

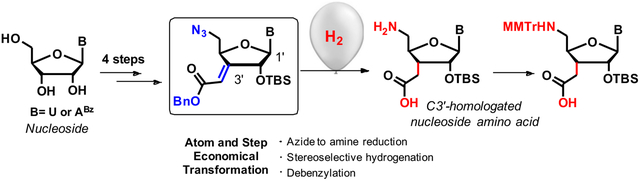
Abstract
Internucleoside amide linkages are excellent mimics of phosphodiesters in RNA and may be used to optimize the properties of short interfering RNAs. Herein we report a remarkably straightforward, efficient and step economic synthesis of C3´-homologated uridine and adenosine amino acids starting from nucleosides in six steps (31% overall yield) and eight steps (16% overall yield), respectively. The key enabling step is a one-pot multi-functional group transformation including a stereo selective hydrogenation, termed “Global Hydrogenation” (GH).
Graphical Abstract
RNA interference (RNAi) has become a broadly used and powerful tool in functional genomics.1 With more than 20 currently active clinical trials, RNAi also has intriguing potential to become a new therapeutic approach.2–4 For therapeutic and other in vivo applications, short interfering RNAs (siRNAs) need chemical modifications to optimize target specificity, enzymatic stability, cellular uptake, biodistribution and pharmacokinetics.5–7 Phosphate backbone modifications are a remarkably successful example of such modifications. Antisense drugs recently approved by FDA contain phosphorothioate (Fomivirsen and Mipomersen8) and neutral phosphorodiamidate morpholino (Eteplirsen9) linkages that demonstrate the cogency of backbone modifications. Another example of highly successful backbone modification is peptide nucleic acid (PNA) that entails the replacement of the entire sugar-phosphate moiety with a neutral N-(2-aminoethyl)glycine backbone, which makes PNA highly resistant to nucleases and proteases.10, 11
Amide linkages have been explored as non-ionic replacements for phosphodiesters in antisense deoxyoligonucleo-tides.12 Isolated amide linkages (such as those in Figure 1b) were well accommodated in DNA-RNA heteroduplexes.13–17 Rozners and co-workers18, 19 showed that amide linkages were excellent structural mimics of phosphodiesters in RNA and did not significantly change the structure, thermal stability and hydration of short all-RNA duplexes.
Figure 1.
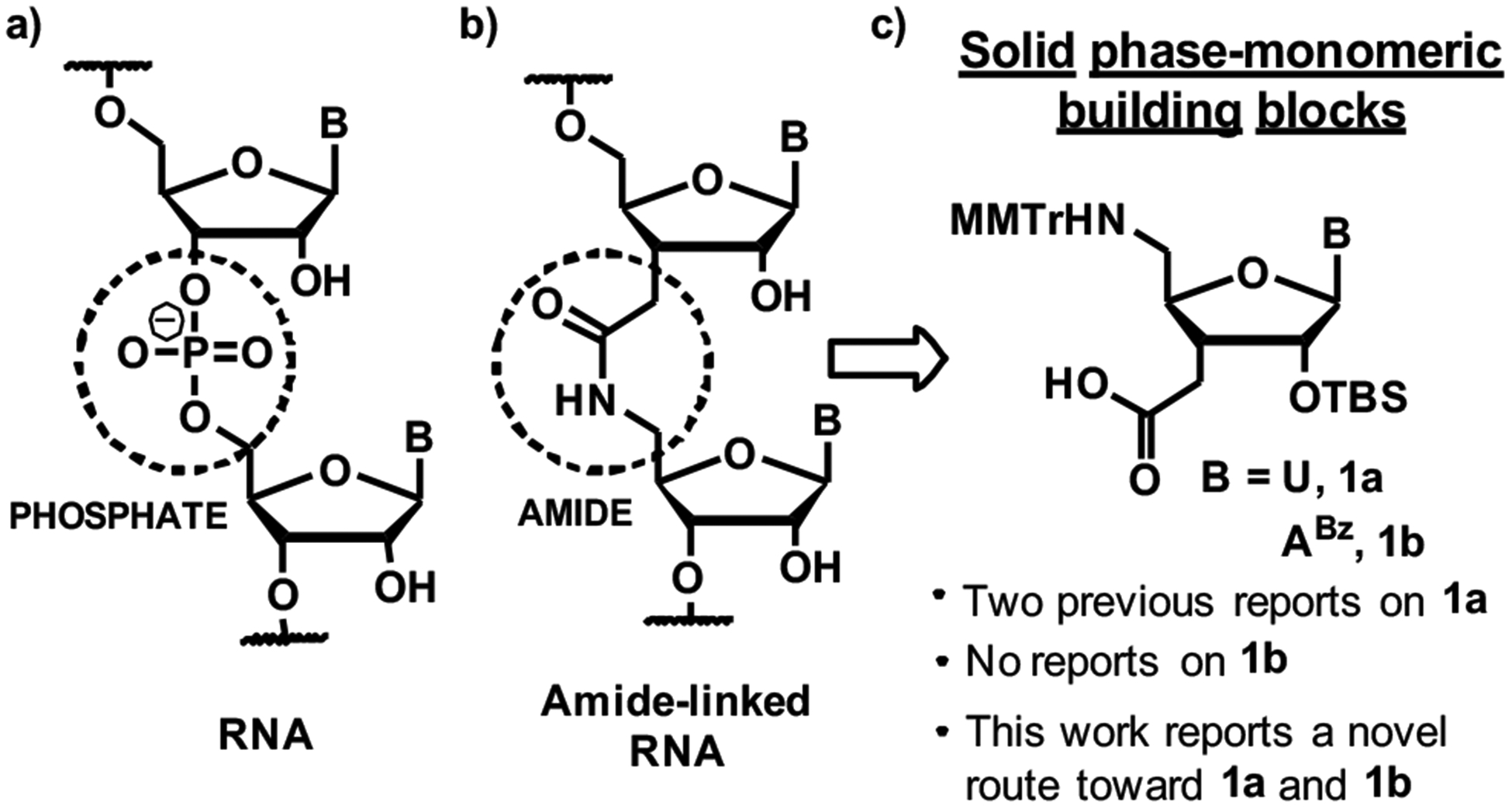
Structure of amide-linked RNA and its synthetic precursors, C3´-homologated nucleoside amino acids
Initial tests of amide linkages as backbone modifications in siRNAs have given promising results. Iwase and co-workers20–22 modified the 3´-overhangs of a siRNA with two amide linkages, which resulted in increased enzymatic stability and no significant change in RNAi activity. Recently, our group showed that isolated amide linkages were well tolerated at internal positions of both guide and passenger strands of siRNAs and increased the silencing activity when placed near the 5´-end of the passenger strand and in the middle of the guide strand.23 Even three consecutive amide linkages at the 5´-end of the passenger strand did not significantly decrease the siRNA activity.24 These results were encouraging for future studies on amide-linked siRNAs and also suggested that amide linkages may be useful modifications for the optimization of other emerging RNA-based gene control technologies such as CRISPR-Cas.25–27 However, the problem for future studies on amide-modified siRNAs and CRISPR-Cas RNAs is the complex, lengthy and low yielding synthesis of C3´-homologated nucleoside amino acids 1 (Figure 1c), the monomers required for introduction of consecutive amide linkages in RNA.
The first synthesis of C3´-homologated ribonucleoside amino acids, monomers required for the preparation of amide-linked RNA was developed by Peterson et al.28 The route started from d-xylose, required 11 steps and produced a 5´-azido precursor of uridine 1a in ~10% yield. This route was used by the same authors to prepare short amide-linked oligouri-dines29 and by Iwase and co-workers to prepare 3´-amide-modified siRNAs.20–22 We developed two synthetic routes to all four C3´-homologated nucleoside amino acids 1 having the 2´-O-acetyl protection.30, 31 However, the 2´-O-acetyl group was later found incompatible with solid-phase synthesis of RNA strands having consecutive amide linkages.24 While it was possible to modify our synthesis to produce 2´-O-TBS protected uridine, which was compatible with synthesis of consecutive amide linkages, the route required 18 steps and gave 1a in only ~4% yield.24, 30, 31 Hence, the laborious and low yielding syntheses of monomers is a major limitation for future studies on amide linkages in RNA based gene control technologies.
In this communication, we report a short and efficient synthesis of C3´-homologated uridine and adenosine amino acids from commercially available nucleosides. The synthesis requires only six steps for 1a (31% overall yield) and eight steps for 1b (16% overall yield). The key step enabling our route is a remarkable one-pot concurrent hydrogenation of three different functional groups, termed “Global Hydrogenation” (GH), that transforms azide to amine, stereo selectively hydrogenates an exocyclic C=C bond and cleaves a benzyl ester protecting group. The use of benzyl protecting group for carboxylic acid avoids the troublesome basic hydrolysis of alkyl (Me or Et) esters and is critical for preparation of compounds having base labile N-protecting groups (e.g., N6-benzoyladenosine). In contrast to previous syntheses, the new route is atom- and step economic and does not require a separate deprotection reaction to reach the final product.
Our synthetic route (Scheme 1) started from uridine and N6-benzoyladenosine32 and followed the C3´-homologation strategy developed by Peterson et al.33 The synthesis began with installation of the 5´-azido group using an Appel reaction34 followed by protection of the 2´-OH as a TBS ether. The large difference in yields of 2a and 2b was somewhat puzzling, but consistent with the original procedure.34 The desired 2´-O-TBS products were separated from the undesired 3´-O-TBS isomers using silica gel column chromatography. The identity of the 2´- and 3´-O-TBS isomers was confirmed by 1H and 2D NMR, see Supporting Information (SI). To increase the efficiency, the undesired 3´-O-TBS compounds were isomerized to mixtures of 2´- and 3´-O-TBS compounds,35 which could be separated again to yield more of the desired 2´-O-TBS isomer. Recycling of the 3´-O-TBS isomers increased the yields of 3a and 3b from 47% and 40% to 66% and 55%, respectively, for experimental details, see SI. The 3´-OH of 3a and 3b were oxidized to their corresponding ketones, using Dess-Martin periodinane (DMP) in dry dichloromethane (DCM). Using fresh DMP reagent with a minute amount of added water36 provided very clean oxidation and the resulting ketones did not require purification. The initial homologation attempts using a Wittig reagent, generated in situ from triphenyl phosphine and benzylbromoacetate, gave low yields (15–40%). Horner-Wadsworth-Emmons reaction with benzyldiethylphosphonoacetate led to decomposition of the ketone. After some optimization, efficient Wittig alkenylation was accomplished using commercially available benzyl(triphenylphosphoranylidene) acetate, furnishing the key advanced intermediates 4a and 4b, substrates for the GH reaction (Scheme 1). The stereochemistry of the Wittig products was not determined because both isomers would give the same product in the next hydrogenation step.
Scheme 1.
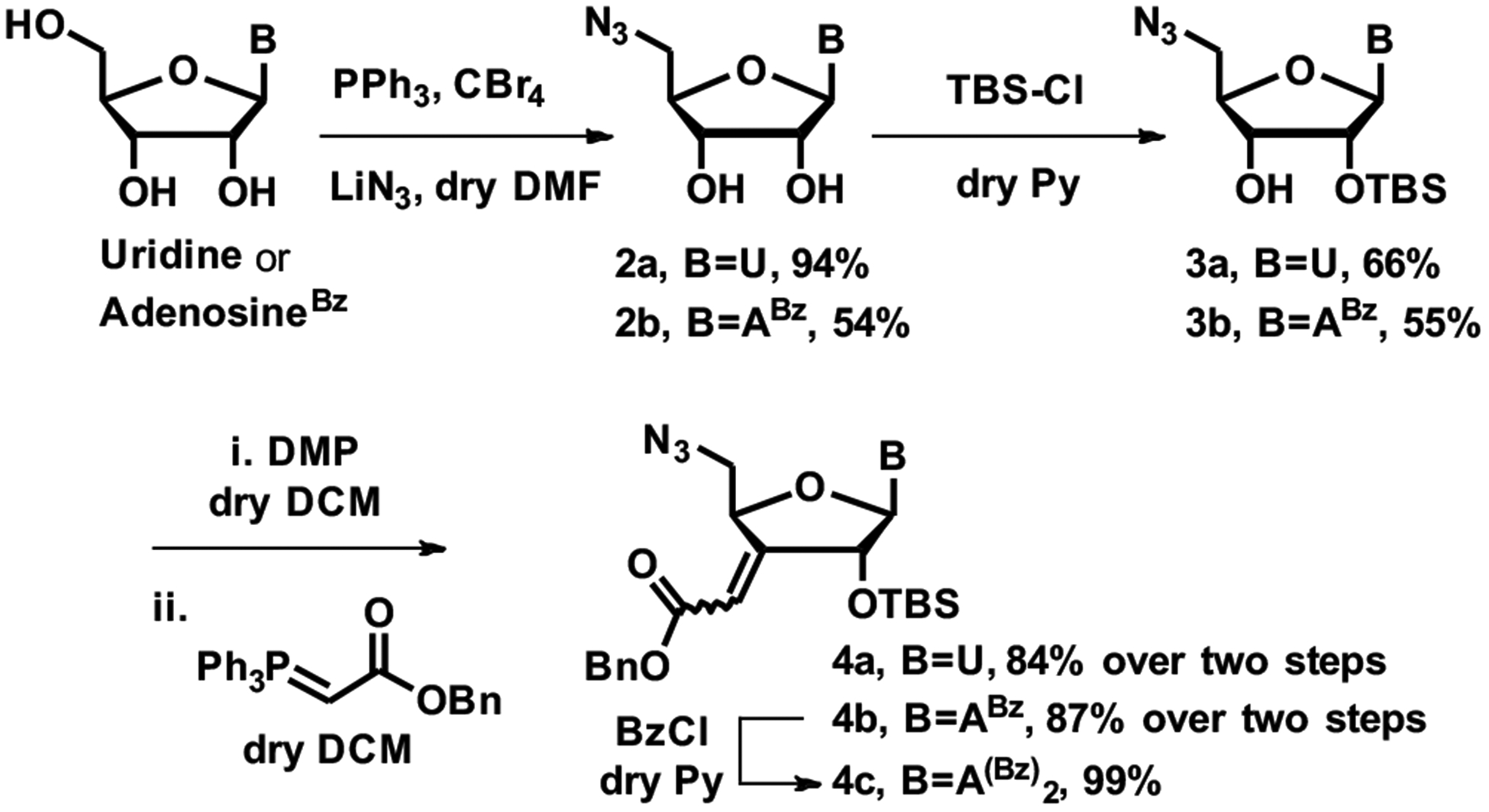
Synthesis of advanced intermediates for global hydrogenation
Our first attempts at GH (Scheme 2) of 4a using 10% Pd/C (40% w/w) and hydrogen gas (delivered from a rubber balloon) in a mixture of MeOH:EtOAc (1:1) furnished the desired amino acid 5a in ~50% yield along with a side product that was assigned as the over-reduced 5,6-dihydrouracil derivative (7, Scheme 2 and Table 1, entry 1) based on NMR analysis of crude reaction mixtures. Using the same conditions for the GH of adenosine 4b (Scheme 2) led to the isolation of the partially reduced compound 6b having an unreacted exocyclic C=C as the sole product. Attempts to promote reduction of the alkene in 4b by increasing the amount of Pd to 100% w/w, extending the reaction time or adding acetic acid, were not successful. Alternative approaches, such as transfer hydrogenation and conjugate reduction of 4b using NiCl2/NaBH4 or Red-Al/CuBr, failed to give the desired product. Our attempts at conjugate reduction of the α,β-unsaturated carboxylic acid 6b were also unproductive. Recognizing that the only chemical difference between the two substrates 4a and 4b was their different nucleobases, uracil and N6-benzoyladenine, respectively, we speculated that the latter might complex with Pd as an electron-rich ligand, reducing the activity of catalyst. We hypothesized that decreasing the electron density of adenine by introducing a second benzoyl group, as in 4c, could block the putative complexation and restore the catalyst activity. Treatment of 4b with benzoyl chloride in dry pyridine (Scheme 1) gave the dibenzoyl-protected derivative 4c as a new GH substrate. Hydrogenation of this new substrate using 10% Pd/C (100% w/w) in MeOH:EtOAc (1:1) over 72 hours yielded the desired GH product 5b in 68% isolated yield (Table 1, entry 8). Consistent with the pioneering studies of Peterson et al.,33 we observed complete stereoselectivity of the hydrogenation of the exocyclic double bond in 4a and 4c, most likely caused by the bulky α−2´-O-TBS group. We obtained the desired α-C3´-homologated products 5a and 5b (Scheme 2) as sole stereoisomers.
Scheme 2.
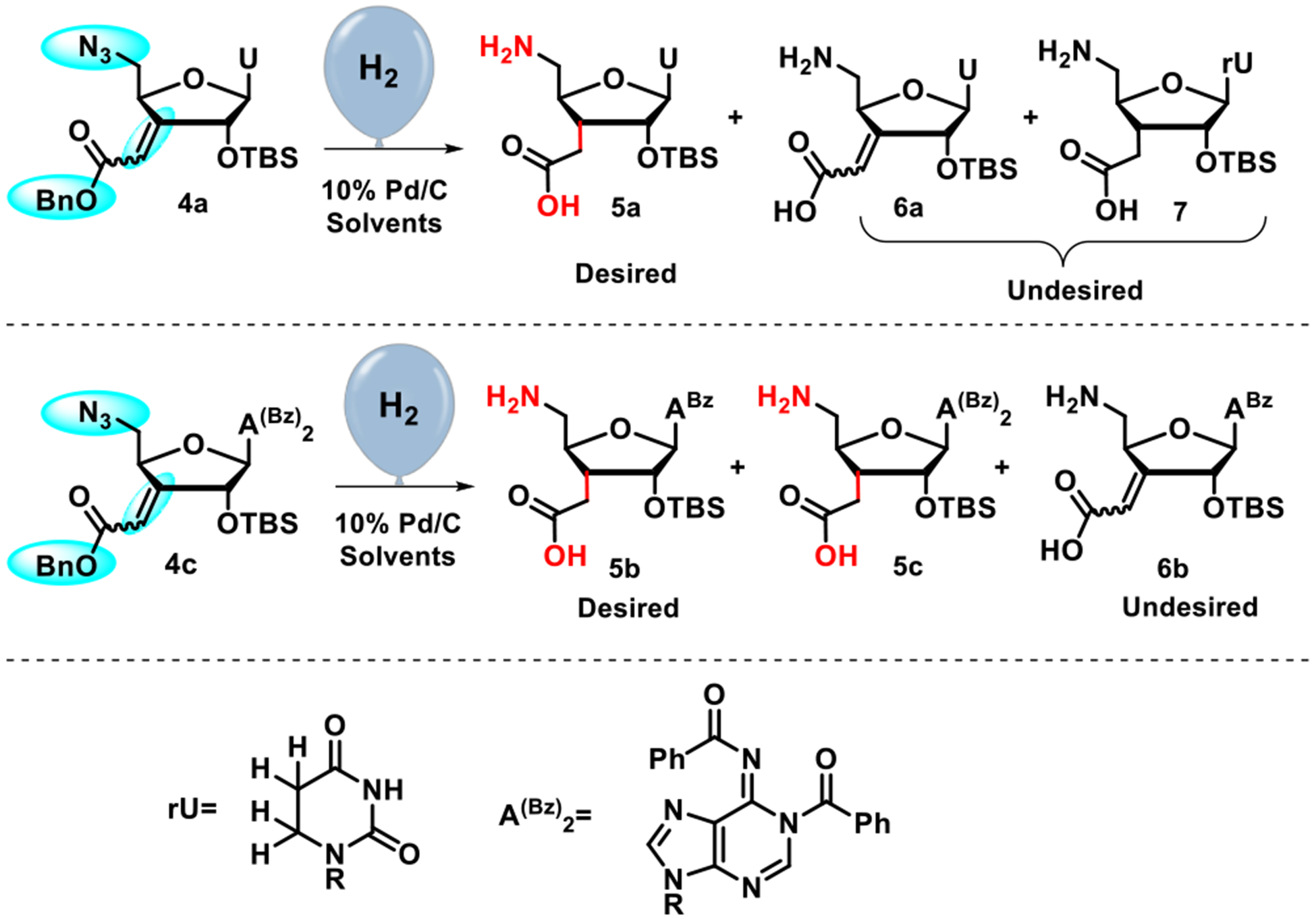
Global hydrogenation of advanced intermediates
Table 1.
Optimization of global hydrogenation conditions
| s. no | GH substrate | Pd% (w/w) | solvent | time (h) | product ratioa (5a:6a:7) or (5b:6b) | yield % of 1a/1b (over 2 steps) |
|---|---|---|---|---|---|---|
| 1 | 4a | 40 | MeOH:EtOAc (1:1) | 20 | 61:0:39 | ndc |
| 2 | 4a | 40 | MeOH:EtOAc (1:1) | 4 | incomplete reaction | ndc |
| 3 | 4a | 40 | MeOH:H2O (1:1) | 4 | 50:0:50 | ndc |
| 4 | 4a | 40 | t-BuOH:H2O (1:1) | 4 | 67:0:33 | ndc |
| 5 | 4a | 30 | t-BuOH:H2O (1:1) | 1.5 | 56:44:0 | ndc |
| 6 | 4a | 30 | t-BuOH:H2O (1:1) | 3.0 | 90:10:0 | 60% |
| 7 | 4a | 30 | t-BuOH:H2O (1:1) | 3.5 | 83:0:17 | ndc |
| 8 | 4c | 100 | MeOH:EtOAc (1:1) | 72 | 68%b | ndc |
| 9 | 4c | 100 | tBuOH:H2O (1:1) | 16 | 17:83 | ndc |
| 10 | 4c | 100 | MeOH:H2O (5:1) | 24 | 95:5 | 61% |
| 11 | 4c | 50 | MeOH:H2O (5:1) | 72 | 90:10 | ndc |
based on 1H NMR analysis of crude reaction mixture filtered over celite;
isolated yield;
nd –not determined, the compound was not used to make 1a/1b.
Replacing EtOAc with water in the mixed solvent system accelerated the reaction and enabled clean and quantitative conversions in shorter time periods (Table 1, cf. entries 1 vs. 3, and 8 vs. 10). In the uridine series, replacement of MeOH with t-BuOH provided milder and more selective hydrogenation conditions, which was critical for elimination of the over-reduced by-product 7. Further optimizations of the reaction conditions by fine-tuning catalyst loading and reaction time (Table 1, entry 6) achieved quantitative conversion and gave a mixture of the desired product 5a and alkene 6a (9:1).
In the adenosine series, more forceful hydrogenation in MeOH:H2O using 100% w/w of Pd catalyst over 24 hours was required to achieve quantitative conversion (entry 10, Table 1). The GH in aqueous methanol led to partial loss of the more labile second benzoyl group,37 resulting in a ~3:1 mixture of 5b and 5c. After a work-up with ammonia to completely convert 5c to 5b, NMR analysis of crude GH reaction mixture showed the desired product 5b along with ~5% of 6b. Reducing the amount of Pd to 50% w/w while increasing reaction time, directly furnished the desired monobenzoylated adenine compound 5b and eliminated the need for ammonia work-up (Table 1, entry 11), albeit in a less favorable ratio of 5b to 6b. During optimization of the GH reaction, we found that isolation of the polar amino acids was cumbersome. Therefore, we optimized the GH reactions based on 1H NMR analysis of crude products. The crude reaction mixtures of successful hydrogenations (based on 1H NMR analysis) were used directly in the next reaction. The final step of synthesis of the C3´-homologated uridine and adenosine amino acid monomers 1a and 1b involved protection of the 5´-amino group of crude GH products using methoxytrityl chloride (MMTr-Cl) in pyridine (Scheme 3). Compounds 1a and 1b are ready for use in our recently developed solid-phase synthesis of RNA containing consecutive amide linkages.24 Our initial attempts at the global hydrogenation of cytidine substrate in t-BuOH:H2O (4:1) failed to reduce the exocyclic double bond. Replacing t-BuOH by MeOH gave a mixture of the desired C3’-homologated cytidine amino acid along with over-reduced (C5=C6) product. The synthesis of guanosine substrate for the GH reaction is currently in progress.
Scheme 3.
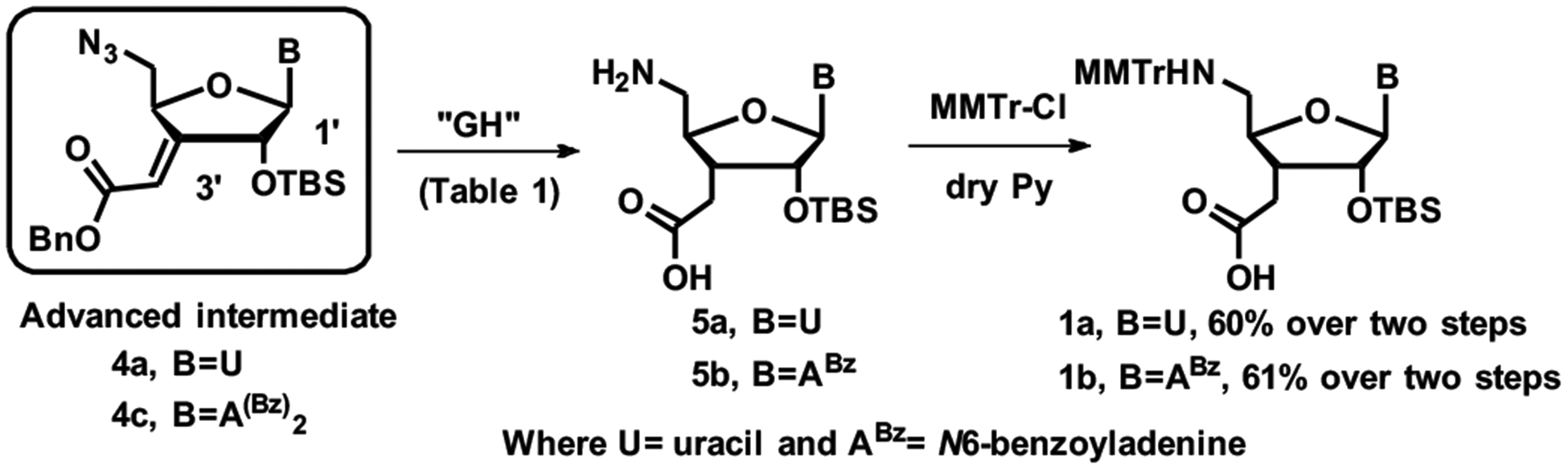
Completion of synthesis of U and A amino acid building blocks
In summary, we have developed a short and efficient synthesis of C3´-homologated uridine and adenosine amino acids 1a and 1b, monomers for solid-phase synthesis of amide-linked RNA. The key innovation and enabling step is a global hydrogenation that achieves three concurrent reactions: transformation of azide to amine, stereoselective hydrogenation of an exocyclic C=C bond and cleavage of a benzyl ester protecting group, all in a one-pot single-step reaction. Compared to previous syntheses, our new route is remarkably efficient and atom- and step economic, achieving 1a in only six steps with 31% overall yield and 1b in eight steps with 16% overall yield from commercially available nucleosides. The new route will open the door to more extensive studies on amide-modified siRNAs that have shown promising results in initial tests.20–24
Supplementary Material
ACKNOWLEDGMENT
We thank NIH (R01 GM071461) for financial support of this research. The Regional NMR Facility (600 MHz instrument) at Binghamton University is supported by NSF (CHE-0922815).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and the supporting spectral data are provided (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Ralph A; Karpilow JM Frontiers in RNAi. Bentham Science; 2014, 1, 268. [Google Scholar]
- (2).Bobbin ML; Rossi JJ Annu. Rev. Pharmacol. Toxicol 2016, 56, 103–122. [DOI] [PubMed] [Google Scholar]
- (3).Zuckerman JE; Davis ME Nat. Rev. Drug Discovery 2015, 14, 843–856. [DOI] [PubMed] [Google Scholar]
- (4).Wittrup A; Lieberman J Nat. Rev. Genet 2015, 16, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Watts JK; Corey DR J. Pathol 2012, 226, 365–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Deleavey GF; Damha MJ Chem. Biol 2012, 19, 937–954. [DOI] [PubMed] [Google Scholar]
- (7).Bramsen JB; Grunweller A; Hartmann RK; Kjems J In Handbook of RNA Biochemistry, Second Edition, Hartmann RK, Schon AB,A, and Westhof E, Ed. Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014, 1243–1277. [Google Scholar]
- (8).Hair P; Cameron F; McKeage K Drugs 2013, 73, 487–493. [DOI] [PubMed] [Google Scholar]
- (9).Lim KRQ; Maruyama R; Yokota T Drug Design, Development and Therapy 2017, 11, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Egholm M; Buchardt O; Christensen L; Behrens C; Freier SM; Driver DA; Berg RH; Kim SK; Norden B; Nielsen PE Nature 1993, 365, 566–568. [DOI] [PubMed] [Google Scholar]
- (11).Nielsen PE; Egholm M; Berg RH; Buchardt O Science 1991, 254, 1497–1500. [DOI] [PubMed] [Google Scholar]
- (12).De Mesmaeker A; Waldner A; Wendeborn S; Wolf RM Pure Appl. Chem 1997, 69, 437–440. [Google Scholar]
- (13).Idziak I; Just G; Damha MJ; Giannaris PA Tetrahedron Lett. 1993, 34, 5417–5420. [Google Scholar]
- (14).De Mesmaeker A; Waldner A; Lebreton J; Hoffmann P; Fritsch V; Wolf RM; Freier SM Angew. Chem., Int. Ed. Engl 1994, 33, 226–229. [Google Scholar]
- (15).Lebreton J; Waldner A; Lesueur C; De Mesmaeker A Synlett 1994, 137–140. [Google Scholar]
- (16).De Mesmaeker A; Lesueur C; Bevierre MO; Waldner A; Fritsch V; Wolf RM Angew. Chem., Int. Ed 1996, 35, 2790–2794. [Google Scholar]
- (17).De Mesmaeker A; Lebreton J; Jouanno C; Fritsch V; Wolf RM; Wendeborn S Synlett 1997, 1287–1290. [Google Scholar]
- (18).Rozners E; Katkevica D; Bizdena E; Strömberg RJ Am. Chem. Soc 2003, 125, 12125–12136. [DOI] [PubMed] [Google Scholar]
- (19).Selvam C; Thomas S; Abbott J; Kennedy SD Angew. Chem., Int. Ed 2011, 50, 2068–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Iwase R; Toyama T; Nishimori K Nucleosides, Nucleotides & Nucleic Acids 2007, 26, 1451–1454. [DOI] [PubMed] [Google Scholar]
- (21).Iwase R; Kurokawa R; Ueno J Nucleic Acids Symp. Ser 2009, 53, 119–120. [DOI] [PubMed] [Google Scholar]
- (22).Iwase R; Miyao H; Toyama T; Nishimori K Nucleic Acids Symp. Ser 2006, 175–176. [DOI] [PubMed] [Google Scholar]
- (23).Mutisya D; Selvam C; Lunstad BD; Pallan PS; Haas A; Leake D; Egli M; Rozners E Nucleic Acids Res. 2014, 42, 6542–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tanui P; Kennedy SD; Lunstad BD; Haas A; Leake D; Rozners E Org. Biomol. Chem 2014, 12, 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hendel A; Bak RO; Clark JT; Kennedy AB; Ryan DE; Roy S; Steinfeld I; Lunstad BD; Kaiser RJ; Wilkens AB; Bacchetta R; Tsalenko A; Dellinger D; Bruhn L; Porteus MH Nat. Biotechnol 2015, 33, 985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rahdar M; McMahon MA; Prakash TP; Swayze EE; Bennett CF; Cleveland DW Proc. Natl. Acad. Sci. U. S. A 2015, 112, E7110–E7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Latorre A; Latorre A; Somoza A Angew. Chem., Int. Ed 2016, 55, 3548–3550. [DOI] [PubMed] [Google Scholar]
- (28).Robins MJ; Doboszewski B; Timoshchuk VA; Peterson MA J. Org. Chem 2000, 65, 2939–2945. [DOI] [PubMed] [Google Scholar]
- (29).Robins MJ; Doboszewski B; Nilsson BL; Peterson MA Nucleosides, Nucleotides Nucleic Acids 2000, 19, 69–86. [DOI] [PubMed] [Google Scholar]
- (30).Rozners E; Liu YJ Org. Chem 2005, 70, 9841–9848. [DOI] [PubMed] [Google Scholar]
- (31).Tanui P; Kullberg M; Song N; Chivate Y; Rozners E Tetrahedron 2010, 66, 4961–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ti GS; Gaffney BL; Jones RA J. Am. Chem. Soc 1982, 104, 1316–1319. [Google Scholar]
- (33).Peterson MA; Nilsson BL; Sarker S; Doboszewski B; Zhang W; Robins MJ J. Org. Chem 1999, 64, 8183–8192. [DOI] [PubMed] [Google Scholar]
- (34).Yamamoto I; Sekine M; Hata TJ Chem. Soc., Perkin Trans 1 1980, 306–310. [Google Scholar]
- (35).Ogilvie KK; Beaucage SL; Schifman AL; Theriault NY; Sadana KL Can. J. Chem 1978, 56, 2768–2780. [Google Scholar]
- (36).Meyer SD; Schreiber SL J. Org. Chem 1994, 59, 7549–7552. [Google Scholar]
- (37).Ogilvie KK; Entwistle DW Carbohydr. Res 1981, 89, 203–210. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.