein is digested using any protease, the amino acid tyrosine is liberated along with peptide fragments. The liberated free tyrosine reacts with Folin–Ciocâlteu’s phenol (F-C reagent) to generate a blue-colored chromophore, which shows absorbance at 660 nm. Hence, the amount of free tyrosine is directly proportional to the protease activity.57,58 Casein has multiple Mpro cleavage sites (SAVLQ↓SGFRK; the arrow indicates the cleavage site), which makes it an appropriate substrate to assess the enzyme activity. In order to optimize the concentration of casein to be used in the subsequent reactions with inhibitors to calculate the values of IC50, the activity of Mpro was measured by using 10 μM enzyme with different casein concentrations in a 400 μL reaction in phosphate buffer at pH 7.5 incubated at 37 °C for an hour. The reaction was terminated using 200 μL of 1.7% trichloroacetic acid followed by high-speed centrifugation at 15000 × g for 5 min. The clear supernatant was transferred to a separate vial, and a 200 μL of 0.5 M sodium carbonate solution was added to it followed by immediate addition of 50 μL of 0.5 M freshly diluted F-C reagent. Reaction contents were mixed by swirling and incubated at 37 °C for 30 min for color development. Reaction mixtures were given a short spin, and the absorbance at 660 nm was recorded for each reaction using a Cary 100 BIO spectrophotometer. The enzyme activity was calculated for each concentration of casein, and it was found to be at maximum when the concentration of casein used was 150 μM. For inhibition studies, 10 μM Mpro was incubated with different concentrations of compounds for an hour. Then, it was followed by addition of 150 μM casein in the same 400 μL reaction containing phosphate buffer at pH 7.5 incubated at 37 °C for an hour. The rest of the protocol remains the same for each reaction as mentioned above, and the same steps were repeated for all of them. The increasing concentrations of compounds cobicistat, cangrelor, and denufosol used to study inhibition are 2 × 10–6, 4 × 10–6, 8 × 10–6, 1.6 × 10–5, 3.2 × 10–5, 6.4 × 10–5, 1.28 × 10–4, and 2.56 × 10–4 M; 6 × 10–4, 8 × 10–4, 1 × 10–3, 1.2 × 10–3, 1.4 × 10–3, 1.6 × 10–3, and 1.8 × 10–3 M; and 4 × 10–4, 8 × 10–4, 1.6 × 10–3, 3.2 × 10–3, 6.4 × 10–3, and 1.28 × 10–2 M, respectively. The percentage inhibition of enzyme activity by these compounds were calculated by putting the values in the formula 100 – (AT/AC × 100) where AT and AC are the values of enzyme activities in the presence and absence of the compounds, respectively. The IC50 value for each inhibitor was determined by plotting the percentage inhibition versus log (conc.) curve using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA), and the exact IC50 values were calculated by nonlinear least squares regression and equation solving for IC50: Y = 100/1 + 10[(logIC50 – X) × (hill slope)], where Y is % inhibition and X is the log inhibitor concentration.
Acknowledgments
P.S. and S.S. thank All India Institute of Medical sciences (AIIMS), New Delhi for the intramural grant. V.V. thanks the Department of Science and Technology (DST), SERB for the National Postdoctoral Fellowship. T.P.S. thanks the Department of Science and Technology (DST) for the SERB Distinguished Research Professorship. A.G. thanks the Indian Council of Medical Research for the award of fellowship. The Supercomputing Facility for Bioinformatics & Computational Biology (SCFBio), IIT Delhi is gratefully acknowledged for providing computational support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04808.
Comparison of the results of Glide Docking, Swiss Docking, and optimization using Fire Docking software (PDF)
Author Contributions
# A.G. and C.R. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- CDC Coronavirus disease 2019 (COVID-19). Cent. Dis. Control Prev. 2020. [Google Scholar]
- Myint S. H. Human Coronaviruses: A Brief Review. Rev. Med. Virol. 1994, 4, 35–46. 10.1002/rmv.1980040108. [DOI] [Google Scholar]
- Hui D. S.; Azhar E. I.; Madani T. A.; Ntoumi F.; Kock R.; Dar O.; Ippolito G.; Mchugh T. D.; Memish Z. A.; Drosten C.; Zumla A.; Petersen E. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infec. Diseases 2020, 91, 264–266. 10.1016/j.ijid.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J.; Li F.; Shi Z. L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H. H.; Wang L. Q.; Liu W. L.; An X. P.; Liu Z. D.; He X. Q.; Song L. H.; Tong Y. G. Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chin. Med. J. 2020, 133, 1051–1056. 10.1097/CM9.0000000000000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandeel M.; Al-Nazawi M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 117627. 10.1016/j.lfs.2020.117627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B.; Wang Y.; Wen D.; Liu W.; Wang J.; Fan G.; Ruan L.; Song B.; Cai Y.; Wei M.; Li X.; Xia J.; Chen N.; Xiang J.; Yu T.; Bai T.; Xie X.; Zhang L.; Li C.; Yuan Y.; Chen H.; Li H.; Huang H.; Tu S.; Gong F.; Liu Y.; Wei Y.; Dong C.; Zhou F.; Gu X.; Xu J.; Liu Z.; Zhang Y.; Li H.; Shang L.; Wang K.; Li K.; Zhou X.; Dong X.; Qu Z.; Lu S.; Hu X.; Ruan S.; Luo S.; Wu J.; Peng L.; Cheng F.; Pan L.; Zou J.; Jia C.; Wang J.; Liu X.; Wang S.; Wu X.; Ge Q.; He J.; Zhan H.; Qiu F.; Guo L.; Huang C.; Jaki T.; Hayden F. G.; Horby P. W.; Zhang D.; Wang C. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Liu Q.; Guo D. Emerging Coronaviruses: Genome Structure, Replication, and Pathogenesis. J. Med. Virol. 2020, 92, 418–423. 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgio J. F.; Alsuwat H. S.; Al Otaibi W. M.; Ibrahim A. M.; Almandil N. B.; Al Asoom L. I.; Salahuddin M.; Kamaraj B.; AbdulAzeez S. State-of-the-art tools unveil potent drug targets amongst clinically approved drugs to inhibit helicase in SARS-CoV-2. Arch. Med Sci. 2020, 16, 508–518. 10.5114/aoms.2020.94567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azeez S. A.; Alhashim Z. G.; Al Otaibi W. M.; Alsuwat H. S.; Ibrahim A. M.; Almandil N. B.; Borgio J. F. State-of-the-art tools to identify druggable protein ligand of SARS-CoV-2. Arch. Med. Sci. 2020, 16, 497–507. 10.5114/aoms.2020.94046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J. Molecular biology of severe acute respiratory syndrome coronavirus. Curr. Opin. microbiol. 2004, 7, 412–419. 10.1016/j.mib.2004.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K.; Ziebuhr J.; Wadhwani P.; Mesters J. R.; Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Yang H.; Yang M.; Ding Y.; Liu Y.; Lou Z.; Zhou Z.; Sun L.; Mo L.; Ye S.; Pang H.; Gao G. F.; Anand K.; Bartlam M.; Hilgenfeld R.; Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl Acad. Sci. 2011, 100, 13190–13195. 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Liu Y.; Yang Y.; Zhang P.; Zhong W.; Wang Y.; Wang Q.; Xu Y.; Li M.; Li X.; Zheng M.; Chen L.; Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020, 10, 766–788. 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzini S.; Musso L.; Dallavalle S.; Artali R. Putative SARS-CoV-2 Mpro Inhibitors from an In-House Library of Natural and Nature-Inspired Products: A Virtual Screening and Molecular Docking Study. Molecules 2020, 25, 3745. 10.3390/molecules25163745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahlawat A.; Kumar N.; Kumar R.; Sandhu H.; Singh I. P.; Singh S.; Sjöstedt A.; Garg P.. Structure-based virtual screening to discover potential lead molecules for the SARS-CoV-2 main protease. J. Chem. Inf. Model. 202020, 10.1021/acs.jcim.0c00546. [DOI] [PubMed] [Google Scholar]
- Marinho E. M.; de Andrade Neto J. B.; Silva J.; da Silva C. R.; Cavalcanti B. C.; Marinho E. S.; Júnior H. V. N. Virtual screening based on molecular docking of possible inhibitors of Covid-19 main protease. Microb. Pathog. 2020, 148, 104365. 10.1016/j.micpath.2020.104365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham M. Q.; Vu K. B.; Pham T. N. H.; Huong L. T. T.; Tran L. H.; Tung N. T.; Vu V. V.; Nguyen T. H.; Ngo S. T. Rapid prediction of possible inhibitors for SARS-CoV-2 main protease using docking and FPL simulations. RSC Adv. 2020, 10, 31991–31996. 10.1039/D0RA06212J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sencanski M.; Perovic V.; Pajovic S. B.; Adzic M.; Paessler S.; Glisic S. Drug Repurposing for Candidate SARS-CoV-2 Main Protease Inhibitors by a Novel In Silico Method. Molecules 2020, 25, 3830. 10.3390/molecules25173830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi P. K.; Upadhyay S.; Singh M.; Raghavendhar S.; Bhardwaj M.; Sharma P.; Patel A. K. Screening and evaluation of approved drugs as inhibitors of main protease of SARS-CoV-2. Int. J. Biol. Macromol. 2020, 164, 2622–2631. 10.1016/j.ijbiomac.2020.08.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P.; Vijayan V.; Pant P.; Sharma M.; Vikram N.; Kaur P.; Singh T. P.; Sharma S. Identification of potential drug candidates to combat COVID-19: a structural study using the main protease (mpro) of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–11. 10.1080/07391102.2020.1798286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Guzmán C. A.; Ruiz-Pernía J. J.; Tuñón I. Unraveling the SARS-CoV-2 Main Protease Mechanism Using Multiscale Methods. ACS Catal. 2020, 10, 12544–12554. 10.1021/acscatal.0c03420. [DOI] [PubMed] [Google Scholar]
- Świderek K.; Moliner V. Revealing the Molecular Mechanisms of Proteolysis of SARS-CoV-2 Mproby QM/MM Computational Methods. Chem. Sci. 2020, 11, 10626–10630. 10.1039/D0SC02823A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W.; Zhang B.; Jiang X.-M.; Su H.; Li J.; Zhao Y.; Xie X.; Jin Z.; Peng J.; Liu F.; Li C.; Li Y.; Bai F.; Wang H.; Cheng X.; Cen X.; Hu S.; Yang X.; Wang J.; Liu X.; Xiao G.; Jiang H.; Rao Z.; Zhang L.-K.; Xu Y.; Yang H.; Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro Schrödinger Release 2017–1; Schrödinger, LLC: New York, NY, 2017. [Google Scholar]
- Protein Preparation Wizard, release 2015; Schrödinger, LLC: New York, NY, 2017. [Google Scholar]
- Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; Kaus J. W.; Cerutti D. S.; Krilov G.; Jorgensen W. L.; Abel R.; Friesner R. A. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- LigPrep Schrödinger Release 2017–1; Schrödinger, LLC: New York, NY, 2017. [Google Scholar]
- Grosdidier A.; Zoete V.; Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D.; Inbar Y.; Nussinov R.; Wolfson H. J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth S. A.; Dror R. O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. 10.1016/j.neuron.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L.; Guo X.; Cong Y.; Feng G.; Li Y.; Zhang J. Z. H. Accelerated Molecular Dynamics Simulation for Helical Proteins Folding in Explicit Water. Front. Chem. 2019, 7, 540. 10.3389/fchem.2019.00540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swegat W.; Schlitter J.; Krüger P.; Wollmer A. MD simulation of protein-ligand interaction: formation and dissociation of an insulin-phenol complex. Biophys. J. 2003, 84, 1493–1506. 10.1016/S0006-3495(03)74962-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey K. D.; Tiwari R. K.; Ojha R. P. Recent advances in protein-ligand interactions: molecular dynamics simulations and binding free energy. Curr. Comput.-Aided Drug Des. 2013, 9, 518–531. 10.2174/15734099113096660036. [DOI] [PubMed] [Google Scholar]
- Santhanam V.; Pant P.; Jayaram B.; Ramesh N. G. Design, synthesis and glycosidase inhibition studies of novel triazole fused iminocyclitol-δ-lactams. Org. Biomol. Chem. 2019, 17, 1130–1140. 10.1039/C8OB03084G. [DOI] [PubMed] [Google Scholar]
- Pant P.; Jayaram B. C5′ omitted DNA enhances bendability and protein binding. Biochem. Biophys. Res. Commun. 2019, 514, 979–984. 10.1016/j.bbrc.2019.05.051. [DOI] [PubMed] [Google Scholar]
- Pant P.; Fisher M. DNA triplex with conformationally locked sugar disintegrates to duplex: Insights from molecular simulations. Biochem. Biophys. Res. Commun. 2020, 662. 10.1016/j.bbrc.2020.08.097. [DOI] [PubMed] [Google Scholar]
- Etheve L.; Martin J.; Lavery R. Dynamics and recognition within a protein–DNA complex: a molecular dynamics study of the SKN-1/DNA interaction. Nucleic Acids Res. 2016, 44, 1440–1448. 10.1093/nar/gkv1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasi M.; Maddocks J. H.; Beveridge D.; Bishop T. C.; Case D. A.; Cheatham T. III; Dans P. D.; Jayaram B.; Lankas F.; Laughton C.; Mitchell J.; Osman R.; Orozco M.; Pérez A.; Petkevičiu̅tė D.; Spackova N.; Sponer J.; Zakrzewska K.; Lavery R. μABC: A systematic microsecond molecular dynamics study of tetranucleotide sequence effects in B-DNA. Nucleic Acids Res. 2014, 42, 12272–12283. 10.1093/nar/gku855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror R. O.; Dirks R. M.; Grossman J. P.; Xu H.; Shaw D. E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- Adcock S. A.; McCammon J. A. Molecular Dynamics: Survey of Methods for Simulating the Activity of Proteins. Chem. Rev. 2006, 106, 1589–1615. 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Babin V.; Berryman J. T.; Betz R. M.; Cai Q.; Cerutti D. S.; Cheatham T. E. III; Darden T. A.; Duke R. E.; Gohlke H.; Goetz A. W.; Gusarov S.; Homeyer N.; Janowski P.; Kaus J.; Kolossváry I.; Kovalenko A.; Lee T. S.; LeGrand S.; Luchko T.; Luo R.; Madej B.; Merz K. M.; Paesani F.; Roe D. R.; Roitberg A.; Sagui C.; Salomon-Ferrer R.; Seabra G.; Simmerling C. L.; Smith W.; Swails J.; Walker R. C.; Wang J.; Wolf R. M.; Wu X.; Kollman P. A.. AMBER 14; University of California: San Francisco, 2014. [Google Scholar]
- Cieplak P.; Cornell W. D.; Bayly C.; Kollman P. A. Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995, 16, 1357–1377. 10.1002/jcc.540161106. [DOI] [Google Scholar]
- Vanquelef E.; Simon S.; Marquant G.; Garcia E.; Klimerak G.; Delepine J. C.; Cieplak P.; Dupradeau F.-Y. R.E.D. Server: A Web Service for Deriving RESP and ESP Charges and Building Force Field Libraries for New Molecules and Molecular Fragments. Nucleic Acids Res. 2011, 39, W511–W517. 10.1093/nar/gkr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. 10.1021/ja00124a002. [DOI] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. chem. theory comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung I. S.; Cheatham T. E. III Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 2008, 112, 9020–9041. 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham T. E. III; Miller J. L.; Fox T.; Darden T. A.; Kollman P. A. Molecular Dynamics Simulations on Solvated Biomolecular Systems: The Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, RNA and Proteins. J. Am. Chem. Soc. 1995, 117, 4193–4194. 10.1021/ja00119a045. [DOI] [Google Scholar]
- Roe D. R.; Cheatham T. E. III PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Swindells M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Gohlke H.; Case D. A. Converging free energy estimates: MM-PB(GB)SA studies on the protein protein complex Ras-Raf. J. Comput. Chem. 2004, 25, 238–250. 10.1002/jcc.10379. [DOI] [PubMed] [Google Scholar]
- Onufriev A.; Bashford D.; Case D. A. Modification of the generalized born model suitable for macromolecules. J. Phys. Chem. B. 2000, 104, 3712–3720. 10.1021/jp994072s. [DOI] [Google Scholar]
- Xue X.; Yang H.; Shen W.; Zhao Q.; Li J.; Yang K.; Chen C.; Jin Y.; Bartlam M.; Rao Z. Production of authentic SARS-CoV Mpro with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 2007, 366, 965–975. 10.1016/j.jmb.2006.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anson M. L. The estimation of pepsin, trypsin, papain, and cathepsin with hemoglobin. J. Gen. Physiol. 1938, 22, 79. 10.1085/jgp.22.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folin O.; Ciocalteu V. On tyrosine and tryptophane determinations in proteins. J. Bio. Chem. 1927, 73, 627–650. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.