

Abstract
Background
Ulcerative colitis (UC) carries an increased risk of primary and recurrent Clostridiodes difficile infection (rCDI), and CDI is associated with UC flares. We hypothesized that specific fecal microbial changes associate with UC flare and rCDI.
Methods
We conducted a prospective observational cohort study of 57 patients with UC and CDI, CDI only, and UC only. Stool samples were collected at baseline, at the end of antibiotic therapy, and after reconstitution for 16S rRNA sequencing. The primary outcomes were recurrent UC flare and rCDI. Logistic regression and Lasso models were constructed for analysis.
Results
There were 21 (45.7%) patients with rCDI, whereas 11 (34.4%) developed UC flare. Patients with rCDI demonstrated significant interindividual (P = 0.008) and intraindividual differences (P = 0.004) in community structure by Jensen-Shannon distance (JSD) compared with non-rCDI. Two cross-validated Lasso regression models predicted risk of rCDI: a baseline model with female gender, hospitalization for UC in the past year, increased Ruminococcaceae and Verrucomicrobia, and decreased Eubacteriaceae, Enterobacteriaceae, Lachnospiraceae, and Veillonellaceae (AuROC, 0.94); and a model 14 days after completion of antibiotics with female gender, increased Shannon diversity, Ruminococcaceae and Enterobacteriaceae, and decreased community richness and Faecalibacterium (AuROC, 0.9). Adding JSD between baseline and post-treatment samples to the latter model improved fit (AuROC, 0.94). A baseline model including UC hospitalization in the past year and increased Bacteroidetes was associated with increased risk for UC flare (AuROC, 0.88).
Conclusion
Fecal microbial features at baseline and after therapy predict rCDI risk in patients with and without UC. These results may help risk stratify patients to guide management.
Keywords: gut microbiota, predictive modeling, Clostridiodes difficile infection, ulcerative colitis, Lasso regression
Fecal microbial features at baseline and after therapy predict risk for recurrent Clostridium difficile infection in patients with and without ulcerative colitis. These results may help risk stratify patients to guide management.
INTRODUCTION
Accumulating evidence suggests that disruptions to the gut microbial structure and function contribute to inflammatory bowel disease (IBD).1 Furthermore, patients with IBD, particularly those with ulcerative colitis (UC), are at increased risk for Clostridiodes difficile (previously Clostridium difficile) infection (CDI).2–4 It is possible that alterations in the gut microbial community may predispose IBD patients to CDI. Murine models suggest that shifts in microbial ecology are associated with susceptibility to experimental CDI.5 Notably, IBD patients with CDI are more likely to have recurrent CDI (rCDI)6 while also demonstrating a 2-fold increased risk for subsequent exacerbation of IBD and colectomy and a 5-fold increased risk for mortality compared with those without CDI.3, 7–9
It is not clear how the microbiome disruptions seen in CDI and IBD relate to each other and/or interact. Thus, we aimed to characterize the fecal microbiota in patients with UC and/or CDI longitudinally and investigate whether fecal microbial dynamics may predict future risk for rCDI and recurrent UC flare. We hypothesized that poor reconstitution of the gut microbiome at the end of antibiotics plus 14 days (EOA + 14) would be associated with higher risk for subsequent rCDI and/or UC flare.
MATERIALS AND METHODS
Study Design
We conducted a single-center prospective, observational cohort study, including symptomatic UC patients who tested positive for CDI (cohort 1), non-IBD patients with symptomatic CDI (cohort 2), and patients with UC flare without CDI (cohort 3). All subjects were 18 years or older and provided written informed consent before enrollment. Subjects were excluded if they had presence of an ostomy or previous history of colectomy.
The primary outcomes were subsequent episodes of UC flare and/or rCDI. Ulcerative colitis flare was defined by onset of typical symptoms occurring after enrollment in the study, along with a 6-point Mayo score >2.5 and a fecal calprotectin >150 μg/g in the absence of CDI.10 A gastrointestinal pathogen polymerase chain reaction (PCR) panel and cytomegalovirus PCR were also performed to rule out other common bacterial, viral, and parasitic causes of acute symptoms in UC patients. Clostridiodes difficile infection was diagnosed by presence of diarrhea (≥3 unformed stools in a 24-hour period) and a positive stool test for toxigenic C. difficile (see online supplemental materials). The rCDI was defined by recurrence of symptoms at least 14 days after initial treatment of CDI and positive C. difficile testing.11 Choice of antibiotic therapy was left up to the individual provider but followed a standardized protocol at the University of Michigan.12 Patients with recurrent or severe CDI, defined as white blood cell (WBC) ≥15 K/μL, Cr ≥1.5x baseline, age 65 years or older, absolute neutrophil count ≤500 K/μL, or albumin ≤2.5 g/dL, were treated with 125 mg of vancomycin by mouth 4 times daily for 10 to 14 days. Patients with mild-moderate CDI were treated with 500 mg of metronidazole by mouth 3 times daily for 10 to 14 days.
Subjects in cohorts 1 and 2 were contacted, and their medical records were reviewed every 60 days for up to 180 days after enrollment to determine recurrence of UC flare and/or rCDI. Due to the small sample sizes in each cohort, patients in cohorts 1 and 2 were analyzed collectively to determine the rate of rCDI while adjusting for UC status. Patients in cohort 1 were also followed to determine the rate of subsequent UC flare. As subjects in cohort 3 (UC flare only) were experiencing an exacerbation of their UC on enrollment and were only followed for 30 days, it was not possible to differentiate between an ongoing vs recurrent UC flare. As a result, patients in cohort 3 were excluded from meeting the primary endpoint of UC flare but were used to adjust models for UC status.
Based on previous data using fecal microbiota to predict development of CDI,13 we expected to require a minimum of 12 cases to build an accurate model. Prior data suggested a 30% rCDI rate in patients with UC and CDI.9 Thus, we conservatively estimated that a sample size of 50 subjects providing 150 stool samples would provide ≥80% power to detect differences across a full range of possible recurrence rates.
Stool Collection
For cohorts 1 and 2, stool samples were collected at baseline (before initiation of antibiotics or medical therapy, day 0), at the end of antibiotics (EOA, approximately day 14), and EOA plus 14 days (approximately day 30; Supplemental Fig. 1). For cohort 3, stool samples were collected at baseline and at day 30 after clinical remission was achieved.
16S Sequence Analysis
After DNA extraction, 16S rRNA genes were sequenced and processed through the computational pipeline mothur14 as previously described (see online supplemental materials).15 The following microbiome metrics were generated: Shannon diversity, Jensen-Shannon distance (JSD) relative to baseline and subsequent samples, community type based on JSD,16 and relative abundance of individual operational taxonomic units (OTUs). Other variables based on taxonomic class were analyzed, including the microbiome health index (MHI), defined as the proportion of Bacteroidia and Clostridia compared with the proportion of Gammaproteobacteria and Bacilli.17
Statistical Analysis
Subjects were included for analysis if they had complete follow-up data for 180 days and provided all fecal samples (Supplemental Fig. 2). Continuous data were compared using 1-way ANOVA or Kruskal-Wallis test when appropriate. Comparisons of proportions were performed using Fisher exact test. All data were analyzed using R version 3.5.2 (R Foundation for Statistical Computing, Vienna, Austria). A 2-tailed P value <0.05 was considered significant for all analyses.
Unadjusted and adjusted logistic regression analyses were performed to identify clinical and microbial variables associated with subsequent UC flare and rCDI. Corrections for multiple comparisons were performed using the Benjamini-Hochberg method.18 Only OTUs that were present in ≥10% of samples were included in the analysis. Redundancy analysis, an ordination technique, was used to compare differences in microbial communities followed by permutational, multivariate ANOVA (PERMANOVA) for significance testing, using the R package vegan version 2.5–4.19
Two predictive models using different techniques were performed. The first method included variables with P values <0.20 for the association with either rCDI or subsequent UC flare based on logistic regression results. A backward stepwise regression method was used to select predictors in the final multivariable model, and interactions among the variables in the final model were assessed. This modeling strategy helped quantify the magnitude, strength, and statistical significance of individual predictors while accounting for confounding. However, it is not ideal for avoiding overfitting and maximizing the generalizability of models.
A second approach using Lasso (least absolute shrinkage and selection operator) regression with cross validation was also employed to generate models that minimized overfitting. Models were built in a stepwise regression fashion, and the optimal model was automatically selected using a 3-fold cross-validation that minimized the penalty term as implemented in the glmnet package version 2.0–16.20 This stepwise modeling strategy was simulated 1000 times, and variables that appeared most frequently were selected for inclusion in the final model. The area under the receiver operator characteristic curve (AuROC) was calculated for each model using the R package pROC version 1.14.0.21
To assess longitudinal associations between clinical and microbial variables of interest with the primary outcomes, generalized estimating equations with an exchangeable or autoregressive correlation structure, generalized linear mixed-effects models, and generalized additive models were utilized using the R packages geepack version 1.2–1,22lme4 version 1.1–21,23 and mgcv version 1.8–28,24 respectively.
ETHICAL CONSIDERATIONS
The study was approved by the institutional review board at the University of Michigan.
RESULTS
Baseline Clinical Characteristics
A total of 57 subjects were enrolled in this study (32 with UC/CDI, 14 with CDI only, and 11 with UC only). Patients with CDI only were older compared with those with UC/CDI and UC only (P = 0.001; Table 1). Patients with UC only were more likely to receive steroids as their initial treatment for UC flare compared with those subjects with UC and CDI (90.9% vs 31.2%; P = 0.001). Patients with UC only were less likely to have received antibiotics in the past year compared with patients with UC and CDI or patients with CDI only (P = 0.006). Patients with UC and CDI were more likely to have CDI confirmed by a positive PCR compared with patients with CDI only (P = 0.001). No other clinical variables were different between the groups.
TABLE 1.
Baseline Demographic Variables for Patients with Ulcerative Colitis and/or Clostridiodes difficile Infection
| UC/CDI (n = 32) | CDI Only (n = 14) | UC Only (n = 11) | P | |
|---|---|---|---|---|
| Age, median (range) | 40.4 (18.1–85.7) | 66.5 (21.2–83.2) | 45.0 (21.5–71.1) | 0.001 |
| Gender, n (%) | ||||
| Female | 11 (34.4) | 7 (50.0) | 5 (45.5) | 0.58 |
| Male | 21 (65.6) | 7 (50.0) | 6 (54.5) | |
| White, n (%) | 29 (90.6) | 13 (92.9) | 10 (90.9) | >0.99 |
| History of Tobacco use, n (%) | 13 (40.6) | 6 (42.9) | 5 (50.0) | 0.93 |
| Extent of Disease Activity in UC, n (%) | ||||
| Proctitis | 0 (0) | NA | 1 (9.1) | 0.30 |
| Left-sided colitis | 5 (15.6) | 3 (27.3) | ||
| Extensive colitis | 1 (3.1) | 0 (0) | ||
| Pan-colitis | 26 (81.2) | 7 (63.6) | ||
| Maintenance Therapy, n (%) | ||||
| 5-ASA | 24 (75.0) | NA | 8 (72.7) | >0.99 |
| Immunomodulators | 15 (46.9) | 5 (45.5) | >0.99 | |
| Biologics | 15 (46.9) | 3 (27.3) | 0.67 | |
| Probiotic use, n (%) | 7 (21.9) | 5 (38.5) | 4 (44.4) | 0.26 |
| Acid suppressive medications, n (%) | 7 (22.6) | 6 (42.9) | 2 (20.0) | 0.67 |
| Prior history of CDI, n (%) | 15 (46.9) | 8 (57.1) | NA | >0.99 |
| Prior use of antibiotics in the past year, n (%) | 25 (80.6) | 12 (85.7) | 3 (30.0) | 0.006 |
| Biochemical parameters, median (range) | ||||
| WBC, K/μL | 7.0 (2.3–21.4) | 10.6 (3.6–18.7) | 9.5 (6.0–15.1) | 0.39 |
| Albumin, g/dL | 3.9 (2.8–5.1) | 3.8 (2.6–4.4) | 4.0 (2.8–4.9) | 0.44 |
| BUN, mg/dL | 13.0 (5.0–21.0) | 12.0 (4.0–57.0) | 10.0 (6.0–25.0) | 0.06 |
| Creatinine, mg/dL | 0.8 (0.6–1.5) | 0.7 (0.5–2.4) | 0.8 (0.7–1.2) | 0.27 |
| Confirmation of CDI, n (%) | ||||
| GDH/EIA positive | 3 (9.4) | 8 (57.1) | NA | 0.001 |
| PCR positive | 29 (90.6) | 6 (42.9) | ||
| Diagnosis of CDI Upon Study Enrollment, n (%) | ||||
| Non-recurrent CDI | 30 (93.8) | 12 (85.7) | NA | 0.60 |
| Recurrent CDIa | 2 (6.2) | 2 (14.3) | ||
| Primary Outcomes | ||||
| CDI Recurrence, n (%) | 14 (43.8) | 7 (50.0) | NA | 0.63 |
| UC Flare, n (%) | 11 (34.4) | NA | NA | NA |
| Steroid Administered for Treatment of UC flare, n (%) | 10 (31.2) | NA | 10 (90.9) | 0.001 |
| Initial Antibiotic Treatment for CDI Recurrence | ||||
| Vancomycin, n (%) | 23 (71.9) | 8 (57.1) | NA | 0.63 |
| Metronidazole, n (%) | 9 (28.1) | 6 (42.9) |
aPatients with diagnosis of CDI upon study entry that occurred within 8 weeks of previous CDI infection were classified as recurrent CDI. These patients could still meet the primary outcomes of recurrent CDI and/or UC flare.
5-ASA, 5-aminosalicylic acid; BUN, blood urea nitrogen; EIA, enzyme immunoassay for the presence of TcdA/TcdB toxin; GDH, glutamate dehydrogenase antigen; PCR, Real-time polymerase chain reaction for the detection of the tcdB gene.
Primary Outcomes
A total of 21 out of 46 subjects (45.7%) met the primary endpoint for rCDI, whereas 11 of 32 subjects (34.4%) developed a subsequent UC flare. For patients who developed recurrent exacerbation of their UC, the median 6-point Mayo score was 4 (range 3–5), and the median fecal calprotectin level was 383.5 μg/g (range 311–692 μg/g).
Patients With rCDI Exhibited a Distinct Community Structure Compared With Non-rCDI
We next performed redundancy analysis to explore differences in microbial communities across populations with rCDI and UC flare as variables (cohorts 1 and 2). There were significant differences in the microbial community structure between patients who subsequently developed rCDI vs non-rCDI (P = 0.008 by PERMANOVA), Fig. 1A). No significant differences were seen between patients with vs without subsequent UC flare (P = 0.44; Fig. 1B).
FIGURE 1.

Patients with recurrent Clostridiodes difficile infection show broad differences in microbial community structure compared with non-rCDI. A, Principal coordinates analysis (PCoA) based on redundancy analysis demonstrated significant differences in the community structure between patients who subsequently developed rCDI (red) compared with patients without rCDI (blue; P = 0.008 by PERMANOVA). B, PCoA plot showing no differences between community structures in patients who subsequently developed UC flare (red) vs non-UC flare (blue; P = 0.44).
Patients With rCDI Showed Greater Intraindividual Microbial Variability Over Time
We evaluated intraindividual community changes in cohorts 1 and 2 by Jensen-Shannon distance (ranging from 0, indicating complete similarity relative to baseline samples, to 1 or complete dissimilarity with baseline samples) over time. At the end of antibiotics, there were no differences in JSD between patients with and without subsequent rCDI (P = 0.41; Fig. 2A). However, 14 days after completion of antibiotics, those patients who subsequently developed rCDI demonstrated greater dissimilarity to baseline samples in community structure compared with non-rCDI patients (P = 0.004; Fig. 2B). There were no differences in JSD at EOA (P = 0.75) or at EOA plus 14 days (P = 0.22) in patients with and without subsequent UC flare.
FIGURE 2.

Patients with recurrent Clostridiodes difficile infection (rCDI) show significant perturbations of the fecal microbiota longitudinally compared with non-rCDI patients. Jensen-Shannon distance, a measure of intraindividual microbial community dissimilarity compared with baseline samples, was calculated over time. A, At the end of antibiotics, there were no differences in JSD between patients who subsequently developed rCDI (red) and non-rCDI (blue; P = 0.41). B, However, 14 days after completion of antibiotics, patients who subsequently developed rCDI (red) demonstrated a significantly elevated JSD (greater community dissimilarity relative to baseline samples) compared with non-rCDI patients (blue; P = 0.004). These results suggest that patients with ongoing perturbations of the fecal microbiota 2 weeks after completion of antibiotics were at higher risk for rCDI.
Baseline Clinical and Microbial Variables Are Predictive for rCDI
Unadjusted variables
We performed logistic regression to determine baseline variables that were associated with risk for rCDI (cohorts 1 and 2) while controlling for UC status (cohorts 1 and 3; Supplemental Table 1). Female gender (odds ratio [OR], 2.5; P = 0.05) and history of antibiotic use in the past year (OR, 3.3; P < 0.10) were associated with an increased risk for rCDI. There was no association between initial antibiotic choice (P = 0.47) or duration of antibiotic use (P = 0.50) and risk of rCDI. An increased relative abundance of Lachnospiraceae was protective against rCDI (OR, 0.52 for every 10% increase; P = 0.02).
Multivariable baseline model for rCDI
Using backward stepwise regression, 5 baseline variables were significantly associated with rCDI even after adjusting for UC status (Table 2). Female gender was associated with an increased risk for rCDI (OR, 16.2; P = 0.005). In contrast, increased OTU richness (OR, 0.86 per every increase of 10 taxa; P = 0.02) and increased relative abundance of Enterobacteriaceae (OR, 0.29 per every 10% increase; P = 0.004), Lachnospiraceae (OR, 0.17 per every 10% increase; P = 0.002), and Veillonellaceae (OR, 0.17 per every 10% increase; P = 0.05) were protective against rCDI (Fig. 3A–D). This final model had excellent fit characteristics (AuROC, 0.91; Fig. 3E).
TABLE 2.
Multivariable Logistic Regression Models of Clinical and Microbial Variables at Baseline Associated With Recurrent Clostridiodes difficile Infection
| OR | 95% CI | P | |
|---|---|---|---|
| Clinical Variables Associated with Increased Risk for rCDI | |||
| Female gender | 16.2 | 2.77–155.62 | 0.005 |
| Microbial Variables Protective Against rCDI | |||
| OTU Richness (per every increase of 10 taxa) | 0.86 | 0.73–0.96 | 0.02 |
| Enterobacteriaceae (per every 10% increase) | 0.29 | 0.09–0.56 | 0.004 |
| Lachnospiraceae (per every 10% increase) | 0.17 | 0.04–0.44 | 0.002 |
| Veillonellaceae (per every 10% increase) | 0.17 | 0.02–0.75 | 0.05 |
Effect sizes are presented after adjustment for UC status.
FIGURE 3.

Identification of microbial variables at baseline associated with increased or decreased risk for recurrent Clostridiodes difficile infection. Three bacterial taxa and community richness were identified by backward stepwise regression as being associated with subsequent rCDI. The relative abundances for these 3 taxa in addition to community richness are shown grouped by whether rCDI occurred. Data are shown as violin plots with accompanying boxplot and whiskers indicating median, interquartile range (IQR) and 1.5 × IQR of the median for the following variables: (A) OTU richness, (B) Enterobacteriaceae, (C) Lachnospiraceae, and (D) Veillonellaceae. Note that OTU richness, Enterobacteriaceae, and Veillonellaceae are shown in logarithmic scale. E, Receiver operating characteristic (ROC) curve for this model identified by backward stepwise regression is shown (AuROC, 0.91). F, ROC for model selected by Lasso regression, which included variables for female gender, hospitalization for UC in the previous year, and increased relative abundances of Ruminococcaceae and Verrucomicrobia, and decreased Eubacteriaceae, Enterobacteriaceae, Veillonellaceae, and Lachnospiraceae (AuROC, 0.94).
Three-fold cross-validated Lasso regression with 1000 simulations identified hospitalization for UC in the past year, female gender, and increased relative abundances of Verrucomicrobia and Ruminococcaceae as being associated with increased risk for rCDI after adjusting for UC status (Supplemental Table 2). Conversely, an increase in Eubacteriaceae, Lachnospiraceae, Veillonellaceae, and Enterobacteriaceae at baseline were protective against subsequent risk for rCDI. A final Lasso regression model including these baseline variables demonstrated excellent predictive capabilities for rCDI (AuROC, 0.94; Fig. 3F).
Longitudinal Models Utilizing Microbial Variables Predict Increased Risk for rCDI
We used microbial data from all time points in cohorts 1 and 2 to model rCDI by fitting generalized estimating equations (GEEs) and generalized linear mixed models (GLMMs) while adjusting for UC status (cohorts 1 and 3). However, models using both GEEs and GLMMs either did not converge or resulted in singular fits likely related to excessive collinearity of variables, overdispersion/excessive variability over time, and/or nonlinearity in the shape of the data. Since the data did not follow a linear pattern (Supplemental Fig. 3A–D), we fit generalized additive models (GAMs), which identified female gender, decreasing OTU richness, and increasing Ruminococcaceae longitudinally as being associated with risk of rCDI while adjusting for UC status (AuROC, 0.87; data not shown).
Clinical and Microbial Variables at Time Point 3 Also Predict Risk for rCDI
Unadjusted variables
We identified by logistic regression that increased relative abundance of Gammaproteobacteria (OR, 1.63 per every 10% increase; P = 0.04), Enterobacteriaceae (OR, 1.78 per every 10% increase; P = 0.03), and Jensen-Shannon distance at time point 3 (EOA plus 14 days) relative to baseline (OR, 1.80 per every 0.1 increase; P = 0.01) were associated with increased rCDI risk (cohorts 1 and 2) after controlling for UC status (cohorts 1 and 3; Supplemental Table 3). Conversely, an increase in Shannon diversity (OR, 0.34; P = 0.04) and OTU richness (OR, 0.85 per every increase of 10 taxa; P = 0.008) and an increased relative abundance of Ruminococcaceae (OR, 0.40 per every 10% increase; P = 0.04), Faecalibacterium (OR, 0.10 per every 10% increase; P = 0.04), Bacteroidetes (OR, 0.60 per every 10% increase; P = 0.05), Lachnospiraceae (OR, 0.62 per every 10% increase; P = 0.06), and MHI (OR, 0.78 per every 1% increase; P = 0.05) were all protective against future risk for rCDI.
Multivariable model selection for rCDI
Model selection by backward stepwise regression identified female gender (OR, 5.69; P = 0.03) and increased relative abundance of Ruminococcaceae (OR, 1.43 per every 1% increase; P = 0.03) at time point 3 as being associated with increased risk for rCDI after adjusting for UC status (Table 3). In contrast, an increase in OTU richness (OR, 0.83 per every increase of 10 taxa; P = 0.03) and increased relative abundance of Faecalibacterium (OR, 0.47 per every 1% increase; P = 0.02) were protective against rCDI (Fig. 4A–C). This model showed good fit characteristics (AuROC, 0.87) for predicting rCDI. When Jensen-Shannon distance at time point 3 relative to baseline was added to the model, it had a statistically significant effect by likelihood ratio testing (P = 0.007) and improved the model fit (AuROC, 0.90).
TABLE 3.
Multivariable Logistic Regression Models of Clinical and Microbial Variables at Time Point 3 (14 Days After Completion of Antibiotics) Associated With Recurrent Clostridiodes difficile Infection
| OR | 95% CI | P | |
|---|---|---|---|
| Clinical and Microbial Variables Associated with Increased Risk for rCDI | |||
| Female gender | 5.69 | 1.36–29.71 | 0.03 |
| Ruminococcaceae (per every 1% increase) | 1.43 | 1.08–2.07 | 0.03 |
| Jensen Shannon Divergence (per every 0.1 increase) | 2.59 | 1.28–6.15 | 0.01 |
| Microbial Variables Protective Against Risk for rCDI | |||
| OTU Richness (per every increase of 10 taxa) | 0.83 | 0.66–0.96 | 0.03 |
| Faecalibacterium (per every 1% increase) | 0.47 | 0.23–0.75 | 0.02 |
Effect sizes are presented after adjustment for UC status.
FIGURE 4.

Fecal microbial characteristics at end of antibiotics plus 14 days are associated with future risk for recurrent CDI. Several microbial variables 14 days after completion of antibiotics were identified as being associated with risk for future rCDI. The relative abundances for representative taxa, Shannon diversity, and community richness are shown grouped by whether rCDI occurred. Data are shown as violin plots with accompanying boxplot and whiskers indicating median, interquartile range (IQR), and 1.5 × IQR of the median for the following variables: (A) OTU richness, (B) Ruminococcaceae, (C) Faecalibacterium, (D) Enterobacteriaceae, and (E) Shannon diversity over time, including at baseline, end of antibiotics, and end of antibiotics plus 14 days (EOA + 14d). Note that OTU richness, Ruminococcaceae, Faecalibacterium, and Enterobacteriaceae are shown in logarithmic scale. F, ROC curve displaying good fit characteristics of model identified by Lasso regression containing variables of female gender, increased Shannon diversity, increased relative abundances of Ruminococcaceae and Enterobacteriaceae, decreased OTU richness and Faecalibacterium, and Jensen-Shannon distance (relative to baseline) at end of antibiotics plus 14 days for risk of future rCDI (AuROC, 0.94).
Similarly, cross-validated Lasso regression simulations identified increased relative abundance of Ruminococcaceae, decreased OTU richness, and decreased Faecalibacterium at time point 3 as being associated with increased risk for rCDI after adjusting for UC status (Supplemental Table 4). Lasso regression also identified additional variables associated with increased rCDI risk, including female gender, increased Enterobacteriaceae (Fig. 4D), and increased Shannon diversity (Fig. 4E). When these variables were included in a final Lasso regression model, this model showed excellent predictive capabilities for identifying rCDI (AuROC, 0.90). When Jensen-Shannon distance at time point 3 relative to baseline was added, the model showed increased fit characteristics (AuROC, 0.94; Fig. 4F).
Baseline Variables Associated with Risk for UC Flare
Unadjusted variables
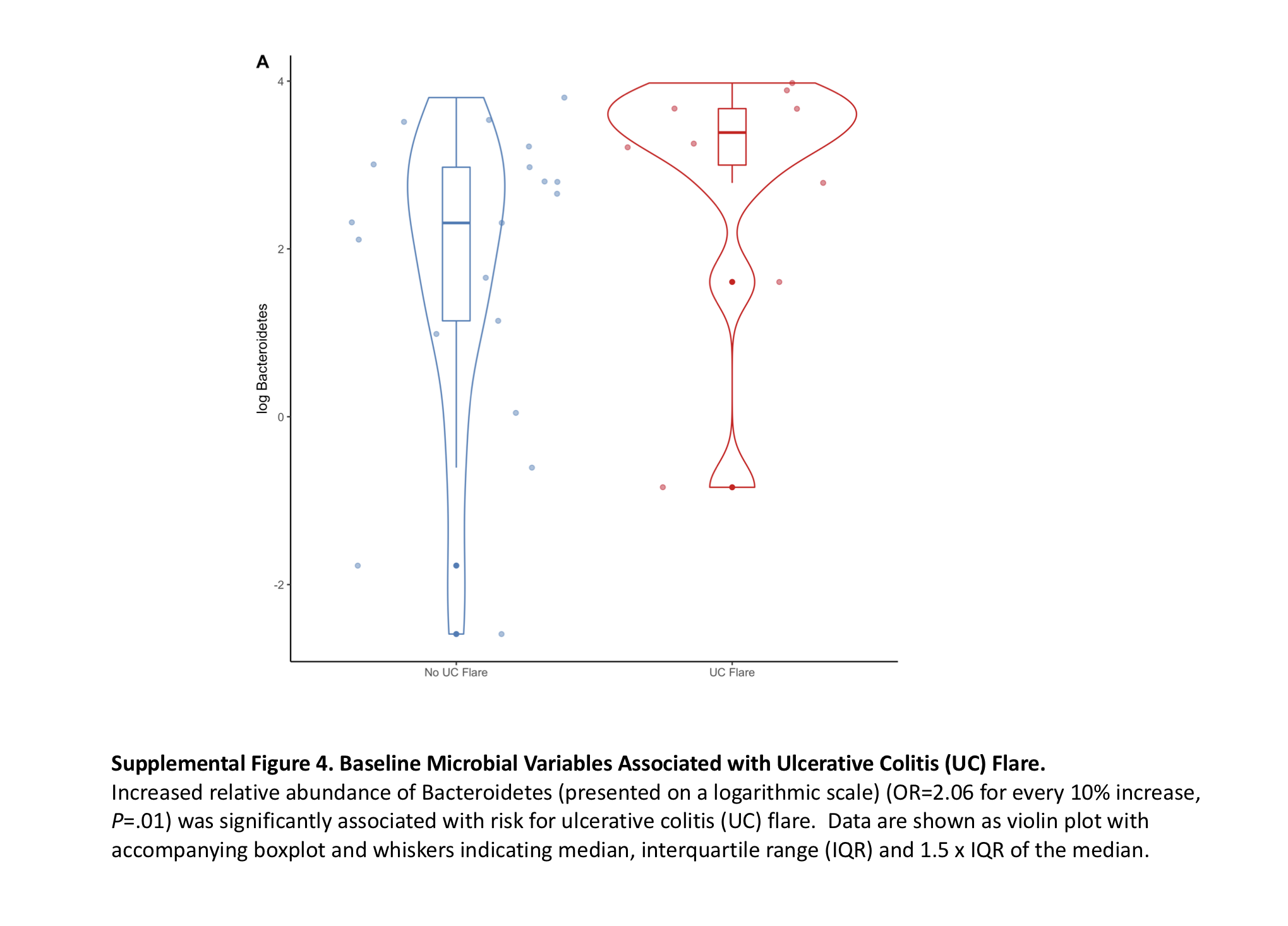
Prior hospitalization for UC in the past year (OR, 16.0; P = 0.003), steroid use for initial treatment for UC exacerbation (OR, 10.5; P = 0.008; Supplemental Table 1), and increased relative abundance of Bacteroidetes (OR, 2.06 for every 10% increase; P = 0.01; Supplemental Fig. 4) were associated with increased risk for UC flare (cohort 1).
Multivariable model for UC flare
Using backward stepwise regression, hospitalization for UC in the previous year (OR, 17.70; P = 0.008) and increased relative abundance of Bacteroidetes at baseline (OR, 1.07; P = 0.03) were associated with increased risk for UC flare (AuROC, 0.88).
Clinical and Microbial Variables Did Not Associate With UC Flare at Time Point 3
In contrast to the results with recurrent CDI, we did not identify microbial variables at time point 3 in cohort 1 that were associated with risk for UC flare.
DISCUSSION
In this prospective, longitudinal study, we have shown that specific microbial characteristics, either at baseline or upon reconstitution of the microbiome, are predictive of patients at high risk for future episodes of rCDI and UC flare. Specifically, we demonstrated that (1) patients who subsequently develop rCDI possess a distinct microbial community structure compared with non-rCDI patients; (2) there are unique microbial characteristics at baseline which can identify patients at risk for rCDI with 94% accuracy; (3) those patients with ongoing perturbations of their fecal microbiota 2 weeks after completion of antibiotics are at increased risk for rCDI; and (4) patients with history of hospitalization for UC in the previous year and increased Bacteroidetes at baseline had increased risk for subsequent UC flare.
Patients with UC and CDI represent a high-risk patient population with significant risk for rCDI and poor outcomes. Identifying those patients with higher risk for recurrence of disease would represent a major advance in the field. Our model of key baseline clinical and microbial variables, including hospitalization for UC in the past year, female gender, increased Verrucomicrobia and Ruminococcaceae, and decreased Eubacteriaceae, Lachnospiraceae, Veillonellaceae, and Enterobacteriaceae demonstrated excellent discriminative ability with 94% accuracy to predict future occurrences of rCDI. Notably, these variables were identified in baseline samples, which suggests that high-risk patients can be identified at the time of CDI diagnosis. It is possible but unproven that these patients may benefit from more aggressive therapy, such as prolonged antibiotic tapers, an antibiotic with lower risk of rCDI such as fidaxomicin,25 fecal microbiota transplantation (FMT), or even a rationally designed probiotic (based on data from the current study) containing Eubacteriaceae, Lachnospiraceae, Enterobacteriaceae, and Veillonellaceae.
Widespread changes in the gut microbiota, including lower diversity and decreased abundance of Lachnospiraceae, have been demonstrated in both UC and CDI.26, 27 Lachnospiraceae is a primary producer of butyrate, which is known to inhibit C. difficile in vitro28 and enhance intestinal epithelial barrier function.29 Furthermore, both C. difficile and Lachnospiraceae are taxonomically classified within the Clostridiales order. Thus, Lachnospiraceae may be protective against rCDI by occupying a similar ecological niche and competing for similar resources as C. difficile. Furthermore, a probiotic containing 33 different bacterial species, including Lachnospiraceae, Ruminococcaceae, and Eubacteriaceae, was protective against rCDI for up to 6 months post-treatment.30
Although there is a paucity of data regarding association between microbial characteristics and rCDI, the available data show some differences from our results. Khanna et al reported that increased abundance of Veillonella, Enterobacteriaceae, and Lachnospiraceae in pretreatment fecal samples were associated with rCDI,31 whereas our results show the opposite directionality for these taxa. However, Khanna and colleagues diagnosed rCDI retrospectively by review of the electronic medical record. As Enterobacteriaceae has classically been considered a marker of dysbiosis in the GI tract, our findings showing a protective effect of Enterobacteriaceae is somewhat surprising. However, it is possible that prior studies in rCDI are not generalizable to patients with concomitant IBD and rCDI, as IBD itself is characterized by widespread changes to the gut microbial community. Furthermore, Enterobacteriaceae is one of the most taxonomically diverse families of bacteria.32 Longitudinal studies using culture-based and culture-independent methods have demonstrated that Enterobacteriaceae, particularly E. coli, are common members of the healthy human gut microbiota.33 Future studies may examine if and identify which species of Enterobacteriaceae are potentially protective factors in rCDI in UC patients.
There are only 2 published studies examining longitudinal changes in the gut microbiota in rCDI. Seekatz et al demonstrated patients with rCDI were more likely to show greater intraindividual similarity in their gut microbiota longitudinally compared with non-rCDI patients, which is discordant with our results.15 However, patients were retrospectively designated as having rCDI vs recovery by chart review. Additionally, the number of and interval between samples collected was highly variable, with samples collected up to 800 days after initial CDI diagnosis. Thus, our respective studies may have captured different trajectories in gut microbial changes between perturbation and reconstitution resulting in rCDI or recovery. Furthermore, our model is more likely to be clinically relevant given the ability to risk stratify patients at baseline, or shortly after completion of antibiotics, rather than weeks or months after collection of the index sample.
In the first prospective longitudinal study, Pakpour et al identified decreased Shannon diversity, depletion of Bacteroidetes, and reduced abundance of Veillonella dispar as being predictive of rCDI.34 However, the predictive capability of their model was poor (AuROC, 0.68). Furthermore, these authors identified only baseline microbial characteristics as being associated with rCDI. It is important to note key demographic and methodologic differences that may explain the discordance between our respective studies. First, the majority of patients enrolled by Pakpour et al identified as being of Afro-Caribbean descent, and IBD patients were specifically excluded. Secondly, Pakpour and colleagues collected 3 stool samples shortly after initiating antibiotic administration, and a final sample was collected 4 days after completion of antibiotic treatment. It is possible that the gut microbiota may have been significantly disturbed by ongoing antibiotic administration, which precluded using microbial characteristics longitudinally to discriminate between patients who recurred vs those who recovered.
In contrast, we identified distinct temporal changes in the gut microbiota that were associated with rCDI. We identified specific microbial changes 14 days after completion of antibiotics that were predictive for risk of future rCDI. Specifically, patients whose fecal microbiota showed greater dissimilarity to their baseline samples were at highest risk for rCDI. This suggests that patients with ongoing disruptions in the gut microbial ecosystem have a permissive state for recurrence of CDI, whereas patients who do not develop rCDI start to develop recovery of their microbial community 14 days after completion of antibiotics. However, these measures of dissimilarity compare fecal microbiota to baseline samples, which can be affected by a myriad of different factors, including presence of inflammation related to UC or CDI, prior antibiotic use, and dietary factors. As such, validation of these findings in larger prospective cohorts is required before they can be translated into clinical use. Additionally, we identified increased Ruminococcaceae and increased Enterobacteriaceae were associated with increased risk for rCDI, whereas an increase in community richness and Faecalibacteria were protective against rCDI even after adjusting for UC status. Similar to Lachnospiraceae, Faecalibacteria are in the Clostridiales order, produce butyrate, and are associated with decreased risk for CDI.35 Expansion of Enterobacteriaceae, which normally occupy only a small fraction of the distal gut microbiota in healthy subjects, can be seen during periods of gut inflammation, such as in IBD or CDI.36, 37
Prior IBD studies have identified different microbial features that are associated with worse outcomes. Shaw et al identified 2 genera, including Coprococcus and Adlercreutzia, which showed fair predictive ability to identify responders to therapy in IBD (AuROC, 0.75).38 We identified prior hospitalization for UC in the previous year and increased Bacteroidetes at baseline as showing good predictive ability to identify patients at higher risk for subsequent UC flare in the next 180 days (AuROC, 0.88). It is notable that we were not able to develop a model identifying microbial characteristics longitudinally with risk for UC flare. This may reflect differences in the pathogenesis of these diseases, where UC may be driven more by host factors, and CDI is dependent on disruptions to the microbial ecological network. However, this study could have been underpowered for microbial differences in UC, and a larger cohort with longer duration of sampling could be required to develop more robust microbial-driven models of UC exacerbation given the wide variability of the IBD microbiome.39
There are several notable strengths of our study, including the longitudinal study design with complete microbiome and clinical outcome data for up to 6 months, repeated sample collection, robust demographic and clinical data, and well-defined study end points. However, there are limitations to our study. First, due to the small sample size, we were unable to verify our model using a test data set that was separate from the training set. Second, there were only a small number of patients who developed UC flare, which likely limited our ability to develop a predictive model for exacerbation of UC. Third, we only sampled the fecal microbiota, and the mucosal-associated microbiota may provide more relevant information regarding host-microbial interactions in UC and CDI. Furthermore, we acquired stool samples during onset of either CDI or UC flare, which likely affected our microbial results. Future studies concentrating on longitudinal sampling before onset of CDI or UC flare may be more predictive in phenotyping risk for future disease. Fourth, 16S sequencing analysis does not allow for the required resolution to accurately classify down to the species or occasionally even the genus level. Although our analyses mainly focused on higher level taxonomic classification, this also minimized the risk for type 1 errors. Future studies may determine whether better resolution provided by metagenomic sequencing analysis will improve model fit characteristics. Fifth, because analyses for rCDI included patients from cohorts 1 and 2, we were unable to determine whether microbial features predicting rCDI are different in patients with vs without UC. Finally, a longer follow-up time may be required for other important adverse clinical outcomes, such as those requiring colectomy.
CONCLUSION
We have identified unique temporal changes in the gut microbiota associated with increased risk for subsequent rCDI in a cohort of patients with UC and/or CDI. Although our findings require validation, these results could have important clinical implications. The novel ability to identify patients at high risk for rCDI with over 90% accuracy using a single stool sample, either at baseline before initiation of antibiotics or at 14 days after completion of antibiotics, could affect future clinical decision-making. Inclusion of changes in microbial diversity over time adds accuracy to the model but could be less clinically useful, as this requires sampling at 2 different time points. Clinicians could utilize this microbial-derived information to escalate preventive therapy in higher-risk patients if prospective studies validate a risk-stratified strategy of preventive therapy. Future work will focus on the mechanisms of how shifts in the gut microbiota predispose patients to rCDI and on the rational design of preventive probiotics to reduce recurrence of Clostridioides difficile infection in high-risk patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Glossary
Abbreviations
- AuROC
area under the receiver operating characteristic curve
- CDI
Clostridiodes difficile infection
- EOA
end of antibiotics
- FMT
fecal microbiota transplantation
- IBD
inflammatory bowel disease
- Lasso
least absolute shrinkage and selection operator
- JSD
Jensen-Shannon distance
- MHI
microbiome health index
- OTU
operational taxonomic unit
- PERMANOVA
permutational multivariate analysis of variance
- rCDI
recurrent Clostridiodes difficile infection
- UC
ulcerative colitis
Author Contribution: AL collected clinical data, participated in analysis and interpretation of the data, participated in statistical analysis, and drafted the manuscript. KR participated in analyzing the data, interpreting the results, and the statistical analysis and helped draft the manuscript. JL participated in acquisition of clinical data and sample processing. MG participated in acquisition of data, sample processing, and data analysis. BM participated in acquisition of clinical data and sample processing. VBY participated in study design, interpreting the results, and drafting the manuscript. PDRH conceived the study, participated in subject recruitment, data acquisition, interpretation of the results, and drafting the manuscript. All authors were involved in critical revision of the manuscript and approved the final manuscript.
Supported by: This study was funded by a research grant from the Crohn’s and Colitis Foundation of America, awarded to PH.
Conflicts of interest: PH receives support from the NIH and the University of Michigan. AAL receives support from the National Center for Advancing Translational Sciences of the NIH (KL2TR002241). VBY and KR receive support from the NIH (AI124255).
REFERENCES
- 1. Nagao-Kitamoto H, Shreiner AB, Gillilland MG 3rd, et al. Functional characterization of inflammatory bowel disease-associated gut dysbiosis in gnotobiotic mice. Cell Mol Gastroenterol Hepatol. 2016;2:468–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rodemann JF, Dubberke ER, Reske KA, et al. Incidence of Clostridium difficile infection in inflammatory bowel disease. Clin Gastroenterol Hepatol. 2007;5:339–344. [DOI] [PubMed] [Google Scholar]
- 3. Nguyen GC, Kaplan GG, Harris ML, et al. A national survey of the prevalence and impact of Clostridium difficile infection among hospitalized inflammatory bowel disease patients. Am J Gastroenterol. 2008;103:1443–1450. [DOI] [PubMed] [Google Scholar]
- 4. Lawson PA, Citron DM, Tyrrell KL, et al. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) Prévot 1938. Anaerobe. 2016;40:95–99. [DOI] [PubMed] [Google Scholar]
- 5. Reeves AE, Theriot CM, Bergin IL, et al. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes. 2011;2:145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Razik R, Rumman A, Bahreini Z, et al. Recurrence of Clostridium difficile infection in patients with inflammatory bowel disease: the RECIDIVISM Study. Am J Gastroenterol. 2016;111:1141–1146. [DOI] [PubMed] [Google Scholar]
- 7. Ananthakrishnan AN, McGinley EL, Binion DG. Excess hospitalisation burden associated with Clostridium difficile in patients with inflammatory bowel disease. Gut. 2008;57:205–210. [DOI] [PubMed] [Google Scholar]
- 8. Ananthakrishnan AN, McGinley EL, Saeian K, et al. Temporal trends in disease outcomes related to Clostridium difficile infection in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2011;17:976–983. [DOI] [PubMed] [Google Scholar]
- 9. Kelsen JR, Kim J, Latta D, et al. Recurrence rate of Clostridium difficile infection in hospitalized pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2011;17:50–55. [DOI] [PubMed] [Google Scholar]
- 10. Lewis JD, Chuai S, Nessel L, et al. Use of the noninvasive components of the Mayo score to assess clinical response in ulcerative colitis. Inflamm Bowel Dis. 2008;14:1660–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis. 2018;66:e1–e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rao K, Higgins PD. Epidemiology, diagnosis, and management of Clostridium difficile infection in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2016;22:1744–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De La Cochetière MF, Durand T, Lalande V, et al. Effect of antibiotic therapy on human fecal microbiota and the relation to the development of Clostridium difficile. Microb. Ecol. 2008;56:395–402. [DOI] [PubMed] [Google Scholar]
- 14. Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seekatz AM, Rao K, Santhosh K, et al. Dynamics of the fecal microbiome in patients with recurrent and nonrecurrent Clostridium difficile infection. Genome Med. 2016;8:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arumugam M, Raes J, Pelletier E, et al. ; MetaHIT Consortium Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blount K, Jones C, Deych E, et al. 1966. Evaluating a prototype microbiome health index (MHI) as a measure of microbiome restoration using data derived from a published study of fecal microbiota transplant (FMT) to treat recurrent Clostridium difficile infections (rCDI). Open Forum Infect Dis. 2018;5:S570. [Google Scholar]
- 18. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- 19. Oksanen J, Blanchet G, Friendly M, et al. vegan: Community ecology package. R package version 2.5–4. 2019. https://CRAN.R-project.org/package=vegan.
- 20. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 21. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Højsgaard S, Halekoh U, Yan J. The R package geepack for generalized estimating equations. J Stat Softw. 2005;15:1–11. [Google Scholar]
- 23. Bates D, Mächler M, Bolker B, et al. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1–48. [Google Scholar]
- 24. Wood SN. Stable and efficient multiple smoothing parameter estimation for generalized additive models. J Am Stat Assoc. 2004;99:673–686. [Google Scholar]
- 25. Crook DW, Walker AS, Kean Y, et al. ; Study ¾ Teams Fidaxomicin versus vancomycin for Clostridium difficile infection: meta-analysis of pivotal randomized controlled trials. Clin Infect Dis. 2012;55(Suppl 2):S93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schirmer M, Denson L, Vlamakis H, et al. Compositional and temporal changes in the gut microbiome of pediatric ulcerative colitis patients are linked to disease course. Cell Host Microbe. 2018;24:600–610.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schubert AM, Rogers MA, Ring C, et al. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. Mbio. 2014;5:e01021–e01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rolfe RD. Role of volatile fatty acids in colonization resistance to Clostridium difficile. Infect Immun. 1984;45:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamer HM, Jonkers D, Venema K, et al. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27:104–119. [DOI] [PubMed] [Google Scholar]
- 30. Petrof EO, Gloor GB, Vanner SJ, et al. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome. 2013;1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khanna S, Montassier E, Schmidt B, et al. Gut microbiome predictors of treatment response and recurrence in primary Clostridium difficile infection. Aliment Pharmacol Ther. 2016;44:715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adeolu M, Alnajar S, Naushad S, S Gupta R. Genome-based phylogeny and taxonomy of the ‘enterobacteriales’: proposal for Enterobacterales ord. nov. divided into the families enterobacteriaceae, erwiniaceae fam. nov., pectobacteriaceae fam. nov., yersiniaceae fam. nov., hafniaceae fam. nov., morganellaceae fam. nov., and budviciaceae fam. nov. Int J Syst Evol Microbiol. 2016;66:5575–5599. [DOI] [PubMed] [Google Scholar]
- 33. Martinson JNV, Pinkham NV, Peters GW, et al. Rethinking gut microbiome residency and the enterobacteriaceae in healthy human adults. Isme J. 2019;13:2306–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pakpour S, Bhanvadia A, Zhu R, et al. Identifying predictive features of Clostridium difficile infection recurrence before, during, and after primary antibiotic treatment. Microbiome. 2017;5:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Milani C, Ticinesi A, Gerritsen J, et al. Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Sci Rep. 2016;6:25945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seekatz AM, Young VB. Clostridium difficile and the microbiota. J Clin Invest. 2014;124:4182–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shaw KA, Bertha M, Hofmekler T, et al. Dysbiosis, inflammation, and response to treatment: a longitudinal study of pediatric subjects with newly diagnosed inflammatory bowel disease. Genome Med. 2016;8:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Halfvarson J, Brislawn CJ, Lamendella R, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. 2017;2:17004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.