

Abstract
Evolution of poorly differentiated chordoma from conventional chordoma has not been previously reported. We encountered a case of a poorly differentiated chordoma with evidence of whole-genome doubling arising from a SMARCB1-deficient conventional chordoma. The tumor presented as a destructive sacral mass in a 43-year-old man and was comprised of a highly cellular poorly differentiated chordoma with small, morphologically distinct nodules of conventional chordoma accounting for <5% of the total tumor volume. Immunohistochemistry revealed both components were strongly reactive for brachyury and lacked normal staining for INI1. Single nucleotide polymorphism (SNP) array analysis identified multiple genomic imbalances in the conventional component, including deletions of 1p, 3p, and 22q (involving SMARCB1) and loss of chromosomes 5 and 15, while the poorly differentiated component exhibited the same aberrations at a more profound level with additional loss of chromosome 4, low level focal deletion of 17p (involving TP53), and tetraploidy. Homozygous deletion of SMARCB1 was present in both components. Fluorescence in situ hybridization (FISH) analysis confirmed the relevant deletions in both components as well as genome doubling in the poorly differentiated tumor. This case suggests that SMARCB1 loss is an early event in rare conventional chordomas that could potentially evolve into poorly differentiated chordoma through additional genomic aberrations such as genome doubling. Further studies with additional patients will be needed to determine if genome doubling is a consistent pathway for evolution of poorly differentiated chordoma.
Keywords: poorly differentiated chordoma, conventional chordoma, SMARCB1, whole-genome doubling
1 |. INTRODUCTION
Chordomas are rare malignant neoplasms that exhibit notochordal differentiation. With an overall incidence of <1 per 100,000, chordomas occur over a broad age range with most arising after the fourth decade of life. Comprising 2–4% of all primary skeletal malignancies, they by and large develop in the midline axial skeleton with only very rare occurrences in the appendicular skeleton or extraosseous sites.1,2 Indicative of their notochordal differentiation, chordomas possess an immunophenotype that includes reactivity for the embryonic nuclear transcription factor T, also known as brachyury.1,3 Chordomas are classified into three histologic subtypes: conventional/chondroid, dedifferentiated, and poorly differentiated.1,4
The conventional subtype, accounting for approximately 75% of chordomas,4 is slow-growing and well-known for its distinctive morphology of notochord-like, monomorphic, epithelioid cells with conspicuous, clear vacuoles (conferring a bubble-like, “physaliferous” appearance) arranged in cords and clusters within an abundant myxoid extracellular matrix. This matrix, in some cases, can assume an appearance remarkably similar to that of hyaline cartilage, which defines the chondroid variant of conventional chordoma1 that most frequently arises in the clivus.4 Genomic studies have found that conventional chordomas exhibit recurrent, large, nonrandom copy number losses of 3, 9p, 1p, 14, 10, and 13.5–9 Aberrations of 3p include two minimum deleted regions affecting SETD2, BAP1, and PBRM1, while aberrations of 9p include a minimum deleted region affecting CDKN2A/CDKN2B.8 Copy number gains are less common but, when present, most frequently involve chromosome 7.5–9 Complex chromothripsis-like genomic rearrangements have also been observed.8 Conventional chordomas consistently exhibit low mutational burdens without apparent recurrent oncogene mutations.5,7,8
Dedifferentiated chordoma, the rarest and most rapidly lethal subtype, is defined by its biphasic morphology of conventional chordoma juxtaposed to a high-grade sarcomatous component. The sarcomatous components of these lesions have been shown to consistently lack brachyury expression and exhibit diminished reactivity for other immunophenotypic markers of conventional chordoma, such as cytokeratin.1 While the molecular basis for dedifferentiation in these tumors is uncertain, it has been speculated that inactivating mutations of tumor suppressor genes, particularly TP53, may contribute.4
A more recently described entity, the poorly differentiated subtype, which tends to occur in pediatric and young adult patients and carries a poor prognosis, morphologically appears as a highly cellular proliferation of haphazardly arranged, atypical, epithelioid to rhabdoid cells lacking the hallmark bubble-like vacuolization and myxohyaline matrix of the conventional subtype. While poorly differentiated chordoma expresses brachyury and may express other immunophenotypic markers expected of conventional chordoma,1,4,9–15 it is essentially defined by loss of hSNF5/INI1/BAF47 expression, which has been attributed to both inactivating mutations of SMARCB1 and recurrent, isolated copy number losses involving 22q11.23.1,4,9,11–15 Distinguishing the poorly differentiated from the dedifferentiated subtype is important given their differing genetic characteristics, biological behavior, age distribution, and the potential for targeted therapy options for poorly differentiated chordomas.1,4,11,12
Although very rare, instances of histologically typical conventional chordomas with loss of hSNF5/INI1/BAF47 expression by immunohistochemistry and/or deletions or mutations of SMARCB1 have been reported predominantly in the clival region and in pediatric age groups.7,8,12,14,15 A single report of a conventional chordoma of the sacrum with dedifferentiation seen in a lung metastasis demonstrated loss of hSNF5/INI1/BAF47 by immunohistochemistry in both its conventional and dedifferentiated sarcomatous components and also exhibited complete loss of brachyury expression in the dedifferentiated component distinguishing it from poorly differentiated chordoma.4 In a comparison of a single case of poorly differentiated chordoma with SMARCB1 deletion with an independent atypical conventional chordoma without SMARCB1 deletion by array comparative genomic hybridization (aCGH) analysis, it was found that poorly differentiated chordoma did not have the complex chromosomal pattern of conventional chordoma, and it was suggested that poorly differentiated chordoma is a molecularly distinct entity.11 Thus far, there have been no reports of conventional chordoma evolving into poorly differentiated chordoma. Herein, we report a definitive case of such, with molecular profiling, providing evidence of a poorly differentiated chordoma arising from a conventional chordoma.
2 |. MATERIALS AND METHODS
This study was conducted under Institutional Review Board approval (protocol 17–067).
2.1 |. Immunohistochemistry (IHC)
IHC studies were performed on 4 μm thick sections cut from non-decalcified, formalin-fixed, paraffin-embedded tissue blocks. Labeling for brachyury monoclonal rabbit antibody (1:500; clone EPR18113, Cambridge, MA) was performed on a BOND-III autostaining system following standard protocols with pretreatment using BOND Epitope Retrieval Solution 2 and detection performed with a BOND Polymer Refine detection kit (Leica Biosystems, Buffalo Grove, IL). Labeling for BAF47/INI1 monoclonal mouse antibody (1:200; clone 25, BD Biosciences, San Jose, CA) was performed on a Ventana BenchMark ULTRA autostaining system following standard protocols with pretreatment using ULTRA Cell Conditioning Solution 1 and detection performed with an OptiView detection kit (Roche Diagnostics, Indianapolis, IN).
2.2 |. Single Nucleotide Polymorphism (SNP) Array Analysis
Non-decalcified, formalin-fixed, paraffin-embedded tissue blocks were selected for analysis. Macrodissection was performed on two different blocks for selection of areas with conventional chordoma and poorly differentiated chordoma morphology.
Genomic DNA was extracted from formalin-fixed, paraffin-embedded tumor tissues using a magnetic bead-based chemagic FFPE DNA kit (PerkinElmer, Waltham, MA) on a Hamilton chemagic STAR liquid handling system (Hamilton Company, Reno, NV). Genome-wide DNA copy number alterations and allelic imbalances were analyzed by OncoScan CNV Assay (Thermo Fisher Scientific, Waltham, MA), which enables the detection of genome-wide copy number alterations such as gain and loss, allele specific changes including copy neutral loss of heterozygosity (cnLOH), ploidy, mosaicism, clonal heterogeneity, and chromothripsis. For each sample, 80 ng of genomic DNA were used. Processing of samples was performed according to manufacturer guidelines. OncoScan SNP array data were analyzed by the software couple of OncoScan Console ChAS 4.0 (Thermo Fisher Scientific, Waltham, MA) and Nexus Copy Number 10 (BioDiscovery, El Segundo, CA) using Affymetrix TuScan algorithm (Thermo Fisher Scientific, Waltham, MA). All array data were also manually reviewed for subtle alterations not automatically called by the software.16
2.2 |. Fluorescence in situ hybridization (FISH)
Non-decalcified, formalin-fixed, paraffin-embedded, 4 μm thick tissue sections with tumor areas marked were used for FISH analysis following standard protocols. To confirm homozygous deletions of SMARCB1 and genomic doubling revealed by SNP array tests, FISH probes for SMARCB1 (22q11.23) (Agilent, Santa Clara, CA) in combination with an internal control probe (22q11.12) (Empire Genomics, Williamsville, NY), 1p36 and 1q25, CDKN2A/CDKN2B and CEP9, and TP53 and CEP17 (Abbott Molecular, Des Plaines, IL) were used. Signal analysis was performed in combination with morphology correlation, and 100 interphase cells within the marked tumor area were evaluated and imaged using a Zeiss fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany) coupled with MetaSystems ISIS software (MetaSystems Hard & Software GmbH, Altlußheim, Germany).
3 |. RESULTS – CASE REPORT
3.1 |. Clinical History, Imaging, and Pathology
The patient, an otherwise healthy 43-year-old man, presented to our institution with a painful, enlarging mass of the lower back after having undergone a cryoablation procedure for a presumed pilonidal cyst. Imaging revealed a 9.8 cm multinodular destructive mass at the level of S5 with obliteration of the coccyx, invasion of the left S5 sacral foramen, and extension to the skin at the superior aspect of the intergluteal cleft. Several bilateral lung nodules measuring up to 0.6 cm were suspicious for a metastatic process. A biopsy performed by the referring institution revealed a highly cellular malignant neoplasm composed of vaguely nested, atypical, epithelioid to rhabdoid cells without extracellular matrix. Immunohistochemical studies revealed the tumor to be reactive for pan-cytokeratin, CAM5.2, vimentin, and brachyury, negative for CK7, CK20, PAX8, and S100 protein, and lack normal nuclear staining for INI1. A diagnosis of poorly differentiated chordoma was finalized, and the patient underwent sacrococcygectomy by en-bloc resection at the level of S2-S3, including the surrounding involved bilateral gluteal soft tissues, after anterior laparoscopic dissection. Anterior dissection was prompted by appreciable anterior bulging of the tumor toward the rectum and by the presence of multiple adenopathy, which were excised and found to be reactive. Other than the previously performed cryoablation, no additional preoperative therapy was performed.
Microscopically, the tumor predominately consisted of an infiltrative, multinodular, highly cellular proliferation of vaguely nested and corded, atypical, epithelioid to rhabdoid cells without extracellular matrix consistent with poorly differentiated chordoma as seen in the biopsy sample (Figure 1A). Additionally identified near the destroyed sacral elements and accounting for <5% of the tumor volume were small nodules of cords and clusters of monomorphic, vacuolated, “physaliferous” cells, without significant atypia, embedded within a myxoid matrix distinctive of conventional chordoma (Figure 1B). These nodules appeared discrete from the surrounding poorly differentiated component. Intermixing of the two components was minimal. The poorly differentiated component exhibited a mitotic rate of 18 per 10 high-power fields (hpf), approximately 50% necrosis, and lymphovascular invasion. The extent to which necrosis was intrinsic to the tumor or the effect of the prior cryoablation could not be determined. The conventional component exhibited a mitotic rate of <1 per 10 hpf and no apparent necrosis or lymphovascular invasion. Immunohistochemical studies revealed diffuse and strong reactivity for brachyury (Figure 1C–D) and uniform loss of INI1 expression (Figure 1E–F) in both the poorly differentiated and conventional components.
FIGURE 1.
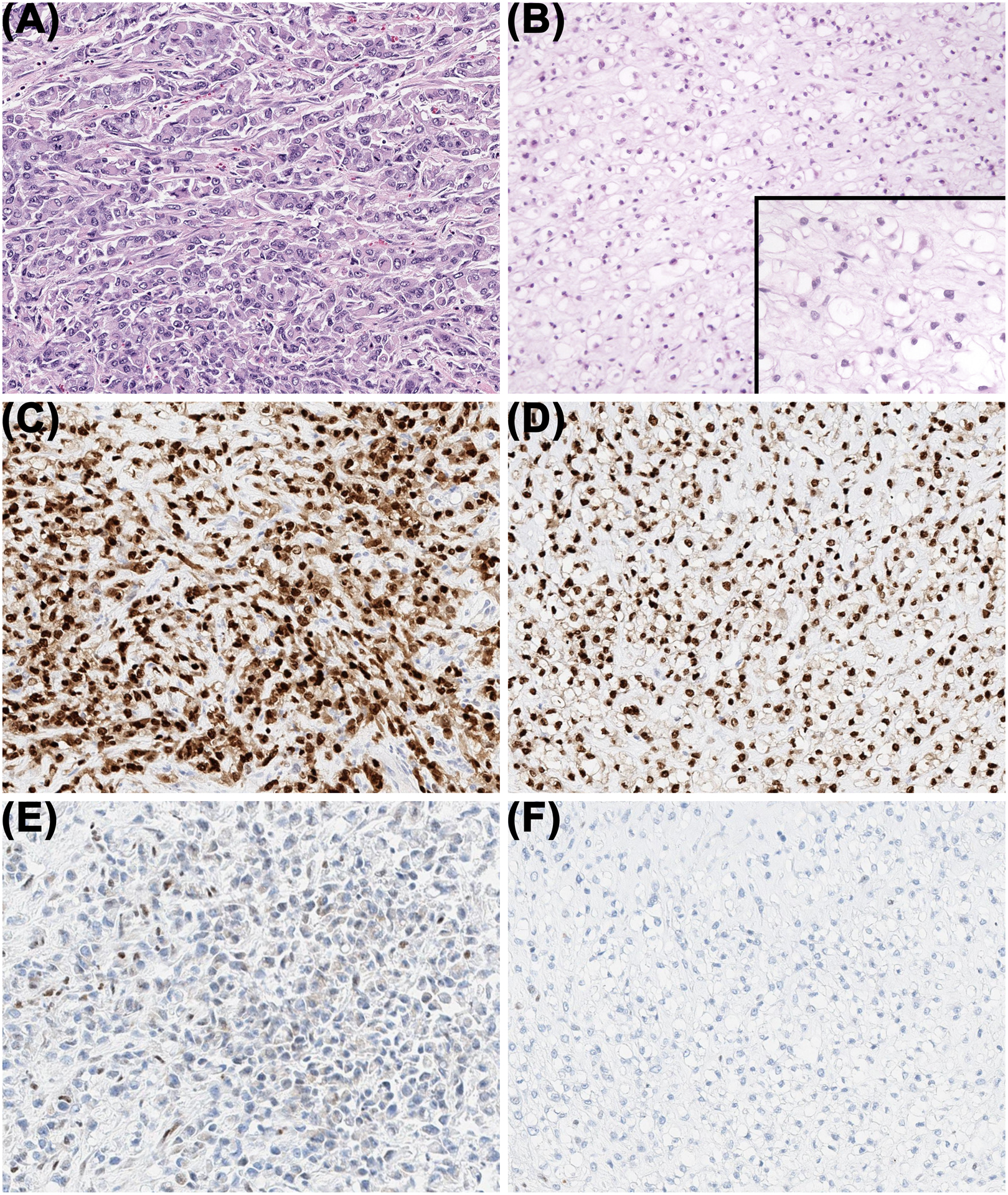
Two discrete chordoma morphologies were observed. A, The poorly differentiated component was composed of a high cellularity proliferation of atypical, mitotically-active epithelioid to rhabdoid cells tightly arranged in vague nests and cords without cytoplasmic vacuolization or an extracellular matrix. B, The conventional component was composed of a lower cellularity proliferation of monomorphic epithelioid cells with large, clear cytoplasmic vacuoles and small nuclei arranged in cords and clusters within a myxoid matrix. Diffuse immunoreactivity for brachyury was present in both the poorly differentiated (C) and conventional (D) components, and nuclear immunoreactivity for INI1 was absent in both the poorly differentiated (E) and conventional (F) components.
3.2 |. SNP Array Analysis and FISH Validation
OncoScan array analysis of the conventional component revealed an imbalanced genomic profile with low level deletion of 1p, 3p (focal, involving SETD2, BAP1, and PBRM1), 22q12 (involving SMARCB1), and loss of chromosomes 5 and 15. Array analysis of the poorly differentiated chordoma revealed a tetraploid genome with the same genomic alterations more pronounced than in the conventional chordoma, supported by allelic differences, with additional loss of chromosome 4 and low level focal deletion of 17p (involving TP53). In both components, a homozygous deletion of SMARCB1 was observed (Figure 2A).
FIGURE 2.
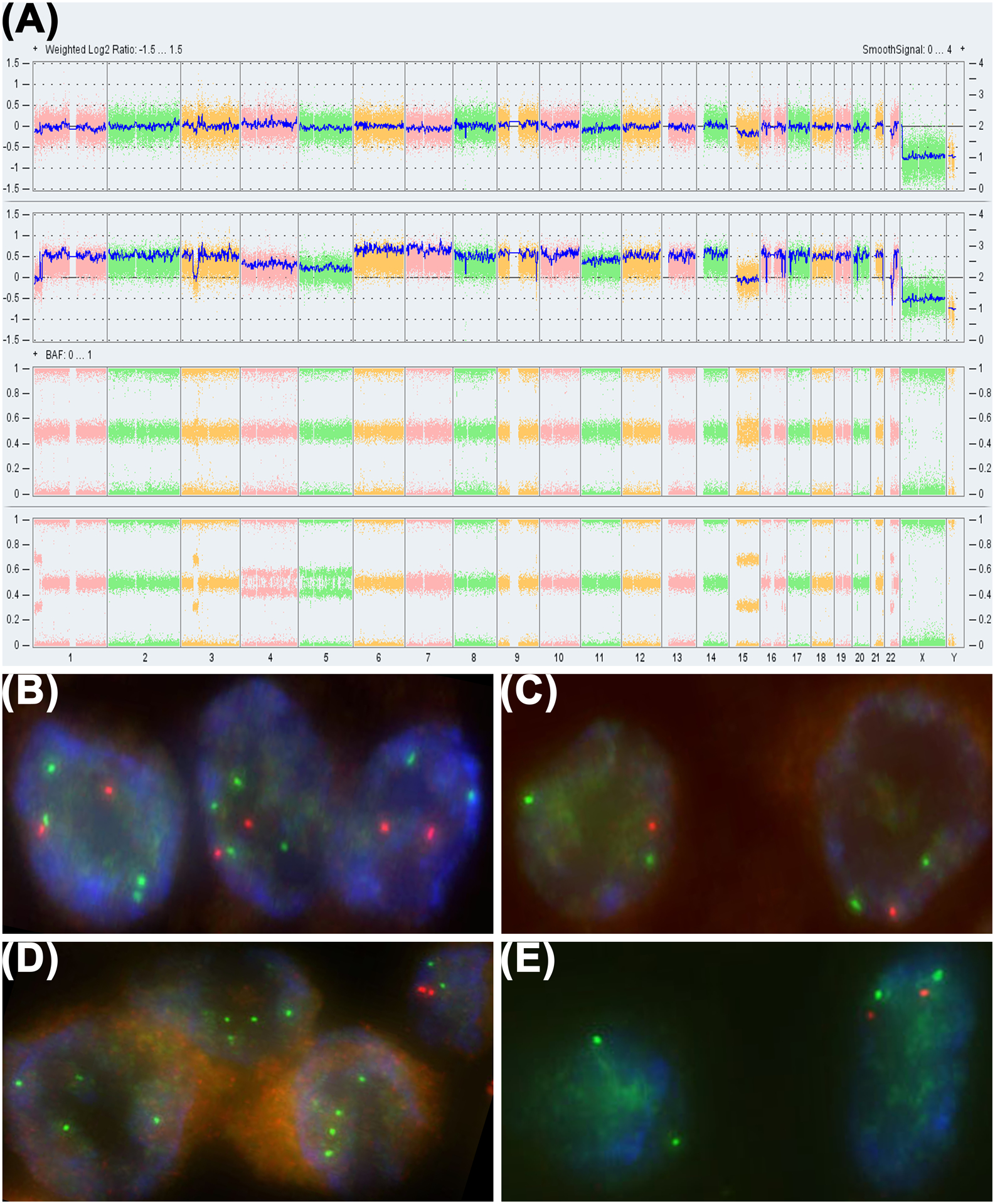
OncoScan SNP array and FISH. A, OncoScan SNP array revealed multiple genomic imbalances including deletions of 1p, 3p, 22q (involving SMARCB1) and loss of chromosomes 5 and 15 (arrows) in the conventional chordoma (top row). These aberrations were also observed in the poorly differentiated tumor (2nd row) at a more profound level with additional low level focal deletion of 17p (involving TP53), loss of chromosome 4, and a doubling of the genome as demonstrated by the disparate B allele frequencies between the two components (lower two rows). Homozygous deletion of 22q11.2 involving SMARCB1 was present in both components (arrows). FISH confirmed the relevant deletions and doubled genome. Two copies of 1p36 (orange) and four copies of 1q25 (green) were observed in 53% of cells in the poorly differentiated component (B), while one copy of 1p36 and two copies of 1q25 were observed in 62% of cells in the conventional component (C). Four signals for an internal control probe (green) and no signals for the SMARCB1 locus (orange) were observed in the poorly differentiated tumor (D), while two signals for the internal control probe and no signals for SMARCB1 were observed in the conventional component (E). Adjacent normal cells (shown in the top right corner of D) displayed two signals each for both the internal control probe and SMARCB1.
FISH analysis confirmed the array findings with evidence of genomic doubling in the poorly differentiated tumor. Two copies of 1p36 and four copies of 1q25 were detected in 53% of cells in the poorly differentiated tumor (Figure 2B) and one copy of 1p36 and two copies of 1q25 were detected in 62% of cells in the conventional component (Figure 2C). In addition, FISH studies for CDKN2A/CDKN2B (9p21) with CEP9 and TP53 (17p13.1) with CEP17 revealed three to four copies in over 40% of cells in the poorly differentiated tumor but not in the conventional component. These results were consistent with a tetraploid genome in the poorly differentiated tumor resulting from a whole-genomic doubling mechanism supported by allelic differences.
A homozygous deletion of SMARCB1 was confirmed in both the conventional and poorly differentiated components. FISH analysis of the poorly differentiated component revealed 31% and 67% of cells with two and four internal control probe signals and no signals for SMARCB1 (Figure 2D), while 90% of cells in the conventional component exhibited two signals for the internal control probe and no signals for SMARCB1 (Figure 2E).
4 |. DISCUSSION
Clinical management of chordomas has historically been difficult. Conventional chordomas have proven to be insensitive to standard cytotoxic chemotherapies,17,18 and while case reports have suggested some effectiveness in dedifferentiated chordomas,19 insufficient evidence exists to recommend traditional agents. Gross total surgical resection with possible adjuvant radiotherapy is the current mainstay of chordoma treatment.17,18 However, for conventional chordomas, local recurrence is frequent, particularly in cases where complete resection is impossible, and metastasis occurs in approximately 30–40% of cases.1,2,17,18 These events have negative implications for long-term survival with a median overall of 6 to 7 years.1,2 Poorly differentiated and dedifferentiated chordomas behave aggressively with bleak clinical outcomes. The median overall survival is approximately 46 months for poorly differentiated chordoma12 and only 20 months for dedifferentiated chordoma.4
Multiple studies have explored genetic aberrations in chordoma with aim towards development of targeted therapeutic agents.17,18 SMARCB1 inactivation is believed to contribute to oncogenesis through loss of inhibition of methyltransferase EZH2 leading to dysregulation of epigenetically-based gene silencing.20 EZH2-targeting agents have shown activity in preclinical trials against malignant rhabdoid tumors,21,22 and clinical trials utilizing EZH2-targeting agents in patients with SMARCB1-deficient neoplasms, including malignant rhabdoid tumor, epithelioid sarcoma, and poorly differentiated chordoma, are ongoing (Clinicaltrials.gov NCT02601937 and NCT02601950).23,24
Whole-genome doubling, observed in the poorly differentiated component of this case, is a common genomic event across all cancer types due to stochastic errors in cell division and is associated with poor prognosis. It is a large-scale evolutionary step in neoplastic progression that tends to arise early following an initial oncogenic driver mutation and is characterized by the formation of a tetraploid genome with subsequent heterozygous copy number losses. As this expansion of copy number alterations contributes to further genomic instability over the course of the disease, whole-genome doubling by itself is considered a portent of tumor aggression.25 Additionally, deletion of TP53, detected by array analysis in the poorly differentiated component in this case, may also contribute to progression in conjunction with whole-genome doubling.
This case suggests that SMARCB1 loss is an early event in rare conventional chordomas that could potentially evolve into a poorly differentiated chordoma through additional genomic aberrations such as doubling of the genome. Immunohistochemical screening of conventional chordomas for loss of hSNF5/INI1/BAF47 expression may be useful to identify cases with a potential for progression to poorly differentiated chordoma. Additional studies with a larger group of patients are needed to confirm if genome doubling is a consistent pathway for evolution of poorly differentiated chordoma.
ACKNOWLEDGMENT
This work was supported by the Department of Pathology at Memorial Sloan Kettering Cancer Center Internal Research Fund and in part by the National Institutes of Health (NIH)/ National Cancer Institute (NCI) Cancer Center Support Grant (CCSG) under award P30 CA008748.
Footnotes
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.WHO Classification of Tumours Editorial Board, ed. WHO Classification of Tumours 5th Edition: Soft Tissue and Bone Tumours. 5th ed Geneva: WHO Press; 2020. [Google Scholar]
- 2.McMaster ML, Goldstein AM, Bromley CM, Ishibe N, Parry DM. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12(1):1–11. [DOI] [PubMed] [Google Scholar]
- 3.Vujovic S, Henderson S, Presneau N, et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209(2):157–165. [DOI] [PubMed] [Google Scholar]
- 4.Hung YP, Diaz-Perez JA, Cote GM, et al. Dedifferentiated chordoma: clinicopathologic and molecular characteristics with integrative analysis [published online ahead of print, 2020 May 14]. Am J Surg Pathol. [DOI] [PubMed] [Google Scholar]
- 5.Le LP, Nielsen GP, Rosenberg AE, et al. Recurrent chromosomal copy number alterations in sporadic chordomas. PLoS One. 2011;6(5):e18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rinner B, Weinhaeusel A, Lohberger B, et al. Chordoma characterization of significant changes of the DNA methylation pattern. PLoS One. 2013;8(3):e56609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choy E, MacConaill LE, Cote GM, et al. Genotyping cancer-associated genes in chordoma identifies mutations in oncogenes and areas of chromosomal loss involving CDKN2A, PTEN, and SMARCB1. PLoS One. 2014;9(7):e101283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L, Zehir A, Nafa K, et al. Genomic aberrations frequently alter chromatin regulatory genes in chordoma. Genes Chromosomes Cancer. 2016;55(7):591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasselblatt M, Thomas C, Hovestadt V, et al. Poorly differentiated chordoma with SMARCB1/INI1 loss: a distinct molecular entity with dismal prognosis. Acta Neuropathol. 2016;132(1):149–151. [DOI] [PubMed] [Google Scholar]
- 10.Hoch BL, Nielsen GP, Liebsch NJ, Rosenberg AE. Base of skull chordomas in children and adolescents: a clinicopathologic study of 73 cases. Am J Surg Pathol. 2006;30(7):811–818. [DOI] [PubMed] [Google Scholar]
- 11.Shih AR, Chebib I, Deshpande V, Dickson BC, Iafrate AJ, Nielsen GP. Molecular characteristics of poorly differentiated chordoma. Genes Chromosomes Cancer. 2019;58(11):804–808. [DOI] [PubMed] [Google Scholar]
- 12.Yeter HG, Kosemehmetoglu K, Soylemezoglu F. Poorly differentiated chordoma: review of 53 cases. APMIS. 2019;127(9):607–615. [DOI] [PubMed] [Google Scholar]
- 13.Mobley BC, McKenney JK, Bangs CD, et al. Loss of SMARCB1/INI1 expression in poorly differentiated chordomas. Acta Neuropathol. 2010;120:745–753. [DOI] [PubMed] [Google Scholar]
- 14.Yadav R, Sharma MC, Malgulwar PB, et al. Prognostic value of MIB-1, p53, epidermal growth factor receptor, and INI1 in childhood chordomas. Neuro Oncol. 2014;16:372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antonelli M, Raso A, Mascelli S, et al. SMARCB1/INI1 involvement in pediatric chordoma: a mutational and immunohistochemical analysis. Am J Surg Pathol. 2017;41(1):56–61. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Zhou N, Patel T, et al. Evaluation of the Affymetrix OncoScan genome-wide SNP-array analysis platform for solid tumor molecular diagnosis (abstract #TT55). J Mol Diagn. 2014; 16:784. [Google Scholar]
- 17.Yakkioui Y, van Overbeeke JJ, Santegoeds R, van Engeland M, Temel Y. Chordoma: the entity. Biochim Biophys Acta. 2014;1846(2):655–669. [DOI] [PubMed] [Google Scholar]
- 18.Stacchiotti S, Sommer J; Chordoma Global Consensus Group. Building a global consensus approach to chordoma: a position paper from the medical and patient community. Lancet Oncol. 2015;16(2):e71–e83. [DOI] [PubMed] [Google Scholar]
- 19.Fleming GF, Heimann PS, Stephens JK, et al. Dedifferentiated chordoma. Response to aggressive chemotherapy in two cases. Cancer. 1993;72(3):714–718. [DOI] [PubMed] [Google Scholar]
- 20.Wilson BG, Wang X, Shen X, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation [published correction appears in Cancer Cell. 2011 Jan 18;19(1):153]. Cancer Cell. 2010;18(4):316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alimova I, Birks DK, Harris PS, et al. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol. 2013;15(2):149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110:7922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chi SN, McCowage GB, Hoffman L, et al. A phase I study of the EZH2 inhibitor tazemetostat in pediatric subjects with relapsed or refractory INI1-negative tumors or synovial sarcoma. J Clin Oncol. 2016;34:15_suppl, TPS10587–TPS10587. [Google Scholar]
- 24.Stacchiotti S, Schöffski P, Jones R, et al. Safety and efficacy of tazemetostat, a first-in-class EZH2 inhibitor, in patients (pts) with epithelioid sarcoma (ES) (NCT02601950). J Clin Oncol. 2019;37:15_suppl, 11003–11003. [Google Scholar]
- 25.Bielski CM, Zehir A, Penson AV, et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet. 2018;50(8):1189–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]