

Abstract
Stargardt macular degeneration (STGD1) is caused by mutations in the gene encoding ABCA4, an ATP-binding cassette protein that transports N-retinylidene-phosphatidylethanolamine (N-Ret-PE) across photoreceptor membranes. Reduced ABCA4 activity results in retinoid accumulation leading to photoreceptor degeneration. The disease onset and severity vary from severe loss in visual acuity in the first decade to mild visual impairment late in life. We determined the effect of 22 disease-causing missense mutations on the expression and ATPase activity of ABCA4 in the absence and presence of N-Ret-PE. Three classes were identified that correlated with the disease onset in homozygous STGD1 individuals: Class 1 exhibited reduced ABCA4 expression and ATPase activity that was not stimulated by N-Ret-PE; individuals homozygous for these variants had an early disease onset (≤ 13 yr); Class 2 showed reduced ATPase activity with limited stimulation by N-Ret-PE; these correlated with a moderate disease onset (14 – 40 yr); Class 3 displayed high expression and ATPase activity that was strongly activated by N-Ret-PE; these were associated with a late disease onset (> 40 yr). Based on our results, we introduce a functionality index for gauging the effect of missense mutations on STGD1 severity. Our studies support the mild phenotype exhibited by the p.Gly863Ala, p.Asn1868Ile, and p.Gly863Ala/p.Asn1868Ile variants.
Keywords: ABCA4, Stargardt macular degeneration, Missense mutations, Hypomorphic variants, ATP-binding cassette transporters, ATPase activity
Graphical Abstract
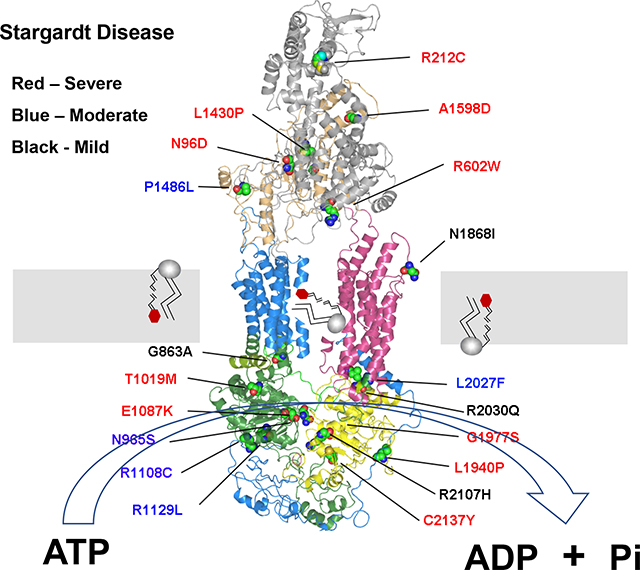
Autosomal recessive Stargardt disease (STGD1; MIM# 248200) is the most common inherited macular degeneration with a prevalence of 1:8,000–10,000 (Cremers, Lee, Collin, & Allikmets, 2020; Fishman, Farber, Patel, & Derlacki, 1987; Tanna, Strauss, Fujinami, & Michaelides, 2017). It is typically characterized by loss in central vision, progressive bilateral atrophy of the macula including the underlying retinal pigment epithelial (RPE) cells, impaired color vision, delayed dark adaptation, and accumulation of fluorescent yellow-white flecks around the macula and midretinal periphery. Affected individuals typically experience a loss in visual acuity in the first or second decade of life and progressive visual deterioration over time with severe cases often diagnosed as cone-rod dystrophy (Fishman et al., 2003; Klevering et al., 2002). In some cases, however, the disease onset occurs later in life with more mild visual impairment (Lambertus et al., 2016; Westeneng-van Haaften et al., 2012; Zernant et al., 2017).
STGD1 is caused by mutations in ABCA4, a gene encoding a 2273 amino acid ATP binding cassette (ABC) transporter (Allikmets et al., 1997; Nasonkin et al., 1998). Like other members of the A subfamily of ABC transporters, ABCA4 is organized as two nonidentical tandem halves with each half containing a transmembrane domain (TMD) consisting of 6 membrane spanning segments, a nucleotide binding domain (NBD), a large exocytoplasmic domain (ECD), and an extended C-terminal segment (Fig 1) (Bungert, Molday, & Molday, 2001). The high-resolution structure of ABCA1, a protein that shares 50% identity with ABCA4, has confirmed the overall structural organization of ABCA proteins and has further revealed the presence of intracellular transverse helices also referred to as coupling helices, V-shaped α-helical hairpin between membrane spanning segments TM5 and TM6 and membrane spanning segments TM11 and TM12, and a distinctive folding pattern of the TMDs and ECDs (Qian et al., 2017).
Figure 1:
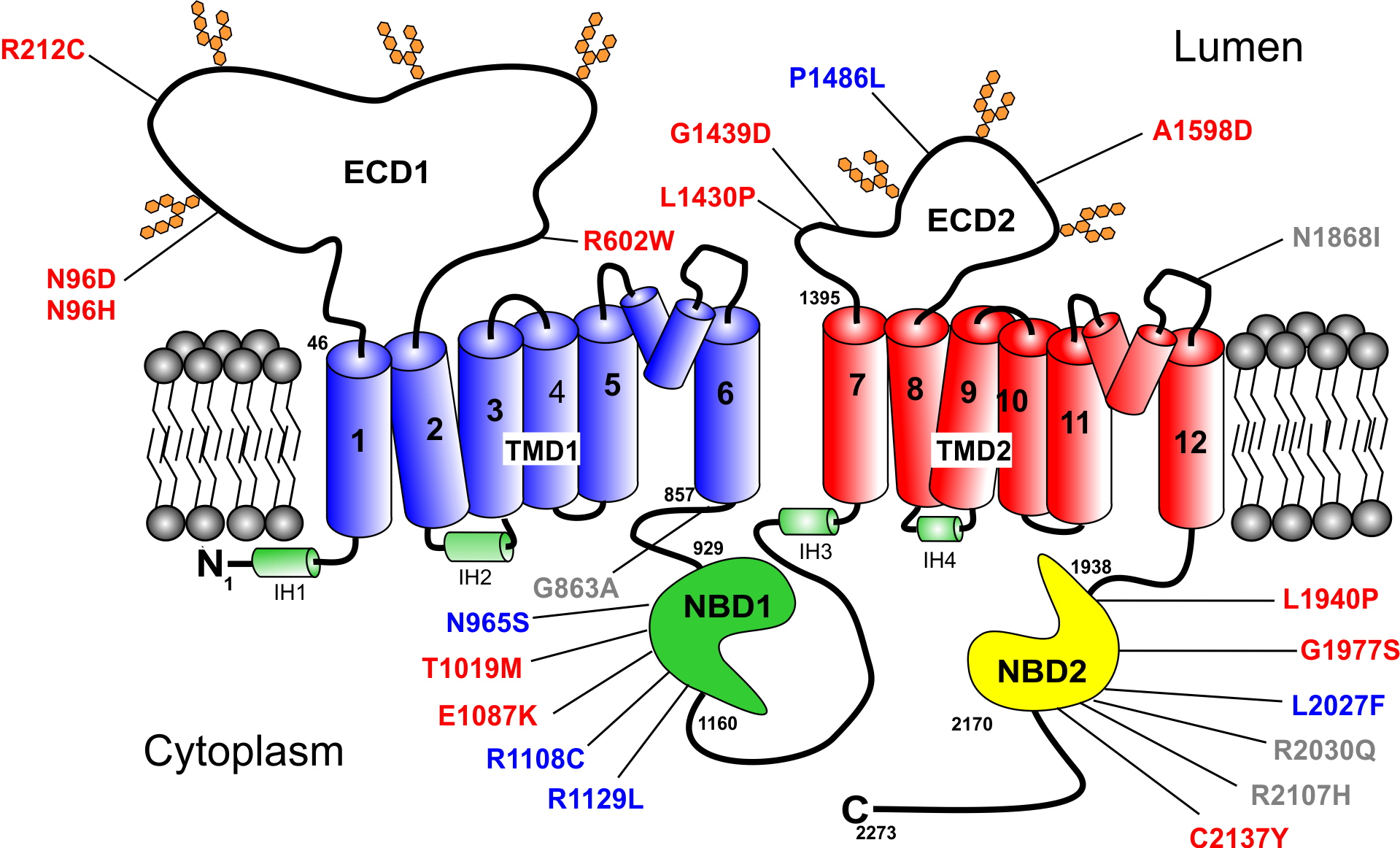
Topological model of ABCA4 showing the organization of the domains and the location of the missense mutations analyzed in this study. ECD1 and ECD2 – exocytoplasmic domains 1 and 2; TMD1 and TMD2 – transmembrane domains 1 and 2. The 12 transmembrane segments are numbered. NBD1 and NBD2 – nucleotide binding domains 1 and 2. Green – proposed intracellular α-helices (IH); two short hairpin α-helices between membrane spanning segments 5 and 6 and 11 and 12 are shown. The missense mutations are colored to indicate their severity class as defined in this study (see Table I) with Class 1 (severe) in red; Class 2 (moderate) in blue; and Class 3 (mild) in grey.
ABCA4 is primarily expressed in rod and cone photoreceptor cells where it is distributed along the rim and incisures of outer segment disks (Illing, Molday, & Molday, 1997; Molday, Rabin, & Molday, 2000; Papermaster, Schneider, Zorn, & Kraehenbuhl, 1978). It is also present at lower levels in photoreceptor inner segments and retinal pigment epithelial (RPE) cells (Lenis et al., 2018; Molday., Wahl, Sarunic, & Molday, 2018). ABCA4 functions as an active lipid importer or flippase, transporting N-retinylidene-phosphatidylethanolamine (N-Ret-PE), the Schiff-base adduct of retinal and phosphatidylethanolamine (PE), from the lumen to the cytoplasmic leaflet of disc membranes (Quazi, Lenevich, & Molday, 2012). N-Ret-PE transport by ABCA4 facilitates the removal of all-trans retinal released from photoactivated rhodopsin and 11-cis retinal not needed for the regeneration of rhodopsin, thereby preventing the buildup of toxic bis-retinoid compounds (Molday, 2015; Quazi & Molday, 2014).
Over 1000 mutations in ABCA4 are known to cause STGD1 (Cornelis et al., 2017; Cremers et al., 2020; Tanna et al., 2017). These include frameshifts, truncations, small nontruncating deletions and insertions, splice site mutations, deep intron alterations, and missense and complex mutations with over 50% being missense mutations. Loss of function mutations in ABCA4 typically result in the accumulation of retinal derivatives and the production of bisretinoids. These compounds including A2E accumulate in RPE cells of STGD1 patients as fluorescent lipofuscin deposits upon phagocytosis of photoreceptor outer segments leading to the degeneration of RPE cells and photoreceptors (Mata, Weng, & Travis, 2000; Sparrow & Boulton, 2005). Phenotypic characteristics of STGD1 patients including an increase in autofluorescence and accumulation of A2E in RPE cells are also observed in Abca4 knockout mice, transgenic mice homozygous for STGD1-disease causing missense mutations, and a Labrador retriever with a frameshift mutation in ABCA4 (Boyer et al., 2012; Charbel Issa et al., 2013; Makelainen et al., 2019; Molday. et al., 2018; Weng et al., 1999; Zhang et al., 2015).
Although extensive genetic screens have identified ABCA4 disease-causing variants in various geographical and ethnic groups (Chacon-Camacho, Granillo-Alvarez, Ayala-Ramirez, & Zenteno, 2013; Cornelis et al., 2017; Hu et al., 2019; Riveiro-Alvarez et al., 2013; Schulz et al., 2017; Zernant et al., 2014), few studies have been carried out to determine the extent to which specific mutations affect the structure and function of ABCA4 at a protein level and how these properties may relate to the age of onset or disease severity (Garces et al., 2018; Sun, Smallwood, & Nathans, 2000; Wiszniewski et al., 2005). Correlating missense mutations with disease severity has been particularly problematic since most STGD1 patients are compound heterozygous for nontruncating variants making it difficult to assign the disease phenotype to one or both variants. However, a number of STGD1 patients have been identified that are homozygous for a given missense mutation and their clinical features including their age of onset have been reported (Burke, Allikmets, Smith, Gouras, & Tsang, 2010; Cornelis et al., 2017; Fakin et al., 2016; Fujinami et al., 2013; Garces et al., 2018; Riveiro-Alvarez et al., 2013; Zernant et al., 2014). Several missense mutations have been characterized as hypomorphic such that they exhibit disease-associated characteristics only under certain conditions (Zernant et al., 2017). In the present study we have evaluated the level of expression and functional properties of over 20 ABCA4 variants found in STGD1 patients homozygous for these variants, and further analyzed the functional activity of several proposed hypomorphic variants. On the basis of these studies, we have developed a scale to gauge the contributions of the residual function of ABCA4 variants on the age of disease onset.
Results
ABCA4 disease-associated variants characterized in this study
The locations of ABCA4 homozygous and hypomorphic missense mutations characterized in the present study are shown in Fig 1. These mutations are distributed throughout ABCA4 with 4 in ECD1 (p.Asn96Asp, p.Asn96His, p.Arg212Cys, p.Arg602Trp), 4 in ECD2 (p.Leu1430Pro, p.Gly1439Asp, p.Pro1486Leu, p.Ala1598Asp), 6 within or close to NBD1 (p.Gly863Ala, p.Asn965Ser, p.Thr1019Met, p.Glu1087Lys, p.Arg1108Cys, p.Arg1129Leu), and 6 within NBD2 (p.Leu1940Pro, p.Gly1977Ser, p.Leu2027Phe, p.Arg2030Gln, p.Arg2107His, p.Cys2137Tyr). The p.Asn1868Ile mutation is present in a loop connecting the V-shaped α-helical hairpin with membrane spanning segment 12 (TM12). Several mutations occur in well-defined motifs crucial for the binding and hydrolysis of ATP. The p.Asn965Ser and p.Glu1087Lys mutations occur in the Walker A (Gly-His-Asn-Gly-Ala-Gly-Lys-Thr) and Walker B (Val-Ile-Leu-Asp-Glu) motifs of NBD1, respectively, and the p.Gly1977Ser mutation is present in the Walker A (Gly-Val-Asn-Gly-Ala-Gly-Lys-Thr) motif of NBD2. The p.Arg2030Gln variant reported to display a mild disease phenotype in compound heterozygous STGD1 patients has been included in our analysis although this variant has yet to be found in a homozygous state.
Expression levels of detergent solubilization of ABCA4 disease variants
The ABCA4 variants were individually expressed in HEK293T cells and their level of expression was quantified in detergent-solubilized cell lysates by Western blotting for comparison with normal ABCA4, referred here as wild-type (WT) ABCA4 (Fig 2AB). Most variants solubilized at greater than 50% of WT ABCA4 levels (Table 1). Exceptions were p.Thr1019Met, p.Arg1108Cys and p.Leu2027Phe which solubilized at levels between 17% and 37% suggesting that a major fraction of these variants are highly misfolded and either irreversibly aggregated or degraded by the endoplasmic reticulum-associated protein degradation (ERAD) pathway of the cell. Three variants, p.Leu1430Pro, p.Pro1486Leu, and p.Arg2107His expressed at >80% WT levels after detergent solubilization indicating that a major fraction of these variants folded into a native-like conformation amenable to mild detergent solubilization.
Figure 2:
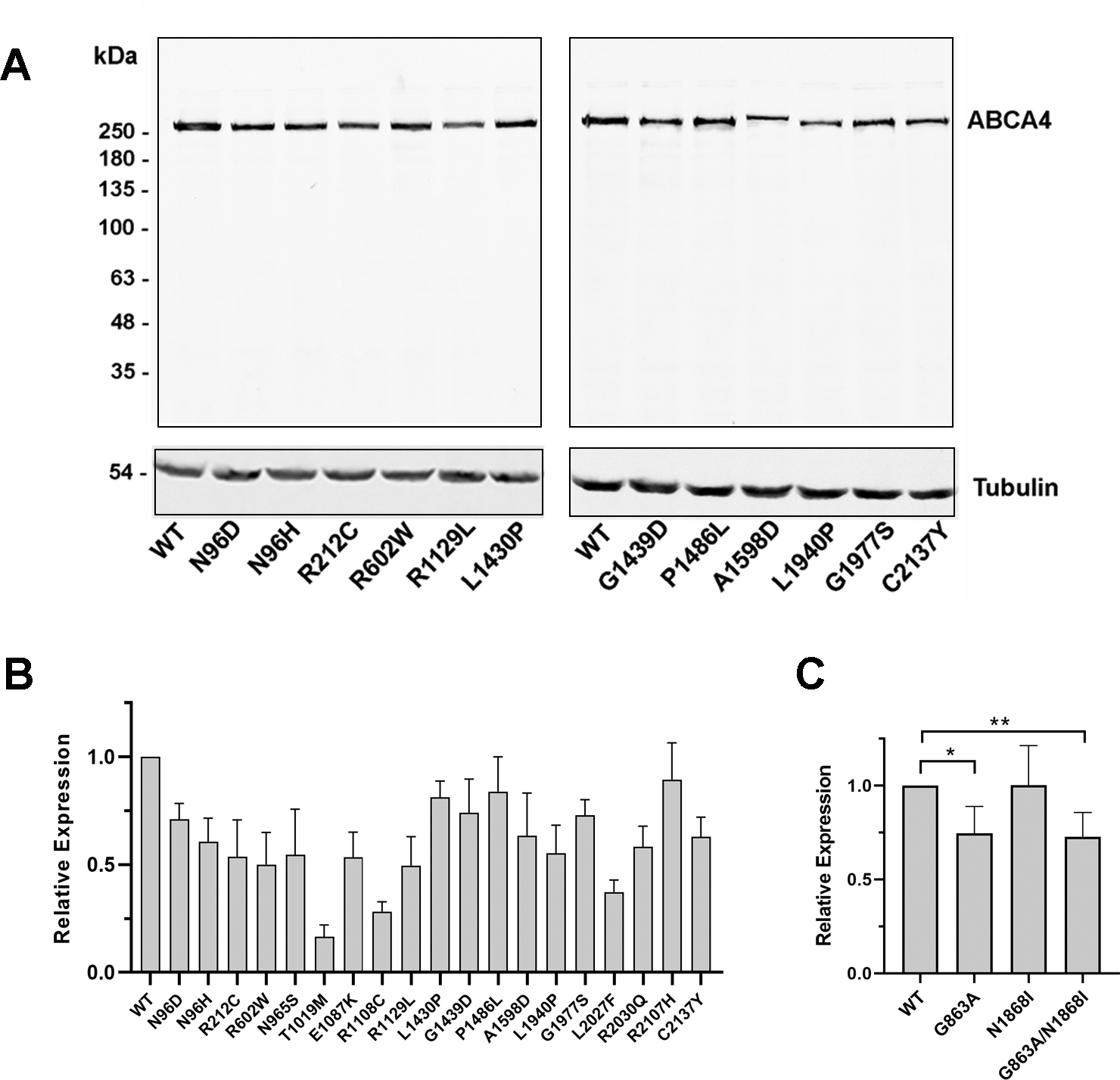
Expression of ABCA4 variants. HEK293T cells were transfected with normal ABCA4 (WT) or variants harboring missense mutations, solubilized in CHAPS detergent and analyzed on Western blots labeled for ABCA4 with the Rim-3F4 monoclonal antibody. A. Representative profile of western blots used for quantification of expression. Tubulin was used as a loading control. B,C. Quantification of expression of ABCA4 variants relative to WT ABCA4. Data are presented as the mean ± SD for n ≥ 3. For hypomorphic variants, the difference between WT ABCA4 and p.Asn1868Ile was not significant; p – values between WT and p.Gly863Ala (*) and WT and p.Gly863Ala/p.Asn1868Ile (**) were 0.0041 and 0.0025, respectively.
Table 1:
Properties of ABCA4 Variants and Age of Onset of STGD1 Patients
| Protein Variant (DNA Variant) | Relative Expression | Relative ATPase Activity ATPase–ATR +ATR | Function Index | Predicted Severity Class | Age of Onset Yr or decade [mean] | Reference |
|---|---|---|---|---|---|---|
| WT | 1.0 | 1.0 1.87±11 | 1.0 | Normal | ||
| p.Asn96Asp (c.286A>G) | 0.71±0.07 | 0.55±0.02 0.57±0.02 | 0.02 | Severe 1 | 23,14 8 [15] | (Salles et al., 2018) (Lambertus et al., 2015) |
| p.Asn96His (c.286A>C) | 0.61±0.17 | 0.23±0.08 0.19±0.08 | 0 | Severe 1 | 20 | (Simonelli et al., 2005) |
| p.Arg212Cys (c.634C>T) | 0.54±0.17 | 0.47±0.13 0.49±0.14 | 0.01 | Severe 1 | 11,11 8,10 [10] | (Fujinami et al., 2013) (Simonelli et al., 2000) |
| p.Arg602Trp (c.1804C>T) | 0.50±0.15 | 0.37±0.07 0.28±0.06 | 0 | Severe 1 | 8 15 [11.5] | (Ortube et al., 2014) (Riveiro-Alvarez et al., 2013)} |
| p.Asn965Ser (c.2894A>G) | 0.55±0.21 | 0.70±0.19 1.02±0.33 | 0.20 | Moderate 2 | 13,13,17,18 2nd decade [16] | (Rosenberg, Klie, Garred, & Schwartz, 2007) (Lee et al., 2017) (Riveiro-Alvarez et al., 2013) |
| p.Thr1019Met (c.3056C>T) | 0.17±0.06 | 0.26±0.04 0.31±0.06 | 0.01 | Severe 1 | 10, 10 [10] | (Fumagalli et al., 2001) |
| p.Glu1087Lys (c.3259G>A) | 0.54±12 | 0.37±0.23 0.31±0.18 | 0 | Severe 1 | <10 | (Fakin et al., 2016) |
| p.Arg1108Cys (c.3322C>T) | 0.28±0.05 | 0.40±0.03 0.75±0.21 | 0.11 | Moderate 2 | 10 | (Fakin et al., 2016) |
| p.Arg1129Leu (c.3386G>T) | 0.50±0.13 | 0.25±0.16 0.49±0.18 | 0.14 | Moderate 2 | 12,19,19, 31 20.6 [20.2] | (Valverde et al., 2006) (Riveiro-Alvarez et al., 2013) |
| p.Leu1430Pro (c.4289T>C) | 0.81±0.08 | 0.38±0.05 0.35±0.13 | 0 | Severe 1 | ~12 | (Rivera et al., 2000) |
| p.Gly1439Asp (c.4316G>A) | 0.74±0.16 | 0.41±0.03 0.46±0.11 | 0.04 | Severe 1 | 8 | (Lewis et al., 1999) |
| p.Pro1486Leu (c.4457C>T) | 0.84±0.16 | 0.56±0.06 0.87±0.33 | 0.30 | Moderate 2 | 25 | (Downes et al., 2012) |
| p.Ala1598Asp (c.4793C>A) | 0.64±0.19 | 0.48±0.21 0.32±0.12 | 0 | Severe 1 | 13 | (Burke et al., 2010) |
| p.Leu1940Pro (c.5819T>C) | 0.55±0.13 | 0.19±0.07 0.19±0.11 | 0 | Severe 1 | 6 | (Riveiro-Alvarez et al., 2013) |
| p.Gly1977Ser (c.5929G>A) | 0.73±0.07 | 0.13±0.08 0.15±0.02 | 0.02 | Severe 1 | 10 | (Riveiro-Alvarez et al., 2013) |
| p. Leu2027Phe (c.6079C>T) | 0.37±0.06 | 0.38±0.02 0.68±0.13 | 0.13 | Moderate 2 | 28 44 [36] | (Downes et al., 2012) (Fujinami et al., 2013) |
| p.Arg2030Gln (c.6089G>A) | 0.58±0.10 | 0.56±0.12 1.13±0.35 | 0.38 | Mild 3 | 52* | (Yatsenko, Shroyer, Lewis, & Lupski, 2001) (Fakin et al., 2016) |
| p.Arg2107His (c.6320G>A) | 0.90±0.17 | 0.66±0.13 1.23±0.17 | 0.59 | Mild 3 | 7th decade | (Zernant et al., 2014) |
| p.Cys2137Tyr (c.6410G>A) | 0.63±0.09 | 0.45±0.03 0.32±0.12 | 0 | Severe 1 | 7 | (Riveiro-Alvarez et al., 2013) |
| p.Gly863Ala (c.2588G>C) | 0.75±0.14 | 0.95±0.22 1.60±0.23 | 0.62 | Mild 3 | >50 not causal in homo-zygosity | (Westeneng-van Haaften et al., 2012) |
| p.Asn1868Ile (c.5603A>T) | 1.00±0.21 | 0.91±0.11 1.68±0.23 | 0.89 | Mild 3 | ND | |
| p.Gly863Ala/p.Asn1868Ile c.2588G>C/c.5603A>T | 0.73±0.13 | 0.80±0.15 1.50±0.27 | 0.59 | Mild 3 | ≥45 | (Zernant et al., 2017) |
Class 1 (Severe) F-index of 0 – 0.05; Class 2 (Moderate) F-index of 0.06 – 0.35; Class 3 (Mild) F-index > 0.35.
For a compound heterozygous patient with a null allele in trans. No homozygous patients reported. ND – No Data available
The levels of the hypomorphic variants were also analyzed after detergent solubilization (Fig 2C). The p.Asn1868Ile variant expressed at the same level as WT ABCA4. The p.Gly863Ala variant and the p.Gly683Ala/p.Asn1868Ile complex variant were also present at high levels indicating that the replacement of the glycine at position 863 for alanine results in only minor alteration in the ABCA4 conformation and/or stability.
ATPase Activities of ABCA4 Variants
Previous studies have shown that the ATPase activity of ABCA4 is stimulated by its substrate N-Ret-PE (Ahn & Molday, 2000; Beharry, Zhong, & Molday, 2004; Sun, Molday, & Nathans, 1999). This functional property mirrors the ATP-dependent flipping of N-Ret-PE across membranes (Quazi et al., 2012). To evaluate the effect of the disease-associated missense mutations on the ATPase activity of ABCA4, the variants were first purified from detergent-solubilized membrane extracts of transfected HEK293T cells by immunoaffinity chromatography. All disease-associated variants were over 90% pure as observed on representative Coomassie blue stained SDS gels shown in Fig 3A.
Figure 3.
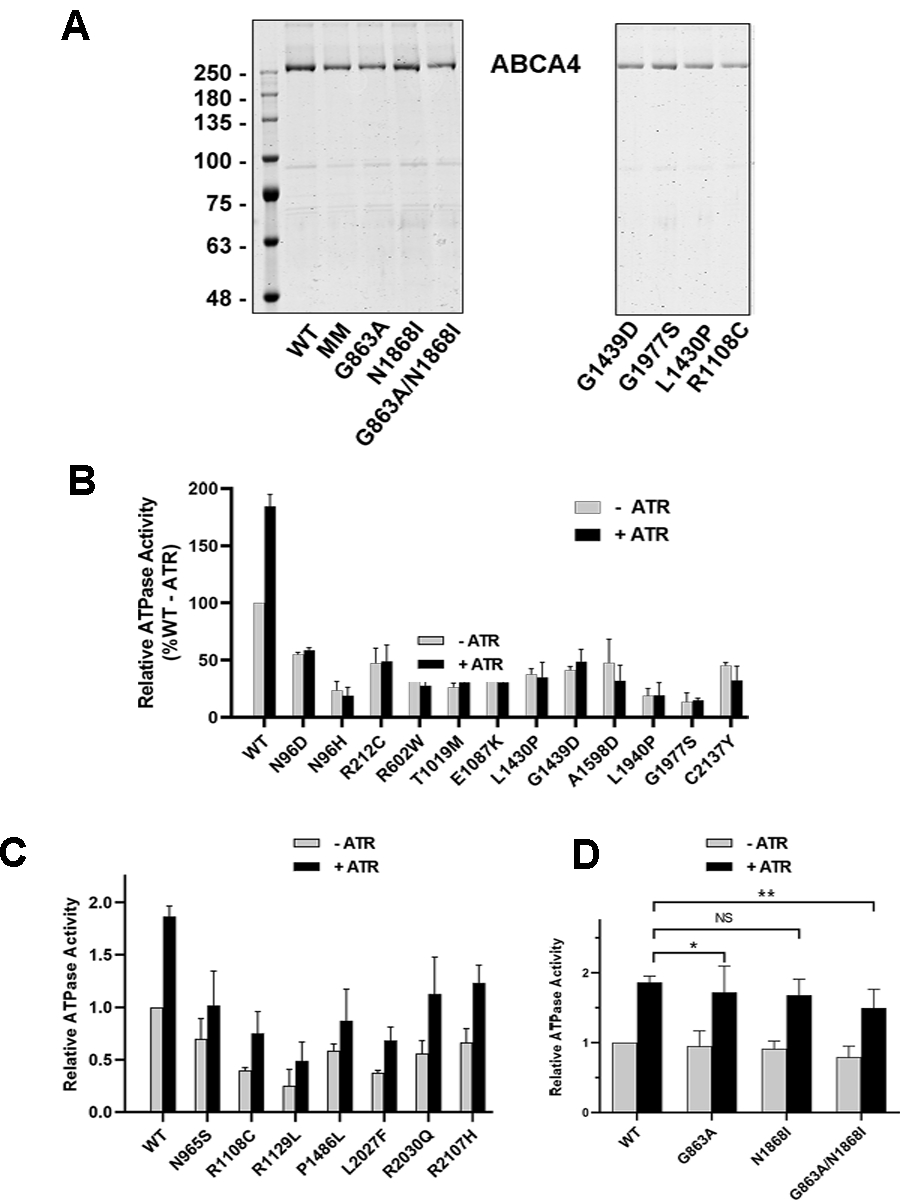
Purification and ATPase activity of ABCA4 Variants. ABCA4 variants were purified on a Rim3F4-Sepharose immunoaffinity matrix. A. Coomassie blue stained SDS gels of several purified ABCA4 variants. The MM variant is the ATPase deficient variant. B,C. ATPase activity of ABCA4 variants in the absence (-ATR) and presence of 40 μM all-trans retinal (+ATR). B. The ATPase activities of this series of variants were not stimulated by the addition of ATR. C. The ATPase activities of this series of variants were stimulated by the addition of ATR. D. ATPase activity of hypomorphic and complex variants. NS – not significant; * P value = 0.037; **P value = 0.049. Data are presented as the mean +/− SD for n≥3 independent experiments.
The ATPase activities of the purified ABCA4 variants were determined in the presence of phosphatidylethanolamine (PE) to obtain the basal ATPase activity (-ATR) and in the presence of both PE and saturating all-trans retinal (+ATR) concentration used to generate the N-Ret-PE substrate. Figures 3B – D show the basal and N-Ret-PE stimulated ATPase activity profiles of the variants at the same protein concentration relative to the basal activity of WT ABCA4. The basal ATPase activity varied with most ABCA4 variants showing basal activity below 50% of WT ABCA4 activity. Addition of 40 μM ATR resulted in a 1.8–2.0-fold increase in the ATPase activity of WT ABCA4 as previously reported (Garces et al., 2018). The variants showed mixed results. Twelve disease-associated variants displaying a marked reduction in basal activity were not activated by the addition of ATR (Fig 3B). Indeed, the ATPase activity of some of these variants was decreased in the presence of ATR. Seven variants (p.Asn965Ser, p.Arg1108Cys, p.Arg1129Leu, p.Pro1486Leu, p.Leu2027Phe, p.Arg2030Gln, p.Arg2107His) displaying decreased basal ATPase activity, however, showed significant stimulation by ATR (Fig 3C).
In contrast, the basal and substrate activated ATPase activities of p.Gly863Ala and p.Asn1868Ile were comparable to WT ABCA4 (Fig. 3D), while the p.Gly863Ala/p.Asn1868Ile complex variant consistently showed a small but significant reduction in substrate-stimulated ATPase activity (P-value of 0.049) when compared to that for WT ABCA4 (Fig 3D).
Binding of N-Ret-PE to ABCA4 Variants
Previous studies have shown that ABCA4 has a high affinity binding site for its substrate N-Ret-PE (Beharry et al., 2004). In the presence of ATP or its nonhydrolyzable derivative AMP-PNP, ABCA4 undergoes a conformational change resulting in the loss of N-Ret-PE binding (Beharry et al., 2004; Tsybovsky, Orban, Molday, Taylor, & Palczewski, 2013). We have investigated the capacity of several disease-associated ABCA4 variants to bind N-Ret-PE in the absence and presence of ATP. As shown in Fig 4, the p.Gly863Ala variant bound N-Ret-PE in the absence and presence of ATP at levels comparable to WT ABCA4. The other variants bound N-Ret-PE in the absence of ATP at reduced levels, indicating that these mutations affected the overall conformation of ABCA4. The p.Leu1940Pro variant unlike the other variants showed only a modest reduction in binding in the presence of ATP suggesting that this variant fails to undergo the transition to a lower substrate affinity binding site in the presence of nucleotide.
Figure 4:
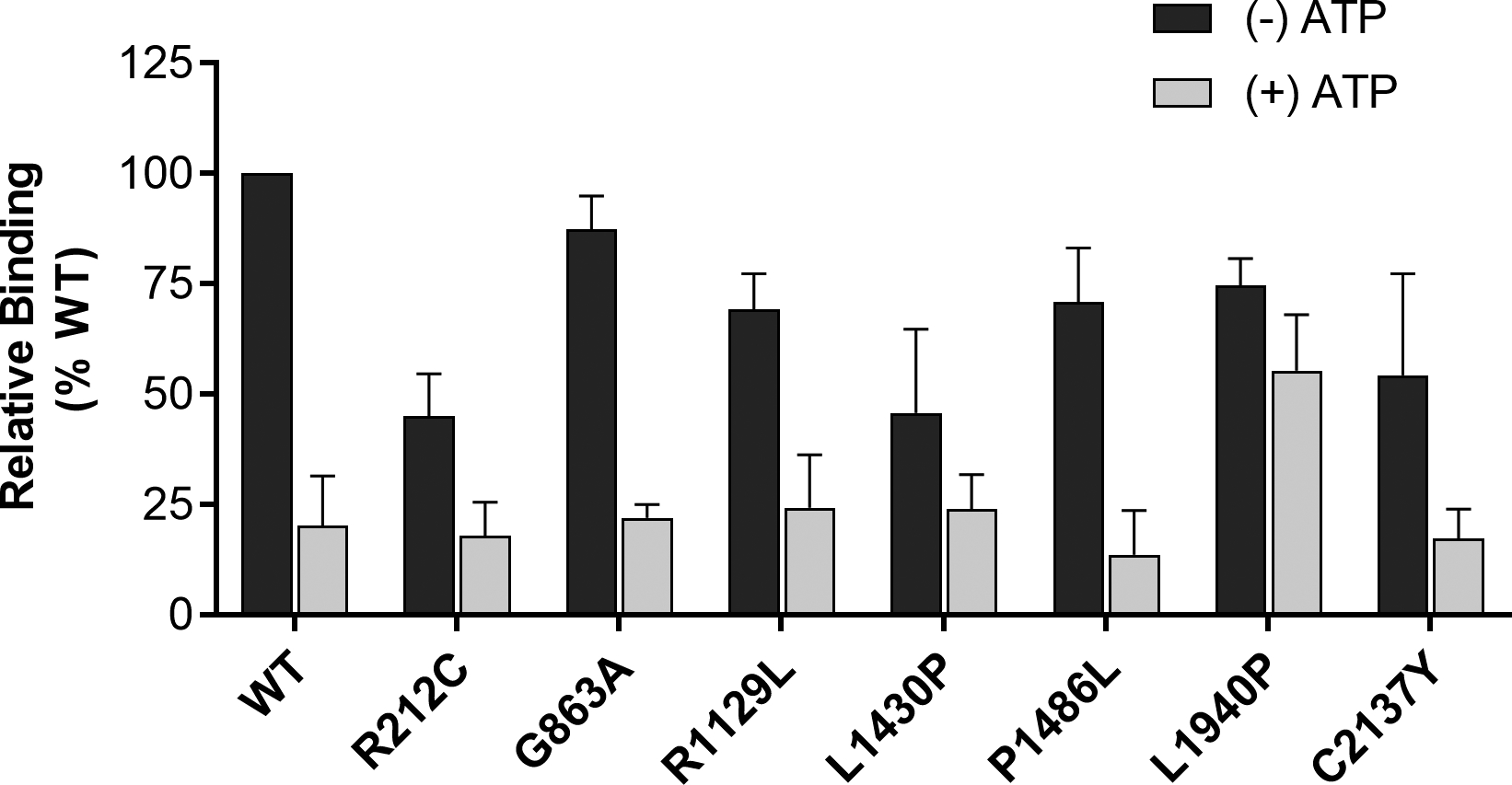
Binding of N-Ret-PE to ABCA4 variants. [3H]-retinal was added to ABCA4 variants that were immobilized on an affinity matrix in buffer containing DOPE in the absence (− ATP) or presence of 1 mM ATP (+ ATP). The samples were eluted with ethanol and the [3H] retinoid was measured in a liquid scintillation counter. The data are normalized to WT ABCA4 in the absence of ATP. Data are presented as the mean +/− SD for n≥3 independent experiments.
Discussion
In this study we examined the biochemical properties of twenty-two ABCA4 variants harboring missense mutations reported to cause STGD1. Amino acid sequence alignments from a range of vertebrates including humans, mouse, chicken, zebrafish, and Xenopus tropicalis indicate that these amino acid substitutions occur in residues that are evolutionarily conserved. An exception is alanine at position 1598 (p.Ala1598) which presents with some conserved substitutions in various vertebrates. Furthermore, with the exception of p.Arg212, p.Ala1598, and p.Asn1868, these residues are conserved in ABCA1, a member of the ABCA subfamily associated with cholesterol efflux and Tangiers disease, and ABCA7, a member of this subfamily that is genetically linked to Alzheimer’s disease, supporting the importance of these residues in the structure-function relationships of these lipid transporters (Coleman, Quazi, & Molday, 2013; Dean, Hamon, & Chimini, 2001).
The disease-associated ABCA4 variants examined in this study can be divided into three main classes. Class 1 includes 12 variants which show diminished expression when compared to WT ABCA4 and low basal ATPase activity that is not stimulated by N-Ret-PE (Fig 3B); Class 2 consists of 5 variants with diminished expression and reduced basal ATPase activity that show modest N-Ret-PE stimulated ATPase activity (Fig 3C); Class 3 contains 5 variants that have relatively high expression and basal ATPase activity that is strongly activated by N-Ret-PE (Fig 3C,D). The latter class includes the hypomorphic variants, p.Gly863Ala and p.Asn1868Ile, and the complex variant p.Gly863Ala/p.Asn1868Ile.
These classes generally correlate well with the age of onset for STGD1 individuals that are bi-allelic for these mutations with few exceptions (Table 1). STGD1 patients harboring Class 1 variants (p.Asn96Asp, p.Arg212Cys, p.Arg602Trp, p.Thr1019Met, p.Glu1087Lys, p.Leu1430Pro, p.Gly1439Asp, p.Ala1598Asp, p.Leu1940Pro, p.Gly1977Ser, and p.Cys2137Tyr) exhibited an early age of disease onset (≤ 13 yr). This range of disease onset is similar to the age of onset for patients with two null alleles, most often within the 1st decade of life (Cornelis et al., 2017; Fakin et al., 2016). An exception is p.Asn96His. In our study, this variant lacks significant N-Ret-PE-activated ATPase activity, but a patient homozygous for this variant has been reported to have a disease onset of 20 yr (Table 1). The p.Asn96Asp variant also shows considerable variation in the age of disease onset from 8 yr in one study to 23 yr in another. The molecular properties of two variants in Class 1, p.Gly1977Ser and p.Arg602Trp, have been previously investigated. The p.Gly1977Ser variant was shown to exhibit diminished basal activity that was not activated by its substrate in agreement with our analysis (Sun et al., 2000). In another study, the p.Arg602Trp variant was reported to mislocalize to the inner segments of Xenopus laevis tadpoles and display low basal ATPase activity indicative of significant protein misfolding (Wiszniewski et al., 2005). Substrate activation was not carried out in the latter study.
Class 2 variants (p.Asn965Ser, p.Arg1108Cys, p.Arg1129Leu, p.Pro1486Leu, and p.Leu2027Phe) are generally associated with an age of onset between 13 and 40 years for homozygous individuals (Table 1). Apparently, the residual substrate transport activity of these mutants results in the removal of significant amounts of retinoid resulting in a later age of onset and more moderate disease phenotype than Class 1 variants. Consistent with our studies, the p.Asn965Ser variant has been shown to have a modest increase in substrate stimulated ATPase activity in earlier in vitro experiments (Quazi et al., 2012; Sun et al., 2000) and to partially localize to photoreceptor outer segments in a transgenic mouse model containing the p.Asn965Ser variant (Molday. et al., 2018). In the latter study N-Ret-PE activation of ATPase activity of p.Asn965Ser was not observed. However, more recent studies using the modified ATPase assay described here show measurable substrate activated activity for this variant. An exception in this class is p.Arg1108Cys which has been reported to be associated with an age of onset of about 10 yr in one study. However, this variant has been categorized as moderate to severe based on clinical studies (Fakin et al., 2016) in line with our data.
Class 3 variants (p.Arg2030Gln, p.Arg2107His, p.Gly863Ala, p.Asn1868Ile, and p.Gly863Ala/p.Asn1868Ile), displaying significant expression levels and basal and N-Ret-PE-stimulated ATPase activities, are associated with a late disease onset (> 40 yr) and more mild visual impairment. For example, a STGD1 patient homozygous for the p.Arg2107His variant was reported to have an age of onset in the 7th decade of life (Zernant et al., 2014). The p.Arg2030Gln variant has also been reported to be associated with a mild disease phenotype when in trans with a null allele (Fakin et al., 2016). Class 3 includes the proposed hypomorphic variants p.Gly863Ala and p.Asn1868Ile and the complex variant p.Gly863Ala/p.Asn1868Ile. These variants showed expression levels and ATPase activities broadly comparable to WT ABCA4 in our studies. The p.Asn1868Ile variant was previously reported to display WT-like ATPase activity in an earlier study (Sun et al., 2000), but in this and other studies the p.Gly863Ala had limited activity in contrast to our current studies (Quazi et al., 2012; Sun et al., 2000). The reason for the discrepancy in the p.Gly863Ala variant is unclear although there were differences in the experimental protocols between these studies. In particular, in our current assay, the ATPase activity was measured on the detergent solubilized protein right after purification, whereas in the earlier studies the ATPase activities were measured after reconstitution into liposomes. It is possible that the p.Gly863Ala variant is relatively unstable in detergent and fails to reconstitute efficiently into liposomes. This remains to be studied in detail.
In order to more quantitatively assess the effect of the mutations on the expression and functional activity of ABCA4, we introduce two experimentally determined factors: the relative expression level of a variant (E-factor = variant expression divided by WT expression) and the relative substrate-stimulated ATPase activity of a variant (S-factor = variant N-Ret-PE-activated ATPase activity minus the variant basal ATPase activity divided by the WT N-Ret-PE-activated ATPase activity minus WT basal ATPase activity). These factors can be combined to produce a functional index (F-index = E-factor × S-factor) that estimates the extent to which the missense mutation will alter the expression and function of ABCA4 and hence may serve as an inverse measure of the severity of STGD1 cases due to these mutations. This is given in Table 1 for each variant examined here along with literature values for the age of onset for STGD1 patients homozygous for these variants. An important factor is the N-Ret-PE-activated ATPase activity since this is a measure of the capacity of the variant to facilitate the removal of retinoid compounds from photoreceptors. In the absence of significant N-Ret-PE-stimulated ATPase activity, the F-index of 0 (nominally < 0.05) is indicative of a severe phenotype similar to that for two null alleles. In some cases, this is negative due to inhibition of the ATPase activity of some variants by ATR, a finding also observed previously (Sun et al., 2000). A relatively high F-index greater than 0.35 is associated a mild variant. In this scale, WT ABCA4 has a F = 1. Variants with a F-index above 0.7 would likely display sufficient activity to be borderline in conferring a mild vs a non-disease phenotype. Variants with intermediate F-index (0.05 – 0.35) are predicted to display a moderate disease phenotype. This class can be further subdivided into moderate-severe at the lower end of the scale and moderate-mild at the upper end. Figure 5 displays the relationship between the functionality index and the age of onset for STGD1 patients homozygous for the variants studied here.
Figure 5:
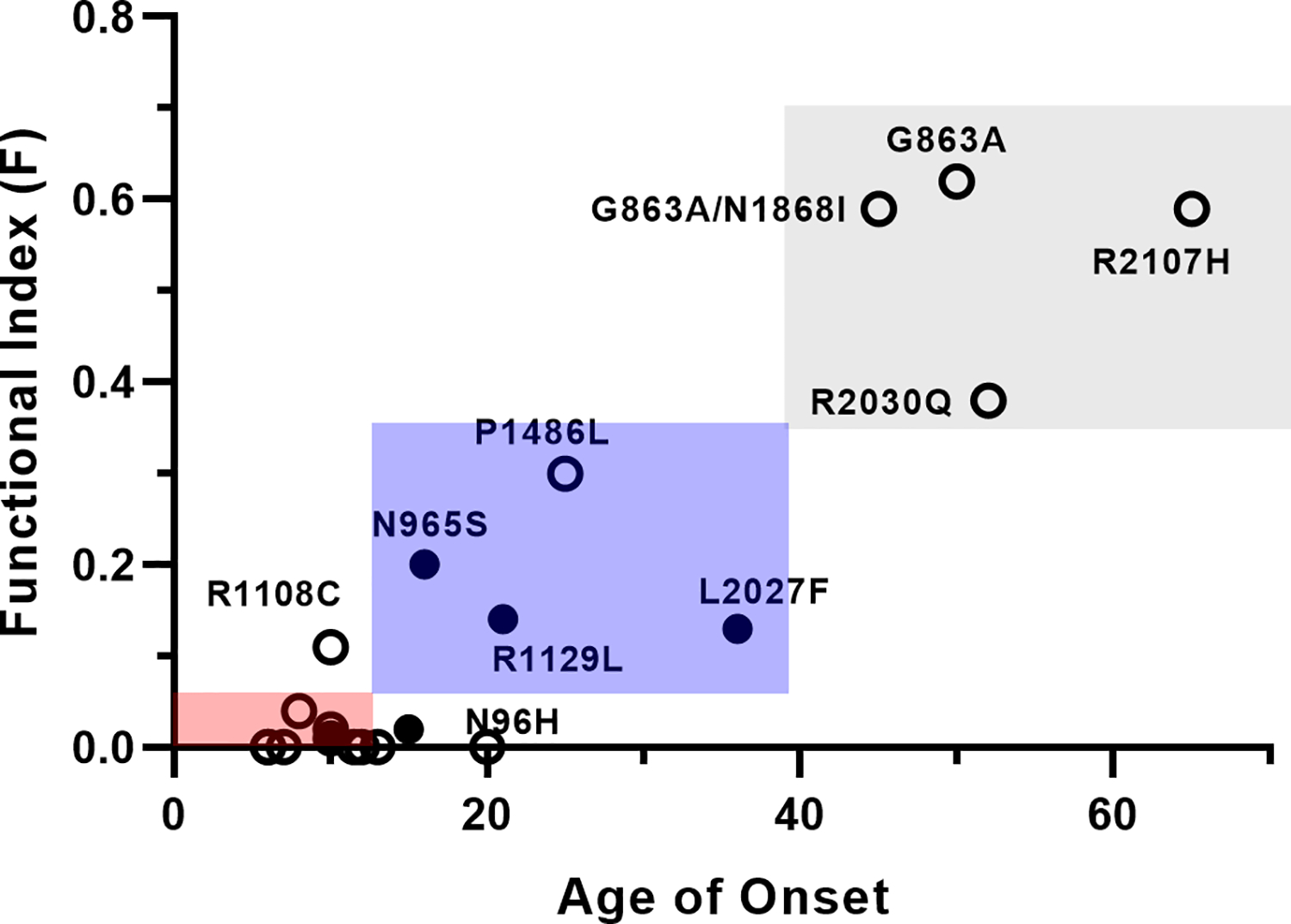
Graph showing the relationship of the functional index (F) to the age of onset for ABCA4 variants found in homozygous STGD1 patients. The red square (lowest) shows the relationship between the range of age of onset and functional index classified as severe (Class 1); blue square (middle) is classified as moderate (Class 2); and the grey (upper) is classified as mild (Class 3). Fill circles are the average age of onset found for multiple patients as reported in the literature; the open circles are for a single patient. The severe patients with a F-index at or close to zero are not labeled, but can be found in Table I.
There has been some controversy as to whether the p.Gly863Ala and p.Asn1868Ile variants are disease-associated variants based on genetic penetrance and frequency of individuals harboring these mutations (Cremers et al., 2020; Runhart et al., 2018; Zernant et al., 2017). However, a recent study, has shown that at least some individuals with a p.Asn1868Ile mutation in trans with a deleterious mutation present with a late disease onset and milder phenotype (Runhart et al., 2018; Zernant et al., 2017). Moreover, individuals homozygous for the complex variant (p.Gly863Ala/p.Asn1868Ile) display a late disease onset (Zernant et al., 2017). Our studies showing a small, but significant decrease in expression and ATPase activity of this complex variant relative to WT ABCA4 are consistent with this variant conferring a mild disease phenotype. The p.Gly863Ala and p.Asn1868Ile mutations are located on the opposite sides of membrane in current ABCA4 model (Fig 1) generated from the ABCA1 structure and therefore are unlikely to directly interact. Instead, we suggest that each mutation modestly affects the function of ABCA4 such that the two mutations in cis reduce the activity of the protein carrying this complex variant sufficiently to cause a late onset, mild disease phenotype.
There are some limitations in correlating expression and residual ATPase activity of disease-causing variants with the age of onset. Our current ATPase activity assays require the use of a mild detergent to solubilize and purify the ABCA4 variants. In most cases this procedure has not affected the activity of ABCA4 variants. However, the p.Gly1961Glu variant has been reported to lack ATPase and ATP-dependent N-Ret-PE flippase activity in multiple in vitro studies despite the fact that this variant is generally associated with a relatively mild phenotype in STGD1 patients. We have suggested that this variant is stable and partially active when retained in membranes, but undergoes protein denaturation during detergent solubilization resulting in a loss in activity (Garces et al., 2018). This remains to be investigated using a strategy that does not require detergent solubilization for assessing the activity of this variant. Nevertheless, none of the variants in the current study (with the possible exception of p.Asn96His) appear to fall into this category. Our studies also do not consider the effect of mutations on the affinity of ABCA4 for its substrate. The level of expression of the variants was also determined in HEK293T cells. This level of expression may be marginally different in photoreceptors cells owing to differences in chaperone proteins.
The age of disease onset is another variable. For example, individuals homozygous for the p.Gly1961Glu mutation, show a range in age of disease onset spanning the 2nd to 5th decade of life (Burke et al., 2010; Cornelis et al., 2017; Cremers et al., 2020). This variation in age of onset may result from other factors such as modifier genes or the age of diagnosis. It is also possible that the age of disease onset does not reflect the severity of the disease. Fakin et al., however, have used electroretinography and fundus autofluorescence to evaluate retinal structure and function in hemizygous STGD1 patients (Fakin et al., 2016). Interestingly, with regard to the variants investigated here, they showed that STGD1 patients harboring the p.Glu1087Lys mutant had a severe phenotype characteristic of a null allele in agreement with our classification. Furthermore, they showed that p.Arg212Cys, p.Arg1108Cys, and p.Leu2027Phe have an intermediate or moderate phenotype, and p.Arg2030Gln has a milder phenotype. With the exception of p.Arg212Cys, their classification was in line with that based on expression and ATPase activity measurements reported here. Another limitation is the number of patients that have been identified and clinically characterized who are homozygous for a given variant. In many cases, data is available for only a single patient. More data is needed to define that range of age onset and clinical severity for these patients.
In summary our studies show that there is a good correlation between substrate stimulated ATPase activity and the severity of STGD1 based on the functional index developed here. Disease variants that show no substrate stimulated activity typically are associated with an early onset and severe phenotype often resembling cone-rod dystrophy; variants that show substrate stimulated ATPase activity, but reduced expression and overall activity are associated with an intermediate or moderate phenotype; and variants with substantial expression and basal and substrate stimulated ATPase activity are associated with a later disease onset and milder phenotype. Accordingly, the functional activity of disease variants is an important factor in gaging the severity of STGD1. The biochemical data and functional index developed here should prove useful in assessing the severity of STGD1 individuals who are compound heterozygous for the variants analyzed in this study. Knowledge of the residual function of disease variants is important in guiding clinical trials focusing on gene and drug based therapeutic treatments for STGD1.
Materials and Methods
Reagents:
The Rho-1D4 and Rim-3F4 monoclonal antibodies were generated in house (Illing et al., 1997; MacKenzie, Arendt, Hargrave, McDowell, & Molday, 1984) and are available from MilliporeSigma (Oakville, ON). β-tubulin antibody was from Abcam (Toronto, ON). Phospholipids including 1, 2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and brain polar lipids (BPL) were obtained from Avanti Polar lipids (Alabaster, AL). ATP and all-trans retinal was from MilliporeSigma and 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) was from Anatrace (Maumee, OH). Protease Inhibitor cocktail (MilliporeSigma, St. Louis, MO), and 1D4 and 3F4 peptides were from Celtek Peptides (Franklin, TN)
Generation of ABCA4 plasmids and site-directed mutagenesis
The cDNA of human ABCA4 (NCBI: NP_000341.2) engineered to contain a C-terminal 9 amino acid 1D4 tag (Thr-Glu-Thr-Ser-Gln-Val-Ala-Pro-Ala-COOH) was subcloned into pCEP4 vectors and human ABCA4 without the tag was subcloned into pcDNA3 as previously described (Ahn & Molday, 2000; Zhong, Molday, & Molday, 2009). Previous studies have shown that the presence of the C-terminal tag does not affect the activity of ABCA4 (Zhong et al., 2009). Missense mutations were generated by PCR-based site-directed mutagenesis (Garces et al., 2018). All DNA constructs were verified by Sanger DNA sequencing. Missense mutations examined in this study are: c.286A>G, p.Asn96Asp (p.N96D); c.286A>C, p.Asn96His (p.N96H); c. 634C>T, p.Arg212Cys (p.R212C); c1804C>T;p.Arg602Trp (p.R602W); c.2894A>G, p.Asn965Ser (p.N965S); c3056C>T, p.Thr1019Met (p.T1019M); c.3259G>A, p.Glu1087Lys (p.E1087K); c.3322C>T, p.Arg1108Cys (p.R1108C); c.3386G>T, p.Arg1129Leu (p.R1129L); c.4289T>C, p.Leu1430Pro (p.L1430P); c.4316G>A, p.Gly1439Asp (p.G1439D); c.4457C>T, p.Pro1486Leu (p.P1486L); c.4793C>A, p.Ala1598Asp (p.A1598D); c.5819T>C, p.Leu1940Pro (p.L1940P); c.5929G>A, p.Gly1977Ser (p.G1977S); c.6089G>A, p.Arg2030Gln (p.R2030Q); c.6320G>A, p.Arg2107His (p.R2107H); c.6410G>A, p.Cys2137Tyr (p.C2137Y); c.2588G>C, p.Gly863Ala (p.G863A); c.5603A>T, p.Asn1868Ile (p.N1868I); c.2588G>C/c.5603A>T, p.Gly863Ala/p.N1868Ile (p.G863A/p.N1868I).
HEK293T cell transfections and protein expression
HEK293T cells were typically transfected with plasmid DNA (5 μg per 10 cm plate) using 1mg/ml Polyethylenimine (PEI) MAX (Polysciences, Warrington, PA) at a ratio PEI(μl):DNA(μg) 3:1. Cells were grown in a humidified incubator (5% CO2) at 37 °C in DMEM supplemented with 8% bovine growth serum, 200 μM L-glutamine and 1x antibiotic-antimycotic (Gibco). After 24 h or 48 h post-transfection, cells were solubilized in Buffer A (25 mM HEPES, pH 7.4, 0.15 mg/ml BPL and 0.07mg/ml DOPE, 150 mM NaCl, 1 mM MgCl2, 10% glycerol, 1 mM dithiothreitol (DTT) containing 18 mM CHAPS and Protease Inhibitor cocktail by slowly adding one plate of cells resuspended in 100 μl of Buffer A to 0.5 ml solubilization buffer. The solution was stirred for 30 min at 4°C and subsequently centrifuged for 10 min at 100,000 g in a Beckman Optima TL centrifuge using a TLA110.4 rotor. The supernatant was applied to a 9% SDS gel and the expression levels of the ABCA4 variants were determined on western blots labeled with the Rim 3F4 antibody and β-tubulin as a loading control as previously described (Garces et al., 2018). Quantification was carried out using a LiCor imager. Three or more independent experiments were performed except where indicated. Data are presented as the mean ± Standard Deviation (SD).
ABCA4 purification, ATPase activity measurements, and retinoid binding assays
The immunoaffinity matrix was prepared by coupling the Rim-3F4 or Rho-1D4 antibody to CNBr activated Sepharose as previously described (Illing et al., 1997). CHAPS detergent-solubilized supernatant from cell lysates were mixed with 50 μl of Rim-3F4 Sepharose 2B or Rho-1D4 Sepharose 2B immunoaffinity matrix for 1 h at 4°C. The matrix was washed six times in Buffer A containing 10 mM CHAPS. The protein was eluted two times with 60 μl each in the same buffer containing 0.5 mg/ml 3F4 or 1D4 peptide at 18°C for 60 min.
An aliquot was analyzed on Coomassie blue stained SDS-gels for quantification of protein levels using bovine serum albumin as standards. The remaining sample was directly used without reconstitution into liposomes (Ahn & Molday, 2000) for ATPase activity assays using the ADP-Glo Kinase Assay luminescence detection protocol (Promega, Madison, WI). Briefly, a 20 μl ATPase reaction was performed by incubating 20–40ng of protein with either 1 μl of 0.8mM all-trans retinal (ATR) or buffer for 15 min followed by adding 4 μl of 1mM ultrapure ATP for 40 min at 37°C. Each reaction was done in triplicate. Six μl of each reaction was then added to a well of a 384 Corning white polystyrene microtiter plate, followed by 6 μl of ADP-Glo reagent and 12 μl of kinase detection reagent. The luminescence signal was detected using on a Molecular Device SpectraMax M3 spectrometer equipped with SoftMax Pro 5.4 software. The ATP-deficient mutant ABCA4-MM in which the lysine residues in the Walker A motif of NBD1 and NBD2 were substituted for methionine, p.Lys969Met and p.Lys1978Met, respectively, was used to subtract background luminescence (Ahn, Beharry, Molday, & Molday, 2003).
The binding of N-Ret-PE to immobilized ABCA4 in the presence and absence of ATP was carried out using [3H] all-trans retinal as previously described (Garces et al., 2018).
Data Analysis
In all cases, three or more independent experiments were carried out. Data are presented as the mean values with ± SD. Unpaired T-test was used to analyze differences between WT and several variants. Statistical analysis was carried out using Graphpad Prism 8.2.
Acknowledgments
We thank Dr. Stephanie Cornelis for valuable discussions on the age of onset of Stargardt patients. These studies were funded by grants from the Canadian Institutes of Health Research Grant PJT 148649 and National Institutes of Health Grant EY002422.
Footnotes
Conflict of Interest:
All authors confirm that they have no conflict of interest
Data Availability:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Uncategorized References
- Ahn J, Beharry S, Molday LL, & Molday RS (2003). Functional interaction between the two halves of the photoreceptor-specific ATP binding cassette protein ABCR (ABCA4). Evidence for a non-exchangeable ADP in the first nucleotide binding domain. J Biol Chem, 278(41), 39600–39608. doi: 10.1074/jbc.M304236200 [DOI] [PubMed] [Google Scholar]
- Ahn J, & Molday RS (2000). Purification and characterization of ABCR from bovine rod outer segments. Methods Enzymol, 315, 864–879. doi: 10.1016/s0076-6879(00)15887-2 [DOI] [PubMed] [Google Scholar]
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, … Lupski JR (1997). A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet, 15(3), 236–246. doi: 10.1038/ng0397-236 [DOI] [PubMed] [Google Scholar]
- Beharry S, Zhong M, & Molday RS (2004). N-retinylidene-phosphatidylethanolamine is the preferred retinoid substrate for the photoreceptor-specific ABC transporter ABCA4 (ABCR). J Biol Chem, 279(52), 53972–53979. doi: 10.1074/jbc.M405216200 [DOI] [PubMed] [Google Scholar]
- Boyer NP, Higbee D, Currin MB, Blakeley LR, Chen C, Ablonczy Z, … Koutalos Y (2012). Lipofuscin and N-retinylidene-N-retinylethanolamine (A2E) accumulate in retinal pigment epithelium in absence of light exposure: their origin is 11-cis-retinal. J Biol Chem, 287(26), 22276–22286. doi: 10.1074/jbc.M111.329235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bungert S, Molday LL, & Molday RS (2001). Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: identification of N-linked glycosylation sites. J Biol Chem, 276(26), 23539–23546. doi: 10.1074/jbc.M101902200 [DOI] [PubMed] [Google Scholar]
- Burke TR, Allikmets R, Smith RT, Gouras P, & Tsang SH (2010). Loss of peripapillary sparing in non-group I Stargardt disease. Exp Eye Res, 91(5), 592–600. doi: 10.1016/j.exer.2010.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon-Camacho OF, Granillo-Alvarez M, Ayala-Ramirez R, & Zenteno JC (2013). ABCA4 mutational spectrum in Mexican patients with Stargardt disease: Identification of 12 novel mutations and evidence of a founder effect for the common p.A1773V mutation. Exp Eye Res, 109, 77–82. doi: 10.1016/j.exer.2013.02.006 [DOI] [PubMed] [Google Scholar]
- Charbel Issa P, Barnard AR, Singh MS, Carter E, Jiang Z, Radu RA, … MacLaren RE (2013). Fundus autofluorescence in the Abca4(−/−) mouse model of Stargardt disease--correlation with accumulation of A2E, retinal function, and histology. Invest Ophthalmol Vis Sci, 54(8), 5602–5612. doi: 10.1167/iovs.13-11688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JA, Quazi F, & Molday RS (2013). Mammalian P4-ATPases and ABC transporters and their role in phospholipid transport. Biochim Biophys Acta, 1831(3), 555–574. doi: 10.1016/j.bbalip.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis SS, Bax NM, Zernant J, Allikmets R, Fritsche LG, den Dunnen JT, … Cremers FP (2017). In Silico Functional Meta-Analysis of 5,962 ABCA4 Variants in 3,928 Retinal Dystrophy Cases. Hum Mutat, 38(4), 400–408. doi: 10.1002/humu.23165 [DOI] [PubMed] [Google Scholar]
- Cremers FPM, Lee W, Collin RWJ, & Allikmets R (2020). Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog Retin Eye Res, 100861. doi: 10.1016/j.preteyeres.2020.100861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, Hamon Y, & Chimini G (2001). The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res, 42(7), 1007–1017. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/11441126 [PubMed] [Google Scholar]
- Fakin A, Robson AG, Chiang JP, Fujinami K, Moore AT, Michaelides M, … Webster AR (2016). The Effect on Retinal Structure and Function of 15 Specific ABCA4 Mutations: A Detailed Examination of 82 Hemizygous Patients. Invest Ophthalmol Vis Sci, 57(14), 5963–5973. doi: 10.1167/iovs.16-20446 [DOI] [PubMed] [Google Scholar]
- Fishman GA, Farber M, Patel BS, & Derlacki DJ (1987). Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology, 94(7), 809–814. doi: 10.1016/s0161-6420(87)33533-x [DOI] [PubMed] [Google Scholar]
- Fishman GA, Stone EM, Eliason DA, Taylor CM, Lindeman M, & Derlacki DJ (2003). ABCA4 gene sequence variations in patients with autosomal recessive cone-rod dystrophy. Arch Ophthalmol, 121(6), 851–855. doi: 10.1001/archopht.121.6.851 [DOI] [PubMed] [Google Scholar]
- Fujinami K, Sergouniotis PI, Davidson AE, Mackay DS, Tsunoda K, Tsubota K, … Webster AR (2013). The clinical effect of homozygous ABCA4 alleles in 18 patients. Ophthalmology, 120(11), 2324–2331. doi: 10.1016/j.ophtha.2013.04.016 [DOI] [PubMed] [Google Scholar]
- Garces F, Jiang K, Molday LL, Stohr H, Weber BH, Lyons CJ, … Molday RS (2018). Correlating the Expression and Functional Activity of ABCA4 Disease Variants With the Phenotype of Patients With Stargardt Disease. Invest Ophthalmol Vis Sci, 59(6), 2305–2315. doi: 10.1167/iovs.17-23364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu FY, Li JK, Gao FJ, Qi YH, Xu P, Zhang YJ, … Wu JH (2019). ABCA4 Gene Screening in a Chinese Cohort With Stargardt Disease: Identification of 37 Novel Variants. Front Genet, 10, 773. doi: 10.3389/fgene.2019.00773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illing M, Molday LL, & Molday RS (1997). The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J Biol Chem, 272(15), 10303–10310. doi: 10.1074/jbc.272.15.10303 [DOI] [PubMed] [Google Scholar]
- Klevering BJ, Blankenagel A, Maugeri A, Cremers FP, Hoyng CB, & Rohrschneider K (2002). Phenotypic spectrum of autosomal recessive cone-rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Invest Ophthalmol Vis Sci, 43(6), 1980–1985. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/12037008 [PubMed] [Google Scholar]
- Lambertus S, Lindner M, Bax NM, Mauschitz MM, Nadal J, Schmid M, … Foveal sparing Atrophy Study, T. (2016). Progression of Late-Onset Stargardt Disease. Invest Ophthalmol Vis Sci, 57(13), 5186–5191. doi: 10.1167/iovs.16-19833 [DOI] [PubMed] [Google Scholar]
- Lenis TL, Hu J, Ng SY, Jiang Z, Sarfare S, Lloyd MB, … Radu RA (2018). Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc Natl Acad Sci U S A, 115(47), E11120–E11127. doi: 10.1073/pnas.1802519115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie D, Arendt A, Hargrave P, McDowell JH, & Molday RS (1984). Localization of binding sites for carboxyl terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry, 23(26), 6544–6549. doi: 10.1021/bi00321a041 [DOI] [PubMed] [Google Scholar]
- Makelainen S, Godia M, Hellsand M, Viluma A, Hahn D, Makdoumi K, … Bergstrom TF (2019). An ABCA4 loss-of-function mutation causes a canine form of Stargardt disease. PLoS Genet, 15(3), e1007873. doi: 10.1371/journal.pgen.1007873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata NL, Weng J, & Travis GH (2000). Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A, 97(13), 7154–7159. doi: 10.1073/pnas.130110497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday RS (2015). Insights into the Molecular Properties of ABCA4 and Its Role in the Visual Cycle and Stargardt Disease. Prog Mol Biol Transl Sci, 134, 415–431. doi: 10.1016/bs.pmbts.2015.06.008 [DOI] [PubMed] [Google Scholar]
- Molday LL, Rabin AR, & Molday RS (2000). ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet, 25(3), 257–258. doi: 10.1038/77004 [DOI] [PubMed] [Google Scholar]
- Molday LL, Wahl D, Sarunic MV, & Molday RS (2018). Localization and functional characterization of the p.Asn965Ser (N965S) ABCA4 variant in mice reveal pathogenic mechanisms underlying Stargardt macular degeneration. Hum Mol Genet, 27(2), 295–306. doi: 10.1093/hmg/ddx400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasonkin I, Illing M, Koehler MR, Schmid M, Molday RS, & Weber BH (1998). Mapping of the rod photoreceptor ABC transporter (ABCR) to 1p21-p22.1 and identification of novel mutations in Stargardt’s disease. Hum Genet, 102(1), 21–26. doi: 10.1007/s004390050649 [DOI] [PubMed] [Google Scholar]
- Papermaster DS, Schneider BG, Zorn MA, & Kraehenbuhl JP (1978). Immunocytochemical localization of a large intrinsic membrane protein to the incisures and margins of frog rod outer segment disks. J Cell Biol, 78(2), 415–425. doi: 10.1083/jcb.78.2.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Zhao X, Cao P, Lei J, Yan N, & Gong X (2017). Structure of the Human Lipid Exporter ABCA1. Cell, 169(7), 1228–1239 e1210. doi: 10.1016/j.cell.2017.05.020 [DOI] [PubMed] [Google Scholar]
- Quazi F, Lenevich S, & Molday RS (2012). ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun, 3, 925. doi: 10.1038/ncomms1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quazi F, & Molday RS (2014). ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc Natl Acad Sci U S A, 111(13), 5024–5029. doi: 10.1073/pnas.1400780111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveiro-Alvarez R, Lopez-Martinez MA, Zernant J, Aguirre-Lamban J, Cantalapiedra D, Avila-Fernandez A, … Ayuso C (2013). Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: retrospective analysis in 420 Spanish families. Ophthalmology, 120(11), 2332–2337. doi: 10.1016/j.ophtha.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runhart EH, Sangermano R, Cornelis SS, Verheij J, Plomp AS, Boon CJF, … Cremers FPM (2018). The Common ABCA4 Variant p.Asn1868Ile Shows Nonpenetrance and Variable Expression of Stargardt Disease When Present in trans With Severe Variants. Invest Ophthalmol Vis Sci, 59(8), 3220–3231. doi: 10.1167/iovs.18-23881 [DOI] [PubMed] [Google Scholar]
- Schulz HL, Grassmann F, Kellner U, Spital G, Ruther K, Jagle H, … Stohr H (2017). Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients From a Multicenter German Cohort-Impact of Selected Deep Intronic Variants and Common SNPs. Invest Ophthalmol Vis Sci, 58(1), 394–403. doi: 10.1167/iovs.16-19936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow JR, & Boulton M (2005). RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res, 80(5), 595–606. doi: 10.1016/j.exer.2005.01.007 [DOI] [PubMed] [Google Scholar]
- Sun H, Molday RS, & Nathans J (1999). Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J Biol Chem, 274(12), 8269–8281. doi: 10.1074/jbc.274.12.8269 [DOI] [PubMed] [Google Scholar]
- Sun H, Smallwood PM, & Nathans J (2000). Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genet, 26(2), 242–246. doi: 10.1038/79994 [DOI] [PubMed] [Google Scholar]
- Tanna P, Strauss RW, Fujinami K, & Michaelides M (2017). Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol, 101(1), 25–30. doi: 10.1136/bjophthalmol-2016-308823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsybovsky Y, Orban T, Molday RS, Taylor D, & Palczewski K (2013). Molecular organization and ATP-induced conformational changes of ABCA4, the photoreceptor-specific ABC transporter. Structure, 21(5), 854–860. doi: 10.1016/j.str.2013.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, & Travis GH (1999). Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell, 98(1), 13–23. doi: 10.1016/S0092-8674(00)80602-9 [DOI] [PubMed] [Google Scholar]
- Westeneng-van Haaften SC, Boon CJ, Cremers FP, Hoefsloot LH, den Hollander AI, & Hoyng CB (2012). Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology, 119(6), 1199–1210. doi: 10.1016/j.ophtha.2012.01.005 [DOI] [PubMed] [Google Scholar]
- Wiszniewski W, Zaremba CM, Yatsenko AN, Jamrich M, Wensel TG, Lewis RA, & Lupski JR (2005). ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum Mol Genet, 14(19), 2769–2778. doi: 10.1093/hmg/ddi310 [DOI] [PubMed] [Google Scholar]
- Zernant J, Collison FT, Lee W, Fishman GA, Noupuu K, Yuan B, … Allikmets R (2014). Genetic and clinical analysis of ABCA4-associated disease in African American patients. Hum Mutat, 35(10), 1187–1194. doi: 10.1002/humu.22626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zernant J, Lee W, Collison FT, Fishman GA, Sergeev YV, Schuerch K, … Allikmets R (2017). Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J Med Genet, 54(6), 404–412. doi: 10.1136/jmedgenet-2017-104540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Tsybovsky Y, Kolesnikov AV, Rozanowska M, Swider M, Schwartz SB, … Palczewski K (2015). Protein misfolding and the pathogenesis of ABCA4-associated retinal degenerations. Hum Mol Genet, 24(11), 3220–3237. doi: 10.1093/hmg/ddv073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Molday LL, & Molday RS (2009). Role of the C terminus of the photoreceptor ABCA4 transporter in protein folding, function, and retinal degenerative diseases. J Biol Chem, 284(6), 3640–3649. doi: 10.1074/jbc.M806580200 [DOI] [PMC free article] [PubMed] [Google Scholar]