

There are few metagenome studies that have explored soil consortia maintained on a complex hydrocarbon substrate after the community interrelationships were formed. A soil bacterial consortium maintained on diesel fuel was utilized as a practical model to investigate bacterial community relationships through metagenomics analyses, consortium member isolation, growth assays, and metabolite identification, which supported the linkage of genomic data and functionality. Two pioneering genera were responsible for the biodegradation of aromatics and alkanes by initiating biotransformation and thereby created specialized niches that were populated by other members. A model that represents these relationships was constructed, which contributes to our understanding of the complex ecological relationships that evolve during prokaryotic hydrocarbon pollutant biodegradation.
KEYWORDS: diesel fuel biodegradation, soil bacterial community, metagenomics
ABSTRACT
A soil bacterial consortium that was grown on diesel fuel and consisted of more than 10 members from different genera was maintained through repetitive subculturing and was utilized as a practical model to investigate a bacterial community that was continuously exposed to petroleum hydrocarbons. Through metagenomics analyses, consortium member isolation, growth assays, and metabolite identification which supported the linkage of genomic data and functionality, two pioneering genera, Sphingobium and Pseudomonas, whose catabolic capabilities were differentiated, were found to be responsible for the creation of specialized ecological niches that were apparently occupied by other bacterial members for survival within the consortium. Coexisting genera Achromobacter and Cupriavidus maintained their existence in the consortium through metabolic dependencies by utilizing hydrocarbon biotransformation products of pioneer metabolism, which was confirmed through growth tests and identification of biotransformation products of the isolated strains. Pioneering Sphingobium and Pseudomonas spp. utilized relatively water-insoluble hydrocarbon parent compounds and facilitated the development of a consortium community structure that resulted in the creation of niches in response to diesel fuel exposure which were created through the production of more-water-soluble biotransformation products available to cocolonizers. That these and other organisms were still present in the consortium after multiple transfers spanning 15 years provided evidence for these ecological niches. Member survival through occupation of these niches led to robustness of each group within the multispecies bacterial community. Overall, these results contribute to our understanding of the complex ecological relationships that may evolve during prokaryotic hydrocarbon pollutant biodegradation.
IMPORTANCE There are few metagenome studies that have explored soil consortia maintained on a complex hydrocarbon substrate after the community interrelationships were formed. A soil bacterial consortium maintained on diesel fuel was utilized as a practical model to investigate bacterial community relationships through metagenomics analyses, consortium member isolation, growth assays, and metabolite identification, which supported the linkage of genomic data and functionality. Two pioneering genera were responsible for the biodegradation of aromatics and alkanes by initiating biotransformation and thereby created specialized niches that were populated by other members. A model that represents these relationships was constructed, which contributes to our understanding of the complex ecological relationships that evolve during prokaryotic hydrocarbon pollutant biodegradation.
INTRODUCTION
Oil spills are recognized as a global environmental pollution issue because many of the petroleum hydrocarbon components such as alkanes, aromatic hydrocarbons, and, especially, polycyclic aromatic hydrocarbons (PAHs) may cause acute toxicity as well as genotoxicity to the surrounding organisms in both aquatic and terrestrial ecosystems (1, 2). Microorganisms that are capable of biodegrading and utilizing petroleum hydrocarbons as carbon and energy sources have thus been attracting interest for their potential roles in bioremediation. It has been proposed from numerous previous studies that utilized isolated organisms that diverse bacterial taxa, including Actinobacteria, Proteobacteria (Alpha-, Beta-, and Gammaproteobacteria), Bacteroidetes, and Firmicutes (see reference 3 and references therein), are representative of bacteria that are capable of petroleum hydrocarbon biodegradation. A single microbial species, however, may only be able to biodegrade a fraction of complex oil components, and it is therefore more likely that cooperative activities across multiple microbial groups are required to effectively and/or completely biodegrade complex oil pollutant mixtures (4, 5). It remains a challenge to fully understand the complex microbial roles and interactions that develop in microbial ecosystems during oil exposure, especially if only pure culture-based approaches are conducted.
As a powerful tool to overcome these challenges, recent studies have applied metagenomics approaches to oil-polluted environments to investigate the indigenous microbial communities that developed as a response to petroleum hydrocarbon exposure (6). Indeed, metagenomic analyses of microbial community structure and functional genes, as well as metatranscriptomic analyses of hydrocarbon-enriched microbial communities, have enabled the identification of potential key hydrocarbon degraders, including uncultivated species, as well as the evaluation of those metabolic capabilities that drive petroleum hydrocarbon biodegradation (7–10). However, these culture-independent approaches often only provide indirect evidence of their metabolic capabilities and interactions among the coexisting microbial members. To further understand the structural complexity of hydrocarbon-degrading microbial ecosystems, investigations that combine microbial cultivation-based direct proof of their metabolic functions in conjunction with in-depth (meta)genomic characterization are required.
In ongoing studies, a hydrocarbon-degrading bacterial consortium that was previously obtained from soil (11) has been maintained with diesel fuel as the sole carbon and energy source, and it represented a dynamic microecosystem model of a mature hydrocarbon-degrading soil bacterial community. This consortium provided a unique opportunity to study soil consortium community structural characteristics after community interrelationships were formed by repetitive transfers. It was studied with the aims to understand community structural dynamics by qualitative analyses and to elucidate the roles of bacterial members during hydrocarbon biodegradation. For these purposes, 16S rRNA gene amplicon sequencing was applied to investigate community structures in the consortium in addition to metagenomic functional gene analyses to characterize the potential metabolic capabilities of major bacterial members. At the same time, the growth and hydrocarbon biotransformation capabilities of bacterial genera isolated from the consortium were evaluated to link genes with function.
RESULTS
Growth and biodegradation of petroleum hydrocarbons by the consortium.
The mature soil consortium that was maintained on diesel fuel for 15 years was grown on different carbon sources for 4 days, and growth was observed in all cultures whether the carbon source was diesel fuel, a 4-component synthetic hydrocarbon nonaqueous-phase liquid mixture (NAPL), n-hexadecane, phenanthrene, naphthalene, or toluene. Growth was confirmed by turbidity and/or biofilm/floc formation in the medium. Macroscopic whitish/brownish bacterial flocs were observed in most cultures within 4 days. Diesel fuel biodegradation was monitored by extraction and gas chromatography analyses, whereby comparison of extracts from the start of the incubation period (Fig. 1A) with abiotic controls (Fig. 1B) and inoculated flasks (Fig. 1C) showed that all n-alkanes up to at least nC23 were undetectable after only 4 days. Diesel components up to nC14 were volatilized (Fig. 1B). Branched alkanes such as pristine and phytane and other unknown compounds were slightly biodegraded or not at all after 4 days (Fig. 1C).
FIG 1.
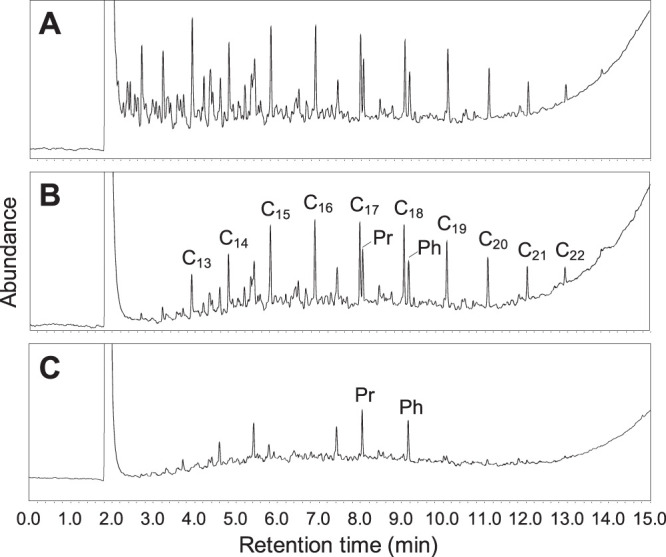
Gas chromatograms showing the biodegradation of diesel fuel by the bacterial consortium. Chromatograms of the start of the incubation period (A), abiotic control after 4 days (B), and with bacteria after 4 days (C). The peaks of n-alkanes (C13 to C22), pristane (Pr; 2,6,10,14-tetramethylpentadecane), and phytane (Ph; 2,6,10,14-tetramethylhexadecane) are indicated.
Microbial communities that grew on petroleum hydrocarbons.
Analyses of results from bacterial 16S rRNA gene amplicon sequencing showed that 15 bacterial operational taxonomic units (OTUs) and at least 12 bacterial genera were present when the consortium was grown on diesel fuel as the sole source of carbon and energy (Fig. 2A). All of these abundant bacterial genera were detected as 1 or 2 OTUs, which revealed low phylogenetic diversity within each bacterial genus (Fig. 2A; see also Table S1 in the supplemental material). Pseudomonas was the most dominant (70.7% relative abundance), followed by Sphingobium (12.3%), Ochrobactrum (6.1%), Achromobacter (4.6%), Cupriavidus (2.2%), Parvibaculum (1.1%), Olivibacter (1.0%), Bradyrhizobium (0.7%), Rhodanobacter (0.5%), Dokdonella (0.4%), Mesorhizobium (0.2%), and Sphingomonas (0.1%). The amplicons that originated from the six major bacterial genera in the consortium (Pseudomonas, Sphingobium, Ochrobactrum, Achromobacter, Cupriavidus, and Parvibaculum) accounted for greater than 97% of the total amplicons sequenced from the culture that grew on diesel fuel. When the consortium was cultivated on 4-component NAPL, Pseudomonas (79.2%) and Sphingobium (10.1%) remained dominant, while relative abundances of Ochrobactrum (0.6%) and Cupriavidus (0.6%) decreased, Parvibaculum was almost undetectable (<0.1%), and Mycobacterium (1.8%) became detectable. When the culture was grown exclusively on n-hexadecane, relative abundances of Sphingobium, Cupriavidus, and Parvibaculum all decreased to less than 0.1% and Pseudomonas was predominant (84.2%). In contrast, when the consortium was separately exposed to phenanthrene, naphthalene, or toluene as the sole source of carbon, Sphingobium grew most dominantly in all cases, accounting for 53.4%, 42.6%, or 53.5%, respectively, of the total number of bacterial amplicons under each condition. Achromobacter also conspicuously increased in the cultures exposed exclusively to aromatic hydrocarbons—18.5% on phenanthrene, 26.1% on naphthalene, and 16.1% on toluene—but not in cultures exposed to n-hexadecane. Interestingly, Cupriavidus was enriched only in the presence of toluene (18.9%). Archaeal sequences were not detected in any of the cultures. These results are summarized in Fig. 2A and Table S1.
FIG 2.

Major bacterial genera detected in the consortium. (A) Relative abundances (percent total number of bacterial amplicons) of the major OTUs when the consortium was grown on different hydrocarbon sources: diesel fuel (diesel-1 and -2; duplicates), 4-component NAPL (NAPL), n-hexadecane (HEX), phenanthrene (PHE), naphthalene (NAP), and toluene (TOL). Amplicons were detected by 16S rRNA gene amplicon sequencing. Bacterial genus of each OTU is shown in parentheses. (B) Number of metagenome reads mapped to the assembled full-length 16S rRNA genes of major bacterial genera in the consortium grown on diesel fuel.
Through shotgun sequencing and subsequent de novo assembly from the consortium when it was grown on diesel fuel, a total of 83,291,099 bp of contigs were obtained (see Table S2). The full-length 16S rRNA genes of nine major bacterial genera were assembled, and their abundances were quantified (Fig. 2B). These assembled 16S rRNA genes are identical to the major OTUs detected by the amplicon sequencing and were further characterized phylogenetically (Fig. 3; Table 1). Consistent with the amplicon sequencing results, Pseudomonas was detected as the most abundant genus, followed by Sphingobium as the second most abundant; Cupriavidus, Achromobacter, Ochrobactrum, Rhodanobacter, Dokdonella, Parvibaculum, and Olivibacter were also detected as major bacterial genera (Fig. 2B). These major genera were identical to or had high identity (>99%) with representative known bacterial strains, with the exception of Parvibaculum, which had <96% similarity with Parvibaculum lavamentivorans DS-1 (CP000774) and higher similarities with uncultured bacterial clones detected from a sewage system (AB255109), oyster shell (EU369117), lake water (DQ015855), hydrocarbon-contaminated sediment (KJ955653), and a benzene, toluene, ethylbenzene, and xylene (BTEX)-contaminated aquifer (JF800700), for example.
FIG 3.

Neighbor-joining phylogenetic tree of full-length 16S rRNA genes assembled from the metagenome of the consortium when it was grown on diesel fuel, including the 16S rRNA gene sequences of bacterial isolates obtained from the consortium (bold font) and compared with phylogenetically related bacterial type strains as references. The tree was created with 1,000 bootstrap iterations, and values below 50% are not reported. NCBI accession numbers are reported in parentheses.
TABLE 1.
Phylogenetic characterization for the major bacterial genera detected from the consortium
| Bacterial genus | MAG |
16S rRNA gene assembled from metagenomic reads |
||||||
|---|---|---|---|---|---|---|---|---|
| Genome size (bp) | Genome completeness (%) | Contamination (%) | Closest bacterial genome (ANI [%]) | Length (bp) | Identical 16S rRNA gene amplicon | Closest bacterial strain (NCBI accession no. [% homology]) | Closest bacterial isolate from the consortium (% homology) | |
| Pseudomonas | 6,686,507 | 99.07 | 0.60 | Pseudomonas aeruginosa DSM 500071 (99.43) | 1,536 | OTU-1 | Pseudomonas aeruginosa DSM 500071 (CP012001 [100]) | KK6 (100) |
| Sphingobium | 4,974,394 | 99.89 | 1.65 | Sphingobium barthaii KK22 (99.98) | 1,485 | OTU-2 | Sphingobium barthaii KK22 (NR_137223 [100]) | KK22 (100) |
| Ochrobactrum | 4,842,250 | 96.96 | 0.43 | Ochrobactrum anthropi ATCC 49188 (97.50) | 1,481 | OTU-3 | Ochrobactrum anthropi ATCC 49188 (CP000758 [100]) | |
| Achromobacter | 6,329,203 | 96.26 | 0.99 | Achromobacter insolitus DSM 23807 (99.03) | 1,511 | OTU-4 | Achromobacter denitrificans PR1 (CP020917 [100]) | KK8 (99.18) |
| Achromobacter insolitus DSM 23807 (CP019325 [99.67]) | ||||||||
| Cupriavidus | 7,560,176 | 96.44 | 0.69 | Cupriavidus necator N-1 (99.41) | 1,530 | OTU-5 | Cupriavidus necator N-1 (CP002877 [100]) | KK10 (99.51) |
| Parvibaculum | 3,950,696 | 99.57 | 0.87 | Parvibaculum lavamentivorans DS-1 (80.62) | 1,486 | OTU-6 | Parvibaculum lavamentivorans DS-1 (CP000774 [95.37]) | |
| Olivibacter | 1,520 | OTU-7 | Olivibacter jilunii 14-2A (NR_109321 [99.46]) | |||||
| Dokdonella | 1,491 | OTU-9 | Dokdonella ginsengisoli Gsoil 191 (NR_041369 [100]) | |||||
| Rhodanobacter | 1,503 | OTU-10 | Rhodanobacter rhizosphaerae CR164 (NR_156938 [100]) | |||||
Reconstruction of metagenome-assembled genomes and bacterial functional gene analyses.
From the metagenomic reads, almost complete (>96% completeness) high-quality (<2% contamination) genomes of six major bacterial genera (Pseudomonas, Sphingobium, Ochrobactrum, Achromobacter, Cupriavidus, and Parvibaculum) were reconstructed (Table 1). The Pseudomonas, Sphingobium, Ochrobactrum, Achromobacter, and Cupriavidus species metagenome-assembled genomes (MAGs) revealed high (>97%) average nucleic acid identity (ANI) values compared to those of reference genomes of the closest bacterial strains; however, the Parvibaculum MAG showed only 80.6% ANI with the closest genome (P. lavamentivorans DS-1), indicating that any phylogenetically related strains of this Parvibaculum strain have not been reported.
As potential marker genes for the key step of aromatic hydrocarbon degradation, several types of aromatic ring-hydroxylating dioxygenase (ARHD) were detected in the metagenome reads. These ARHDs may be classified into the following three classes according to Chakraborty et al. (12) (Fig. 4): class A, annotated as naphthalene 1,2-dioxygenase (nahAcAd; EC 1.14.12.12), benzene/toluene 1,2-dioxygenase (bnzAB-todC1C2; EC 1.14.12.3), or 3-phenylpropanoate dioxygenase (hcaEF; EC 1.14.12.19); class B, annotated as benzoate/toluate 1,2-dioxygenase (benAB/xylXY; EC 1.14.12.10) or anthranilate 1,2-dioxygenase (antAB; EC 1.14.12.1); and class C, annotated as salicylate 5-hydroxylase (nagGH; EC 1.14.13.172) or anthranilate 1,2-dioxygenase. The Sphingobium MAG encoded seven sets of ARHD large- and small subunits, which were previously characterized as ahdA1A2a, -b, -c, -d, -e, and -f (bphA1A2a to -f) and xylXY in other sphingomonads genomes (13, 14). These seven gene sets covered all three ARHD classes: class A, ahdA1A2a, ahdA1A2b, and ahdA1A2f; class B, xylXY; and class C, ahdA1A2c, ahdA1A2d, and ahdA1A2e (Fig. 4). Other bacterial MAGs (Pseudomonas, Achromobacter, Cupriavidus, and Parvibaculum) also possessed class B and class C ARHDs; however, class A ARHDs were not detected in these MAGs except for the Parvibaculum MAG, which harbored an ARHD gene set distantly clustered in class A and for which the function is uncertain (Fig. 4; Table 2). No ARHD gene set was found in the Ochrobactrum MAG.
FIG 4.

Neighbor-joining phylogenetic tree of concatenated amino acid sequences of ARHD large and small subunits obtained from the metagenome of the consortium grown on diesel fuel (bold font, colored) and the reference enzymes. Enzymes are classified into class A, B, and C ARHDs according to Chakraborty et al. (12). The tree was created with 1,000 bootstrap iterations, and values below 50% are not reported.
TABLE 2.
Numbers of aromatic hydrocarbon- and alkane-degrading genes harbored in the major bacterial MAGs detected in the consortium
| Enzyme | No. of genes in genus: |
|||||
|---|---|---|---|---|---|---|
| Pseudomonas | Sphingobium | Ochrobactrum | Achromobacter | Cupriavidus | Parvibaculum | |
| Aromatic ring-hydroxylating dioxygenase | ||||||
| Class A (benzene/naphthalene/PAH-dioxygenases) | 0 | 3 | 0 | 0 | 0 | 1 |
| Class B (benzoate/anthranilate-dioxygenases) | 2 | 1 | 0 | 2 | 1 | 1 |
| Class C (anthranilate dioxygenase/salicylate hydroxylase) | 0 | 3 | 0 | 1 | 2 | 0 |
| Aromatic ring-cleaving dioxygenase | ||||||
| 1,2-Dihydroxynaphthalene dioxygenase (nahC) [EC 1.13.11.56] | 0 | 1 | 0 | 1 | 0 | 1 |
| Catechol 1,2-dioxygenase (catA) [EC 1.13.11.1] | 1 | 1 | 0 | 1 | 1 | 0 |
| Catechol 2,3-dioxygenase (xylE) [EC 1.13.11.2] | 0 | 2 | 1 | 1 | 1 | 0 |
| Protocatechuate 3,4-dioxygenase (pcaGH) [EC 1.13.11.3] | 1 | 0 | 1 | 0 | 1 | 0 |
| Gentisate 1,2-dioxygenase (nagI) [EC 1.13.11.4] | 1 | 1 | 0 | 2 | 2 | 1 |
| Aromatic ring-monooxygenase | ||||||
| Phenol/toluene 2-monooxygenase (dmpKLMNOP) [EC 1.14.13.243] | 0 | 1 | 0 | 0 | 1 | 0 |
| Toluene methyl-monooxygenase (xylM) [EC 1.14.15.26]a | 0 | 1 | 0 | 0 | 0 | 0 |
| Alkane monooxygenase | ||||||
| Alkane 1-monooxygenase (alkB) [EC 1.14.15.3] | 2 | 1 | 0 | 0 | 0 | 0 |
| Putative CYP153 family alkane monooxygenase | 0 | 1 | 0 | 0 | 0 | 5 |
| Putative flavin-binding alkane monooxygenase (almA) | 2 | 0 | 0 | 0 | 1 | 2 |
Oxidation of the methyl group on the aromatic ring.
Class A ARHDs of Sphingobium (ahdA1A2a, -b, and -f) in the consortium are predicted to be responsible for the initial biotransformation steps of phenanthrene, naphthalene, and toluene, i.e., the addition of two oxygen atoms to the aromatic rings (15) (step 1 in Fig. 5). Genes for ferredoxin and ferredoxin reductase (ahdA3A4) (16) were also identified in the Sphingobium MAG. At the same time, the nahB (bphB) gene, which encodes aromatic dihydrodiol dehydrogenase for the subsequent reaction, was also found in the Sphingobium and Parvibaculum MAGs. Only Sphingobium possessed all of the genes necessary for downstream biotransformation of both 3,4-dihydroxyphenanthrene and 1,2-dihydroxynaphthalene (nahC [bphC], nahD, and nahE) (step 2 in Fig. 5). A potential homologue of the nahF gene, which catalyzes the subsequent step after nahE, was not found in all MAGs. Alternatively, the xylC gene of Sphingobium, which encodes benzaldehyde dehydrogenase, was likely involved in this step (step 2) (17). Class C ARHDs of Sphingobium (ahdA1A2c, -d, and -e) transformed 1-hydroxy-2-naphthoic acid to 1,2-dihydroxynaphthalene, as well as salicylic acid to catechol (13), in addition to salicylate 1-hydroxylase encoded by the nahG gene of Achromobacter and Cupriavidus (step 3 in Fig. 5). Sphingobium also potentially biotransformed 1,2-dihydroxynaphthalene through an intradiol ring cleavage pathway via phthalic acid to benzoic acid; however, the genes involved in these steps are unknown (step 4 in Fig. 5) (16).
FIG 5.

Distributions of key aromatic hydrocarbon degradation genes harbored in six high-quality MAGs for Sphingobium, Pseudomonas, Ochrobactrum, Achromobacter, Cupriavidus, and Parvibaculum spp. reconstructed from the consortium grown on diesel fuel. Arrows labeled as different colors indicate functional genes found in each MAG. (Step 1) Initial biotransformation step of phenanthrene, naphthalene, and toluene by class A ARHDs. (Step 2) Transformation of 3,4-dihydroxyphenanthrene and 1,2-dihydroxynaphthalene via extradiol ring cleavage pathways. (Step 3) Decarboxylation of 1-hydroxy-2-naphthoic acid and salicylic acid. (Step 4) Transformation of 1,2-dihydroxynaphthalene via intradiol ring cleavage pathway. (Step 5) Methyl-monooxygenation of toluene. (Step 6) Ring-monooxygenation of toluene. (Step 7) Transformation of benzoic acid to catechol. (Step 8) Transformation of phthalic acid to protocatechuic acid. (Step 9) Transformation of salicylic acid to gentisic acid. (Step 10) Degradation of gentisic acid. (Steps 11 and 12) Degradation of catechol via intradiol or extradiol ring cleavage pathways. (Step 13) Degradation of protocatechuic acid.
Genes for toluene methyl monooxygenase (xylMABC) were found only in the Sphingobium MAG, which were likely responsible for biotransformation of toluene to benzoic acid (step 5 in Fig. 5). Toluene was also likely biotransformed to 3-methylcatechol by phenol/toluene 2-monooxygenase encoded by the dmpKLMNP genes found in Sphingobium and Cupriavidus MAGs (step 6 in Fig. 5) or by class A ARHDs of Sphingobium (ahdA1A2a, -b, and -f). Benzoic acid was likely biotransformed to catechol by benzoate/toluate 1,2-dioxygenase encoded by the xylXYZ and xylL genes that were found in multiple consortium member MAGs, including Sphingobium, Pseudomonas, Cupriavidus, and Parvibaculum (xylL not detected), as well as the functionally uncertain class B ARHDs of Achromobacter (step 7 in Fig. 5).
Genes for biotransformation of phthalic acid to protocatechuic acid (pht2345) were exclusively found in Cupriavidus MAG (step 8 in Fig. 5). Sphingobium, Achromobacter, and Cupriavidus possessed the genes for biodegradation of salicylic acid through the tricarboxylic acid (TCA) cycle via gentisic acid (nag genes) (steps 9 and 10 in Fig. 5) or via catechol that was further biodegraded through intradiol (cat genes) (step 11 in Fig. 5) or extradiol (xyl genes) (step 12 in Fig. 5) ring cleavage pathways; the xylJK genes were not found in Achromobacter MAG. Genes for gentisic acid biodegradation (nagILK; step 10) were found in all MAGs except for the Ochrobactrum MAG, and genes for protocatechuic acid biodegradation (pca gene) (step 13 in Fig. 5) were found in Pseudomonas, Ochrobactrum, and Cupriavidus MAGs (Fig. 5; see also Table S3).
As a well-known functional marker gene for alkane degradation, alkane 1-monooxygenase (alkB; EC 1.14.15.3) was detected in Pseudomonas and Sphingobium MAGs (Table 2). On the other hand, the Parvibaculum MAG possessed multiple (at least five) copies of the putative homologue gene for another known alkane monooxygenase, cytochrome P450 CYP153 family monooxygenase (18). The Parvibaculum MAG also harbored putative flavin-binding long-chain alkane monooxygenase (almA) homologues (19–21), which were also found in Pseudomonas and Cupriavidus MAGs (Table 2). Genes for the downstream pathways of alkane chain biodegradation, such as those encoding alcohol dehydrogenase (EC 1.1.1.1), aldehyde dehydrogenase (EC 1.2.1.3), and acyl coenzyme A (acyl-CoA) synthetase (EC 6.2.1.3), were conserved in all bacterial MAGs (Table S3).
Growth tests of major bacterial genera isolated from the consortium.
Four bacterial isolates that represented four different major genera in the consortium were obtained in pure cultures (Pseudomonas sp. strain KK6, Sphingobium barthaii strain KK22, Achromobacter sp. strain KK8, and Cupriavidus sp. strain KK10) (Table 1 and Fig. 3), and their growth on different hydrocarbon substrates was tested (Table 3). Among these isolates, strain KK6 exclusively grew on n-hexadecane, and only strain KK22 grew on phenanthrene, naphthalene, or toluene as the sole source of carbon and energy (16, 22). As representative biotransformation products of these aromatic hydrocarbons, 1-hydroxy-2-naphthoic acid, 1,2-dihydroxynaphthalene, salicylic acid, gentisic acid, phthalic acid, protocatechuic acid, and benzoic acid were utilized in growth tests of the isolates. Only strain KK22 was confirmed to have the potential to grow on 1-hydroxy-2-naphthoic acid and 1,2-dihydroxynaphthalene. Strains KK22, KK8, and KK10 were found to grow on salicylic acid and phthalic acid, although growth of strain KK22 on phthalic acid was weak. Strain KK6 showed growth on gentisic acid in addition to strains KK8 and KK10, while strain KK22 did not. Only strain KK6 was confirmed to grow on protocatechuic acid. All four strains grew on benzoic acid. Strains KK6 and KK22 produced macroscopic biofilms during growth. Furthermore, these two strains showed hemolytic clearing, which indicated that these strains possessed biosurfactant production capabilities. Results of all growth tests are summarized in Table 3.
TABLE 3.
Summary of the growth test results of bacterial isolates obtained from the consortium
| Parameter | Pseudomonas sp. KK6 | Sphingobium barthaii KK22 | Achromobacter sp. KK8 | Cupriavidus sp. KK10 |
|---|---|---|---|---|
| Bacterial growth on hydrocarbonsa | ||||
| n-Hexadecane | + | − | − | − |
| Phenanthrene | − | +b | − | − |
| Naphthalene | − | +b | − | − |
| Toluene | − | +b | − | − |
| 1-Hydroxy-2-naphthoic acid | − | +b | − | − |
| 1,2-Dihydroxynaphthalene | − | +/− | − | − |
| Salicylic acid | − | +b | +b | +b |
| Gentisic acid | + | − | + | + |
| Phthalic acid | − | +/− | + | + |
| Protocatechuic acid | + | − | − | − |
| Benzoic acid | +b | +b | +b | +b |
| Biosurfactant production (hemolysis)c | + | + | − | − |
Bacterial growth was evaluated (+ or −) by visual inspection of turbidity and/or floc formation after 10 days.
Presence of biotransformed products in the culture was confirmed by LC/ESI(−)-MS/MS.
Biosurfactant production was evaluated (+ or −) through a hemolytic test using sheep blood agar.
Identification of biotransformation products of the bacterial isolates.
With the aims to link biodegradation genes to function, the catabolic capabilities of the four isolated bacterial strains were investigated by extraction, separation, and identification of biotransformation products from culture medium by liquid chromatography electrospray ionization-tandem mass spectrometry in negative ionization mode [LC/ESI(−)-MS/MS] when these strains were individually exposed to aromatic substrates as the sole sources of carbon and energy (Table 3; see also Table S4). 1,2-Dihydroxynaphthalene and salicylic acid were determined to be metabolites of pathway convergence when strain KK22 was grown on phenanthrene, naphthalene, or 1-hydroxy-2-naphthoic acid from these chemical analyses (see Fig. S1). When grown on toluene, benzoic acid production was confirmed from strain KK22 (Fig. 5; see also Fig. S2). Because catechol was also predicted to be a biotransformation product of pathway convergence (Fig. 5), it was targeted in chemical analyses as a biotransformation product of salicylic acid and benzoic acid by all four strains (see Fig. S3). Analyses revealed that catechol was detected in all cultures that grew on salicylic acid (strains KK22, KK8, and KK10) or benzoic acid (all four strains). Additionally, gentisic acid, which was predicted to occur through biotransformation of salicylic acid (Fig. 5), was only detected in salicylic acid-grown cultures of strain KK8 (see Fig. S4).
DISCUSSION
Diesel fuel component-degrading bacterial consortium.
The consortium grown on diesel fuel was found to comprise six major bacterial genera (>1% relative abundances) (Fig. 2A) and at least six minor bacterial genera in a hydrocarbon-degrading community that formed after natural competitive and cooperative relationships were established. The consortium ecosystem may be considered to be—relative to the soil ecosystem from whence it came—a selectively enriched bacterial guild that utilized diesel fuel components as sources of carbon and energy whereby the members detected had survived in the consortium through numerous transfers by outcompeting other organisms. The 16S rRNA gene-based bacterial community analyses indicated that the consortium, when grown on diesel fuel, was largely dominated by Pseudomonas (>70%), followed by Sphingobium as the second most-abundantly detected genus (Fig. 2). These two genera were also dominant when grown on a less-complex synthetic 4-component NAPL mixture. When the carbon sources were reduced to single hydrocarbon substrates, however, Pseudomonas dominated on n-hexadecane and outcompeted Sphingobium, while Sphingobium was highly enriched on aromatic hydrocarbon sources (Fig. 2A). These results indicated that these two genera were largely responsible for alkane or aromatic hydrocarbon degradation, respectively. Achromobacter and Cupriavidus were also considered to be candidate aromatic hydrocarbon degraders based upon bacterial community analysis, whereas Cupriavidus was only enriched on toluene. Relative abundances of Ochrobactrum did not significantly change among different carbon sources, which suggested that it was capable of degrading both alkane and aromatic hydrocarbons or, more likely, that it grew on the products of alkane and aromatic compound biodegradation by other hydrocarbon degraders such as Pseudomonas and Sphingobium. Previous work showed that an alkane-degrading bacterium Rhodococcus formed bioflocs in association with non-alkane-degrading Ochrobactrum and Bacillus spp., which utilized the biotransformation products (organic acids) provided by Rhodococcus, and it was concluded overall that this multispecies association enhanced alkane degradation (23). Parvibaculum was one of the major bacterial genera in the diesel fuel-grown culture (1.2%); however, it appeared to have been outcompeted in cultures grown on simplified hydrocarbon sources. These results may indicate that Parvibaculum preferred to grow on carbon sources other than n-hexadecane, phenanthrene, naphthalene, or toluene and likely occupied a different niche from those of other major bacteria in the diesel fuel-grown culture.
Multispecies aromatic hydrocarbon biodegradation ignited by Sphingobium.
Consistent with the 16S rRNA gene amplicon sequencing results, metagenomic functional gene analyses of the consortium revealed that the metabolic potential of Sphingobium lead to the biodegradation of aromatic substrates, which was also supported by the results of growth tests of the isolates (Table 3). Among the six bacterial MAGs reconstructed from the consortium, the Sphingobium MAG was highlighted as the potential key aromatic hydrocarbon degrader, because it harbored three gene sets of class A ARHDs (ahdA1A2, -a, -b, and -f) and toluene/xylene methyl monooxygenase genes (xylMABC) as well as gene clusters involved in the downstream degradation pathways (Fig. 5; see also Table S3 in the supplemental material). Sphingomonad bacteria such as Sphingomonas, Sphingobium, Novosphingobium, and Sphingopyxis, which are found in various environments, are known as efficient degraders of aromatic hydrocarbons, including a variety of PAHs, and the metabolic functions of some sphingomonad isolates were previously characterized (24). ARHD gene sets harbored by these sphingomonad genomes were also conserved in S. barthaii KK22 (14) and the Sphingobium MAG in the consortium. Among the three gene sets of class A ARHDs of sphingomonads, ahdA1A2b was previously functionally characterized as being capable of biotransforming ethyl/propylbenzene, cumene, p-cymene, and some PAHs, while ahdA1A2f was suggested to be more responsible for biotransforming 2-, 3-, 4-, and 5-ring PAHs (15). Based upon this evidence, it was hypothesized that the Sphingobium in the consortium studied here triggered the biodegradation of various aromatic hydrocarbon substrates by catalyzing the initial reactions by utilizing the class A ARHDs. During the utilization and acquisition of carbon and energy by Sphingobium via these reactions, other consortium members likely utilized the biotransformation products produced.
According to the genomic evidence, five other major genera potentially utilized less-complex more-biodegradable forms of aromatic substrates such as benzoic acid, salicylic acid, gentisic acid, and protocatechuic acid through different types of aromatic ring-cleaving dioxygenases (Fig. 5; Table 2). Among these five genera, Pseudomonas aeruginosa strains, Ochrobactrum spp., Achromobacter spp., and Cupriavidus spp. have been reported in the literature as being capable of biodegrading PAHs (25–28); however, isolates of these genera obtained from the consortium studied here did not show clear growth on phenanthrene, naphthalene, or toluene as the sole source of carbon and energy (Table 3). Growth tests of these isolates followed by the identification of biotransformation products suggested that Sphingobium in the consortium exclusively biodegraded and grew on multiring aromatic hydrocarbons and toluene and that coexisting Pseudomonas, Achromobacter, and Cupriavidus were involved in the downstream reactions. Combined with the bacterial community analysis results of the consortium, Pseudomonas was predicted to be more adapted to participate in the downstream pathways of phenanthrene and naphthalene biodegradation via gentisic acid or protocatechuic acid, while Cupriavidus likely competed with Pseudomonas by involvement in the toluene biodegradation pathways. Finally, Achromobacter was involved in both the multiring aromatic and toluene biodegradation pathways. Interestingly, although S. barthaii strain KK22 and Sphingobium in the consortium were found to possess genes for the biodegradation of salicylic acid via gentisic acid (nag genes) (steps 9 and 10 in Fig. 5), gentisic acid was not detected from the biodegradation of multiring aromatics by S. barthaii strain KK22 (16). Moreover, strain KK22 did not grow on gentisic acid (Table 3), which suggested that biodegradation of salicylic acid via gentisic acid in the consortium was likely driven by Achromobacter and Pseudomonas and not by Sphingobium.
Phenol/toluene 2-monooxygenase genes (dmpKLMNOP) in the Cupriavidus MAG were found to be well conserved in other Cupriavidus strains and therefore expected to catalyze the first step of toluene biodegradation (29), although Cupriavidus sp. strain KK10 was not confirmed to grow on toluene. Previous reports showed that this strain biodegraded two-ring aromatic compounds; however, biotransformation was demonstrated under circumstances whereby a second organic carbon source, (i.e., cosubstrate) aided biodegradation (30, 31). Cometabolic relationships are considered to be relatively common during PAH biodegradation and were likely occurring in the consortium, adding to the challenges of studying these types of microbial ecosystems (32).
Interestingly, the Parvibaculum MAG possessed a putative class A ARHD gene set, which was clustered with biphenyl degradation genes (bphBCD). Downstream of bphBCD, degradation genes (bphEFG) were found as a cluster in this MAG; it was separated from the class A ARHD-bphBCD cluster by a scaffold break but likely belonged to the same gene cluster. These gene clusters were exclusively found in Parvibaculum MAG; therefore, it was likely capable of growth on biphenyl and alkylbiphenyls (33) which are present in diesel fuel (34) but not the synthetic NAPL (Fig. 2A). Interestingly, because Parvibaculum possessed xylXY but not xylL, it was not likely capable of biodegrading the benzoic acid products of biphenyl biodegradation. These results suggested that Parvibaculum may have occupied a specialized niche in the diesel fuel-grown community. Uncultivated Parvibaculum-related strains were previously discovered in biphenyl-enriched soil bacterial communities (35, 36); however, their metabolic functions were not characterized in detail.
Overall, results of this investigation strongly suggested that Sphingobium acted as a pioneer aromatic hydrocarbon-degrading organism in the consortium, which initiated aromatic hydrocarbon biotransformation and the production of specialized niches that enabled other members’ survival.
Coexistence of Pseudomonas and Parvibaculum which preferred different-chain-length alkanes.
Different types of known alkane monooxygenase genes were found in the consortium metagenome. The most typical, alkane 1-monooxygenase (alkB; EC 1.14.15.3), was encoded in the Pseudomonas and Sphingobium MAGs. Putative cytochrome P450 CYP153 family alkane monooxygenase homologues were also revealed in the Parvibaculum and Sphingobium MAGs, while putative flavin-binding long-chain alkane monooxygenase (almA) homologues were revealed in the Pseudomonas, Parvibaculum, and Cupriavidus MAGs (Table 2). The alkane biodegradation capabilities of Pseudomonas aeruginosa and some species of Parvibaculum were previously reported (37–40), while Sphingobium and Cupriavidus have not been reported to biodegrade alkanes to the best of our knowledge. In growth tests of bacterial isolates on n-hexadecane as a representative alkane in diesel fuel, only Pseudomonas sp. strain KK6 grew; however, Parvibaculum has not been isolated from the consortium. Consistent with these results, Pseudomonas was highly enriched in the consortium grown on n-hexadecane, while Sphingobium, Cupriavidus, and Parvibaculum were apparently outcompeted (Fig. 2A). These results suggested that Parvibaculum preferred or was dependent upon alkane substrates other than n-hexadecane and was outcompeted by Pseudomonas when only n-hexadecane was present. Parvibaculum was first reported for its capability of degrading a commercial surfactant alkylbenzene sulfonate (41), and the presence of CYP153 alkane monooxygenase genes in addition to almA genes in Parvibaculum genomes has been reported (21, 42). According to the literature, although alkB-type alkane monooxygenase is capable of hydroxylating alkanes with chain lengths up to C32, CYP153 is responsible for biodegradation of short- and medium-chain-length alkanes (<C12) (18), and the almA type biodegrades long-chain-length alkanes (C32 or longer) (21). Therefore, the lack of an alkB gene in the Parvibaculum genome may explain why it was not detected in the culture grown exclusively on n-hexadecane. Parvibaculum in the consortium may have been responsible for the biodegradation of short- and middle-chain-length alkanes; however, it shall require confirmation.
All six bacterial MAGs harbored genes for the downstream reactions of alkane chain biodegradation such as alcohol dehydrogenase and aldehyde dehydrogenase, and consistent with this, Ochrobactrum and Achromobacter spp. were enriched in the consortium grown on n-hexadecane, potentially, by utilizing the by-products released from alkane-degrading Pseudomonas. In contrast, Sphingobium, Cupriavidus, and Parvibaculum spp. unlikely grew in association with Pseudomonas and were therefore outcompeted (Fig. 2A). Thus, specific interspecies associations among these major bacterial genera may have been occurring in the consortium, which may be further investigated by bacterial cocultivation experiments using selected isolates and/or through microscopic inspection of their spatial distributions in the consortium. Finally, the actinomycete Mycobacterium sp. was detected as the only Gram-positive organism in the consortium when it was grown on the 4-component NAPL or n-hexadecane; its niche in the consortium during hydrocarbon biodegradation is uncertain and shall require further investigation.
Coexistence and interactions among the diesel fuel-degrading pioneers and cocolonizers.
Results of our combined metagenomics and pure culture-based growth tests indicated that several major bacterial genera in the consortium were responsible for diesel fuel biodegradation. In particular, the two dominant genera in the consortium, Sphingobium and Pseudomonas, likely initiated and led biotransformation of aromatic and alkane hydrocarbons, respectively. Notably, these two genera generated macroscopic biofilms while growing on petroleum hydrocarbons and also showed biosurfactant activities. Biofilm formation and biosurfactant production that enable bacteria to increase the bioavailabilities of hydrophobic hydrocarbons are known as typical strategies for the acquisition of carbon and energy from hydrophobic organic compounds (43–46). Interestingly, flagellar formation and chemotaxis genes were well conserved in all bacterial MAGs reconstructed (see Table S5), which may enable these bacteria to access favorable growth sources. Hypothetically, when the consortium was exposed to diesel hydrocarbons, pioneering Sphingobium and Pseudomonas spp. first attached to the hydrophobic NAPL and started utilizing NAPL components by forming biofilms (bioflocs), and this served as the “ignition” step for subsequent hydrocarbon biodegradation and utilization by the consortium. Next, their biofilm matrices (extracellular polymeric substances [EPS]) adsorbed surrounding hydrocarbons and facilitated other bacterial genera to cocolonize. The cocolonizers then utilized the hydrocarbon biotransformation products derived from pioneer catabolism and/or their secondary metabolites, (e.g., EPS). Although the multispecies biodegradation of alkanes and aromatic hydrocarbons took place in the consortium, it required the pioneering organisms, Sphingobium and Pseudomonas, to initiate hydrocarbon biotransformation, which supported the ecosystem as a whole through the production of niches specific for other members (Fig. 6). Furthermore, the coexistence of the cocolonizers in the consortium likely contributed to community ecosystem dynamics by regulating biofilm formation (47), accelerating hydrocarbon biodegradation (23), and/or reducing downstream potentially toxic products (48).
FIG 6.

Hypothetical model of multispecies bacterial diesel fuel biodegradation by the consortium. Hydrophobic oil droplets are first colonized by the pioneer aromatic and alkane hydrocarbon-degrading bacteria, Sphingobium and Pseudomonas, which initiate biotransformation of complex hydrocarbons by forming biofilms. The hydrocarbon biotransformation products and secondary metabolites such as biofilm matrices produced by the pioneers, which have relatively higher bioavailability, are thus utilized by other bacterial cocolonizers. These cocolonizers mainly consisted of Ochrobactrum, Achromobacter, and Cupriavidus, and their coexistence may enhance further biofilm formation and accelerate hydrocarbon biodegradation. Another major bacterial genus, Parvibaculum, was potentially responsible for biodegradation of short- and middle-chain-length alkanes and/or biphenyl-related compounds.
Considering that the presence of more than 10 consortium members was confirmed, each member must have found and exploited a niche(s) that allowed for their survival in the overall consortium ecosystem, and their survival through occupation of specific niches (e.g., pioneers and cocolonizers) therefore led to robustness of each group within the multispecies diesel fuel-degrading bacterial community. Coexistence of a select few pioneer hydrocarbon degraders and a multispecies cocolonizer cohort may be a common trait in natural ecosystems exposed to petroleum hydrocarbon pollution (4, 49). Further experiments using isolated bacterial strains are expected to reveal the potential bacterial interspecies interactions between the pioneers and cocolonizers, such as metabolic cross-feeding or cell coaggregation systems (50). The results of this investigation provided insight into how hydrocarbon-degrading microorganisms may interact with each other to develop communities, support specialized niches, and maintain community structural integrity during exposure to petroleum hydrocarbon pollution in the environment.
MATERIALS AND METHODS
Chemicals.
Naphthalene (99% purity), catechol (99%), salicylic acid (>99% purity), benzoic acid (>99% purity), 2,5-dihydroxybenzoic acid (gentisic acid; 98% purity), phthalic acid (98% purity), 3,4-dihydroxybenzoic acid (protocatechuic acid; >95% purity), n-hexadecane (>97% purity), toluene (high-performance liquid chromatography [HPLC] grade), N,N-dimethylformamide (DMF; >99% purity), methanol (LC-MS grade), ethylacetate (HPLC grade), and chloroform (HPLC grade) were purchased from Wako Chemical (Osaka, Japan). 1-Hydroxy-2-naphthoic acid (>97% purity) was purchased from Kanto Chemical Co. (Tokyo, Japan). Phenanthrene (98% purity) was purchased from Sigma-Aldrich (St. Louis, MO, USA). 1,2-Dihydroxynaphthalene (>95% purity) and pristane (2,6,10,14-tetramethylpentadecane; >95% purity) were from Tokyo Chemical Industries (Tokyo, Japan).
Growth conditions of hydrocarbon-degrading bacterial consortium.
A hydrocarbon-degrading bacterial consortium was originally recovered from soil (11, 32, 51) and was maintained on 0.2% (wt/vol) diesel fuel (filtered sterilized, 0.22 μm) (52) in Stanier’s basal medium (SBM) for approximately 15 years, during which it was routinely transferred to fresh medium approximately once per month. In this study, the consortium was transferred to and grown on different carbon sources in 300-ml-volume conical flasks that contained 100 ml of SBM as follows: (i) with 0.1% (wt/vol) diesel fuel, (ii) with a four-component synthetic hydrocarbon nonaqueous-phase liquid mixture (4-component NAPL) that consisted of n-hexadecane (final concentration 700 mg liter−1), phenanthrene (50 mg liter−1), naphthalene (50 mg liter−1), and toluene (200 mg liter−1) as representative alkane and aromatic hydrocarbon compounds found in diesel fuel, and (iii) with single hydrocarbon substrates, (a) n-hexadecane (700 mg liter−1), (b) phenanthrene (50 mg liter−1), (c) naphthalene (50 mg liter−1), or (d) toluene (200 mg liter−1). The 4-component NAPL was prepared by addition of 0.25 g of phenanthrene and 0.25 g of naphthalene to a mixture of 4.52 ml of n-hexadecane and 1.15 ml of toluene at room temperature and shaken to dissolve the components. The NAPL was added to SBM at a concentration of 0.1% (wt/vol). When phenanthrene or naphthalene was added as a single hydrocarbon substrate, it was solubilized in DMF and added to SBM to a final concentration of 50 mg liter−1. After subculturing the consortium three times under each of the described conditions, it was grown for 4 days on the respective carbon sources by rotary shaking at 150 rpm in the dark at 30°C and subjected to microbial genomic DNA extraction and chemical analyses as described below. For potentially volatile individual carbon sources, e.g., naphthalene and toluene, silicone stoppers were utilized.
Analyses of diesel fuel biodegradation by the consortium.
In 300-ml-volume conical flasks, the consortium was exposed to 0.1% (wt/vol) diesel fuel as the sole carbon and energy source in a total volume of 100 ml of SBM under the incubation conditions described above. At the start of the experiment and after 4 days, whole-flask extractions were conducted with equal volumes of chloroform by rotary shaking in darkness at 200 rpm and 20°C for 24 h. Abiotic controls consisted of 0.1% (wt/vol) diesel fuel without cells and were treated identically to exposed cell cultures. One-milliliter volumes of chloroform extracts were separated from the aqueous phase, transferred to glass LC vials, and treated with less than 20 mg of anhydrous sodium sulfate.
Samples were analyzed by gas chromatography with flame ionization detection (model GC-2014; Shimadzu, Kyoto, Japan) that was equipped with a capillary column (ZB-5; Phenomenex, CA, USA) (30 m, inside diameter [i.d.] of 0.25 mm, 0.25 μm). The carrier gas was helium and the flow rate was 1.0 ml per min. The injector temperature was 280°C and the detector temperature was 300°C. The temperature program was as follows: 150°C for 2 min followed by continuous increase of 10°C per min to 280°C, and a final holding temperature of 280°C for 10 min for a 25-min total run time. Sample injection volumes were 1 μl each and were applied manually by a 10-μl glass microsyringe fitted with a Chaney adaptor (Hamilton, Reno, NV, USA). Peak areas and retention times were determined by GC solution software version 2.4 (Shimadzu). Retention times of naphthalene, n-hexadecane, pristane, and phenanthrene were confirmed to be 3.4, 6.9, 8.0, and 9.3 min each after preparation in chloroform.
DNA extraction.
Bacterial cells were collected by vacuum filtration of cultures through 0.22-μm polyvinylidene difluoride (PVDF) membrane filters (Durapore; Merck Millipore, Darmstadt, Germany) and stored at 20°C. Bacterial genomic DNA was extracted using the DNeasy PowerWater DNA isolation kit (Qiagen, Hilden, Germany) and stored at −20°C for downstream analyses. Biological duplicates were prepared for the cultures grew on diesel fuel for the subsequent amplicon sequencing analyses.
16S rRNA gene amplicon sequencing.
Bacterial 16S rRNA gene amplicon sequencing was performed using the Illumina MiSeq platform at the Bioengineering Lab Co., Ltd. (Sagamihara, Japan), and the procedure is summarized as follows: the V4 region of the bacterial and archaeal 16S rRNA gene was amplified from the extracted bacterial DNA by PCR using the primer set 515f/806r connected with an adapter sequence for the second PCR (53). The first PCR was performed by using TaKaRa Ex Taq DNA polymerase (TaKaRa-Bio, Kusatsu, Japan) according to the following protocol: 10 ng of DNA as the template, initial denaturing at 94°C for 2 min, 25 cycles of denaturing at 94°C for 30 s, annealing at 50°C for 30 s, and extension at 72°C for 30 s, followed by final extension at 72°C for 5 min. The PCR products were purified using AMPure XP (Beckman Coulter, Brea, CA, USA) and subjected to the second PCR using TaKaRa Ex Taq HS DNA polymerase (TaKaRa-Bio) according to the following protocol: initial denaturing at 94°C for 2 min, 12 cycles of denaturing at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s, followed by final extension at 72°C for 5 min. After purification (AMPure XP) and quality check of the PCR products, PCR amplicons were sequenced using the Illumina MiSeq platform (2 × 300 bp). After quality filtering of the raw sequence reads using the Fastx toolkit (fastq_barcode_splitter, v.0.0.14), primer sequence trimming, chimera removal, denoising, and OTU clustering were carried out using the DADA2 plugin in QIIME2 (v.2020.2) (54).
Metagenomic sequencing and genome reconstruction of major bacterial genera.
Metagenomic shotgun sequencing was conducted using the DNBSEQ-G400 platform (MGI Tech Co., Ltd., Shenzhen, China) at the Bioengineering Lab Co., Ltd., and the procedure was summarized as follows: extracted DNA samples were fragmented (400 bp), and the shotgun libraries were prepared with the MGIEasy universal DNA library prep Set. After the circular single-stranded DNA (ssDNA) was created using the MGIEasy circularization kit, DNA Nanoballs (DNB) were generated using an MGISEQ-200RS high-throughput sequencing set. Created DNB were sequenced using the DNBSEQ-G400 (2 × 150 bp). Raw sequence reads were filtered using sickle (v. 1.33) (55) and assembled using SPAdes (v. 3.13.2) (56). Full-length 16S rRNA genes originating from major bacterial genera were assembled and their abundances were determined using phyloFlash (v. 3.3b2) (57).
The quality reads were mapped to the assembled contigs using Bowtie2 (58) with default parameters set for coverage calculation, and genome binning was performed using MetaBAT2 (59) with default parameters. Metagenome-assembled genomes (MAGs) of major bacterial genera were reconstructed using the binned genomes and further taxonomic assignment of unbinned contigs. Genome completeness and contamination of these MAGs were evaluated using CheckM (60). Functional genes were annotated using Prokka (61). Average nucleic acid identities (ANIs) between each MAG and the reference bacterial genomes were calculated using the ANI calculator provided by the Konstantinidis lab at the Georgia Institute of Technology (enve-omics.ce.gatech.edu/ani/index).
Phylogenetic analyses.
Phylogenetic trees were created based on neighbor-joining using MEGA (v. 7.0.26) with 16S rRNA gene sequences or amino acid sequences of functional enzymes. For these analyses, relevant sequences obtained from metagenomes were aligned with the reference sequences using ClustalW in MEGA software, and the trees were generated using a Tamura-Nei model (nucleic acid) or Jones-Taylor-Thornton model (amino acid) with 1,000 bootstrap iterations.
Bacterial growth and biosurfactant production assays.
Bacteria were isolated from the consortium grown on diesel fuel by serial dilution or colony separation on Noble agar (Becton, Dickinson Biosciences, San Jose, CA) that was supplied with selected hydrocarbons or on complex media such as nutrient agar in this study and in previous studies; bacteria were phylogenetically identified through 16S rRNA gene sequencing (Macrogen Japan Co., Kyoto, Japan) (22, 30, 62).
Growth of bacterial isolates on n-hexadecane, phenanthrene, naphthalene, toluene, 1-hydroxynaphthoic acid, 1,2-dihydroxynaphthalene, salicylic acid, gentisic acid, phthalic acid, protocatechuic acid, and benzoic acid was assayed by transferring diesel fuel-grown cultures of each isolate to 100-ml-volume conical flasks that each contained 20 ml of SBM and 25 to 200 mg liter−1 of selected hydrocarbons. Carbon sources were added to flasks in DMF (≤0.1% [wt/vol]), and cultures were incubated as described above. Bacterial growth was evaluated by visual inspection of turbidity and/or floc formation for 10 days of incubation and compared to that of biotic (cells only) and abiotic (carbon source only) controls.
Biosurfactant production was evaluated by streaking bacterial isolates onto 5% sheep blood agar (Eiken Chemical Co., Tokyo), followed by incubation at 30°C for up to 5 days. Agar plates were examined for hemolytic clearing surrounding bacterial colonies (63).
Analyses of biotransformation products of bacterial isolates by LC/ESI(−)-MS/MS.
Under conditions that were identical to those described for bacterial growth assays, cells were exposed to single carbon compounds for 12 to 72 h and extracted at neutral pH with equal volumes of ethyl acetate, after which, culture fluids were reextracted in an identical manner after acidification to pH 2 to 3 with HCl. Organic layers were recovered, treated with anhydrous sodium sulfate, and evaporated to dryness under a gentle nitrogen gas stream at room temperature. Residues were resuspended in methanol and transferred into 1.5-ml-volume brown glass vials.
Extracts were analyzed by liquid chromatography electrospray ionization-tandem mass spectrometry in negative ionization mode [LC/ESI(−)-MS/MS] by using a Waters 2690 Separations module delivery system in line with a Waters model 2998 photodiode array detector (Waters Corp., Milford, MA, USA). The LC system was interfaced with a Quattro Ultima triple-stage quadrupole mass spectrometer (Waters-Micromass, Manchester, UK). Generally, 20-μl-volume sample aliquots were applied to an XSelect CSH C18 column (4.6 mm i.d. by 150 mm, 3.5-μm particle size; Waters) that was in line with a Security Guard cartridge system precolumn fitted with a wide-pore C18 cartridge (Phenomenex, Torrance, CA, USA) via autoinjection. LC was operated at a flow rate of 0.3 ml/min, where the mobile phase consisted of 77% methanol and 23% water and was administered to the mass spectrometer by electrospray which utilized nitrogen gas as the nebulizing gas. The ion source temperature was 130°C, the desolvation temperature was 350°C, and cone voltage was 35 V. Nitrogen gas was used as the desolvation gas, at 600 liters/h, and cone gas, at 60 liters/h. Argon gas (99.9999%) was used as the collision cell gas. Results from full-scan analyses were examined to determine the putative mass ions of interest by comparing the results of analyses of extracts from cells exposed to a particular compound with the results of analyses of extracts from unexposed cells and abiotic controls.
Putative ions of interest were identified and targeted for tandem mass product ion scan analyses which were conducted in negative ionization mode by using collision induced dissociation (CID) to generate spectra to be used in the determination of the molecular structures of unknown biotransformation products. Collision energies in the range of 8 to 20 eV were utilized.
Data availability.
Microbial 16S rRNA gene amplicon sequence data and metagenomics data obtained in this study were deposited at NCBI under BioProject PRJNA634975. Assembled metagenomic contigs were further uploaded to the Integrated Microbial Genome and Microbiomes (IMG/MER) database (Joint Genome Institute, IMG metagenome identifier [ID] 3300038749). The draft genome of S. barthaii KK22 was previously reported (64) (BioProject PRJDB1457 and IMG/MERGenome ID 2571042868).
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by a Japanese Society for the Promotion of Science (JSPS) KAKENHI grant (19K15738) and a Sasakawa Scientific Research Grant from The Japan Science Society (2019-4019) to J.F.M. and a JSPS KAKENHI grant (26505010) to R.A.K.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Blumer M. 1971. Scientific aspects of the oil spill problem. Boston Coll Environ Aff Law Rev 1:4. [Google Scholar]
- 2.Chaîneau CH, Yepremian C, Vidalie JF, Ducreux J, Ballerini D. 2003. Bioremediation of a crude oil-polluted soil: biodegradation, leaching and toxicity assessments. Water Air Soil Pollut 144:419–440. doi: 10.1023/A:1022935600698. [DOI] [Google Scholar]
- 3.Varjani SJ. 2017. Microbial degradation of petroleum hydrocarbons. Bioresour Technol 223:277–286. doi: 10.1016/j.biortech.2016.10.037. [DOI] [PubMed] [Google Scholar]
- 4.Head IM, Jones DM, Röling WFM. 2006. Marine microorganisms make a meal of oil. Nat Rev Microbiol 4:173–182. doi: 10.1038/nrmicro1348. [DOI] [PubMed] [Google Scholar]
- 5.Colores GM, Macur RE, Ward DM, Inskeep WP. 2000. Molecular analysis of surfactant-driven microbial population shifts in hydrocarbon-contaminated soil. Appl Environ Microbiol 66:2959–2964. doi: 10.1128/aem.66.7.2959-2964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Techtmann SM, Hazen TC. 2016. Metagenomic applications in environmental monitoring and bioremediation. J Ind Microbiol Biotechnol 43:1345–1354. doi: 10.1007/s10295-016-1809-8. [DOI] [PubMed] [Google Scholar]
- 7.Handley KM, Piceno YM, Hu P, Tom LM, Mason OU, Andersen GL, Jansson JK, Gilbert JA. 2017. Metabolic and spatio-taxonomic response of uncultivated seafloor bacteria following the Deepwater Horizon oil spill. ISME J 11:2569–2583. doi: 10.1038/ismej.2017.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dombrowski N, Donaho JA, Gutierrez T, Seitz KW, Teske AP, Baker BJ. 2016. Reconstructing metabolic pathways of hydrocarbon-degrading bacteria from the Deepwater Horizon oil spill. Nat Microbiol 1:16057. doi: 10.1038/nmicrobiol.2016.57. [DOI] [PubMed] [Google Scholar]
- 9.Tremblay J, Fortin N, Elias M, Wasserscheid J, King TL, Lee K, Greer CW. 2019. Metagenomic and metatranscriptomic responses of natural oil degrading bacteria in the presence of dispersants. Environ Microbiol 21:2307–2319. doi: 10.1111/1462-2920.14609. [DOI] [PubMed] [Google Scholar]
- 10.Dubinsky EA, Conrad ME, Chakraborty R, Bill M, Borglin SE, Hollibaugh JT, Mason OU, Piceno YM, Reid FC, Stringfellow WT, Tom LM, Hazen TC, Andersen GL. 2013. Succession of hydrocarbon-degrading bacteria in the aftermath of the Deepwater Horizon oil spill in the Gulf of Mexico. Environ Sci Technol 47:10860–10867. doi: 10.1021/es401676y. [DOI] [PubMed] [Google Scholar]
- 11.Kanaly R, Bartha R, Fogel S, Findlay M. 1997. Biodegradation of [14C]benzo[a]pyrene added in crude oil to uncontaminated soil. Appl Environ Microbiol 63:4511–4515. doi: 10.1128/AEM.63.11.4511-4515.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakraborty J, Ghosal D, Dutta A, Dutta TK. 2012. An insight into the origin and functional evolution of bacterial aromatic ring-hydroxylating oxygenases. J Biomol Struct Dyn 30:419–436. doi: 10.1080/07391102.2012.682208. [DOI] [PubMed] [Google Scholar]
- 13.Pinyakong O, Habe H, Omori T. 2003. The unique aromatic catabolic genes in sphingomonads degrading polycyclic aromatic hydrocarbons (PAHs). J Gen Appl Microbiol 49:1–19. doi: 10.2323/jgam.49.1. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Q, Yue S, Bilal M, Hu H, Wang W, Zhang X. 2017. Comparative genomic analysis of 26 Sphingomonas and Sphingobium strains: dissemination of bioremediation capabilities, biodegradation potential and horizontal gene transfer. Sci Total Environ 609:1238–1247. doi: 10.1016/j.scitotenv.2017.07.249. [DOI] [PubMed] [Google Scholar]
- 15.Khara P, Roy M, Chakraborty J, Ghosal D, Dutta TK. 2014. Functional characterization of diverse ring-hydroxylating oxygenases and induction of complex aromatic catabolic gene clusters in Sphingobium sp. PNB. FEBS Open Bio 4:290–300. doi: 10.1016/j.fob.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maeda AH, Nishi S, Hatada Y, Ohta Y, Misaka K, Kunihiro M, Mori JF, Kanaly RA. 2020. Chemical and genomic analyses of polycyclic aromatic hydrocarbon biodegradation in Sphingobium barthaii KK22 reveals divergent pathways in soil sphingomonads. Int Biodeterior Biodegradation 151:104993. doi: 10.1016/j.ibiod.2020.104993. [DOI] [Google Scholar]
- 17.Kim E, Zylstra GJ. 1999. Functional analysis of genes involved in biphenyl, naphthalene, phenanthrene, and m-xylene degradation by Sphingomonas yanoikuyae B1. J Ind Microbiol Biotechnol 23:294–302. doi: 10.1038/sj.jim.2900724. [DOI] [PubMed] [Google Scholar]
- 18.van Beilen JB, Funhoff EG, van Loon A, Just A, Kaysser L, Bouza M, Holtackers R, Röthlisberger M, Li Z, Witholt B. 2006. Cytochrome P450 alkane hydroxylases of the CYP153 family are common in alkane-degrading eubacteria lacking integral membrane alkane hydroxylases. Appl Environ Microbiol 72:59–65. doi: 10.1128/AEM.72.1.59-65.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Throne-Holst M, Wentzel A, Ellingsen TE, Kotlar H-K, Zotchev SB. 2007. Identification of novel genes involved in long-chain n-alkane degradation by Acinetobacter sp. strain DSM 17874. Appl Environ Microbiol 73:3327–3332. doi: 10.1128/AEM.00064-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wentzel A, Ellingsen TE, Kotlar H-K, Zotchev SB, Throne-Holst M. 2007. Bacterial metabolism of long-chain n-alkanes. Appl Microbiol Biotechnol 76:1209–1221. doi: 10.1007/s00253-007-1119-1. [DOI] [PubMed] [Google Scholar]
- 21.Wang W, Shao Z. 2012. Diversity of flavin-binding monooxygenase genes (almA) in marine bacteria capable of degradation long-chain alkanes. FEMS Microbiol Ecol 80:523–533. doi: 10.1111/j.1574-6941.2012.01322.x. [DOI] [PubMed] [Google Scholar]
- 22.Maeda AH, Kunihiro M, Ozeki Y, Nogi Y, Kanaly RA. 2015. Sphingobium barthaii sp. nov., a high molecular weight polycyclic aromatic hydrocarbon-degrading bacterium isolated from cattle pasture soil. Int J Syst Evol Microbiol 65:2919–2924. doi: 10.1099/ijs.0.000356. [DOI] [PubMed] [Google Scholar]
- 23.Sayavedra-Soto LA, Chang W-N, Lin T-K, Ho C-L, Liu H-S. 2006. Alkane utilization by Rhodococcus strain NTU-1 alone and in its natural association with Bacillus fusiformis L-1 and Ochrobactrum sp. Biotechnol Prog 22:1368–1373. doi: 10.1021/bp060100u. [DOI] [PubMed] [Google Scholar]
- 24.Stolz A. 2009. Molecular characteristics of xenobiotic-degrading sphingomonads. Appl Microbiol Biotechnol 81:793–811. doi: 10.1007/s00253-008-1752-3. [DOI] [PubMed] [Google Scholar]
- 25.Nie M, Yin X, Ren C, Wang Y, Xu F, Shen Q. 2010. Novel rhamnolipid biosurfactants produced by a polycyclic aromatic hydrocarbon-degrading bacterium Pseudomonas aeruginosa strain NY3. Biotechnol Adv 28:635–643. doi: 10.1016/j.biotechadv.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arulazhagan P, Vasudevan N. 2011. Biodegradation of polycyclic aromatic hydrocarbons by a halotolerant bacterial strain Ochrobactrum sp. VA1. Mar Pollut Bull 62:388–394. doi: 10.1016/j.marpolbul.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 27.Janbandhu A, Fulekar MH. 2011. Biodegradation of phenanthrene using adapted microbial consortium isolated from petrochemical contaminated environment. J Hazard Mater 187:333–340. doi: 10.1016/j.jhazmat.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 28.Kuppusamy S, Thavamani P, Megharaj M, Lee YB, Naidu R. 2016. Polyaromatic hydrocarbon (PAH) degradation potential of a new acid tolerant, diazotrophic P-solubilizing and heavy metal resistant bacterium Cupriavidus sp. MTS-7 isolated from long-term mixed contaminated soil. Chemosphere 162:31–39. doi: 10.1016/j.chemosphere.2016.07.052. [DOI] [PubMed] [Google Scholar]
- 29.Pérez-Pantoja D, De la Iglesia R, Pieper DH, González B. 2008. Metabolic reconstruction of aromatic compounds degradation from the genome of the amazing pollutant-degrading bacterium Cupriavidus necator JMP134. FEMS Microbiol Rev 32:736–794. doi: 10.1111/j.1574-6976.2008.00122.x. [DOI] [PubMed] [Google Scholar]
- 30.Fukuoka K, Tanaka K, Ozeki Y, Kanaly RA. 2015. Biotransformation of indole by Cupriavidus sp. strain KK10 proceeds through N-heterocyclic- and carbocyclic-aromatic ring cleavage and production of indigoids. Int Biodeterior Biodegradation 97:13–24. doi: 10.1016/j.ibiod.2014.11.007. [DOI] [Google Scholar]
- 31.Fukuoka K, Ozeki Y, Kanaly RA. 2015. Aerobic biotransformation of 3-methylindole to ring cleavage products by Cupriavidus sp. strain KK10. Biodegradation 26:359–373. doi: 10.1007/s10532-015-9739-0. [DOI] [PubMed] [Google Scholar]
- 32.Kanaly RA, Bartha R. 1999. Cometabolic mineralization of benzo[a]pyrene caused by hydrocarbon additions to soil. Environ Toxicol Chem 18:2186–2190. doi: 10.1002/etc.5620181010. [DOI] [PubMed] [Google Scholar]
- 33.Furukawa K, Fujihara H. 2008. Microbial degradation of polychlorinated biphenyls: biochemical and molecular features. J Biosci Bioeng 105:433–449. doi: 10.1263/jbb.105.433. [DOI] [PubMed] [Google Scholar]
- 34.Liang F, Lu M, Keener TC, Liu Z, Khang SJ. 2005. The organic composition of diesel particulate matter, diesel fuel and engine oil of a non-road diesel generator. J Environ Monit 7:983–988. doi: 10.1039/b504728e. [DOI] [PubMed] [Google Scholar]
- 35.Macedo AJ, Timmis KN, Abraham W-R. 2007. Widespread capacity to metabolize polychlorinated biphenyls by diverse microbial communities in soils with no significant exposure to PCB contamination. Environ Microbiol 9:1890–1897. doi: 10.1111/j.1462-2920.2007.01305.x. [DOI] [PubMed] [Google Scholar]
- 36.Luo W, D'Angelo EM, Coyne MS. 2008. Organic carbon effects on aerobic polychlorinated biphenyl removal and bacterial community composition in soils and sediments. Chemosphere 70:364–373. doi: 10.1016/j.chemosphere.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Z, Hou Z, Yang C, Ma C, Tao F, Xu P. 2011. Degradation of n-alkanes and polycyclic aromatic hydrocarbons in petroleum by a newly isolated Pseudomonas aeruginosa DQ8. Bioresour Technol 102:4111–4116. doi: 10.1016/j.biortech.2010.12.064. [DOI] [PubMed] [Google Scholar]
- 38.Rosario-Passapera R, Keddis R, Wong R, Lutz RA, Starovoytov V, Vetriani C. 2012. Parvibaculum hydrocarboniclasticum sp. nov., a mesophilic, alkane-oxidizing alphaproteobacterium isolated from a deep-sea hydrothermal vent on the East Pacific Rise. Int J Syst Evol Microbiol 62:2921–2926. doi: 10.1099/ijs.0.039594-0. [DOI] [PubMed] [Google Scholar]
- 39.Lai Q, Wang L, Liu Y, Yuan J, Sun F, Shao Z. 2011. Parvibaculum indicum sp. nov., isolated from deep-sea water. Int J Syst Evol Microbiol 61:271–274. doi: 10.1099/ijs.0.021899-0. [DOI] [PubMed] [Google Scholar]
- 40.Schleheck D, Cook AM. 2005. ω-Oxygenation of the alkyl sidechain of linear alkylbenzenesulfonate (LAS) surfactant in Parvibaculum lavamentivoransT. Arch Microbiol 183:369–377. doi: 10.1007/s00203-005-0002-7. [DOI] [PubMed] [Google Scholar]
- 41.Schleheck D, Tindall BJ, Rosselló-Mora R, Cook AM. 2004. Parvibaculum lavamentivorans gen. nov., sp. nov., a novel heterotroph that initiates catabolism of linear alkylbenzenesulfonate. Int J Syst Evol Microbiol 54:1489–1497. doi: 10.1099/ijs.0.03020-0. [DOI] [PubMed] [Google Scholar]
- 42.Nie Y, Chi C-Q, Fang H, Liang J-L, Lu S-L, Lai G-L, Tang Y-Q, Wu X-L. 2014. Diverse alkane hydroxylase genes in microorganisms and environments. Sci Rep 4:4968. doi: 10.1038/srep04968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnsen AR, Karlson U. 2004. Evaluation of bacterial strategies to promote the bioavailability of polycyclic aromatic hydrocarbons. Appl Microbiol Biotechnol 63:452–459. doi: 10.1007/s00253-003-1265-z. [DOI] [PubMed] [Google Scholar]
- 44.Dasgupta D, Ghosh R, Sengupta TK. 2013. Biofilm-mediated enhanced crude oil degradation by newly isolated Pseudomonas species. ISRN Biotechnol 2013:250749. doi: 10.5402/2013/250749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grimaud R. 2010. Biofilm development at interfaces between hydrophobic organic compounds and water, p 1491–1499. In Timmis KN. (ed), Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, Germany. [Google Scholar]
- 46.Perfumo A, Smyth TJP, Marchant R, Banat IM. 2010. Production and roles of biosurfactants and bioemulsifiers in accessing hydrophobic substrates, p 1501–1512. In Timmis KN. (ed), Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, Germany. [Google Scholar]
- 47.Isaac P, Alessandrello MJ, Macedo AJ, Estévez MC, Ferrero MA. 2017. Pre-exposition to polycyclic aromatic hydrocarbons (PAHs) enhance biofilm formation and hydrocarbon removal by native multi-species consortium. J Environ Chem Eng 5:1372–1378. doi: 10.1016/j.jece.2017.02.031. [DOI] [Google Scholar]
- 48.Moons P, Michiels CW, Aertsen A. 2009. Bacterial interactions in biofilms. Crit Rev Microbiol 35:157–168. doi: 10.1080/10408410902809431. [DOI] [PubMed] [Google Scholar]
- 49.Yakimov MM, Timmis KN, Golyshin PN. 2007. Obligate oil-degrading marine bacteria. Curr Opin Biotechnol 18:257–266. doi: 10.1016/j.copbio.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 50.Mori JF, Ueberschaar N, Lu S, Cooper RE, Pohnert G, Küsel K. 2017. Sticking together: inter-species aggregation of bacteria isolated from iron snow is controlled by chemical signaling. ISME J 11:1075–1086. doi: 10.1038/ismej.2016.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanaly RA, Bartha R, Watanabe K, Harayama S. 2000. Rapid mineralization of benzo[a]pyrene by a microbial consortium growing on diesel fuel. Appl Environ Microbiol 66:4205–4211. doi: 10.1128/aem.66.10.4205-4211.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanaly RA, Watanabe K. 2004. Multiple mechanisms contribute to the biodegradation of benzo[a]pyrene by petroleum-derived multicomponent nonaqueous-phase liquids. Environ Toxicol Chem 23:850–856. doi: 10.1897/03-191. [DOI] [PubMed] [Google Scholar]
- 53.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang K, Bin Keefe CR, Keim P, Kelley ST, Knights D, et al. . 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joshi NA, Fass JN. 2011. sickle: a sliding-window, adaptive, quality-based trimming tool for FASTQ files (version 1.33). https://github.com/najoshi/sickle.
- 56.Nurk S, Bankevich A, Antipov D, Gurevich A, Korobeynikov A, Lapidus A, Prjibelsky A, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, McLean J, Lasken R, Clingenpeel SR, Woyke T, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling genomes and mini-metagenomes from highly chimeric reads, p 158–170. In Deng M, Jiang R, Sun F, Zhang X (ed). Research in computational molecular biology. Springer, Berlin, Germany. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gruber-Vodicka HR, Seah BKB, Pruesse E. phyloFlash: rapid SSU rRNA profiling and targeted assembly from metagenomes. mSystems, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z. 2019. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 62.Kanaly RA, Harayama S, Watanabe K. 2002. Rhodanobacter sp. strain BPC1 in a benzo[a]pyrene-mineralizing bacterial consortium. Appl Environ Microbiol 68:5826–5833. doi: 10.1128/aem.68.12.5826-5833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mulligan CN, Cooper DG, Neufeld RJ. 1984. Selection of microbes producing biosurfactants in media without hydrocarbons. J Ferment Technol 62:311–314. [Google Scholar]
- 64.Maeda AH, Nishi S, Ozeki Y, Ohta Y, Hatada Y, Kanaly RA. 2013. Draft genome sequence of Sphingobium sp. strain KK22, a high-molecular-weight polycyclic aromatic hydrocarbon-degrading bacterium isolated from cattle pasture soil. Genome Announc 1:e00911-13. doi: 10.1128/genomeA.00911-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microbial 16S rRNA gene amplicon sequence data and metagenomics data obtained in this study were deposited at NCBI under BioProject PRJNA634975. Assembled metagenomic contigs were further uploaded to the Integrated Microbial Genome and Microbiomes (IMG/MER) database (Joint Genome Institute, IMG metagenome identifier [ID] 3300038749). The draft genome of S. barthaii KK22 was previously reported (64) (BioProject PRJDB1457 and IMG/MERGenome ID 2571042868).