

Abstract
The aim of this study (NCT04343053) is to investigate the relationship between platelet activation, myocardial injury, and mortality in patients affected by Coronavirus disease 2019 (COVID-19). Fifty-four patients with respiratory failure due to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection were enrolled as cases. Eleven patients with the same clinical presentation, but negative for SARS-CoV-2 infection, were included as controls. Blood samples were collected at three different time points (inclusion [T1], after 7 ± 2 days [T2] and 14 ± 2 days [T3]). Platelet aggregation by light transmittance aggregometry and the circulating levels of soluble CD40 ligand (sCD40L) and P-selectin were measured. Platelet biomarkers did not differ between cases and controls, except for sCD40L which was higher in COVID-19 patients (p = .003). In COVID-19 patients, P-selectin and sCD40L levels decreased from T1 to T3 and were higher in cases requiring admission to intensive care unit (p = .004 and p = .008, respectively). Patients with myocardial injury (37%), as well as those who died (30%), had higher values of all biomarkers of platelet activation (p < .05 for all). Myocardial injury was an independent predictor of mortality. In COVID-19 patients admitted to hospital for respiratory failure, heightened platelet activation is associated with severity of illness, myocardial injury, and mortality.
ClinicalTrials.gov number: NCT04343053.
Keywords: COVID-19, myocardial injury, platelet aggregation, P-selectin, soluble CD40 ligand
Introduction
Pathophysiology of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is still largely unknown. Abnormalities in the thrombotic state in Coronavirus disease 2019 (COVID-19) patients have been highly debated [1–4]. Observational studies have suggested that pro-thrombotic alterations may be a significant driver of the pathological disease process [1–4]. Heightened D-dimer levels have been independently associated with the risk of mortality [2]. Consistently, autopsy reports revealed the presence of several pulmonary microthrombi [3]. Elevated levels of von Willebrand factor and soluble thrombomodulin were associated with lower rates of hospital discharge [4]. Platelets are involved in both hemostasis and immune response [5]. Baseline platelet levels and changes were associated with mortality in COVID-19 patients [1]. Platelets from COVID-19 patients were more activated, aggregated faster, and have increased expression of monocyte tissue factor [6,7]. Functional assays showed that platelets from COVID-19 patients are hyperresponsive, and sensitized to release inflammatory cytokines, and to adhere more efficiently [8]. Taken together, the data suggest that platelets may have the potential to contribute to the thrombo-inflammation in COVID-19 [5]. However, if these abnormalities in platelet aggregation of COVID-19 patients are associated with myocardial injury and then mortality, also after correction for potential confounding factors, is still unclear.
For these reasons, ex-vivo platelet activation was investigated in COVID-19 patients by measuring both plasmatic markers (soluble CD40 ligand and P-selectin) and light transmittance aggregometry. The association between these parameters and the occurrence of myocardial injury and mortality was finally assessed.
Methods
Study Design
The “Pro-thrombotic status in patients with SARS-Cov-2 infection” (ATTAC-Co) study is an investigator-initiated, prospective, single-center study recruiting consecutive patients admitted to hospital because of respiratory failure due to COVID-19 between April and May 2020. The patients were recruited at the Respiratory and Intensive Care Units specifically dedicated to the management of COVID-19 patients (COVID-19 Units) of the Azienda Ospedaliera Universitaria di Ferrara (Cona [FE], Italy). An electronic CRF was used to collect demographic characteristics, previous medical history, comorbid conditions, vital parameters (including arterial blood gas test), laboratory and imaging data, detailed information related to oxygen supplementation and concomitant treatments. In-hospital occurrence of adverse events was daily monitored, whereas after hospital discharge follow-up visits were performed every 30 days (last update June 30th, 2020). Three different blood samplings (at inclusion [T1], after 7 ± 2 days [T2] and 14 ± 2 days [T3]) for the evaluation of several biomarkers of platelet activation, coagulation cascade activation, fibrinolysis, endothelial dysfunction, and inflammation were withdrawn. Here we report the main findings related to platelet biomarkers (namely light transmittance aggregometry, P-selectin and soluble CD40 ligand). The protocol was approved by the corresponding Ethics Committee (Comitato Etico di Area Vasta Emilia Centro, Bologna, Italy). All patients gave their written informed consent. In case of unconsciousness, the informed consent was signed by the next of kin or legal authorized representative. The study is registered at www.clinicaltrials.gov with the identifier NCT04343053.
Study Population
Inclusion criteria were: i) age >18 years; ii) confirmed SARS-CoV-2 infection; iii) hospitalization for respiratory failure; iv) need for invasive or noninvasive mechanical ventilation or only oxygen support. A group of patients admitted to hospital between April and May 2020 with similar clinical characteristics in term of respiratory failure presentation, but negative for SARS-CoV-2 infection was also included as controls. Cases and controls were excluded from the study if any of the following criteria applied: prior administration of P2Y12 inhibitor (clopidogrel, ticlopidine, prasugrel, ticagrelor) or anticoagulant drugs (warfarin or novel oral anticoagulants), known disorder of coagulation or platelet function and/or chronic inflammatory disease. SARS-CoV-2 infection was confirmed by reverse-transcriptase-polymerase-chain-reaction assay (Liaison MDX, Diasorin, Saluggia, Italy) from nasopharyngeal swab specimen. Respiratory failure was defined as a PaO2/FiO2 (P/F) ratio ≤200 mmHg. As predefined, the cases were categorized according to severity of illness in those requiring admission to the intensive care unit (ICU, ICU cases) vs. others (non-ICU cases, admitted to COVID-19 respiratory unit). Clinical management was in accordance with current guidelines and specific recommendations for COVID-19 pandemic by Health Authorities and Scientific Societies.
Blood Samples
Study blood samplings were performed from an antecubital vein using a 21-gauge needle or from central venous line at T1, T2 and T3. In the control group one single blood sample at T1 was performed. All patients underwent blood sampling in the early morning, at least 12 hours after last administration of low weight molecular heparin (LWMH). The first 2 to 4 mL of blood was discarded, and the remaining blood was collected in sodium citrate tubes for determination of platelet aggregation and in tubes for serum/plasma collection. The serum and plasma samples were stored at −80°C.
Platelet Aggregation Assays
Platelet aggregation (PA) was assessed using light transmittance aggregometry and by measuring circulating values of soluble CD40 ligand (sCD40L) and P-selectin [9–12]. In brief, PA was measured at 37°C with AggRAM Advanced Modular System light transmittance aggregometer (Helena Laboratories, Beaumont, TX, USA) according to standard protocols [9–11]. Platelet-rich plasma (PRP) was obtained as a supernatant after centrifugation of citrated blood at 200 g for 10 min. The platelet count in PRP was adjusted to the range of 150.000 to 300.000/µL by dilution with autologous plasma when out of range. Platelet-poor plasma (PPP) was obtained by a second centrifugation of the blood fraction at 1500 g for 15 min. Light transmission was adjusted to 0% with PRP and to 100% for PPP for each measurement. PA was assessed after challenge with arachidonic acid (AA, 1 mM), adenosine diphosphate (ADP, 5 and 20 μmol/L) and thrombin receptor-activating peptide (TRAP, 5 μmol/L). Curves were recorded for 6 min, and maximal aggregation was measured. The plasma levels of soluble CD40 ligand (sCD40L) and P-selectin were determined with a bead-based multiplex immunoassay (EMD Millipore Burlington, MA, USA). These latter laboratory analyses were done from June 8 to June 30, 2020 in the Translational Research Center of the Maria Cecilia Hospital, Cotignola (RA), Italy.
Aims of the Study
The ATTAC-Co study primarily aims to evaluate the levels of the tested biomarkers in COVID19 patients presenting with respiratory failure and to explore the correlations between these biomarkers and the severity and outcomes of the disease. For the purposes of the present study the biomarkers were compared between: i) cases vs. controls ii) ICU vs. non-ICU cases, iii) cases developing myocardial injury vs. not, and iv) cases who died vs. alive. Myocardial injury was defined in presence of at least one value of elevated cardiac troponin above the upper reference limit (high sensitivity troponin I, URL 20 ng/L and 12 ng/L for man and woman, respectively) [13]. GC, MC and SS had full access to all the data in the study and take responsibility for its integrity and data analysis.
Statistical Analysis
The ATTAC-Co is a proof-of-concept study. Therefore, conducting a formal sample size computation was not possible. We assumed that 50 patients would be sufficient for our purpose. Continuous variables with normal distribution were expressed as mean ± SD. Continuous variables with a non-normal distribution were expressed as median and interquartile range. Normal distribution of the variables was tested with the Kolmogorov–Smirnov test. The variables normally distributed were compared by t test and 1-way ANOVA; otherwise the Mann-Whitney U and Kruskal-Wallis tests were used. Categorical variables were summarized in terms of numbers and percentages and were compared by using the two-sided Fisher’s exact test. To model continuous response variable represented by serial measurements over time within subject we set up a linear mixed effect model taking into account for correlations between measurements on the same subject to have optimal model fits and honest inference. Parameter estimation was performed by restricted maximum likelihood, random intercept with fixed mean. For each response variable a full model for overall population and stratified by ICU vs. non-ICU, myocardial injury vs. non myocardial injury, and non-survivors vs. survivors containing potential confounders was built. Potential confounders were identified as the variables with a p-value <0.1 for the comparison of subgroups in Table I, supplemental Table 2 and supplemental Table 4, respectively. Backward stepwise selection was carried out to remove poor informative variable evaluating Akaike’s information criterion (AIC) and log-likelihood ratio test significance. Once models were optimized, estimated marginal effect were calculated and plotted as response variable predicted values against time. Myocardial injury was tested using Cox regression as predictor of mortality. Variables showing a p-value <0.1 in the supplemental Table 2 were included in the multivariable model. Independent predictors were selected with backward stepwise modeling approach. Variables remaining significant at a threshold p-value ≤0.05 were retained as final predictors. For all comparisons, a p-value of <0.05 was considered statistically significant. When appropriate, 95% confidence intervals (CIs) were calculated. All analyses were performed with R version 3.5.1 by an independent statistician (MM).
Table I.
Baseline characteristics and values of platelet biomarkers in controls vs. cases
| Controls (n = 11) |
Cases (n = 54) |
p1 | Non-ICU Cases (n = 17) |
ICU Cases (n = 37) |
p2 | |
|---|---|---|---|---|---|---|
| Age, (years) | 70 [66–80] | 65 [57–73] | 0.244 | 73 [69–81] | 64 [57–70] | 0.008 |
| Male sex, no. (%) | 8 (73) | 40 (74) | 0.999 | 9 (53) | 31 (84) | 0.023 |
| BMI (kg/m2) | 24.3 [21.7–27.0] | 26.4 [24.2–30.2] | 0.169 | 24.5 [22.9–28.7] | 27.7 [25.5–30.9] | 0.076 |
| Comorbidities, no. (%) | ||||||
| Hypertension | 5 (45) | 30 (55) | 0.741 | 10 (58) | 20 (54) | 0.776 |
| Dyslipidemia | 5 (45) | 11 (20) | 0.120 | 6 (35) | 5 (13) | 0.080 |
| Active smoker | 3 (27) | 0 (0) | 0.003 | 0 (0) | 0 (0) | 0.999 |
| Former smoker | 4 (36) | 16 (30) | 0.725 | 5 (29) | 11 (29) | 0.999 |
| Diabetes | 3 (27) | 7 (13) | 0.353 | 2 (11) | 5 (13) | 0.999 |
| Prior MI | 0 (0) | 3 (6) | 0.999 | 2 (11) | 1 (3) | 0.230 |
| Prior coronary revascularization | 0 (0) | 3 (6) | 0.999 | 2 (11) | 1 (3) | 0.314 |
| Prior CVA | 1 (9) | 2 (4) | 0.432 | 2 (11) | 0 (0) | 0.095 |
| Peripheral artery disease | 2 (18) | 8 (15) | 0.673 | 5 (29) | 3 (8) | 0.092 |
| COPD | 4 (36) | 5 (9) | 0.037 | 1 (6) | 4 (11) | 0.999 |
| Home medical therapy, no. (%) | ||||||
| Aspirin | 1 (9) | 5 (9) | 0.999 | 2 (12) | 3 (8) | 0.645 |
| ACE inhibitors | 4 (36) | 21 (39) | 0.999 | 7 (41) | 14 (38) | 0.999 |
| Beta-blockers | 2 (18) | 12 (22) | 0.999 | 6 (35) | 6 (16) | 0.161 |
| Calcium channel blockers | 1 (9) | 9 (17) | 0.999 | 1 (6) | 8 (22) | 0.244 |
| Statins | 2 (18) | 9 (17) | 0.999 | 5 (29) | 4 (11) | 0.120 |
| Laboratory data at inclusion | ||||||
| WBC, (u x103/L) | 12.1 [9.6–15.1] | 9.1 [5–15] | 0.121 | 7.1 [6–10.5] | 9.6 [7.6–12.7] | 0.106 |
| Lymphocytes, (u x 103/L) | 0.86 [0.71–1.75] | 0.80 [0.52–1.06] | 0.217 | 0.87 [0.47–0.99] | 0.74 [0.54–1.16] | 0.648 |
| Hemoglobin, (g/dl) | 12.1 [11.4–13.7] | 10.7 [9.6–12.3] | 0.071 | 11.7 [11.2–14.3] | 10.5 [9.1–11.6] | 0.001 |
| Creatinine clearance (mg/dl) | 87.2 [45.6–108.6] | 88.1 [74.1–106.2] | 0.391 | 80.4 [58.9–89.9] | 97.6 [76.1–109.5] | 0.045 |
| Platelets, (u x103/L) | 253.5 [162–277] | 293 [245–382] | 0.072 | 310 [181–401] | 291 [247–373] | 0.754 |
| Fibrinogen, (mg/dl) | 464 [452–802] | 708 [576–823] | 0.275 | 632 [439–745] | 726.5 [609–862] | 0.088 |
| D-dimer (mg/L FEU) | 0.85 [0.70–1.7] | 2.3 [1.2–4.2] | 0.064 | 1 [0.50–1.75] | 2.9 [1.6–4.3] | 0.002 |
| C-reactive protein (mg/dl) | 5.7 [3.4–10.3] | 12.2 [5.4–20.5] | 0.087 | 5.4 [1.4–11.6] | 14.4 [10.4–21.4] | 0.001 |
| HS troponin I (ng/L) | 9 [3–18] | 15.5 [8–52] | 0.001 | 10.5 [7–15.5] | 18.5 [9–60] | 0.096 |
| Platelet biomarkers | ||||||
| AA-induced max PA, % | 43 [31–48] | 39 [30–54] | 0.879 | 31.5 [28.5–47.5] | 41 [33–56] | 0.128 |
| ADP 5-induced max PA, % | 38 [35–39] | 44 [36–53] | 0.109 | 38 [32–46] | 48 [40–55] | 0.023 |
| ADP 20-induced max PA, % | 53 [33–61] | 58 [45–65] | 0.234 | 48 [42–63] | 61 [51–69] | 0.040 |
| TRAP-induced max PA, % | 57 [45–71] | 57.5 [46–76] | 0.680 | 51 [43–58] | 66 [47–78] | 0.053 |
| P-selectin, (ng/ml) | 75.7 [63.1–89.6] | 74 [50–113] | 0.944 | 50.1 [37.9–78.8] | 81.2 [55.2–134.9] | 0.009 |
| sCD40L, (pg/ml) | 84.7 [46.4–244.6] | 398.2 [167.3–859.6] | 0.003 | 231.2 [97.4–415.5] | 432.5 [264.9–1008.5] | 0.007 |
p1: for the comparison between controls vs. cases.
p2: for the comparison between non-ICU vs ICU cases
BMI: body mass index. MI: myocardial infarction. CVA: cerebrovascular accident. COPD: chronic obstructive disease. ACE: angiotensin converting enzyme. WBC: white blood cells. FEU: fibrinogen equivalent unit. HS: high sensitivity. AA: arachidonic acid. ADP: adenosine diphosphate. TRAP: thrombin receptor agonist peptide. sCD40L: soluble CD40 ligand.
Results
Overall, 65 patients were included in the final study cohort (Table I). The cases (COVID-19 patients) were 54. The median time from symptom’s onset to hospital admission was 9 [5–11] days. The median simplified acute physiology score (SAPS) II and sequential organ failure assessment (SOFA) values were 27 [21–26] and 3 [2–5], respectively. Median hospital stay was 35 [20–50] days. Seventeen (31%) patients were admitted and treated in the COVID-19 pneumological unit, whereas 37 (69%) required admission to the COVID-19 ICU (Table I). Invasive mechanical ventilation was required in 27 (50%) cases, whereas 16 (30%) needed noninvasive mechanical ventilation. Patients admitted to ICU were younger, pre-dominantly male, with lower hemoglobin values, but greater d-dimer and C-reactive protein values (Table I). Median stay in ICU was 24 [14–33] days. All patients received 4000–8000 IU enoxaparin twice daily. Eleven patients were included in the control group and their baseline characteristics did not differ from the cases group (Table I and Supplemental Table 1).
Platelet Activation in Controls Vs. Cases
Independently to the agonist used, platelet aggregation values did not differ between cases and controls (Table I). Similarly, we did not observe any significant difference in P-selectin (Table I). On the contrary, sCD40L was significantly higher in COVID-19 patients (p = .003) (Table I).
Platelet Activation in Cases
The values of platelet aggregation were stable overtime from T1 to T3 (Table II). On the contrary, P-selectin and sCD40L values significantly reduced from T1 to T3 (p = .044 and p = .039, respectively) (Table II). Median values of platelet aggregation, P-selectin and sCD40L stratified in ICU vs. non-ICU patients were shown in Table II. After correction for potential confounding factors, the values of platelet aggregation did not differ between ICU vs non-ICU patients (Figure 1). On the contrary, P-selectin and sCD40L values were higher in ICU vs. non-ICU patients (Figure 1).
Table II.
Platelet biomarkers in COVID-19 patients
| T1 | T2 | T3 | p | ||
|---|---|---|---|---|---|
| AA-induced max PA, % | all | 39 [30–54] | 37 [32–45] | 39 [35–46] | 0.824 |
| non-ICU | 31.5 [28.5–47.5] | 28 [25–41] | 35 [30.5–44] | ||
| ICU | 41 [33–56] | 38 [33] | 39 [35–46] | ||
| ADP 5-induced max PA, % | all | 44 [36–53] | 46 [40–51] | 43 [37–48] | 0.321 |
| non-ICU | 38 [32–46] | 38 [33–52] | 39.5 [35–43] | ||
| ICU | 48 [40–55] | 48 [42–50] | 43 [38–48] | ||
| ADP 20-induced max PA, % | all | 58 [45–65] | 60 [48–64] | 52 [47–58] | 0.290 |
| non-ICU | 48 [42–63] | 47 [41–64] | 50 [44–53.5] | ||
| ICU | 61 [54–65] | 61 [54–65] | 56 [47–59] | ||
| TRAP-induced max PA, % | all | 57.5 [46–76] | 60.5 [50–71] | 54 [48–60] | 0.497 |
| non-ICU | 51 [43–58] | 51 [43–58] | 46.5 [44–58.5] | ||
| ICU | 66 [47–78] | 62 [54–71] | 56 [51–64] | ||
| P-selectin, (ng/ml) | all | 74 [50–113] | 69.6 [40.5–104.1] | 63.2 [39.8–87.7] | 0.044 |
| non-ICU | 50.1 [37.8–78.8] | 52.6 [31.2–68.4] | 37.9 [26.8–80.4] | ||
| ICU | 81.2 [55.2–134.9] | 78.8 [58.1–122.8] | 65.8 [45.1–92.1] | ||
| sCD40L, (pg/ml) | all | 398 [167–859.6] | 340.6 [76.1–642.8] | 164.1 [79.7–556.9] | 0.039 |
| non-ICU | 231.1 [97.4–415.5] | 65.9 [23.4–208.1] | 72.9 [46.3–164] | ||
| ICU | 432.4 [264.9–1008.5] | 463.1 [108.8–1028.9] | 192.3 [87.8–756.9] |
The values are reported as median and interquartile range. The p-value is for the trend from T1 to T3.
ICU: intensive care unit. AA: arachidonic acid. ADP: adenosine diphosphate. TRAP: thrombin receptor agonist peptide. sCD40L: soluble CD40 ligand.
Figure 1.
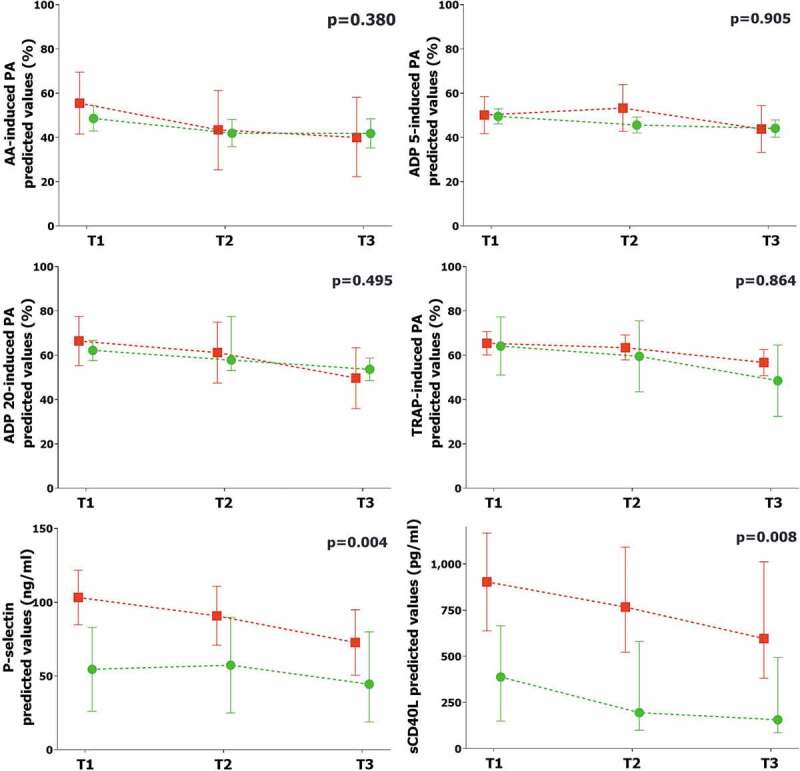
Comparison of platelet biomarkers in ICU vs. non-ICU patients
The graphs represent the marginal effect plots. The squares and the circles represent the marginal effect at the specific time point. The whiskers are the 95%CI. The lines between squares and circles represent the trend over time. The p-value in the upper right portion of each figure represents the overall difference over time between subgroups.Red line and square: ICU patients. Green line and circle: non-ICU patients.ICU: intensive care unit. AA: arachidonic acid. ADP: adenosine diphosphate. TRAP: thrombin receptor agonist peptide. sCD40L: soluble CD40 ligand.
Platelet Activation and Myocardial Injury
Myocardial injury was observed in 20 (37%) patients (18% in non-ICU patients vs. 46% in ICU patients, p = .068). Baseline characteristics stratified according to the occurrence of myocardial injury are shown in Supplemental Table 2. Daily trend of high sensitivity troponin I is reported in Supplemental Figure 1. The values of all platelet aggregation, P-selectin and sCD40L, after correction for potential confounding factors, were higher and differed over time in patients with myocardial injury compared to others (Figure 2).
Figure 2.
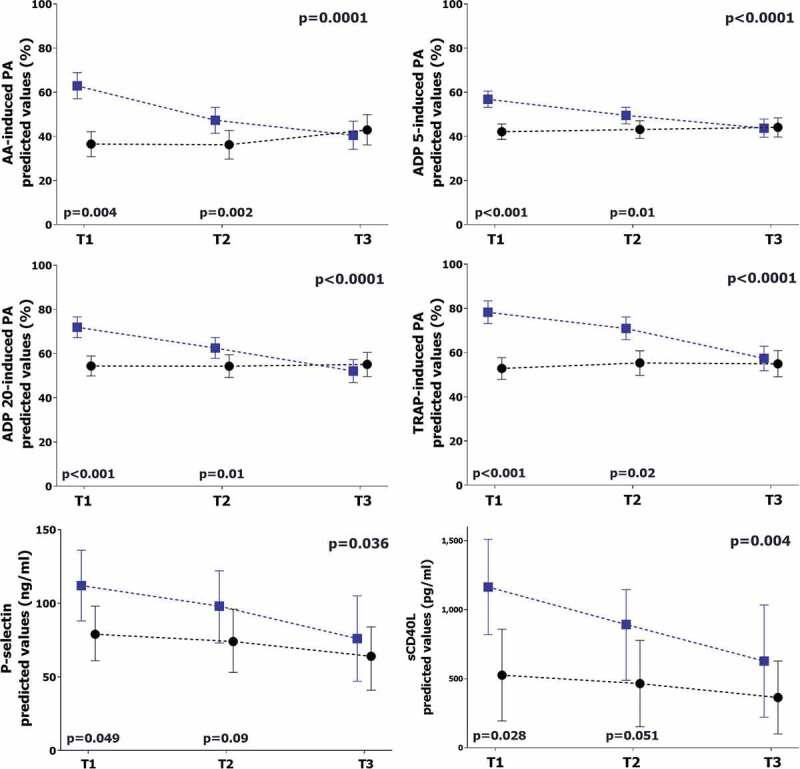
Comparison of platelet biomarkers in patients with or without myocardial injury
The graphs represent the marginal effect plots. The squares and the circles represent the marginal effect at the specific time point. The whiskers are the 95%CI. The lines between squares and circles represent the trend over time. The p-value in the upper right portion of each figure represents the overall difference over time between subgroups. The p-values in the lower portion of each figure represent the difference in the single time point.Blue line and square: patients with myocardial injury. Black line and circle: patients without myocardial injury.AA: arachidonic acid. ADP: adenosine diphosphate. TRAP: thrombin receptor agonist peptide. sCD40L: soluble CD40 ligand.
Platelet Activation and Mortality
During hospitalization, one (2%) patient suffered from an ischemic stroke and 3 (5.5%) patients developed deep venous thrombosis and pulmonary embolism. No patient met the criteria for myocardial infarction. Overall, 16 (30%) patients died (23% in non-ICU patients vs. 32% in ICU patients, p = .749). Baseline characteristics and median values of platelet biomarkers stratified according to occurrence of mortality are shown in Supplemental Tables 4 and 5. After correction for confounding factors, platelet aggregation, P-selectin and sCD40L values were higher and with a different trend over time in patients who died compared to those who survived (Figure 3). Mortality was significantly higher in patients with myocardial injury vs. others (60% vs 11.7%, p < .001) (Supplemental Figure 2). After correction for potential confounding factors, myocardial injury emerged as an independent predictor of mortality (HR 6.28, 95%CI 1.65–23.92, p = .007) (Supplemental Table 6).
Figure 3.
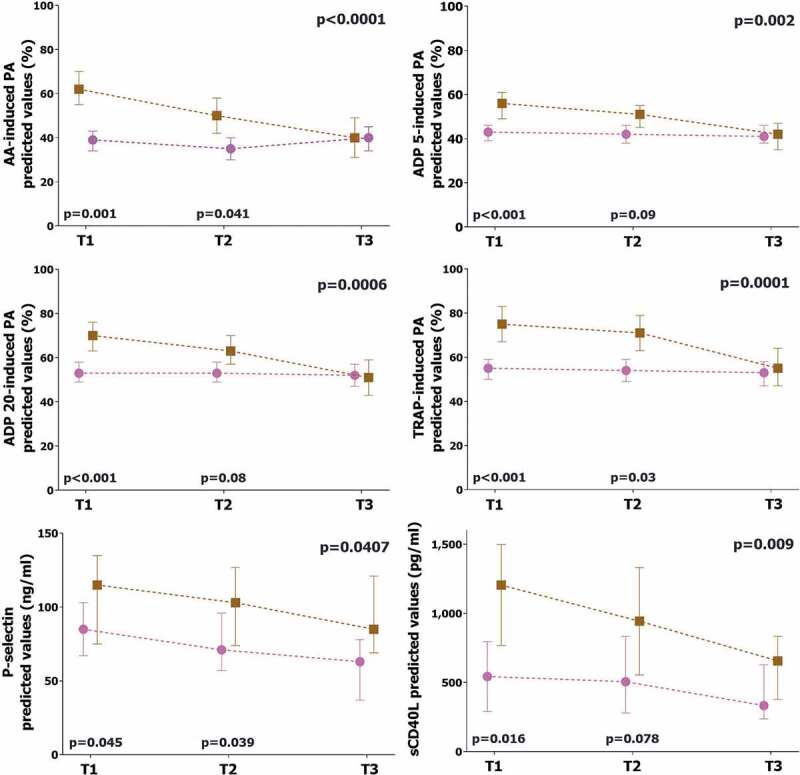
Comparison of platelet biomarkers in survivors vs. non-survivors
The graphs represent the marginal effect plots. The squares and the circles represent the marginal effect at the specific time point. The whiskers are the 95%CI. The lines between squares and circles represent the trend over time. The p-value in the upper right portion of each figure represents the overall difference over time between subgroups. The p-values in the lower portion of each figure represent the difference in the single time point.Brown line and square: non-survivors. Pink line and circle: survivors.AA: arachidonic acid. ADP: adenosine diphosphate. TRAP: thrombin receptor agonist peptide. sCD40L: soluble CD40 ligand.
Discussion
The present analysis of the ATTAC-Co study showed that:
In patients with moderate-severe respiratory failure, there was no difference in biomarkers of platelet activation, except for sCD40L, between those COVID-19 positive and negative.
In COVID-19 patients, platelet aggregation values were stable through the 14-day study period, whereas P-selectin and sCD40Lvalues decreased from T1 to T3.
Platelet aggregation did not vary in relation to the severity of illness, whereas P-selectin and sCD40L values were heightened in patients with more severe disease.
Higher values of platelet aggregation, P-selectin and sCD40L were associated with myocardial injury and mortality.
Prior studies reported significant alterations in platelet activation in patients with hospital acquired pneumonia or severe sepsis [11,12,14]. These alterations were associated with changes in inflammation, coagulation and endothelial function and no different response was generally observed across causative micro-organisms [14]. The infection by SARS-CoV-2 appears to confirm this paradigm. Preliminary studies showed that patients with COVID-19 have an altered and heightened platelet activation compared to healthy subjects [4,6–8]. It has been shown that SARS-CoV-2 and its Spike protein directly stimulated platelets to facilitate the release of coagulation factors, the secretion of inflammatory factors, and the formation of leukocyte-platelet aggregates [15]. Our study confirms the finding that COVID-19 is associated with modification in platelet activation and aggregation. However, these alterations are not specific for COVID-19 patients and are also observed in patients admitted with respiratory failure due to causes other than SARS-CoV-2 infection. The exception is in the values of sCD40L, which is significantly elevated in COVID-19 patients.
Previous studies suggested that alterations in platelet aggregation of COVID-19 patients are correlated to the severity of illness [4,6–8]. Critically ill COVID-19 patients showed higher circulating values of several inflammation and thrombosis markers, including P-selectin and sCD40L [4,6–8]. Regarding platelet aggregation values, we did not find differences between ICU vs. non-ICU COVID-19 patients. However, it should be noted that COVID-19 patients admitted to ICU differed from others in several baseline characteristics (age, sex, BMI, medical history, laboratory data). All these parameters significantly affect the platelet aggregation [16]. Thus, it is not surprising that after correction for confounding factors the differences between ICU and non-ICU COVID-19 patients were not confirmed. On the contrary, P-selectin and sCD40L values were influenced by COVID-19 severity, being heightened in ICU patients. This finding is consistent with previous studies and it could be explained by the fact that P-selectin and sCD40L are not platelet-specific markers but can be released by other cells (i.e. endothelial cells). The CD40-CD40 ligand system is widely distributed on leukocytic and non-leukocytic cells, including endothelial and smooth-muscle cells, and on activated platelets [17]. The sCD40L is proinflammatory for endothelial cells and promotes coagulation by inducing expression of tissue factor on monocytes [17]. The sCD40L is actively released after platelet stimulation, it interacts with the glycoprotein IIb/IIIa receptor, and it is necessary for the stability of arterial thrombi [17]. P-selectin functions as cell adhesion molecule on the surfaces of activated endothelial cells and platelets [17]. During inflammation, P-selectin moves from an internal cell location to surfaces mediating recruitment of leukocytes and platelet aggregation [18]. Accordingly, the possible explanation for the higher P-selectin and sCD40L observed values could be outside from platelet-specific mechanisms. We can hypothesize that platelets act as amplifiers in the COVID-19 inflammatory burden [8]. This could be further supported by the observed over time reduction of sCD40L and P-selectin levels from T1 to T3. Alternatively, the higher values could be explained by a stronger involvement of the endothelium in the COVID-19 disease [4,5,15].
The major strength and novelty of our analysis is the association between heightened platelet activation and myocardial injury. This relationship is strong and consistent with all the biomarkers. From the first reports on COVID-19 patients, myocardial injury has been reported in around 20%-30% of cases, and it was associated with mortality [19]. The pathophysiological mechanisms underlying myocardial injury caused by COVID-19 are still under investigation. Hypotheses include direct damage to the cardiomyocytes, systemic inflammation, myocardial interstitial fibrosis, interferon mediated immune response, exaggerated cytokine response by Type 1 and 2 helper T cells, in addition to coronary plaque destabilization, and hypoxia. Autopsy studies suggested the presence of the viral genome in the myocardial tissue [20]. Particularly, viral infection seems to involve the endothelial compartment and may cause capillary endothelial cell/microvascular dysfunction [4,21]. Previous findings in patients with influenza pneumonia revealed that, during the initial phases, virus replication and dissemination is associated with platelet activation, generation of neutrophil-platelet aggregates, and fibrin/micro-thrombus formation [22,23]. A similar pattern seems to be present in the SARS-CoV-2 infection. We may speculate that SARS-CoV-2 infection may induce endothelial injury and concomitant platelet activation leading to micro-thrombus formation and then myocardial injury. This pro-aggregating status contributes to the observed higher mortality. If confirmed, this hypothesis could have important clinical implications. Ongoing randomized clinical trials (RCTs) are trying to validate the safety and efficacy of higher doses of LMWH [NCT04373707, NCT04401293, NCT04372589]. Our data suggests that antiplatelet agents could be an intriguing alternative, especially in the early phase of infection. Supportive of this hypothesis is the finding that patients admitted to hospital with community-onset pneumonia who were established on chronic aspirin therapy showed lower mortality rates as compared to those who were aspirin-free [24]. The XANTHIPPE (Targeting Platelet-Leukocyte Aggregates in Pneumonia with Ticagrelor) study showed that ticagrelor reduced leukocytes with attached platelets as well as inflammatory biomarkers [25]. Tirofiban significantly improved the ventilation/perfusion ratio in 5 COVID-19 patients with severe respiratory failure [26]. Obviously, this hypothetical benefit should be balanced with the bleeding risk. The later phase of the most severe COVID-19 cases is associated with diffuse alveolar hemorrhage, reduction in platelet count, depletion of coagulation factors, followed by disseminated intravascular coagulation (DIC). In these cases, the bleeding complications overwhelm the thrombotic complications and antiplatelet agents, as well as high dose of LMWH, can be counterproductive. Consequently, the results of ongoing clinical trials are eagerly awaited to answer this unmet clinical need [NCT04365309, NCT04410328, NCT04324463, NCT04409834].
Study Limitations
This is a “proof-of-concept” hypothesis-generating study and several limitations should be considered. This is a single-center study in COVID-19 patients with respiratory failure. Our findings should be confirmed in a larger multi-center scenario. We included COVID-19 patients with respiratory failure. Our data is not applicable to asymptomatic or mild-symptoms COVID-19 patients. Although we performed three different assessments at three different time-points, we did not cover the entire disease’s course and we cannot capture potential alterations before hospital admission. We measured a limited number of platelet biomarkers (light transmittance aggregometry, sCD40L, P-selectin). This selection was based on previous studies on similar topic [12,27], but it should be noted that many other quantitative and functional assays are available for the investigation of platelet activation [4–8]. In addition, it should be remarked that sCD40L and P-selectin can be released from other cells aside from platelets. Future studies are clearly on demand to confirm and expand our findings and to better clarify the mechanisms behind myocardial injury in COVID-19 patients.
Conclusions
In COVID-19 patients admitted to hospital for moderate-severe respiratory failure, higher values of platelet activation were observed in patients with more severe disease, myocardial injury, as well as in non-survivors.
Funding Statement
The study was an investigator-driven clinical trial conducted by the University of Ferrara.
Author Contribution Statement
Conceived and designed the research: Gianluca Campo, Marco Contoli, Savino Spadaro. Acquired the data: Ottavio Zucchetti, Luca Ronzoni, Luca Morandi, Marco Verri, Simone Biscaglia, Rita Pavasini, Luca Di Ienno, Emanuele D’Aniello. Perfomed laboratory analyses: Francesco Vieceli Dalla Sega, Francesca Fortini, Paola Rizzo. Performed statistical analysis: Marco Manfrini. Handled funding and supervision: Roberto Ferrari, Roberto Zoppellari. Drafted the manuscript: Gianluca Campo, Marco Contoli, Savino Spadaro, Alberto Fogagnolo. Made critical revision of the manuscript for key intellectual content: Carlo Alberto Volta, Alberto Papi.
Declaration Of Interest
No authors reported conflict of interest.
DATA AVAILABILITY STATEMENT
The data underlying this article will be shared on reasonable request by contacting the corresponding author.
References
- 1.Liu Y, Sun W, Guo Y, Chen L, Zhang L, Zhao S, Long D, Yu L.. Association between platelet parameters and mortality in coronavirus disease 2019: retrospective cohort study. Platelets 2020;31(4):490–496. doi: 10.1080/09537104.2020.1754383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang L, Yan X, Fan Q, Liu H, Liu X, Liu Z, Zhang Z. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J Thromb Haemost 2020;18(6):1324–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, Rech R, Colombo R, Antinori S, Corbellino M, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis 2020;20(10):1135–1140. doi: 10.1016/S1473-3099(20)30434-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, Baluha A, Bar N, Bona RD, Burns AJ, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 2020;7(8):e575–e582. doi: 10.1016/S2352-3026(20)30216-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinod K, Deppermann C. Immunothrombosis and thromboinflammation in host defense and disease. Platelets 2020. Sep 8:1–11. Epub ahead of print.;.. doi: 10.1080/09537104.2020.1817360 [DOI] [PubMed] [Google Scholar]
- 6.Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben CJ, Petrey AC, Tolley ND, Guo L, Cody MJ, et al. Platelet gene expression and function in COVID-19 patients. Blood 2020;136(11):1317–1329. doi: 10.1182/blood.2020007214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, Righy C, Franco S, Souza TML, Kurtz P, et al. Platelet activation and platelet-monocyte aggregates formation trigger tissue factor expression in severe COVID-19 patients. Blood 2020;136(11):1330–1341. doi: 10.1182/blood.2020007252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, Limami Y, Zaid N, Sadki K, Ben El Haj R, et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res 2020. September 17;127(11):1404–1418. Epub ahead of print. doi: 10.1161/CIRCRESAHA.120.317703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campo G, Pavasini R, Biscaglia S, Ferri A, Andrenacci E, Tebaldi M, Ferrari R. Platelet aggregation values in patients with cardiovascular risk factors are reduced by verbascoside treatment. A randomized study. Pharmacol Res 2015;97:1–6. doi: 10.1016/j.phrs.2015.03.020 [DOI] [PubMed] [Google Scholar]
- 10.Campo G, Valgimigli M, Gemmati D, Percoco G, Tognazzo S, Cicchitelli G, Catozzi L, Malagutti P, Anselmi M, Vassanelli C, et al. Value of platelet reactivity in predicting response to treatment and clinical outcome in patients undergoing primary coronary intervention: insights into the STRATEGY Study. J Am Coll Cardiol 2006;48:2178–2185. doi: 10.1016/j.jacc.2005.12.085 [DOI] [PubMed] [Google Scholar]
- 11.Fogagnolo A, Taccone FS, Campo G, Montanari G, Capatti B, Ferraro G, Erriquez A, Ragazzi R, Creteur J, Volta CA, et al. Impaired platelet reactivity in patients with septic shock: a proof-of-concept study. Platelets 2020;31(5):652–660. doi: 10.1080/09537104.2019.1663807 [DOI] [PubMed] [Google Scholar]
- 12.Cangemi R, Casciaro M, Rossi E, Calvieri C, Bucci T, Calabrese CM, Taliani G, Falcone M, Palange P, Bertazzoni G, et al. Platelet activation is associated with myocardial infarction in patients with pneumonia. J Am Coll Cardiol 2014;64(18):1917–1925. doi: 10.1016/j.jacc.2014.07.985 [DOI] [PubMed] [Google Scholar]
- 13.Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, White HD. Fourth Universal Definition of Myocardial Infarction (2018). J Am Coll Cardiol 2018;72:2231–2264. doi: 10.1016/j.jacc.2018.08.1038 [DOI] [PubMed] [Google Scholar]
- 14.Kinasewitz GT, Yan SB, Basson B, Comp P, Russell JA, Cariou A, Um SL, Utterback B, Laterre PF, Dhainaut JF. Universal changes in biomarkers of coagulation and inflammation occur in patients with severe sepsis, regardless of causative micro-organism [ISRCTN74215569]. Crit Care 2004;8(2):R82–90. doi: 10.1186/cc2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, Liu M, Zhao X, Xie Y, Yang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol 2020;13(1):120. doi: 10.1186/s13045-020-00954-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maree AO, Fitzgerald DJ. Variable platelet response to aspirin and clopidogrel in atherothrombotic disease. Circulation. 2007;115:2196–2207. doi: 10.1161/CIRCULATIONAHA.106.675991 [DOI] [PubMed] [Google Scholar]
- 17.Heeschen C, Dimmeler S, Hamm CW, van den Brand MJ, Boersma E, Zeiher AM, Simoons ML. CAPTURE Study Investigators. Soluble CD40 ligand in acute coronary syndromes. N Engl J Med 2003;348(12):1104–1111. doi: 10.1056/NEJMoa022600 [DOI] [PubMed] [Google Scholar]
- 18.André P. P-selectin in haemostasis. Br J Haematol 2004;126(3):298–306. doi: 10.1111/j.1365-2141.2004.05032.x [DOI] [PubMed] [Google Scholar]
- 19.Lala A, Johnson KW, Januzzi JL, Russak AJ, Paranjpe I, Richter F, Zhao S, Somani S, Van Vleck T, Vaid A, et al. Prevalence and impact of myocardial injury in patients hospitalized with COVID-19 infection. J Am Coll Cardiol 2020;76:533–546. doi: 10.1016/j.jacc.2020.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindner D, Fitzek A, Bräuninger H, Aleshcheva G, Edler C, Meissner K, Scherschel K, Kirchhof P, Escher F, Schultheiss HP, Blankenberg S, Püschel K, Westermann D. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020 Nov 1;5(11):1281-1285. doi: 10.1001/jamacardio.2020.3551.PMID:32730555;PMCID:PMC7385672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fox SE, Li G, Akmatbekov A, Harbert JL, Lameira FS, Brown JQ, Vander Heide RS. Unexpected features of cardiac pathology in COVID-19 infection. Circulation 2020;142(11):1123–1125. doi: 10.1161/CIRCULATIONAHA.120.049465 [DOI] [PubMed] [Google Scholar]
- 22.Lê VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin JL, Adrian I, Errazuriz-Cerda E, Frasquilho S, Antunes L, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med 2015;191(7):804–819. doi: 10.1164/rccm.201406-1031OC [DOI] [PubMed] [Google Scholar]
- 23.Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, Harris ES, Weyrich AS. In vivo platelet activation in critically ill patients with primary 2009 influenza A(H1N1). Chest 2012;141(6):1490–1495. doi: 10.1378/chest.11-2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falcone M, Russo A, Cangemi R, Farcomeni A, Calvieri C, Barillà F, Scarpellini MG, Bertazzoni G, Palange P, Taliani G, et al. Lower mortality rate in elderly patients with community-onset pneumonia on treatment with aspirin. J Am Heart Assoc 2015;4(1):e001595. doi: 10.1161/JAHA.114.001595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sexton TR, Zhang G, Macaulay TE, Callahan LA, Charnigo R, Vsevolozhskaya OA, Li Z, Smyth S. Ticagrelor reduces thromboinflammatory markers in patients with Pneumonia. JACC Basic Transl Sci 2018;3(4):435–449. doi: 10.1016/j.jacbts.2018.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viecca M, Radovanovic D, Forleo GB, Santus P. Enhanced platelet inhibition treatment improves hypoxemia in patients with severe Covid-19 and hypercoagulability. A case control, proof of concept study. Pharmacol Res 2020;158:104950. doi: 10.1016/j.phrs.2020.104950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chandler AB, Earhart AD, Speich HE, Kueter TJ, Hansen J, White MM, Jennings LK. Regulation of CD40L (CD154) and CD62P (p-selectin) surface expression upon GPIIb-IIIa blockade of platelets from stable coronary artery disease patients. Thromb Res 2010;125(1):44–52. doi: 10.1016/j.thromres.2009.04.017 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data underlying this article will be shared on reasonable request by contacting the corresponding author.