

Abstract
We present a supercomputer-driven pipeline for in-silico drug discovery using enhanced sampling molecular dynamics (MD) and ensemble docking. Ensemble docking makes use of MD results by docking compound databases into representative protein binding-site conformations, thus taking into account the dynamic properties of the binding sites. We also describe preliminary results obtained for 24 systems involving eight proteins of the proteome of SARS-CoV-2. The MD involves temperature replica exchange enhanced sampling, making use of massively-parallel supercomputing to quickly sample the configurational space of protein drug targets. Using the Summit supercomputer at the Oak Ridge National Laboratory, more than 1 ms of enhanced sampling MD can be generated per day. We have ensemble docked repurposing databases to ten configurations of each of the 24 SARS-CoV-2 systems using AutoDock Vina. We also demonstrate that, using Autodock-GPU on Summit, it is possible to perform exhaustive docking of one billion compounds in under 24 hours. Finally, we discuss preliminary results and planned improvements to the pipeline, including the use of quantum mechanical (QM), machine learning, and artifical intelligence (AI) methods to cluster MD trajectories and rescore docking poses.
Graphical Abstract
Introduction
The need for rapid time-to-solution in drug discovery has become accentuated by the Covid-19 pandemic. Given the urgency of the pandemic, in parallel with vaccine development, both repurposing established antivirals and discovering new drugs are needed. Among the present candidates, apart from antibody treatments the antiviral remdesivir, a nucleoside analog that acts by interfering with RNA synthesis, it is active against SARS-CoV-21–5, shortening recovery from Covid-19 in hospitalized patients.6 Other promising candidates, such as dexamethasone, may modulate the host response7.
When surveying clinical trials for Covid-19, one is struck by the number of trials that are not based on knowledge of the drug interacting with a known target. There are several examples. As one illustration, baloxavir is a specific inhibitor of the cap-snatching endonuclease of influenza virus, which is a member of the PD-(D/E)xK two-metal nuclease superfamily8. Coronaviruses have an endonuclease but of a completely different fold (NendoU) and different active site residues9. NendoU also oligomerizes into a hexamer. Although it would seem unlikely that baloxavir would bind to NendoU, has nevertheless been in clinical trials. Similarly, lopinavir and ritonavir are also undergoing testing, even though they target proteases of the unrelated HIV4. Although in principle, enzyme active sites with similar chemical functions may bind similar ligands, the steric and physicochemical substrates of drug-protein binding are nuanced. The frequent trial of drugs specific for targets known to be absent in SARS-CoV-2 seems to us to be symptomatic of a lack of precision in combating this pandemic. For further information on other ongoing trials on small molecule drugs, biologics, passive immunization with antibodies, and vaccines, the reader is referred to the comprehensive review by Liu et al. 10. The aforementioned challenges, taken together, demonstrate that there exists an unequivocal need for de novo drug discovery campaigns as well as repurposing studies11.
There are a number of events in the SARS-CoV-2 viral replication cycle12 to target for antiviral therapeutic development; from viral entry to membrane fusion, travel to the host endoplasmic reticulum where translation of the viral genome occurs, to formation of the viral replication complex and formation from host membranes of double-membrane vesicles (DMVs) 13, 14, the passage of the replicon through the Golgi and the release of the virion from the cell. Each of these steps involves key viral proteins and occurs in a different compartment of the host cell. For example, the binding of the virus to the ACE2 receptor involves the receptor-binding domain (RBD) of the virus S (spike) protein, pre-fusion cleavage involves the binding of host TMPRSS and furin proteases to the S1/S2 dibasic domain15 formation of the replication complex and the DMVs involves the non-structural proteins (NSPs), and the N protein is required for packaging of the viral genome into the newly assembled virion16. The replication complex is made up of 15 mature NSPs, which are encoded by orf1ab and orf1a genes as the pp1ab and pp1a polyproteins17. Currently, many efforts are targeting the main protease, MPro18, which is required for cleavage of the large viral polypeptide into its functional proteins, the RNA-dependent RNA polymerase (RdRp)19 responsible for the production of new viral RNA, and some efforts target prevention of S cleavage20. In addition, viral proteins also function to impede the host’s defense mechanisms: both proteases have been shown to inhibit the human immune response by interacting with immune proteins in SARS-CoV21. It is, therefore, important to understand regions on these proteins that act as binding sites for both substrates (as in the case of the proteases) and for protein-protein interaction.
In previous work, very early in the pandemic, we combined restrained temperature replica-exchange molecular dynamics (restrained T-REMD) simulations with virtual high-throughput screening in a supercomputer-based ensemble docking campaign to identify well-characterized drugs, metabolites, or natural products that bind to either the S-protein: ACE2 receptor interface or the RBD of the S-protein22. From this ensemble docking campaign, we provided a ranking of the predicted binding affinities of over 8000 drugs, metabolites, and natural products (and their isomers) with regards to the SARS-CoV-2 S-protein and the S-protein: ACE2 receptor complex. The ranked list has been incorporated into experimental testing using a high throughput screen that was implemented in the SARS CoV outbreak, and new compounds will be added as discovered. Three of the top compounds, hypercin (a component of St. John’s Wort), imatinib, and quercetin, identified in the intial S-Protein: ACE2 receptor screen are are now in clinical trials.
Here, we report on an optimized supercomputing pipeline for early-stage drug discovery together with results on 24 systems involving 8 SARS-CoV-2 proteins. The computational approach mimics what happens in nature, using ‘structure-based’ drug discovery. Generally speaking, the availability of many experimental protein structures combined with massive increases in computational power and methodological advances have led to a resurgence of computational studies in which trial compounds are docked into binding sites in three-dimensional models of the protein targets and then ranked according to their strength of binding. Computational docking has been particularly useful in early stages of molecular discovery in order to identify initial hits to be prioritized for experimental validation.
Early docking studies were performed with static target crystal structures and rigid ligands. These were quite successful in some cases, such as in the discovery of antivirals for HIV and influenza 23, 24. Unfortunately, though, at that time, structures for few targets existed, and the process was relatively inefficient: calculations were relatively inaccurate, and computers could dock only ~100 compounds in a reasonable timeframe. Since the 1990s, the power of supercomputers has increased by a factor of a million or so. Rigid docking of over a billion compounds has been performed in a few days. Thus, virtual high-throughput screening has outperformed equivalent experimental high-throughput screening and has been shown to rapidly identify very tightly binding compounds25.
Strictly rigid docking does not often take place in protein: ligand interactions, as both ligands and proteins, undergo thermally-driven internal motions, which lead to fluctuating binding site conformations26. Therefore, a particularly important development has been the recognition that incorporating target flexibility into drug discovery protocols can improve the drug discovery process27. Ensemble docking uses different conformations of the protein targets of interest, and combinatorially performs the docking of databases of compounds against each of the protein target conformations. This process models the conformational selection binding mechanism, as opposed to a more limited induced-fit mechanism. The method requires the generation of an ensemble of protein conformations to be used in the docking calculations.
Ensemble docking of small probe molecules for flexible pharmacophore modeling was introduced in 1999. It was shown that consensus pharmacophore models, based on multiple MD structures or on multiple crystallographic structures, were more successful than models based on single conformations in yielding successful predictions of binding28. In our own labs, ensemble docking has produced experimentally validated hits against each of the 16 protein targets presented to us over the past few years. Our groups have increasingly used an ensemble approach to perform docking22, 29–46. In addition, we have shown that the clustering of protein target MD trajectories usually brings a large improvement in the quality of ensemble docking compared to what is obtained using single structure docking 47.
Ensemble generation using MD and docking both require significant computational power - to perform MD simulations of sufficient duration and to dock large databases of compounds. This combinatorially large computational time requirement essentially limits this approach to high-performance computing (HPC) architectures for large database screenings, even when only a subset of protein conformations is used in docking, for example, following clustering of the target’s MD configurations. HPC involves the use of specialized, large supercomputing systems to perform large calculations that are parallelized over many compute nodes, each consisting of dozens of cores. Traditionally, the use of a high-speed interconnect allows rapid communication between separate compute units and clever parallelization schemes to enable rapid calculations on problems too large to fit on a single compute unit. These schemes have historically involved specialized programs that focus efforts to optimize communication overlap. The use of graphics processing units (GPUs) has helped to accelerate many types of calculations. The Summit supercomputer, housed at the Oak Ridge Leadership Computing Facility (OLCF), is currently the fastest supercomputer in the United States. Summit is an IBM AC922 system consisting of 4608 large nodes, each with six NVIDIA Volta V100 GPUs per node. Each node also contains two POWER9 CPU sockets for a total of 42 cores per node. The GROMACS MD program48–50 is able to make use of all aspects of the Summit supercomputer’s HPC utilities, including the GPUs and the interconnect, providing for both strong and weak scaling, which dramatically decreases time per MD step and increases the size of the system that can be simulated efficiently. The temperature replica exchange molecular dynamics (T-REMD) routine 51–54 which was chosen here for the MD calculations (see below) uses the interconnect not only to allow for parallelization of a single simulated biomolecule, but also to communicate between separate replicas of the system, each carried out at a different temperature, and performs exchanges between replicas to accelerate the conformational sampling of the biomolecule55.
Protein-ligand docking has hitherto not been considered a traditional HPC task, as each docking calculation is short and does not require multiple nodes to complete. In fact, many docking programs can run on a single CPU core. However, the number of cores on a supercomputer or cluster can provide a resource to perform many simultaneous docking calculations that greatly decrease the time-to-solution for screening a large dataset of ligands. Cloud and distributed computing resources also provide this type of completely parallel solution for high-throughput screening56, 57. The use of GPUs has recently been made possible for the widely-used program AutoDock458, 59 resulting in the program AutoDock-GPU, which provides up to 50X speedup over AutoDock4 (available at https://github.com/ccsb-scripts/AutoDock-GPU) 60–64. Thus, the use of leadership HPC facilities for ensemble docking can provide the ability to screen billions of ligands to a full set of conformations generated with HPC-based MD simulations. Quantum mechanical refinement of classical docking ranking based on fragment molecular orbital (FMO) techniques also naturally benefits greatly from massively parallel supercomputer capabilities65.
In this work, we describe our efforts establishing a supercomputing-based pipeline for ensemble docking and preliminary results on its application to discovering therapeutics that target viral proteins of SARS-CoV-2. The pipeline and results presented here represent our contribution to date to the work of the USA HPC Covid-19 Consortium that was created on March 29th, 2020. We describe the choice of 8 targets and the preparation of protein models from experimental data. We report on T-REMD simulations performed for the targets totaling about half a millisecond of simulation time. We have docked repurposing databases to ten configurations of each protein simulated using the popular docking program Autodock Vina. We also describe efforts deploying Autodock-GPU60–62 at scale on Summit that demonstrate the docking of over a billion compounds in 24 hours with full structural optimization of the ligand. Future developments involving the use of AI and quantum chemistry in rescoring and clustering are also outlined. The pipeline described here can also be used in future work to target human proteins66, 67 known to interact with viral proteins, or in disease-causing responses in Covid-19 and more generally in computational structure-based drug discovery.
Methods
Computational methods in drug discovery narrow a vast chemical search space to a tractable set of compounds suitable for experimental testing. Experimental work can involve a variety of tests, including live virus testing as well as target engagement studies, and will not be considered further here. Rather, we discuss the procedures of structural modeling, MD simulation, and docking.
a. Choice of target proteins and generation of structural models from experimental work
Multiple groups have been using structural biology techniques, including X-ray crystallography, small-angle scattering (SAS), and cryogenic electron microscopy (cryoEM), to investigate the structure of proteins and protein complexes from SARS-CoV-2 68–71. However, obtaining a structure from the Protein Data Bank (PDB) or perhaps a revised model from another resource is only the starting point. Often structures obtained from the PDB do not fully resolve all residues, and a determination must be made whether and how to model them. Also, as structural models are rapidly being released to aid in the fight against COVID-19, the potential inclusion of a few structural errors is an unfortunate reality. In particular, the identification of metal cations in protein structures requires careful thought and examination of its local coordination environment.
Even with perfectly assigned and complete experimental structures, it may not be enough to consider viral protein targets as chemically invariant structures for modeling and binding calculations. Large differences in pH in various cell compartments as the virus travels through the host cell 72–74 can qualitatively change the protein’s structure and function. Differences in pH also affect the protonation states of the proteins and the small molecules being tested as drugs, altering drug binding preferences. Finally, the oligomerization states of the target proteins are important to consider as the interactions between protein monomers may influence the shapes of the active sites. Another important factor to consider when performing in silico screens using ensemble docking is the ability to construct a useful model of a particular protein for MD simulation. For instance, certain metal-containing regions of a protein may not have an existing classical mechanics model (force field parameters), or existing models may be inadequate. In addition, highly charged, disordered, and ion-dependent biomolecules have been known to have less accurate force fields and may perform poorly in an MD simulation 75–78.
Proteins chosen for ensemble docking in this study were those that had a crystal structure available with a reasonable resolution, were amenable to accurate simulation with classical MD force fields, and were also known to be important for viral pathogenicity based on either recent studies or those on SARS-CoV. The 24 systems studied comprise nine protein domains. Two of these, RBD of the S (spike) protein and the N-terminal region of the N (nucleocapsid) protein, are domains in structural proteins found attached/within the virion. The N protein is used for packaging the viral genome and is essential for the assembly of the virion79. The remaining seven domains come from non-structural proteins (NSPs) 3, 5, 9, 10, 15, and 16, which form the replication complex and are involved in a number of key tasks leading to the creation of new virus particles80. Two domains come from NSP3 81, the ADP ribose phosphatase (ADRP, also known as macro- or “X”) domain, and the papain-like protease domain (PLPro)82. The ADRP seems to be involved in ADP-ribosylation, which is used in cell signaling and thus may act to inhibit the host immune response83. PLPro cleaves regions of the polyprotein to release non-structural proteins and also is involved in the mechanisms the virus uses to counteract the host immune response, for instance by interaction with host immune proteins 80, 84 Nsp 5 is the main protease (MPro), which self-cleaves and also cleaves other regions along the polyprotein, releasing essential proteins to perform their tasks in the assembly of the replication complex, and is also involved in interacting with and preventing the action of host immune proteins 84, 85. The exact function of NSP 9 is unclear, but has been found in SARS-CoV to be required for viral replication and has been shown to bind to RNA oligonucleotides86, Nsp 15 is an endoribonuclease specific for uridine whose exact function is also unknown, but has been implicated in interfering with host immune response both through direct interaction and by cleaving viral RNA to prevent detection by the host87. NSP 16 is thought to be a methyltransferase that requires NSP 10 as a co-factor, and acts to disguise viral mRNA from the host immune response by adding a methylation onto the RNA cap which host cells use to mark RNA as belonging to “self” versus “pathogen.”88–90.
The explosion in research and literature fueled by the Covid-19 pandemic, together with the need for searching through related literature on other coronaviruses, has created a challenge for researchers needing to understand the structural details and cellular contexts of the SARS-CoV-2 proteins. To help navigate this challenging landscape, we have been developing new tools based on natural language processing for enabling a more robust search for specific questions required for our modeling, simulation, and ligand docking work91–93 featuring targeted filtering and exploiting external resources (e.g., Wikidata, ChEBI, PubChem) to expand our search capability. For example, after we generate a set of related keywords, the service will screen for the terms referring to a chemical substance and fetch the chemical information (e.g., SMILES string) from the PubChem automatically. In addition, using this keyword search enables the ontologies (e.g., Wikidata, ChEBI) to be used to link related chemicals and their properties for document annotations in query results. The main data resource of the system is a collection of scientific papers, which are collated from major publications. The full-text article access and download from the publishers’ archives are performed under the publishers’ agreements, and the internal article corpus in our system is updated on a weekly basis.
To provide a diverse survey of the conformational ensembles of the SARS CoV-2 viral proteome, we performed T-REMD simulations of 24 different model systems listed in Table 1. An additional supplementary table (Table S1) is also provided, which summarizes the PDB entry simulated, complete protonation state choices (where applicable), and the number of replicas used for the T-REMD.
Table 1.
Model Systems Simulated
| Protein/ System Notes | ||
|---|---|---|
| S (Spike) Protein Receptor Binding Domain (RBD) / “Apo” (PDB: 6W41) | S Protein RBD / Complexed with ACE2 (PDB: 6W41) | MPro / monomer, CHARMM-GUI default protonation (PDB: 6Y2E) |
| MPro / dimer, CHARMM-GUI default protonation (PDB: 6Y2E) | MPro / dimer, ‘charged’ protonation variant (PDB: 6WQF) | MPro monomer/ HIE41 protonation variant (PDB: 6WQF) |
| MPro dimer / HIE protonation variant (PDB: 6WQF) | MPro monomer / HID41 protonation variant (PDB: 6WQF) | MPro dimer / HID41 protonation variant (PDB: 6WQF) |
| NSP15 (endoribonuclease) / hexamer (PDB: 6VWW) | NSP15 (Endoribonuclease) / monomer (PDB: 6VWW) | NSP10:NSP16 Complex (Methyltransferase) (PDB: 6W4H) |
| NSP10 / monomer (PDB: 6W4H) | NSP16 / monomer (PDB: 6W4H) | N (nucleocapsid) N-terminus phosphoprotein / monomer (PDB: 6M3M) |
| N (nucleocapsid) N-terminus phosphoprotein / tetramer (PDB: 6M3M) | N (nucleocapsid) N-terminus phosphoprotein / tetramer complexed with Zn (PDB: 6YVO) | N (nucleocapsid) N-terminus phosphoprotein / monomer alternate crystal structure (PDB: 6YVO) |
| NSP9 / monomer (PDB: 6W4B) | NSP9 / dimer (PDB: 6W4B) | NSP3 ADP ribose phosphatase / asymmetric unit (PDB: 6W02) |
| PLPro / monomer ‘charged’ protonation variant (PDB: 6W9C) | PLPro / monomer ‘neutral’ variant (PDB: 6WRH) | NSP3 ADP ribose phosphatase (PDB: 6W02) |
b. Simulation Model Preparation
To engage in the use of MD for the rapid generation of conformational ensembles for drug discovery one requires that the input for MD be generated in a semiautomated fashion by which the atomic coordinates, obtained from experimental or in silico protein structure prediction methods, can be quickly processed into MD input files. To facilitate this semiautomated approach, CHARMM-GUI was used for most model building94. The general system building method used here involves the direct retrieval of structures from the PDB and processing to model missing residues, assign protonation states, add disulfide bonds (where noted in the PDB annotation), add glycosylation (where resolved in the crystal structures of the S-protein receptor binding domain and ACE2), neutralize the charge of the system (using Na+ and Cl- ions), and solvate (with TIP3P water). Many proteins have coordinated ions that serve structural roles, such as the Zn2+ cations in Nsp10, or catalytic roles. Thus, the treatment of Zn-complexes in fixed-charged classical MD force-fields is a challenge, and for some systems, it may result in the failure to maintain Zn-protein coordination95 96 97, and when found necessary (as noted below and also summarized in Table S1) an explicit bond representation was used. All of these considerations mandate an abundance of care when preparing a biologically accurate model for simulation. Below we discuss considerations taken into account when modeling some of the proteins simulated.
S (Spike) Protein (PDB: 6W41)
Presently available structures of the S protein have nine gaps totaling approximately 150 residues, in addition to over 20 and over 100 missing residues at the N- and C-termini, respectively. Current models also lack post-translational modifications, including glycosylation and formation of disulfide bonds. The S protein is heavily glycosylated, with roughly 20% of its mass in glycan chains, yet at most, a few mono or disaccharides are present in the structure.
In our preliminary study22, we made use of a homology model of the entire spike with restraints applied such that only the human ACE2-Spike interface was unconstrained. Here, using crystal structures of the ACE2-S protein complex, simulations of the receptor-binding domain of the S Protein (Spike) both in complex with the human ACE2 receptor and on its own (referred to within the text as the “Apo” RBD) were performed. The viral spike receptor-binding domain was chosen to provide insight into the details of the initial viral-host recognition process. Glycosylation resolved from crystallographic imaging was used, and an annotated disulfide bond was also included.
Main protease. (PDB: 6Y2E & 6WQF)
The main protease, MPro, is an attractive target for the development of antiviral drugs. There is compelling evidence that the enzymatically active species is the dimeric assembly of MPro. A dimer is observed in most crystal structures of CoV MPro, as well as in solution at sufficiently high concentrations. In addition, a linear increase in the enzyme activity at increasing concentration suggests catalytic incompetence of the monomer98. Therefore, the full dimer was considered in the present MD simulations for MPro, using as starting coordinates the apo-homodimer in the crystal structures 6Y2E and 6WQF99, 100.
The crystal structures show that SARS-CoV-2 MPro, similarly to other MPro’s85, 99, 101, 102, is composed of three domains: Domains I (residues 8–101) and II (residues 102–184) are arranged in an antiparallel β-barrel structure, and domain III (residues 201–303) contains five α-helices arranged in a globular cluster. Domain III is a specific feature of CoV MPro proteins and was suggested to be essential in the proteolytic activity by keeping domain II and the long loop connecting domains II and III (residues 185–200) in the proper orientation, and/or by orienting the N-terminal residues that are essential for the dimerization101. Dimerization occurs through interactions between the helical domains of the two monomers and through hydrogen bonding interactions between the N-terminal residues of one monomer and key residues in the other monomer. In particular, the salt bridge between the N-terminal Ser1 of one monomer and Glu166 of the other monomer has been suggested to be essential to maintaining the catalytically competent conformation101, 103. The substrate-binding site is located in a cleft between domains I and II and contains a highly conserved catalytic dyad formed by Cys145 and His41. Comparison among the two apo crystal structures and the crystal structure obtained in the presence of an inhibitor reveals85 only minor structural differences in the position of a few side-chains and no relevant changes in the substrate-binding site, except for the rotation of the side chain of Met165, which is in the proximity of His41.
Although the catalytic mechanism is not fully understood, there is a general agreement in considering that the proteolytic activity of CoV MPros is initiated by activation of the enzyme through a proton transfer reaction in the catalytic dyad, leading to a charge-separated state with a highly reactive thiolate. It has also been suggested that such a proton transfer reaction is induced by the presence of the native substrate104, 105. Therefore, in the present MD simulations of the apoenzyme, Cys145 and His41 were simulated in their neutral state, with His41 protonated at Nδ (i.e., HSD). This choice is based on the observation that the His41-Nε appears to be the best candidate as proton acceptor from Cys145 because in the crystal structures the His41-Nε is closer than the His41-Nδ to the Cys145-S and the His41-Nδ is already involved in a hydrogen bond to a highly conserved water molecule, which is considered the third element of the catalytic site. A recent QM/MM study106 also supports this proton transfer mode and the role of water in catalysis. However, the ε-nitrogen protonation state for His41 (HIE) cannot be decisively ruled out, and MD simulations were performed also considering this alternative, although less probable, protonation state.
The protonation states of two additional His residues, namely His163 and His172, were also highlighted as being crucial for the enzymatic activity of CoV MPro. In particular, the doubly protonated (cationic) state of His163 at pH 6.0 was suggested to modulate relevant conformational variations involving Glu166, Phe140, and His172, leading to a catalytically inactive conformation101. At higher pH values, both His163 and His172 should be uncharged, and, on the basis of the hydrogen-bonding pattern that can be inferred from the crystal structures, the HSE protonation state was used for both His163 and His172 in the present MD simulations. All other His residues were also simulated in their neutral state, assigning the Nδ or Nε protonation state on the basis of their chemical environment and hydrogen-bonding patterns. The selected protonation states are as follows: HSD64, HSD80, HSE164, HSE246.
PLPro (PDB: 6W9C and 6WRH).
For PLPro, the Zn cation was coordinated to C189, C192, C224, and C226. Similar to MPro, the protonation states of the His residues in PLPro were not readily available. Here we pursued two potential protonation state varients, a charged variant and a neutral variant. For the charged variant the protonation states were obtained with the use of the PropKa 3.0 server assuming pH ~5, corresponding to its presumed cellular (lysosome) environment 107, with 6W9C being assigned to pH 5 based on its physiological role in acidic environments. Protonation states for the neutral state were manually assigned using 6WRH as the original coordinate file, with C111S mutation reversed. For both variants, Zn-coordination during temperature-replica exchange was enforced by topological patches applied with CHARMM-compatible tools. TopoGromacs108 was used to convert the system and associated force field to GROMACS format.
NSP15 (PDB: 6VWW).
Large (His) tags present during the recombinant expression processes to purify NSP15 for crystallization; however, these tags were not removed before crystallization. Prior to simulating the monomeric and hexameric forms, the artifactual His tags were removed from NSP15 using MOE2019 and subjected to a “quick prep” with the prepare protein module of MOE to resolve potential issues in the resulting structure. The resulting truncated PDBs were then uploaded to CHARMM-GUI for neutralization and solvation.
NSP10 (PDB: 6W4H).
For NSP10, both in its monomeric and a complexed form with one Zn cation liganded to C4370, C4373, C4381, and C4383, while the other bound Zn was liganded to C4327, C4330, H4336, and C4343. For 6VYO, H59 and H145 were liganded.
c. Molecular Mechanics and Molecular Dynamics System Preparation (Force Fields, Counter-Ions, Energy Minimization, and Equilibration)
All simulations were performed using the GROMACS48, 109 software suite, and the CHARMM36m force field110, which is the default of choice using the CHARMM-GUI. For all systems, the protein was solvated in water-boxes with edge-distances of 1nm, and only neutralizing Na+ and Cl− ions were used. Short-range interactions were treated with a smooth force-switch cutoff of 1.2 nm, and long-range electrostatics were treated using the particle-mesh Ewald (PME) formalism, as implemented in GROMACS111. To facilitate the use of a 2-fs MD timestep, all covalent bonds to hydrogen were restrained with the LINCS algorithm112 in all simulations. Following system preparation, all solvated models generated were subject to steepest-descent energy minimization with a stopping condition of either reaching the force-convergence criteria of 1000 kJ mol−1 nm−1 or a maximum of 5000 iterations. Energy minimization was performed primarily to remove potential clashes between the solvent, ions, and the protein (or protein complex) of interest. Post-clash removal minimization, short (250 ps) NPT relaxation simulations (with default positions restrains generated from CHARMM-GUI) were performed to relax the simulation box dimensions for each replica (at different temperatures) independently (see T-REMD Protocol). For these relaxation simulations, the Berendsen baro/thermostats113 (as implemented in GROMACS) and an integration time step of 1 fs were used.
d. T-REMD Protocol
MD simulations provide a means to study the conformational dynamics of proteins. However, frequently MD becomes ‘trapped,’ resulting in the need for many long simulations to effectively sample a protein’s conformational landscape114. To overcome this sampling challenge, enhanced sampling techniques can be used. For the present work, temperature replica-exchange molecular dynamics (T-REMD) was employed, whereby multiple copies of a target system are simulated simultaneously with each copy (replica) at a different temperature, with periodic coordinate swapping (performed in such a manner as that preserves detailed balance) between the copies52–54. By running at multiple temperatures, with exchanges, the dynamics of the system avoids ‘kinetic traps’ and provides a robust sampling of the protein free-energy landscape, and thus the protein conformational diversity115. T-REMD was chosen for several reasons:
it guarantees an increase in sampling efficiency over straightforward MD116,
it does not require the assignment of reaction coordinates (or collective-variables) a priori to accelerate conformational sampling
it does not require direct modification to the system Hamiltonian117.
T-REMD simulations for each system were performed with the GROMACS simulation suite. A limited temperature range of 310 K to ~350 K was chosen to maintain physiological configuration space. For each protein system, the number of replicas and temperatures for each replica was chosen using the temperature predictor server by Patriksson and van der Spoel118 with a target exchange probability set at 0.2 though the actual exchange was found to be ~0.3 for all systems. All simulations were performed for a total of 750 ns per replicate.
After relaxation, production T-REMD simulations were performed with a frame saving rate of 10 ps and an integration time step of 2 fs. Production simulations were performed, similar to the relaxation simulations, in the NPT ensemble. Unlike the relaxation simulations, the V-rescale (Bussi) thermostat119 and the Parrinello-Rahman barostat120, 121, were used. Regardless of the temperature window, the target pressure for each replicate was set to 1 bar.
e. Trajectory Analysis
For all systems, the measures of the gyration tensor (from which shape anisotropies are derived), solvent-accessible surface area (SASA), and pairwise simulation frame versus simulation frame RMSD matrices, and RMSD based clusters, were obtained using a combination in-house VMD122 scripts, NumPy, and SciPy123. The RMSD clustering specifically only considered the lowest temperature replica (310 K), and rapidly generated the pairwise RMSD matrices using the QCP algorithm124. Clustering was performed using hierarchical clustering with a complete linkage method, as implemented within SciPy. For generality in evaluating structural diversity, clustering was initially performed based on the RMSD of all heavy protein atoms, and where additional diversity of active sites was of interest for subsequent docking, a second round of clustering was performed based on binding site residues and protein-protein interfaces. VMD atom selections for the docking specific clustering are summarized in Table S2.
Although T-REMD is an efficient simulation method, and the 310K data do correspond to a formal statistical mechanical ensemble generated at this temperature, as with other enhanced sampling methods the risk is always present that the enhancement of the sampling takes the system to regions of configurational space beyond that that would be significantly sampled by the protein physiologically; for example, to partially or wholly unfolded states. We, therefore, take care to identify these and to not perform docking screens on such configurations.
f. Docking
Two different docking databases were used.
A smaller database of potential ligands was built merging together the content of the SWEETLEADS125, 126 repurposing database SuperDRUG2127, 128, and the NCI-diversity database 129, yielding 13,757 unique compounds. This database has been ensemble docked to all systems, as listed in Table 2, with noted targeted binding sites. This database was docked using local HPC clusters using Autodock Vina.
Supercomputing docking runs were performed involving billion-plus compound screens of the Enamine database using an accelerated version of Autodock: Autodock-GPU. To date, these runs have been performed on two crystal structures of MPro.
Table 2.
List of proteins and binding sites used for ‘smaller database’ docking. PPI refers to a protein-protein interface. In some cases, FTMap was used to identify potential binding sites (see Table S2)
| Receptor / Binding Site | Receptor / Binding Site |
|---|---|
| MPro monomer / catalytic pocket | NSP15 monomer / catalytic pocket |
| MPro dimer / PPI | NSP15 dimer / PPI |
| NSP9 dimer / FTMap sites | NSP10 monomer / PPI to NSP16 |
| Nucleocapsid phosphoprotein / RNA binding site | NSP16 monomer / PPI to NSP10 |
| Nucleocapsid phosphoprotein / PPI | NSP10:NSP16 / PPI |
| Nucleocapsid tetramer / FTMap sites | NSP3 ADRP domain (asymetric unit, dimer) / active site |
| NSP3 ADRP domain (monomer) active site | NSP9 monomer / PPI |
f.1. Smaller database docking
Data and Protocols
Docking to the target structures obtained from the MD simulations (as listed in Table 1) was performed on various HPC clusters using Vina MPI 130 and MOE. Two sets of structures were used in the ensemble docking. In the first series of docking calculations, only the first 100ns of the T-REMD trajectories were used, and the results of the docking simulations were passed on to collaborators for experimental testing. In the second series of docking, as the MD trajectories were expanded beyond their initial first 100ns, the clustering was performed on the entire 750 ns trajectories, as described in the results section below.
For the VinaMPI130 calculations, the “Exhaustiveness” parameter was kept at its default value of 10. Databases of potential ligands were built merging together the content of the SWEETLEADS125, 126, SuperDRUG2127, 128, and NCI-diversity databases129, yielding 13,757 unique compounds.
Using the program MOE, compounds with more than 49 rotatable bonds were deleted from the database, and only one stereoisomer was included for each compound. Very low molecular weight (<58) compounds (single atoms, ions, very small functional groups). The resulting database included ~9K unique molecules. The compounds were protonated at pH 7 and energy-minimized using the MOE software to obtain low energy 3D structures. The compounds were saved on disk in sdf format and then converted to PDBQT format using AutoDock Tools131, 132.
The ligands were docked to 10 clusters per receptor, each cluster corresponding to a different configuration of the binding pocket. The clusters corresponding to the first 100ns of the MD simulations have been uploaded on the publicly available structure repository https://cmbcovid19.flywheelsites.com/data/additional data, including the complete trajectories from the 750ns T-REMD simulations is forthcoming. The residues used to determine the clusters fall into one of three categories: the protein active site, residues at the protein-protein interfaces (for complexes), and all the protein non-hydrogen atoms. Tables 2 and list the receptors and binding sites we have screened so far.
Binding Sites for Docking
In general, we have two classes of potential binding sites: 1) catalytic pocket or substrate-binding site and 2) PPI. The first aims at identifying potential competitive inhibitors of the viral proteins, and the second aims at finding compounds potentially disrupting a viral protein-protein complex. Binding site definition requires manual intervention and cannot be easily automated. Examples of definitions are listed below for three viral proteins.
In the main protease dimer (PDB: 6WQF), the docking box contains catalytic sites of chain A and PPI residues. The docking box was constructed to align with the peptide-binding groove on either side of the catalytic dyad of chain A, which extends outward to include the S3, S2, S1, S1’, and S2’ catalytic pockets.
In the NSP10-NSP16 complex (PDB: 6W4H), the S-adenosyl methionine (SAM) binding site Asp6928 in NSP16 was considered89. In addition, PPI residues such as Tyr4349, Val4295 to Leu4298 in NSP10, and Gln6885 in NSP16 were included. Tyr4349 and Gln6885 interact with each other in SARS-CoV virus89, and Val4295 to Leu4298 are hot spot residues in the SARS-CoV-2 virus computationally predicted using the crystal structure along with the KFC2 method133, which is based on a machine learning predictive model (https://kfc.mitchell-lab.org). Hot spot residues are the fraction of PPI residues that account significantly for the overall protein-protein binding affinity, and they are typically determined experimentally using alanine scanning mutagenesis134.
In the N-terminal domain of nucleocapsid protein tetramer (PDB: 6VYO), three critical RNA-binding residues on the beta-sheet core were included in docking: Arg88, Arg92, and Arg10771, 135 136.
f.2. Billion-compound supercomputer docking with Enamine Real database
A major aim of this exercise was to see whether it would be possible to dock a billion compounds with full ligand optimization on the OLCF Summit supercomputer in 24 hours of wall-clock time. To perform efficient ensemble docking, we modified AutoDock-GPU60, 62, to enable it to run at peak efficiency on the Summit system. For compatibility, OpenCL kernels were re-written in CUDA, and file input and output were streamlined to enable it to keep up with the GPU’s speed. These modifications, together with the size of the Summit supercomputer, indeed allow over 1 billion compounds to be docked within 24 hours. This capability will enable giga-compound docking for a number of proteins in the viral proteome and beyond.
We performed initial docking tests using this framework on NSP15 (NendoU) and the main protease (MPro). For NSP15 we used a 9,000 compound dataset composed of the SWEETLEADS125 database with additional ligands, and also a trimmed version of this dataset containing only ligands containing less than 11 rotatable bonds, consisting of about 5,000 ligands. For tests with MPro, we used a 90,000 ligand subset of the Enamine REAL database 137. All ligands were prepared with AutoDockTools 132, 138, and the receptor grids were generated with the program autogrid with a grid spacing of 0.375 Å. We tested a set of search box sizes: 40, 25, 20 and 15 Å3, and different settings for the number of runs, nruns, which defines how many separate instances of the genetic algorithm are executed. For the trimmed dataset, we also performed docking with AutoDock Vina with exhaustiveness of 10 to compare results. These results provided us with the confidence to dock over 1 billion compounds from the Enamine real database to two different MPro crystal structures, 5R84 and 6WQF100, with a search space 25 Å large on each side, centered on the active site. The analysis of this dataset is ongoing. Due to the documented inaccuracies of force field-based scoring functions in the task of screening and affinity prediction of compounds,139 rescoring of at least 1 percent of the billion compounds is being performed using the accurate, yet highly computationally efficient machine learning-based rescoring method known as RF-Score-3140. Also, at least 50% of those compounds re-scored with RF-Score-3 will be further filtered using recently developed rescoring described below in Future Directions and Preliminary Results from New Methodologies, subsection Protein-ligand rescoring using machine learning.
Sequence analysis and mutational entropy calculations
We performed an analysis of available sequences of the SAR-CoV-2 virus to look for numbers of mutations and map these locations on the proteins we were using as drug discovery targets. All complete, high-coverage genomes labeled as human host SARS-CoV-2 were downloaded from GISIAID141, 142 on May 5, 2020, yielding a total of 16,252 genomes. Sequences were filtered to remove any genomes with greater than 3% ambiguous (N) nucleotides or were less than 29,000 nucleotides in length, resulting in 14,284 genomes. Multiple sequence alignment of the 14,284 genomes was performed using MAFFT143 v.7.464 with the --addfragments method using NC_045512.2 (EPI_ISL_402125) as the reference genome and removing insertions relative to the reference. Mature protein-coding sequences for each protein were extracted from the alignment using coordinates from the reference genome and translated using FAST144 v1.6, with protein sequences containing internal stop codons discarded from further analyses. Shannon entropy145 was calculated for every column of each protein alignment using a custom script, disregarding ambiguous and gap characters using a custom script. Additionally, the frequency and types of substitutions with respect to the reference were recorded. For visualization of the mutation entropy per residue of the proteins studied in this paper, entropy values were color-coded in protein PDB structures. Known SARS-CoV and SARS-CoV-2 structures were downloaded from the Protein Data Bank, their sequences were aligned with the SARS-CoV-2 reference genome (NC_045512.2) using BLASTP, and the calculated entropy of the sequences was embedded in the PDB file in the place of the B factor column using a custom Python script.
Preliminary QM Refinement Protocol
Along with ML-based approaches, quantum mechanics (QM)-based refinement of classical docking results is being developed here as a tool to narrow down the list of promising inhibitor candidates146. Until recently, the inclusion of QM electronic structure in high-throughput drug screening was deemed computationally intractable due to the enormous computational resources required even for density functional theory (DFT) calculations. The poor scaling of most quantum chemical methods further exacerbates the situation. A viable emergent alternative is the recently developed linear-scaling version of an approximate, yet remarkably accurate DFT method called “fragment molecular orbital density-functional tight-binding (FMO-DFTB) 147. This method is implemented in the widely-utilized GAMESS quantum chemistry code148. We here report preliminary calculations of FMO-DFTB with the so-called polarizable continuum model (PCM) of the solvent 149 for quantum mechanics-based evaluation of potential COVID-19 spike protein inhibitor drugs identified by re-clustering and re-docking to an extended simulation of the S protein, similar to the initial work by Smith & Smith 22. For the PCM calculations, the cavity was calculated using simplified united atomic (SUAHF) radii150which are available for all the chemical elements contained in all ligand compounds. Because the binding side is widely open, the dielectric constant of water ε=78.39 was used. In addition, to improve the accuracy in describing non-covalent interactions, the D3 dispersion correction was employed. To obtain the refined binding energy of a given candidate, its unbound geometry, the unbound protein, and its corresponding complex were optimized using FMODFTB/PCM. While the unbound ligands were completely optimized, only selective residues in the binding pocket of the unbound protein, and in the protein-ligand complexes were locally optimized. The QM-refined binding energy is defined here as the difference between FMO-DFTB/PCM total energy of the complex and the sum of the total energy of unbound protein plus total energy of unbound ligand. In preliminary work, the QM-refinement was carried out for the Vina top-10 best binders of each spike protein cluster. In total, 15 spike protein clusters were investigated, and the binding energies of 150 protein-ligands complexes were refined.
Results
We present here preliminary results obtained for members of the SARS-CoV-2 proteome. Naturally, ongoing refinements of the results are continually being undertaken, and the results are incomplete. However, they give a snapshot report on the state of delivery of the pipeline. At the moment of submission, 24 T-REMD simulations have been performed on nine members of the proteome, in various oligomerization and protonation states, for a total of 0.612 ms of MD aggregated over all replicas and ~17.25μs aggregated overall lowest temperature windows. At present ~2.07M physical docking calculations have been performed with the smaller database and on Summit 2.4 billion docking calculations with the Enamine REAL database. The preliminary results presented are general trends observed in the MD and docking runs and do not describe details of the candidate compounds or dynamical properties of individual proteins, which will be reserved for future publications. Results of MD and docking are available at the website https://coronavirus-hpc.ornl.govand will be updated as new simulations and docking results become available.
a. T-REMD Scaling Performance
Figure 1 shows the performance per replica on Summit of T-REMD simulations for the majority of the simulations performed in this work using GROMACS version 2020.1. A few simulations were run with GROMACS 2018 and/or with different scheduling parameters and achieved only 20–50% of the performance shown above and are not included in the figure. We found that performance was maximized when running all bonded and nonbonded calculations on the GPUs (interatomic and both particle-mesh Ewald and pairwise Lennard-Jones). With the noted choices, performance saturates at around 100 ns/day for 250,000 atoms and above, even if more nodes are allocated per replica, for two reasons. First, the GPU-based fast Fourier transform is limited to a single GPU, and communication latencies between nodes slow down the calculation. However, throughput around 100 ns/day can still be achieved for simulations above 250,000 atoms if nodes are increased proportionately to system size.
Figure 1.

Simulation throughput per replica. Each point represents the performance achieved by replica-exchange MD simulations on a single protein/water system. Run parameters were one replica per node (each node has 6 GPUs), using between 24 and 40 replicas in a given system.
b. T-REMD: Conformational Sampling of SARS-CoV-2 Proteins.
T-REMD simulations were performed with the number of replicas ranging from 20 to 60 for 750 ns each for an aggregate sampling of over 0.6 ms (Table 1 & Table S1). Given the scaling data noted above, for the total 816 replicas simulated, the calculations (if performed simultaneously) used the equivalent of ~18% of the entire Summit supercomputer for ~3 days. The performance, if the entire machine were used to simulate all of the different protein systems at the same time, would thus scales up to ~1 ms/day. For all systems, the replicate temperatures range from 310 K to ~350 K, and the average exchange probabilities were near 0.3.
From the simulations, structural diversity was quantified by calculating, when a binding site is known, the gyration tensor of the binding site residues, the solvent-accessible surface area (SASA) of the binding sites, and the construction of pairwise snapshot-snapshot root-mean-squared deviation (RMSD) matrices for the target temperature replica, i.e., the replica with the temperature set to 310 K (see Methods for calculation details). Additionally, using the gyration tensor, the shape anisotropy of the pockets was also obtained.
Linkage-based RMSD clustering, using the pairwise RMSD matrices was performed to gauge the overall structural diversity of the proteins. Figure 2 provides example conformations and calculated quantities for one example target, the neutral variant of the PL-Protease (PLPro). Similar plots for the other simulated systems are provided as supplemental material and on https://coronavirus-hpc.ornl.gov.
Figure 2.

Configurational variability of PLPro (PDB: 6WRH) with neutral HIS protonation states. (A) Overlay of 26 RMSD aligned structures from the lowest temperature replicate spanning the 750 ns of sampling. (B) Population distribution for shape anisotropy (κ) and solvent accessible surface area (SASA), with redder colors indicating greater occupancy of these kappa-SASA combinations. The distributions are also reflected by one-dimensional histograms above and to the right of the plot, and black dots within the population distribution, which represent position information for 10% of the total snapshots considered. (C) Pairwise RMSD clustering for the lowest temperature replica, with the snapshots ordered according to their cluster. The clusters in this instance were defined using a cutoff of half the maximum RMSD observed within the simulation and are labeled according to color with a color-bar for reference located above the plot. (D) Pairwise RMSD distribution across all snapshots. (E) Population statistics for the clusters introduced in (C).
From Figure 2A (and subfigure A of the Figures S1 through S23), it is clear that the simulations generate a diverse ensemble of states with varied loop structures. For the case of the neutral variant PL-Protease, Figs. 3C and 3E indicate the existence of a number of dominant conformational states. Figs. 3D and 3C further suggest that, although six dominant states exist, these states could be grouped into two ‘super-states,’ which may indicate a switching like behavior or the potential existence of a ‘hinge.’ Finally, subfigure B shows a significant amount of sampling of rod-like geometries (anisotropies near 1); however, there are states that have a correlated reduction in SASA and shape anisotropy, which would correspond with a nearly continuous transition between rod-like structures and spheroid-like structures.
Figure 3.

Configurational variability of the PLPro (PDB: 6WRH) active site region generally bounded by the black dashed lines and the next step in analysis after Figure 3. Each of the differently colored aligned protein models represents the center of a populous cluster, as defined by active site conformation RMSD. Residues such as R164, E165, Y266, Q267, and F302 vary in conformation substantially and highlight the conformational variation within the ensemble created through T-REMD. For clearer visualization, only residues 91 and onward for PLPro are shown, as this selection was used for active site alignment. Within the VMD122 rendering, sidechains are displayed without their hydrogens.
The general conformation variation highlighted by Figs. 2 and S1–22, to some degree, masks the conformational variation within binding sites; however, when for docking to the individual binding sites, clusters within the T-REMD trajectory are identified and demonstrate significant variability within the active site region (Fig. 4). While not specifically active site residues, residue variability at the tip of the loop centered on Y266 and the charged residue pair R164-E165 near the active site imply that accounting for the protein conformational ensemble is essential. Otherwise, the docking calculations would be strongly biased by the rotameric states present in the single static structure used in typical single-structure docking calculations.
Figure 4.

Distribution of the number of identical compounds being found in n-number of target top 500-compounds selection out of 9,014 compounds.
c. Smaller Database Docking
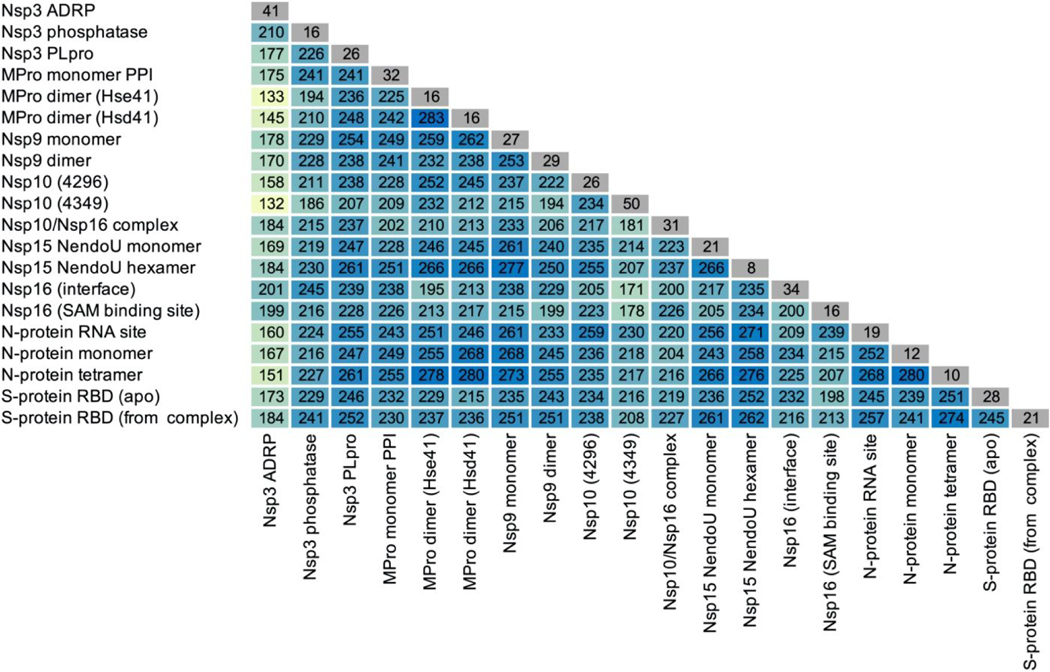
A preliminary analysis was performed of general trends seen in docking the smaller database to the 24 SARA-CoV-2 protein systems. For each protein target, all the docking results from each of the 10 cluster configurations were combined, and the top 500 scoring compounds extracted. The selectivity of the compounds for any given target varies considerably (Table 3) with the number of compounds present on any two different top 500 lists as low as 132 or as high as 283. In comparison, from two random selections of 500 items out of 9,014 items (see Figure S24, 5% percentile = 19 compounds, 95% percentile = 36 compounds), 27 identical compounds would be expected on average. Thus, the high number of identical top-scoring compounds observed between any two targets indicates a non-random selection of these duplicate compounds.
Table 3.
The number of duplicate compounds found in the top 500 lists in specific pairs of proteins. In grey/diagonal: the number of compounds unique to the corresponding target/site in the respective top 500 lists.
|
For any particular target, the number of non-duplicate compounds is relatively low, ranging from 8 to 50 (Table 3). The majority the compounds selected bind to a single target (Figure 4). However, of all the compounds that are found in the top 500 lists, over half are calculated to bind to 3 or more targets. Molecular weight was found to be only weakly related to the number of protein targets a compound is calculated to bind to (See Figure S25 & S26). Therefore, the overlap in the top-scoring compounds is not an artifact of the size of the ligand. In the absence of a systematic experimental assay on each of these compounds, it is difficult to assess the significance of the overlap in the top 500 compounds. It is important to note that the “top compounds” are assembled based on relative docking scores between compounds against the same target, and not based on their absolute docking scores, which could artifactually inflate the number of duplicates. A high number of duplicates between the lists obtained on two different proteins could also indicate a computational bias of some compounds based on other criteria than their good fit to the targets.
On the other hand, such high numbers could correctly identify promiscuous binding sites that do not display marked structural specificity and hence could be indeed targeted by similar compounds. It is outside of the scope of the present work to assertively differentiate between these two possibilities. However, the number of duplicates varies greatly across several pairs of targets, which renders unlikely a systematic bias in the docking (because of, say, molecular weight or other ligand properties independent of the target’s binding site).
Only ~55% of the top 500 compounds were the same in the docking results from the 100 ns and 750 ns clusters. Thus, extending the T-REMD simulation time by a factor of 7.5 nearly doubled the chemical diversity. Future analysis will be needed to indicate if the compounds that are identical in both sets of docking calculations are promiscuous compounds that would bind to many protein structures or if many of the clusters from the MD trajectories end up being selected by the same compounds.
d. Comparison of Docking with Experimental Screening Results
The chemical databases used in the ensemble docking contained compounds from a variety of sources (i.e. SweetLeads, NCI, and Enamine). In a separate experimental screening campaign, 2,900 chemicals have been tested by the National Institutes of Health, National Center for Advancing Translational Sciences (NCATS) and listed on https://opendata.ncats.nih.gov/covid19/databrowser (accessed 11/02/2020). Many of these chemicals are included in our docking databases. Therefore, the computational predictions from docking were compared to positives experimentally identified by by NCATS.
NCATS report results for spike protein (Spike-ACE2 protein-protein interaction (AlphaLISA) and MPro (3CL Enzymatic Activity) assays, which are equivalent to a subset of the docking calculations described here. We determined how many of the experimentally-tested NCATS compounds were in our smaller docking database and identified how many of the experimental positives were in the top ranked lists for each protein (Table 4). We also report the corresponding percentage of true positives, i.e., how many of the top computational-scoring compounds were identified by NCATS as active as a percentage of how many of the top computational-scoring compounds were experimentally tested by NCATS.
Table 4:
Number of top-scoring computationally predicted compoundsa, corresponding NCATS-tested compounds as a subset from first column, percentage of strong and strong+moderately active compounds for the Spike protein (top) and MPro (bottom) targets.
| # of top compounds (docking)a | # of corresponding compounds tested (NCATS) | percentage of NCATS actives (strong) | percentage of NCATS actives (strong+moderate) |
|---|---|---|---|
| Spike | |||
| 673 | 235 | 14.0% | 53.2% |
| 420 | 158 | 17.1% | 57.0% |
| 292 | 108 | 20.4% | 61.1% |
| 149 | 55 | 25.5% | 67.3% |
| 81 | 27 | 33.3% | 77.8% |
| 17 | 4 | 100.0% | 100.0% |
| MPro | |||
| 968 | 359 | - | 7.0% |
| 648 | 221 | - | 6.8% |
| 459 | 156 | - | 7.7% |
| 248 | 86 | - | 9.3% |
| 136 | 45 | - | 8.9% |
| 32 | 7 | - | 14.3% |
Top compounds from docking were obtained from the top 500, 300, 200, 100, and 50 ranked lists that correspond to each of the spike and MPro targets. For both systems multiple docking runs were considered and only unique compounds are reported.
We found that computational prediction is systematically enriched compared to a random selection of compounds. The experimental hit rate for NCATS compounds active in the spike protein is 6.1% for “ strong actives” (NCATS definition) and 28.3% for strong and moderately active compounds (the value of 28.3% being unusually high). In contrast, the computational enrichment is between about twice to four times as high (Table 4 & figure 5)). Out of the 673 unique compounds in the union of our top 500 lists foreach spike protein simulation variant, 235 have been tested experimentally by NCATS. Of these 235 compounds, 33 (14%) are experimentally strong actives and 125 (53%) are strongly or moderately active. Narrowing the ranked lists from docking to the top 10 resulted in 17 unique compounds for which 4 have experimental activity (0.14% of the total NCATS screen). Interestingly, all 4 of the experimentally tested compounds (i.e., 100% of the tested compounds in our top 10 lists) are strongly active.
Figure 5.

Comparison of S-protein (Spike) true-positive rates for strong-actives. Plot shows percentage of experimental NCATS positives in top computational-predicted chemicals as solid line. Dashed-line represents constant NCATS positive rate for comparision.
For the MPro assay, NCATS identifies only 1 strongly active out of 2,675 compounds in the approved drugs collection that were tested experientally. Therefore, we considered the overlap in our database with both the strong and moderately active compounds. Although the enrichment rates obtained through computational docking was not as high as the rates obtained for the spike protein, the computationally-obtained MPro enrichments ranged from 7% to 14% and are still systematically better than the rates obtained exeprientally (5.7%).
Interestingly, for both targets, as the number of ranked compounds considered is decreased, the computational enrichment improves. As expected, the very top screening compounds with the best docking score are often the most likely to have experimental activity and the further we go down the ranked list the lower the computational enrichment. Thus, docking performs better as a tool for identifying a small number of active compounds in a very small subset of a database, rather than a tool to identify many active compounds in a large subset of a database. This result is important when considering the prioritization of compounds from the billion-plus compound screen described below. A threshold of 10% or even 5% of a billion compounds database would still likely be too large a number to screen experientally, but less than 1% would be more amenable to experiental validation. The present results suggest that the best enrichments are indeed obtained for a small or very small subset of the chemical databases used in docking.
e. Billion-Compound Supercomputing Screens.
We found that for the ligands with fewer numbers of rotatable bonds, such as found in the Enamine dataset, a docking calculation using 20 repeated runs could be performed in 0.5–2.5 seconds when using the Summit GPU (Fig. 6). The same set of ligands docked with Vina on Summit’s CPUs showed a large spread of timings, with some ligands requiring nearly five minutes to complete (Fig. 6). In practice, this means that with GPU-enabled docking, it is feasible to flexibly dock a billion compounds in about a day on modern supercomputers, whereas with Vina, a similar calculation would require a multi-year effort on a university cluster. We confirmed that for ligands with less than 11 rotations, the Solis-Wets algorithm in AD-GPU provides equivalent results to the new ADADELTA algorithm61. For the trimmed dataset, the top 5% of scores obtained with AD-GPU using the Solis-Wets algorithm formed an intersection with the top 5% of scores from Vina consisting of 18% of each top 5% set. Analysis of the full billion ligand sets is currently ongoing.
Figure 6.

General benchmarking of Autodock-GPU and Autodock Vina performance against subset of Enamine database.
f. Mutation Analysis
The mutation frequency of the proteins simulated in this study is generally low. It should be noted, however, that it is as yet early in the history of SAR-CoV-2, and thus increased relative variability of residues along the proteome may indicate the propensity of those residues for future mutations. We did find higher variability, given by the entropy values, in other SARS-CoV-2 proteins not included in this study; in particular, the Spike mutation D614G noted in other reports continues to be seen with high frequency since being described in a recent preprint that performed an analysis on GISAID through April 13151. We counted 9107 D614G mutations (up from 3577 found April 13) and calculated entropy of 0.94 for this residue. The NSP12 RdRp protein also shows a large mutation entropy at residue 323, with a mutation entropy of 0.95. This residue, P, has mutated to L 9078 times (and F 3 times). Note that not every protein was represented in all sequences used for entropy calculations. Other regions of the genome with higher entropy values (greater than 0.5) are residues 203 and 204 of orf9 (entropy 0.70 and 0.69, respectively), residue 85 of NSP2 (0.74), 37 of NSP6 (0.57), 57 of orf3a (0.81), and 84 of orf8 (0.56).
The highest entropy found among the structures in this study was in the main protease, with an entropy of 0.13 for residue 15, a glycine. We found 261 G15S mutations and one G15D mutation in our dataset. The MPro also has a number of other residues with relatively high entropies, including residue 90, with entropy 0.07 and 117 K90R mutations, and 266 with entropy 0.04 and 64 A266V mutations. After this, the next highest entropy was 0.06 for the N-terminal region of the N protein and also for NSP15. These are displayed in Figure 7. An entropy of 0.04 was also found for domain X of NSP3 (glycine 76). A lower mutation entropy was found in PLPro, compared to MPro, with the highest value being 0.03. These mutations are important to consider when choosing targets for drug discovery, in that a protein that seems to be more rapidly mutating could potentially lead to an ineffective therapeutic if mutations alter the shape of the drugbinding site. In the case of MPro, the highest entropy mutations were not found in the active site; however, it is possible that they may still affect its conformation indirectly. The reduced mutation entropy for PLPro may indicate that an effort to target a protease could meet with fewer mutation-related problems if targeting PLPro rather than MPro.
Figure 7.

Example mutational entropy analysis. Residues are colored by entropy, with redder colors corresponding to greater entropy.
Future Directions and Preliminary Results from New Methodologies.
As emphasized above, this article is a progress report on an ongoing project. The development of the pipeline is continuing with advances being made in several directions. Notably, we are incorporating artificial intelligence and machine learning into rescoring ligand ranking and clustering the MD trajectories. Further, we are developing methods to rescore docking using quantum chemical approaches. Although these developments have not been incorporated into the pipeline at the time of writing and were not applied to generate the results described above, we report on progress with them here.
a. Clustering MD trajectories using Deep Learning and AI.
The deluge of data generated from simulations such as the T-REMD runs reported here can make traditional approaches of machine learning and clustering approaches (based on measures of similarity in the RMSD-space, or other metrics) quite challenging. Often, practical aspects of computing dictate the use of subsample tracts of the MD data itself or use of prior knowledge about these datasets (e.g., knowing that the ligand binds only in a certain orientation) to filter such datasets. Deep learning techniques can be particularly valuable in ‘sifting’ through large datasets and can be powerful for clustering T-REMD simulations. We are investigating the use of a variational autoencoder with convolutional filters (CVAE), previously developed to cluster protein folding trajectories152, 153, to cluster the T-REMD simulations of NSP15. As shown in Fig. 8 we find that the latent dimensions learned from the simulations indeed cluster the simulation data into a small number of conformational states. These states correspond to transitions observed in the simulations, as seen from various measured observables from the data such as the binding site RMSD, SASA, and the radius of gyration.
Figure 8.

Deep learning clusters T-REMD simulations of the NSP15 hexameric complex into conformational states that are potentially relevant for docking studies. (A) A 3D-representation of the CVAE learned from the T-REMD simulations shows the presence of multiple conformational states. Each conformation from the simulation is painted using the RMSD to the starting structure and shows the presence of distinct directions in the conformational landscape where low- and high-RMSD structures are distributed. To understand this representation better, we use an at-stochastic neighbor embedding (t-SNE) algorithm to embed the data into a low-dimensional space, where we can clearly visualize how the conformational landscape is organized. In this two-dimensional space, we visualize various observables from the simulations, including (B) RMSD to the native structure, (C) SASA, and (D) radius of gyration. In each of these cases, we can observe the presence of at least three dominant sub-states with distinct structural characteristics, which can be further used for docking simulations.
The outcomes from the clustering provide insights into aspects of how the T-REMD simulations have sampled the conformational landscape - for example, in the case of this protein, as observed in Panel C, there is only one conformational state which has sampled a large SASA, indicating a potentially open state (which has only a minor change in the overall RMSD, Panel B). This information can be particularly helpful for selecting conformations and identifying metastable states for docking simulations154.
b. Protein-ligand rescoring using machine-learning.
The computational identification of drug compounds, and small molecules in general, that bind to a protein consists of three distinct tasks: 1) identifying a putative conformation of the protein-ligand complex (the docking problem); 2) given a docked conformation, determining whether or not the ligand is a true binder (the screening problem); and 3) is determined to be a true binder, ascertaining a relative, or better yet, absolute binding affinity (the affinity, or scoring, problem). In principle, one could perform the screening and affinity prediction problems using molecular dynamics techniques such as free energy perturbation, thermodynamic integration, or more approximate methods such as MM/PB(GB)SA (molecular mechanics/Poisson-Boltzmann[generalized Born]-surface area). However, this is computationally intractable for large numbers of compounds, even with supercomputers, and the accuracy can often be poor. Furthermore, these rigorous first principles-based methods assume a putative binding site, and cannot be applied to cases where the binding site is unknown. While the score or energy given by computational docking programs such as AutoDock Vina is reasonably well-suited for docking pose prediction, improvements are possible on the screening and affinity problems, and for this, we use here machine learning.
There is an ongoing need for the development of computationally tractable models that can be easily validated on benchmark docking data sets. To this end, accurate, physics-based, machine-learned models for the docking and affinity have been trained using the PDBbind database, a dataset consisting of experimentally determined protein-ligand complex structures with accurate experimental binding affinities 155–158. On an independent data set, the CASF-2013 benchmark159, 160, affinity prediction (random forest-based) models achieve, at best, a Pearson correlation (R2) of 0.86, and docking pose prediction classifiers achieve an area under the curve of the receiver operator characteristic (AUC of ROC) of 0.91 using support vector machines (Demerdash et al., In Review). While the random forest model trained on unnormalized features achieved the best R2 at 0.86, a range of additional models (trained with random forest, gradient boosted trees, or support-vector machines using normalized or unnormalized features) achieved R2 of 0.81–0.85. Regarding the docking pose prediction, the model used here achieves greater enrichment for native-like structures (78%) than AutoDock Vina (63%) (Demerdash et al., In Preparation).
A model dedicated to virtual screening as a first step in triaging candidate molecules was developed. Once again, as with affinity and pose prediction, this model requires docked structures as input. This model is trained to discriminate between active and inactive compounds, and affinity ranking is performed as a second step only on the true active compounds. To this end, a support-vector machine-based model using the Dataset of Useful Decoys-Enhanced, a database of 102 proteins with experimentally verified active and inactive compounds, has been trained161, 162. Preliminary performance on an independent validation set is encouraging, achieving AUC of ROC of 0.80 and recall of 0.76; that is, 76% of experimentally validated true positives were predicted positively by the model. This model is currently being subjected to further optimized, primarily through the calculation of additional physicochemical descriptors (features) and the optimization of hyperparameters.
Due to the urgent nature of the Covid-19 drug discovery campaign, computational expediency precluded calculating features on all docked structures for a given compound and, in turn, precluded running the docking pose classifier on the output of AutoDock Vina. Therefore, we relied on AutoDock Vina’s ranking and not the machine-learned docking pose classifier, thereby reducing the number of feature calculations that must be performed and increasing the throughput. (Parallelization efforts and code optimization are underway, so that feature calculation on all docked poses and subsequent application of the docking pose classifier becomes less onerous.) The virtual screening model was applied to these top-scoring structures from AutoDock Vina, generating a “binder” vs. “non-binder” classification. Subsequently, affinity prediction models were applied to just those complex structures classified as “binder.” The affinity prediction was performed using the range of high-performing models on docked structures corresponding to each MD cluster representative used in ensemble docking (See Ensemble Docking with HPC Methods for details.). This results in affinity predictions on typically 10 cluster representatives, each with affinity predictions from 5 machine-learned models (1 SVM, 2 boosted tree, and two random forest approaches), resulting in 50 “cases.” For each case, the top-500 ligands in terms of predicted affinity, were obtained. Molecules that appeared in the top 500 in at least 25 of the 50 cases were deemed hits and are presently undergoing experimental testing.
QM analysis of S-protein docking results
In a preliminary evaluation of the accuracy of FMO-DFTB/PCM in describing the interactions between ligands and the S-protein, we compare FMO-DFTB/PCM pair interaction energy (PIE) to that of the higher-level, but more expensive FMO-MP2/PCM method. The PIEs were calculated for ligands binding to the S-protein in the binding pocket. Figure 9 shows that FMO-DFTB/PCM interaction energies agree very well with high-level ab initio FMO-MP2/PCM data with the R correlation coefficient; in this case, it is 0.984. The high correlation between FMO-DFTB/PCM PIE and FMO-MP2/PCM PIE indicates that FMODFTB/PCM may be a fast and reliable QM-based method for interaction energy calculations.
Figure 9.

Pair interaction energy (PIE) decomposition analysis for FMO-DFTB/PCM plotted against FMO-MP2/3–21G/PCM data.
An updated preliminary homology model and T-REMD simulation similar to that reported by Smith & Smith of the S-protein RBD and re-docking to new clusters from followed by a re-evaluation of the top scoring 150 complexes with FMO-DFTB/PCM was performed as follows: 15 protein conformations and their ten strongest binding ligands predicted by AutoDock Vina were selected, and geometry optimizations were performed at the FMO-DFTB3-D3(BJ)/3ob/PCM level of theory. The binding energies of the top-3 best binders ranked by FMO-DFTB/PCM are listed in Table 5. According to our preliminary results, although FMO-DFTB/PCM agrees well with Vina in categorizing the strong binders, with all Vina, top-10 ligands have considerably stronger FMO-DFTB/PCM binding energies (ΔEbind < −12 kcal/mol) and the QM-based ranking is significantly different from the Vina ranking.
Table 5.
The binding energy of the top-3 best ranked by FMO-DFTB/PCM and their binding free energy predicted by Autodock Vina.
| Ligand SWEETLEAD ID | Protein Cluster ID | FMO-DFTB/PCM ΔEbind(kcal/mol) | Vina ΔGbind(kcal/mol) |
|---|---|---|---|
| 4752 | 7 | −67.75 | −5.40 |
| 7055 | 11 | −66.78 | −7.60 |
| 4698 | 12 | −66.41 | −7.60 |
It is important to note that the current FMO-DFTB/PCM energy, which is based on solvent-corrected binding interaction energies is not the binding free energy. Various additional contributions to the binding free energy can be separately evaluated, and work is underway in this regard. For example, an entropic contribution can be estimated from vibrational frequencies once the requisite Hessian matrix is available.
Conclusions
The present manuscript reports on the establishment of a supercomputer-based virtual high-throughput screening ensemble-docking pipeline that takes into account the dynamic properties of protein targets, as well as preliminary results on simulations and docking screens to a number of protein targets from SARS-CoV-2.
The speed at which structural data have been derived experimentally for the SARS-CoV-2 proteome means that several of the simulations reported above were ‘out of date’ almost immediately. By this, we mean that the simulations were performed using models derived from experiments that had been superseded by higher-resolution or more complete data. Examples of these are the S-protein, MPro, N Protein, and NSP9. Clearly, as information on structures increases in quality, simulations will be further repeated. Furthermore, the complexity of the structural models derived is expected to increase. For example, models of the S protein interacting with the viral envelope or extending up to the complete virion can be envisaged and, in principle at least, incorporated into drug screening protocols.
The present results provide comprehensive simulation models for 8 of the viral proteins in 24 molecular systems. T-REMD is well suited for massively parallel supercomputing because many replicas are run simultaneously, and they need to communicate with each other. In the present tests, 350ns/day/replica was obtained for the smallest (NSP3 phophatase/ADRP) system, and this, therefore, scales up to about 1.5ms/day of aggregate MD time, given the hypothetical situation that one had about 100 different proteins to run of roughly the same size. For bigger systems, with 105 atoms, the throughput is lower, about 1.0 ms/day. Nevertheless, it is clear that extensive simulation data can be obtained on many proteins with a short time-to-solution on this machine. As one possible future direction, one might envisage running T-REMD on the 44 drug targets that have been suggested as a minimal screen for the toxicity effects in human drug trials163.
The ensemble docking performed so far mostly involves repurposing databases and therefore is limited to about 10k compounds. Many of these compounds are predicted to be quite promiscuous in binding to the targets. Two of the compounds identified in the top 1% of our preliminary S-protein screen have been reported to be in two registered clinical trials (quercetin and hypericin). Further, several compounds from the screens reported above show activity in reducing live viral infectivity: these results will be reported elsewhere.
The docking results using the smaller database were not run on Summit, because of the fact that for Summit code running on GPUs is preferred. However, as COVID-19 therapeutic research moves beyond repurposing to the discovery of novel compounds, there is a need to quickly screen many more compounds. Therefore, we have installed Autodock-GPU and demonstrated that it is capable of screening 1 billion compounds on Summit in 12 hours when scaled to the whole machine. Although several other groups have reported billion-compound screens, these have been using AI approaches or rigid docking without pose optimization164, 165. The present billion-compound screen calculations, therefore, represent a potential supercomputer-driven paradigm shift in computational drug discovery and can be envisaged to be performed on dozens of proteins in a single day when the exascale era of supercomputing arrives, as planned for 2021.
Supplementary Material
Acknowledgments
This work was made possible in part by a grant of high-performance computing resources and technical support from the Alabama Supercomputer Authority to JB and KB.
JCG was supported by the National Institute of Health under Grant No. NIH R01-AI148740
CJC was supported by a National Science Foundation Graduate Research Fellowship under Grant No. 2017219379.
This research used resources of the Oak Ridge Leadership Computing Facility at the Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC05-00OR22725 and National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility operated under Contract No. DE-AC02-05CH11231.
This research was supported by the Cancer Research Informatics Shared Resource Facility of the University of Kentucky Markey Cancer Center (P30CA177558) and the University of Kentucky’s Center for Computational Sciences (CCS) high-performance computing resources.
Footnotes
Supplemental Materials
Additional tables describing the simulations systems are provided as supplemental material for interested readers. Additionally, descriptive figures showing cluster diversity for each protein system are also provided as supplemental material.
This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05–00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
References
- 1.Agostini ML; Andres EL; Sims AC; Graham RL; Sheahan TP; Lu X; Smith EC; Case JB; Feng JY; Jordan R, Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. MBio 2018, 9, e00221–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown AJ; Won JJ; Graham RL; Dinnon KH 3rd; Sims AC; Feng JY; Cihlar T; Denison MR; Baric RS; Sheahan TP, Broad spectrum antiviral remdesivir inhibits human endemic and zoonotic deltacoronaviruses with a highly divergent RNA dependent RNA polymerase. Antiviral Res 2019, 169, 104541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheahan TP; Sims AC; Leist SR; Schafer A; Won J; Brown AJ; Montgomery SA; Hogg A; Babusis D; Clarke MO; Spahn JE; Bauer L; Sellers S; Porter D; Feng JY; Cihlar T; Jordan R; Denison MR; Baric RS, Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat Commun 2020, 11, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amanat F; Krammer F, SARS-CoV-2 Vaccines: Status Report. Immunity 2020, 52, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Wit E; Feldmann F; Cronin J; Jordan R; Okumura A; Thomas T; Scott D; Cihlar T; Feldmann H, Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc Natl Acad Sci U S A 2020, 117, 6771–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beigel JH; Tomashek KM; Dodd LE; Mehta AK; Zingman BS; Kalil AC; Hohmann E; Chu HY; Luetkemeyer A; Kline S; Lopez de Castilla D; Finberg RW; Dierberg K; Tapson V; Hsieh L; Patterson TF; Paredes R; Sweeney DA; Short WR; Touloumi G; Lye DC; Ohmagari N; Oh MD; Ruiz-Palacios GM; Benfield T; Fatkenheuer G; Kortepeter MG; Atmar RL; Creech CB; Lundgren J; Babiker AG; Pett S; Neaton JD; Burgess TH; Bonnett T; Green M; Makowski M; Osinusi A; Nayak S; Lane HC; Members A-SG, Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med 2020, 383, 1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson RM; Vinetz JM, Dexamethasone in the management of covid −19. BMJ 2020, 370, m2648. [DOI] [PubMed] [Google Scholar]
- 8.Omoto S; Speranzini V; Hashimoto T; Noshi T; Yamaguchi H; Kawai M; Kawaguchi K; Uehara T; Shishido T; Naito A; Cusack S, Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci Rep 2018, 8, 9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim Y; Jedrzejczak R; Maltseva NI; Wilamowski M; Endres M; Godzik A; Michalska K; Joachimiak A, Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020, 29, 1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu C; Zhou Q; Li Y; Garner LV; Watkins SP; Carter LJ; Smoot J; Gregg AC; Daniels AD; Jervey S; Albaiu D, Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent Sci 2020, 6, 315–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim PS; Read SW; Fauci AS, Therapy for Early COVID-19: A Critical Need. JAMA 2020. [DOI] [PubMed] [Google Scholar]
- 12.Jiang S; Hillyer C; Du L, Neutralizing Antibodies against SARS-CoV-2 and Other Human Coronaviruses. Trends Immunol 2020, 41, 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Netherton CL; Wileman T, Virus factories, double membrane vesicles and viroplasm generated in animal cells. Curr Opin Virol 2011, 1, 381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.den Boon JA; Diaz A; Ahlquist P, Cytoplasmic viral replication complexes. Cell Host Microbe 2010, 8, 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffmann M; Kleine-Weber H; Pohlmann S, A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol Cell 2020, 78, 779–784 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagemeijer MC; Rottier PJ; de Haan CA, Biogenesis and dynamics of the coronavirus replicative structures. Viruses 2012, 4, 3245–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu A; Peng Y; Huang B; Ding X; Wang X; Niu P; Meng J; Zhu Z; Zhang Z; Wang J, Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell host & microbe 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X; Zhang L; Duan Y; Yu J; Wang L; Yang K; Liu F; You T; Liu X; Yang X, Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020. [DOI] [PubMed] [Google Scholar]
- 19.Yin W; Mao C; Luan X; Shen D-D; Shen Q; Su H; Wang X; Zhou F; Zhao W; Gao M, Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X; Cao R; Zhang H; Liu J; Xu M; Hu H; Li Y; Zhao L; Li W; Sun X, The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discovery 2020, 6, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lei J; Hilgenfeld R, RNA-virus proteases counteracting host innate immunity. FEBS Lett. 2017, 591, 3190–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith M; Smith J, Repurposing Therapeutics for COVID-19: Supercomputer-Based Docking to the SARS-CoV-2 Viral Spike Protein and Viral Spike Protein-Human ACE2 Interface. 2020. [Google Scholar]
- 23.Kaldor SW; Kalish VJ; Davies JF 2nd; Shetty BV; Fritz JE; Appelt K; Burgess JA; Campanale KM; Chirgadze NY; Clawson DK; Dressman BA; Hatch SD; Khalil DA; Kosa MB; Lubbehusen PP; Muesing MA; Patick AK; Reich SH; Su KS; Tatlock JH, Viracept (nelfinavir mesylate, AG1343): a potent, orally bioavailable inhibitor of HIV-1 protease. J. Med. Chem 1997, 40, 3979–85. [DOI] [PubMed] [Google Scholar]
- 24.von Itzstein M; Wu W-Y; Kok GB; Pegg MS; Dyason JC; Jin B; Van Phan T; Smythe ML; White HF; Oliver SW, Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [DOI] [PubMed] [Google Scholar]
- 25.Gorgulla C; Boeszoermenyi A; Wang ZF; Fischer PD; Coote PW; Padmanabha Das KM; Malets YS; Radchenko DS; Moroz YS; Scott DA; Fackeldey K; Hoffmann M; Iavniuk I; Wagner G; Arthanari H, An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amaro RE; Baudry J; Chodera J; Demir Ö; McCammon JA; Miao Y; Smith JC, Ensemble Docking in Drug Discovery. Biophys. J 2018, 114, 2271–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teague SJ, Implications of protein flexibility for drug discovery. Nature reviews Drug discovery 2003, 2, 527–541. [DOI] [PubMed] [Google Scholar]
- 28.Carlson HA; Masukawa KM; McCammon JA, Method for Including the Dynamic Fluctuations of a Protein in Computer-Aided Drug Design. The Journal of Physical Chemistry A 1999, 103, 10213–10219. [Google Scholar]
- 29.Amaro RE; Baudry J; Chodera J; Demir Ö; McCammon JA; Miao Y; Smith JC, Ensemble Docking in Drug Discovery. Biophys J 2018, 114, 2271–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pi M; Kapoor K; Wu Y; Ye R; Senogles SE; Nishimoto SK; Hwang D-J; Miller DD; Narayanan R; Smith JC; Baudry J; Quarles LD, Structural and Functional Evidence for Testosterone Activation of GPRC6A in Peripheral Tissues. Molecular Endocrinology 2015, 29, 1759–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pi M; Kapoor K; Ye R; Nishimoto SK; Smith JC; Baudry J; Quarles LD, Evidence for osteocalcin binding and activation of GPRC6A in β-cells. Endocrinology 2016, 157, 1866–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evangelista W; Weir RL; Ellingson SR; Harris JB; Kapoor K; Smith JC; Baudry J, Ensemble-based docking: From hit discovery to metabolism and toxicity predictions. Biorg. Med. Chem 2016, 24, 4928–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao Z; Riccardi D; Velazquez HA; Chin AL; Yates CR; Carrick JD; Smith JC; Baudry J; Quarles LD, A computationally identified compound antagonizes excess FGF-23 signaling in renal tubules and a mouse model of hypophosphatemia. Science Signaling 2016, 9, ra113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdali N; Parks JM; Haynes KM; Chaney JL; Green AT; Wolloscheck D; Walker JK; Rybenkov VV; Baudry J; Smith JC; Zgurskaya HI, Reviving Antibiotics: Efflux Pump Inhibitors That Interact with AcrA, a Membrane Fusion Protein of the AcrAB-TolC Multidrug Efflux Pump. ACS Infectious Disease 2017, 3, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dale JB; Smeesters PR; Courtney HS; Penfound TA; Hohn CM; Smith JC; Baudry JY, Structure-based design of broadly protective group a streptococcal M protein-based vaccines. Vaccine 2017, 35, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haynes KM; Abdali N; Jhawar V; Zgurskaya HI; Parks JM; Green AT; Baudry J; Rybenkov VV; Smith JC; Walker JK, Identification and Structure–Activity Relationships of Novel Compounds that Potentiate the Activities of Antibiotics in Escherichia coli. J. Med. Chem 2017, 60, 6205–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Velazquez HA; Riccardi D; Xiao Z; Quarles LD; Yates CR; Baudry J; Smith JC, Ensemble docking to difficult targets in early-stage drug discovery: Methodology and application to fibroblast growth factor 23. Chemical Biology & Drug Discovery 2018, 91, 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao Z; Baudry J; Cao L; Huang J; Chen H; Yates CR; Li W; Dong B; Waters CM; Smith JC, Polycystin-1 interacts with TAZ to stimulate osteoblastogenesis and inhibit adipogenesis. Journal of Clinical Investigation 2018, 128, 157–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pi M; Kapoor K; Ye R; Hwang D-J; Miller DD; Smith JC; Baudry J; Quarles LD, Computationally identified novel agonists for GPRC6A. PloS one 2018, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Darzynkiewicz ZM; Green AT; Abdali N; Hazel A; Fulton RL; Kimball J; Gryczynski Z; Gumbart JC; Parks JM; Smith JC, Identification of binding sites for efflux pump inhibitors of the AcrAB-TolC component AcrA. Biophys. J 2019, 116, 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evangelista Falcon W; Ellingson SR; Smith JC; Baudry J, Ensemble Docking in Drug Discovery: How Many Protein Configurations from Molecular Dynamics Simulations are Needed To Reproduce Known Ligand Binding? Journal of Physical Chemistry B 2019, 123, 5189–5195. [DOI] [PubMed] [Google Scholar]
- 42.Aranha MP; Spooner C; Demerdash O; Czejdo B; Smith JC; Mitchell JC, Prediction of peptide binding to MHC using machine learning with sequence and structure-based feature sets. Biochimica et Biophysica Acta-General Subjects 2020, 129535. [DOI] [PubMed] [Google Scholar]
- 43.Micholas S; Jeremy C,S, Repurposing Therapeutics for COVID-19: Supercomputer-Based Docking to the SARS-CoV-2 Viral Spike Protein and Viral Spike Protein-Human ACE2 Interface. ChemRxiv 2020, 10.26434/chemrxiv.11871402.v4. [DOI] [Google Scholar]
- 44.Parks JM; Smith JC, How to discover antiviral drugs quickly. New England Journal of Medicine 2020. [DOI] [PubMed] [Google Scholar]
- 45.Agarwal R; Bensing BA; Mi D; Vinson PN; Baudry J; Iverson TM; Smith JC, Structure based virtual screening identifies small molecule effectors for the sialoglycan binding protein Hsa. Biochem. J 2020, 477, 3695–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta M; Ha K; Agarwal R; Quarles LD; Smith JC, Molecular dynamics analysis of the binding of human interleukin-6 with interleukin-6 α-receptor. Proteins: Structure, Function, and Bioinformatics n/a. [DOI] [PubMed] [Google Scholar]
- 47.Evangelista Falcon W; Ellingson SR; Smith JC; Baudry J, Ensemble Docking in Drug Discovery: How Many Protein Configurations from Molecular Dynamics Simulations are Needed To Reproduce Known Ligand Binding? The Journal of Physical Chemistry B 2019, 123, 5189–5195. [DOI] [PubMed] [Google Scholar]
- 48.Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- 49.Kutzner C; Páll S; Fechner M; Esztermann A; de Groot BL; Grubmüller H, Best bang for your buck: GPU nodes for GROMACS biomolecular simulations. J. Comput. Chem 2015, 36, 1990–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Der Spoel D; Lindahl E; Hess B; Groenhof G; Mark AE; Berendsen HJC, GROMACS: Fast, flexible, and free. J. Comput. Chem 2005, 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- 51.Earl DJ; Deem MW, Parallel tempering: Theory, applications, and new perspectives. Physical Chemistry Chemical Physics 2005, 7, 3910–3916. [DOI] [PubMed] [Google Scholar]
- 52.Hansmann UHE, Parallel tempering algorithm for conformational studies of biological molecules. Chemical Physics Letters. 1997, 281, 140–150. [Google Scholar]
- 53.Sugita Y; Okamoto Y, Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- 54.Sugita Y; Kitao A; Okamoto Y, Multidimensional replica-exchange method for freeenergy calculations. The Journal of Chemical Physics 2000, 113, 6042–6051. [Google Scholar]
- 55.Bernardi RC; Melo MCR; Schulten K, Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochimica et Biophysica Acta (BBA) - General Subjects 2015, 1850, 872–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsai T-Y; Chang K-W; Chen CY-C, iScreen: world’s first cloud-computing web server for virtual screening and de novo drug design based on TCM database@Taiwan. J. Comput. Aided Mol. Des 2011, 25, 525–531. [DOI] [PubMed] [Google Scholar]
- 57.Dolezal R; Sobeslav V; Hornig O; Balik L; Korabecny J; Kuca K HPC Cloud Technologies for Virtual Screening in Drug Discovery. Cham, 2015; Springer International Publishing: Cham, 2015; pp 440–449. [Google Scholar]
- 58.Huey R; Morris GM; Olson AJ; Goodsell DS, A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem 2007, 28, 1145–1152. [DOI] [PubMed] [Google Scholar]
- 59.Huey R; Goodsell DS; Morris GM; Olson AJ, Grid-based hydrogen bond potentials with improved directionality. Letters in Drug Design & Discovery 2004, 1, 178–183. [Google Scholar]
- 60.El Khoury L; Santos-Martins D; Sasmal S; Eberhardt J; Bianco G; Ambrosio FA; Solis-Vasquez L; Koch A; Forli S; Mobley DL, Comparison of affinity ranking using AutoDock-GPU and MM-GBSA scores for BACE-1 inhibitors in the D3R Grand Challenge 4. J. Comput. Aided Mol. Des 2019, 33, 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Solis-Vasquez L; Santos-Martins D; Koch A; Forli S Evaluating the Energy Efficiency of OpenCL-accelerated AutoDock Molecular Docking. In 2020 28th Euromicro International Conference on Parallel, Distributed and Network-Based Processing (PDP), 2020; IEEE: 2020; pp 162–166. [Google Scholar]
- 62.Santos-Martins D; Solis-Vasquez L; Koch A; Forli S, Accelerating autodock4 with gpus and gradient-based local search. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.LeGrand S; Scheinberg A; Tillack AF; Thavappiragasam M; Vermaas JV; Agarwal R; Larkin J; Poole D; Santos-Martins D; Solis-Vasquez L GPU-Accelerated Drug Discovery with Docking on the Summit Supercomputer: Porting, Optimization, and Application to COVID-19 Research. In Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, 2020; 2020; pp 1–10. [Google Scholar]
- 64.Vermaas JV; Sedova A; Baker M; Boehm S; Rogers D; Larkin J; Glaser J; Smith M; Hernandez O; Smith J, Supercomputing Pipelines Search for Therapeutics Against COVID-19. Computing in Science & Engineering 2020, 1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fedorov DG, The fragment molecular orbital method: theoretical development, implementation in GAMESS and applications. WIREs Computational Molecular Science 2017, 7. [Google Scholar]
- 66.Gordon DE; Jang GM; Bouhaddou M; Xu J; Obernier K; White KM; O’Meara MJ; Rezelj VV; Guo JZ; Swaney DL, A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou H; Fang Y; Xu T; Ni WJ; Shen AZ; Meng XM, Potential therapeutic targets and promising drugs for combating SARS-CoV-2. British Journal of Pharmacology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim Y; Jedrzejczak R; Maltseva NI; Wilamowski M; Endres M; Godzik A; Michalska K; Joachimiak A, Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C, Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 1–5. [DOI] [PubMed] [Google Scholar]
- 70.Lan J; Ge J; Yu J; Shan S; Zhou H; Fan S; Zhang Q; Shi X; Wang Q; Zhang L, Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [DOI] [PubMed] [Google Scholar]
- 71.Kang S; Yang M; Hong Z; Zhang L; Huang Z; Chen X; He S; Zhou Z; Zhou Z; Chen Q, Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharmaceutica Sinica B 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maeda Y; Kinoshita T The acidic environment of the Golgi is critical for glycosylation and transport. In Methods Enzymol.; Elsevier: 2010; Vol. 480, pp 495–510. [DOI] [PubMed] [Google Scholar]
- 73.Wu MM; Grabe M; Adams S; Tsien RY; Moore HP; Machen TE, Mechanisms of pH regulation in the regulated secretory pathway. Journal of Biolological Chemistry 2001, 276, 33027–35. [DOI] [PubMed] [Google Scholar]
- 74.Zumla A; Chan JFW; Azhar EI; Hui DSC; Yuen K-Y, Coronaviruses — drug discovery and therapeutic options. Nature Reviews Drug Discovery 2016, 15, 327–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen AA; Pappu RV, Parameters of Monovalent Ions in the AMBER-99 Forcefield: Assessment of Inaccuracies and Proposed Improvements. The Journal of Physical Chemistry B 2007, 111, 11884–11887. [DOI] [PubMed] [Google Scholar]
- 76.Yoo J; Aksimentiev A, Improved Parametrization of Li+, Na+, K+, and Mg2+ Ions for All-Atom Molecular Dynamics Simulations of Nucleic Acid Systems. The Journal of Physical Chemistry Letters 2012, 3, 45–50. [Google Scholar]
- 77.Ahlstrand E; Schpector JZ; Friedman R, Computer simulations of alkali-acetate solutions: Accuracy of the forcefields in difference concentrations. The Journal of Chemical Physics 2017, 147, 194102. [DOI] [PubMed] [Google Scholar]
- 78.Jing Z; Liu C; Cheng SY; Qi R; Walker BD; Piquemal J-P; Ren P, Polarizable Force Fields for Biomolecular Simulations: Recent Advances and Applications. Annual Review of Biophysics 2019, 48, 371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang C.-k.; Hou M-H; Chang C-F; Hsiao C-D; Huang T.-h., The SARS coronavirus nucleocapsid protein–forms and functions. Antiviral research 2014, 103, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Astuti I; Ysrafil, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 2020, 14, 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lei J; Kusov Y; Hilgenfeld R, Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral research 2018, 149, 58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Báez-Santos YM; St John SE; Mesecar AD, The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral research 2015, 115, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Michalska K; Kim Y; Jedrzejczak R; Maltseva NI; Stols L; Endres M; Joachimiak A, Crystal structures of SARS-CoV-2 ADP-ribose phosphatase (ADRP): from the apo form to ligand complexes. bioRxiv 2020, 2020.05.14.096081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lei J; Hilgenfeld R, RNA-virus proteases counteracting host innate immunity. FEBS Lett. 2017, 591, 3190–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; Duan Y; Yu J; Wang L; Yang K; Liu F; Jiang R; Yang X; You T; Liu X; Yang X; Bai F; Liu H; Liu X; Guddat LW; Xu W; Xiao G; Qin C; Shi Z; Jiang H; Rao Z; Yang H, Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [DOI] [PubMed] [Google Scholar]
- 86.Littler DR; Gully BS; Colson RN; Rossjohn J, Crystal Structure of the SARSCoV-2 Non-structural Protein 9, Nsp9. iScience 2020, 23, 101258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim Y; Jedrzejczak R; Maltseva NI; Wilamowski M; Endres M; Godzik A; Michalska K; Joachimiak A, Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020, 29, 1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosas-Lemus M; Minasov G; Shuvalova L; Inniss NL; Kiryukhina O; Wiersum G; Kim Y; Jedrzejczak R; Enders M; Jaroszewski L; Godzik A; Joachimiak A; Satchell KJF, The crystal structure of nsp10-nsp16 heterodimer from SARS-CoV-2 in complex with S-adenosylmethionine. bioRxiv 2020, 2020.04.17.047498. [Google Scholar]
- 89.Chen Y; Su C; Ke M; Jin X; Xu L; Zhang Z; Wu A; Sun Y; Yang Z; Tien P; Ahola T; Liang Y; Liu X; Guo D, Biochemical and Structural Insights into the Mechanisms of SARS Coronavirus RNA Ribose 2′-O-Methylation by nsp16/nsp10 Protein Complex. PLOS Pathogens 2011, 7, e1002294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Menachery VD; Debbink K; Baric RS, Coronavirus non-structural protein 16: Evasion, attenuation, and possible treatments. Virus Research 2014, 194, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mikolov T; Chen K; Corrado G; Dean J, Efficient Estimation of Word Representations in Vector Space. arXiv 2013, 1301.3781. [Google Scholar]
- 92.Robertson S, The Probabilistic Relevance Framework: BM25 and Beyond. Foundations and Trends® in Information Retrieval 2010, 3, 333–389. [Google Scholar]
- 93.Devlin J; Chang M-W; Lee K; Toutanova K BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. In Association for Computational Linguistics, Minneapolis, Minnesota, 2019; Minneapolis, Minnesota, 2019. [Google Scholar]
- 94.Jo S; Kim T; Iyer VG; Im W, CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem 2008, 29, 1859–65. [DOI] [PubMed] [Google Scholar]
- 95.Zhang J; Yang W; Piquemal J-P; Ren P, Modeling Structural Coordination and Ligand Binding in Zinc Proteins with a Polarizable Potential. Journal of Chemical Theory and Computation 2012, 8, 1314–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Calimet N; Simonson T, CysxHisy–Zn2+ interactions: Possibilities and limitations of a simple pairwise force field. J. Mol. Graphics Modell 2006, 24, 404–411. [DOI] [PubMed] [Google Scholar]
- 97.Wambo TO; Chen LY; McHardy SF; Tsin AT, Molecular dynamics study of human carbonic anhydrase II in complex with Zn2+ and acetazolamide on the basis of all-atom force field simulations. Biophys. Chem 2016, 214–215, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fan K; Wei P; Feng Q; Chen S; Huang C; Ma L; Lai B; Pei J; Liu Y; Chen J, Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem 2004, 279, 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R, Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kneller DW; Phillips G; O’Neill HM; Jedrzejczak R; Stols L; Langan P; Joachimiak A; Coates L; Kovalevsky A, Structural plasticity of SARS-CoV-2 3CL M(pro) active site cavity revealed by room temperature X-ray crystallography. Nature Communications 2020, 11, 3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang H; Yang M; Ding Y; Liu Y; Lou Z; Zhou Z; Sun L; Mo L; Ye S; Pang H, The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proceedings of the National Academy of Sciences 2003, 100, 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang F; Chen C; Tan W; Yang K; Yang H, Structure of Main Protease from Human Coronavirus NL63: Insights for Wide Spectrum Anti-Coronavirus Drug Design. Scientific Reports 2016, 6, 22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Anand K; Palm GJ; Mesters JR; Siddell SG; Ziebuhr J; Hilgenfeld R, Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002, 21, 3213–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paasche A; Zipper A; Schäfer S; Ziebuhr J; Schirmeister T; Engels B, Evidence for substrate binding-induced zwitterion formation in the catalytic Cys-His dyad of the SARSCoV main protease. Biochemistry 2014, 53, 5930–5946. [DOI] [PubMed] [Google Scholar]
- 105.Katarzyna S; Vicent M, Revealing the Molecular Mechanisms of Proteolysis of SARSCoV-2 Mpro from QM/MM Computational Methods. ChemRxiv 2020, 10.26434/chemrxiv.12283967.v1. [DOI] [Google Scholar]
- 106.Świderek K; Moliner V, Revealing the molecular mechanisms of proteolysis of SARSCoV-2 Mpro by QM/MM computational methods. Chemical Science 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dolinsky TJ; Czodrowski P; Li H; Nielsen JE; Jensen JH; Klebe G; Baker NA, PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vermaas JV; Hardy DJ; Stone JE; Tajkhorshid E; Kohlmeyer A, TopoGromacs: Automated Topology Conversion from CHARMM to GROMACS within VMD. Journal of Chemical Information and Modeling . 2016, 56, 1112–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hess B; Kutzner C; van der Spoel D; Lindahl E, GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. Journal of Chemical Theory and Computation 2008, 4, 435–47. [DOI] [PubMed] [Google Scholar]
- 110.Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD, CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nature methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Abraham MJ; Gready JE, Optimization of parameters for molecular dynamics simulation using smooth particle-mesh Ewald in GROMACS 4.5. J Computational Chemistry 2011, 32, 2031–2040. [DOI] [PubMed] [Google Scholar]
- 112.Hess B, P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [DOI] [PubMed] [Google Scholar]
- 113.Berendsen HJC; Postma JPM; Gunsteren W. F. v.; DiNola A; Haak JR, Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics 1984, 81, 3684–3690. [Google Scholar]
- 114.Bernardi RC; Melo MCR; Schulten K, Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim Biophys Acta 2015, 1850, 872–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hansmann UHE, Parallel tempering algorithm for conformational studies of biological molecules. Chemical Physics Letters 1997, 281, 140–150. [Google Scholar]
- 116.Nymeyer H, How Efficient Is Replica Exchange Molecular Dynamics? An Analytic Approach. Journal of Chemical Theory and Computation 2008, 4, 626–636. [DOI] [PubMed] [Google Scholar]
- 117.Yang YI; Shao Q; Zhang J; Yang L; Gao YQ, Enhanced sampling in molecular dynamics. Journal Chemical Physics. 2019, 151, 070902. [DOI] [PubMed] [Google Scholar]
- 118.Patriksson A; van der Spoel D, A temperature predictor for parallel tempering simulations. Physical Chemistry Chemical Physics 2008, 10, 2073–2077. [DOI] [PubMed] [Google Scholar]
- 119.Bussi G; Donadio D; Parrinello M, Canonical sampling through velocity rescaling. J. Chem. Phys 2007, 126. [DOI] [PubMed] [Google Scholar]
- 120.Parrinello M; Rahman A, polymorphic transitions in single-crystalls - a new molecular dynamics method J. Appl. Phys 1981, 52, 7182–7190. [Google Scholar]
- 121.Nosé S; Klein M, Constant pressure molecular dynamics for molecular systems. Mol. Phys 1983, 50, 1055–1076. [Google Scholar]
- 122.Humphrey W; Dalke A; Schulten K, VMD: visual molecular dynamics. Journal of molecular graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 123.Bressert E, SciPy and NumPy: an overview for developers. “ O’Reilly Media, Inc.”: 2012. [Google Scholar]
- 124.Theobald DL, Rapid calculation of RMSDs using a quaternion-based characteristic polynomial. Acta Crystallogr. Sect. A: Found. Crystallogr 2005, 61, 478–480. [DOI] [PubMed] [Google Scholar]
- 125.Novick PA; Ortiz OF; Poelman J; Abdulhay AY; Pande VS, SWEETLEAD: an in silico database of approved drugs, regulated chemicals, and herbal isolates for computer-aided drug discovery. PLoS One 2013, 8, e79568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Novick PA; Ortiz OF; Poelman J; Abdulhay AY; Pande VS, SWEETLEAD: an in silico database of approved drugs, regulated chemicals, and herbal isolates for computer-aided drug discovery. PLoS One 2013, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Goede A; Dunkel M; Mester N; Frommel C; Preissner R, SuperDrug: a conformational drug database. Bioinformatics 2005, 21, 1751–1753. [DOI] [PubMed] [Google Scholar]
- 128.Siramshetty VB; Eckert OA; Gohlke B-O; Goede A; Chen Q; Devarakonda P; Preissner S; Preissner R, SuperDRUG2: a one stop resource for approved/marketed drugs. Nucleic Acids Res. 2018, 46, D1137–D1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Repositories, N., In; 2013.
- 130.Ellingson SR; Smith JC; Baudry J, VinaMPI: Facilitating multiple receptor high-throughput virtual docking on high-performance computers. Journal of Compututational Chemistry 2013, 34, 2212–2221. [DOI] [PubMed] [Google Scholar]
- 131.El-Hachem N; Haibe-Kains B; Khalil A; Kobeissy FH; Nemer G AutoDock and AutoDockTools for protein-ligand docking: Beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) as a case study. In Neuroproteomics; Springer: 2017, pp 391–403. [DOI] [PubMed] [Google Scholar]
- 132.Morris GM; Huey R; Olson AJ, Using autodock for ligand-receptor docking. Current protocols in bioinformatics 2008, 24, 8.14. 1–8.14. 40. [DOI] [PubMed] [Google Scholar]
- 133.Zhu X; Mitchell JC, KFC2: a knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins: Structure, Function, and Bioinformatics 2011, 79, 2671–2683. [DOI] [PubMed] [Google Scholar]
- 134.Macalino SJY; Basith S; Clavio NAB; Chang H; Kang S; Choi S, Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery. Molecules 2018, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lagzian M; Valadan R; Saeedi M; Roozbeh F; Hedayatizadeh-Omran A; Amanlou M; Alizadeh-Navaei R, Repurposing naproxen as a potential antiviral agent against SARS-CoV-2. 2020, DOI: 10.21203/rs.3.rs-21833/v1. [DOI] [PubMed] [Google Scholar]
- 136.Dinesh DC; Chalupska D; Silhan J; Veverka V; Boura E, Structural basis of RNA recognition by the SARS-CoV-2 nucleocapsid phosphoprotein. bioRxiv 2020, 2020.04.02.022194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shivanyuk A; Ryabukhin S; Tolmachev A; Bogolyubsky A; Mykytenko D; Chupryna A; Heilman W; Kostyuk A, Enamine real database: Making chemical diversity real. Chemistry today 2007, 25, 58–59. [Google Scholar]
- 138.Forli W; Halliday S; Belew R; Olson AJ, In; Citeseer: 2012. [Google Scholar]
- 139.Gaillard T, Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J Chemical Infromation and Modeling 2018, 58, 1697–1706. [DOI] [PubMed] [Google Scholar]
- 140.Li H; Leung KS; Wong MH; Ballester PJ, Improving AutoDock Vina Using Random Forest: The Growing Accuracy of Binding Affinity Prediction by the Effective Exploitation of Larger Data Sets. Molecular Informatics 2015, 34, 115–26. [DOI] [PubMed] [Google Scholar]
- 141.GISAID. https://www.gisaid.org/
- 142.GISAID, GISAID-Homepage [Google Scholar]
- 143.Katoh K; Toh H, Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 2010, 26, 1899–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lawrence TJ; Kauffman KT; Amrine KCH; Carper DL; Lee RS; Becich PJ; Canales CJ; Ardell DH, FAST: FAST Analysis of Sequences Toolbox. Frontiers in Genetics 2015, 6, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shannon CE, A Mathematical Theory of Communication. Bell System Technical Journal 1948, 27, 379–423. [Google Scholar]
- 146.Morao I; Heifetz A; Fedorov DG Accurate Scoring in Seconds with the Fragment Molecular Orbital and Density-Functional Tight-Binding Methods. In Quantum Mechanics in Drug Discovery; Springer: 2020, pp 143–148. [DOI] [PubMed] [Google Scholar]
- 147.Nishimoto Y; Fedorov DG; Irle S, Density-functional tight-binding combined with the fragment molecular orbital method. Journal Chemical Theory and Compututation 2014, 10, 4801–4812. [DOI] [PubMed] [Google Scholar]
- 148.Barca GMJ; Bertoni C; Carrington L; Datta D; De Silva N; Deustua JE; Fedorov DG; Gour JR; Gunina AO; Guidez E; Harville T; Irle S; Ivanic J; Kowalski K; Leang SS; Li H; Li W; Lutz JJ; Magoulas I; Mato J; Mironov V; Nakata H; Pham BQ; Piecuch P; Poole D; Pruitt SR; Rendell AP; Roskop LB; Ruedenberg K; Sattasathuchana T; Schmidt MW; Shen J; Slipchenko L; Sosonkina M; Sundriyal V; Tiwari A; Galvez Vallejo JL; Westheimer B; Wloch M; Xu P; Zahariev F; Gordon MS, Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [DOI] [PubMed] [Google Scholar]
- 149.Fedorov DG; Kitaura K; Li H; Jensen JH; Gordon MS, The polarizable continuum model (PCM) interfaced with the fragment molecular orbital method (FMO). Journal of Chemical Theory and Computation. 2006, 27, 976–985. [DOI] [PubMed] [Google Scholar]
- 150.Zhan C-G; Chipman DM, Cavity size in reaction field theory. The Journal of chemical physics 1998, 109, 10543–10558. [Google Scholar]
- 151.Korber B; Fischer W; Gnanakaran SG; Yoon H; Theiler J; Abfalterer W; Foley B; Giorgi EE; Bhattacharya T; Parker MD, Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv 2020. [Google Scholar]
- 152.Bhowmik D; Gao S; Young MT; Ramanathan A, Deep clustering of protein folding simulations. BMC Bioinformatics 2018, 19, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Romero R; Ramanathan A; Yuen T; Bhowmik D; Mathew M; Munshi LB; Javaid S; Bloch M; Lizneva D; Rahimova A; Khan A; Taneja C; Kim S-M; Sun L; New MI; Haider S; Zaidi M, Mechanism of glucocerebrosidase activation and dysfunction in Gaucher disease unraveled by molecular dynamics and deep learning. Proceedings of the National Academy of Sciences 2019, 116, 5086–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee H; Turilli M; Jha S; Bhowmik D; Ma H; Ramanathan A DeepDriveMD: Deep-Learning Driven Adaptive Molecular Simulations for Protein Folding. In 2019 IEEE/ACM Third Workshop on Deep Learning on Supercomputers (DLS), 17–17 Nov. 2019, 2019; 2019; pp 12–19. [Google Scholar]
- 155.Wang R; Fang X; Lu Y; Wang S, The PDBbind database: Collection of binding affinities for protein− ligand complexes with known three-dimensional structures. J. Med. Chem 2004, 47, 2977–2980. [DOI] [PubMed] [Google Scholar]
- 156.Wang R; Fang X; Lu Y; Yang C-Y; Wang S, The PDBbind database: methodologies and updates. J. Med. Chem 2005, 48, 4111–4119. [DOI] [PubMed] [Google Scholar]
- 157.Cheng T; Li X; Li Y; Liu Z; Wang R, Comparative assessment of scoring functions on a diverse test set. Journal of Chemical Information & Modeling 2009, 49, 1079–1093. [DOI] [PubMed] [Google Scholar]
- 158.Li Y; Han L; Liu Z; Wang R, Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. Journal of Chemical Information & Modeling 2014, 54, 1717–1736. [DOI] [PubMed] [Google Scholar]
- 159.Li Y; Han L; Liu Z; Wang R, Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J Chem Inf Model 2014, 54, 1717–36. [DOI] [PubMed] [Google Scholar]
- 160.Li Y; Liu Z; Li J; Han L; Liu J; Zhao Z; Wang R, Comparative assessment of scoring functions on an updated benchmark: 1. Compilation of the test set. Journal of Chemical Information & Modeling 2014, 54, 1700–16. [DOI] [PubMed] [Google Scholar]
- 161.Huang N; Shoichet BK; Irwin JJ, Benchmarking Sets for Molecular Docking. J. Med. Chem 2006, 49, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mysinger MM; Carchia M; Irwin JJ; Shoichet BK, Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem 2012, 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bowes J; Brown AJ; Hamon J; Jarolimek W; Sridhar A; Waldron G; Whitebread S, Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nature Reviews Drug Discovery 2012, 11, 909–922. [DOI] [PubMed] [Google Scholar]
- 164.Ton AT; Gentile F; Hsing M; Ban F; Cherkasov A, Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Molecular informatics 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gorgulla C; Boeszoermenyi A; Wang Z-F; Fischer PD; Coote PW; Das KMP; Malets YS; Radchenko DS; Moroz YS; Scott DA, An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.