

Abstract
Conventional antibody–drug conjugates (ADCs) are heterogeneous mixtures of chemically distinct molecules that vary in both drugs/antibody (DAR) and conjugation sites. Suboptimal properties of heterogeneous ADCs have led to new site-specific conjugation methods for improving ADC homogeneity. Most site-specific methods require extensive antibody engineering to identify optimal conjugation sites and introduce unique functional groups for conjugation with appropriately modified linkers. Alternative nonrecombinant methods have emerged in which bifunctional linkers are utilized to cross-link antibody interchain cysteines and afford ADCs containing four drugs/antibody. Although these methods have been shown to improve ADC homogeneity and stability in vitro, their effect on the pharmacological properties of ADCs in vivo is unknown. In order to determine the relative impact of interchain cysteine cross-linking on the therapeutic window and other properties of ADCs in vivo, we synthesized a derivative of the known ADC payload, MC-MMAF, that contains a bifunctional dibromomaleimide (DBM) linker instead of a conventional maleimide (MC) linker. The DBM-MMAF derivative was conjugated to trastuzumab and a novel anti-CD98 antibody to afford ADCs containing predominantly four drugs/antibody. The pharmacological properties of the resulting cross-linked ADCs were compared with analogous heterogeneous ADCs derived from conventional linkers. The results demonstrate that DBM linkers can be applied directly to native antibodies, without antibody engineering, to yield highly homogeneous ADCs via cysteine cross-linking. The resulting ADCs demonstrate improved pharmacokinetics, superior efficacy, and reduced toxicity in vivo compared to analogous conventional heterogeneous ADCs.
Keywords: antibody–drug conjugate, ADC, homogeneous, bifunctional, linker, site-specific, conjugation, maleimide, dibromomaleimide, MMAF, auristatin, interchain, disulfide, DAR, hinge, cysteine, trastuzumab, Her2, CD98
Graphical Abstract
INTRODUCTION
Antibody–drug conjugates (ADCs) are a promising new class of targeted therapeutic agents for treatment of cancer.1,2 Most ADCs are synthesized by conjugating a cytotoxic compound or “payload” to a tumor specific monoclonal antibody. The payloads are conjugated using amino or sulfhydryl specific linkers that react selectively with lysines or cysteines on the antibody surface. A typical antibody contains over 50 lysines and eight interchain cysteines as potential conjugation sites. The optimal DAR (drugs/antibody ratio) for most ADCs, however, ranges from two to four drugs/antibody.3 ADCs with suboptimal DARs are prone to aggregation, poor solubility, and instability, which often lead to increased toxicity and/or inadequate efficacy in vivo.4 The discrepancy between the number of potential conjugation sites and the desired DAR, combined with the use of linkers that lack site-specificity, results in heterogeneous ADCs that vary in both DAR and the conjugation sites.5 Consequently, most of the ADCs in clinical development for cancer indications contain dozens or more of chemically distinct ADC molecules, each with unique pharmacological properties.6,7
Conjugation through antibody cysteines minimizes ADC heterogeneity relative to lysine conjugation because there are fewer potential conjugation sites.8 The process typically involves partial reduction of four antibody interchain disulfide bonds to generate up to eight reactive cysteine thiol groups, followed by conjugation of payloads containing thiol-specific maleimide linkers.9 The resulting ADCs are composed of dozens of chemically distinct molecules with DARs ranging from zero to eight payloads per antibody. The maleimide linkers typically used for cysteine conjugation result in thio-succinimide linkages between the payload and the antibody known to undergo side reactions such as elimination or thiol exchange, resulting in premature release of the payloads from the ADCs.10
New site-specific conjugation methods have emerged in order to reduce ADC heterogeneity and other undesirable properties associated with conventional methods. Most are recombinant methods focused on modification of the antibody with unique functional groups to enable site-specific conjugation with orthogonally modified linkers.11 For example, cysteine mutations have been introduced into different antibodies to provide free thiol groups for conjugation with payloads containing conventional thiol-specific maleimide linkers.12,13 The process affords homogeneous ADCs containing approximately two drugs/antibody, but additional antibody reduction/oxidation steps are required to obtain mutants suitable for conjugation. Later studies revealed that subtle differences in the ADC microenvironments significantly affect linker stability, which correlates with improved efficacy.14 The combined results indicate that ADC activity is highly dependent upon the conjugation sites and suggest that optimal conjugation sites are likely to be different for each antibody.
Recombinant methods using non-natural amino acids to enable site-specific conjugation have also been reported. For example, stop codon suppression methodology was used to produce antibodies containing phenyl ketone side chains for site-specific conjugation to hydroxylamine linkers.15 The approach was later combined with cell-free antibody expression technology to introduce unique functional groups into over one hundred different conjugation sites in trastuzumab, an anti-Her2 antibody approved for treatment of breast cancer.16 The results were consistent with previous findings in that ADC activity was highly dependent on the conjugation site. Antibody expression and conjugation efficiency were also site-dependent, suggesting that site optimization is required for each ADC.
Alternative semisynthetic methods for site-specific conjugation have been described in which recognition sequences were engineered into different locations on antibodies for subsequent enzymatic modification in order to create unique functional groups for site-specific conjugation. For example, a microbial transglutaminase recognition sequence was introduced into 90 different positions on an anti-EGFR antibody.17 Twelve different sites were found to be suitable for conjugation based on antibody expression and conjugation efficiencies. The mutated antibodies were then enzymatically conjugated to an appropriately modified payload to afford ADCs with approximately two drugs/antibody. Similar methods were used to construct anti-CD30 ADCs with four drugs/antibody which demonstrated an improved therapeutic index in rodent models.18 An alternative semisynthetic approach was used to introduce a formyl-glycine generating enzyme (FGE) recognition sequence at different sites on a tumor-specific antibody. Treatment of the mutant antibodies with FGE resulted in site-specific formation of aldehyde tags that were subsequently conjugated to appropriately modified linkers.19
Nonrecombinant methods for reducing ADC heterogeneity are being developed in order to enable existing antibodies to be used for construction of homogeneous ADCs. For example, Badescu and co-workers used a bis-sulfone linker designed to react with two antibody cysteines to conjugate an auristatin payload (MMAE) to trastuzumab and its Fab fragments.20 The linker contained a protease cleavable self-immolative dipeptide to facilitate payload release and a PEG spacer to improve water solubility and reduce potential aggregation. The conjugation process results in formation of a three-carbon bridge across interchain cysteines to afford an ADC enriched in the fraction containing four drugs/antibody (up to 78%). The ADCs inhibited BT474 tumor growth in vivo at relatively high doses (>10 mg/kg), but conventional heterogeneous ADCs were not included in the study and the contribution of the parent antibody (trastuzumab) to the observed efficacy was not reported. In a follow-up study by Godwin and co-workers, preparative hydrophobic interaction chromatography (HIC) was used to isolate cysteine bridged ADCs with defined DARs ranging from one to four drugs/antibody.21 ADC activity correlated with drug loading, but the potency relative to conventional heterogeneous ADCs was not reported. Nonetheless, the results suggested that appropriately designed bifunctional linkers could be used to improve ADC homogeneity and other pharmacological properties without antibody engineering.
Disubstituted maleimides were previously shown to be highly efficient thiol cross-linking reagents and have been used in a variety of protein conjugation applications including the synthesis of bispecific antibodies and homogeneous ADCs.22-25 For example, Schumacher and co-workers synthesized doxorubicin analogues containing dibromomaleimide linkers and conjugated them to trastuzumab, a commonly used benchmark for evaluating new ADC technology.22 The linkers were designed to cross-link two interchain cysteines on the antibody forming a two-carbon bridge to afford ADCs with approximately four drugs/antibody. The resulting ADCs demonstrated comparable Her2 binding affinity to trastuzumab, but their potency against antigen expressing cells and in vivo pharmacological properties were not reported. A similar approach was used by Maruani et al., who used dibromopyridazinedione linkers to conjugate doxorubicin to trastuzumab via a dual click strategy.23 The resulting ADCs demonstrated improved homogeneity and were rapidly internalized by antigen expressing cells in vitro, but their pharmacological properties in vivo were not reported.
In order to determine the relative effects of interchain cysteine cross-linking on the pharmacological properties of ADCs in vivo, we synthesized a novel derivative of monomethyl auristatin F (MMAF) that contains a bifunctional dibromomaleimide (DBM) linker designed to react with two adjacent cysteine thiol groups on the antibody. The DBM-MMAF derivative is analogous to a well-known auristatin linker-payload (MC-MMAF) except that the conventional maleimide is replaced with a bifunctional dibromomaleimide. ADCs were synthesized via conjugation of the DBM-MMAF derivative with two different antibodies, and their properties were compared with those of analogous ADCs synthesized using the conventional MC-MMAF payload. The results demonstrate that conjugation via interchain cysteine cross-linking with dibromomaleimide (DBM) linkers yields highly homogeneous ADCs containing four drugs per antibody. The ADCs demonstrate improved pharmacokinetic properties, superior efficacy, and reduced toxicity compared to conventional heterogeneous ADCs. Moreover, the methods described here are scalable and can be broadly applied to most antibodies without recombinant engineering.
EXPERIMENTAL METHODS
Materials.
MMAF and MC-MMAF were purchased from Concortis Biosystems (San Diego, CA). All other chemical reagents were purchased from VWR, AK Scientific, or Sigma-Aldrich and used as received. The anti-MMAF antibody was prepared at Genscript (NJ). All human cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA) or the Japanese Collection of Research Bioresources Cell Bank (JCRB; Osaka, Japan) and were maintained as recommended. Other materials and sources are described below individually for each section.
Synthesis of Dibromomaleimide (DBM) Linker.
6-Aminohexanoic acid (0.512 mg, 3.91 mmol) was added to a solution of 3,4-dibromofuran-2,5-dione (1 g, 3.91 mmol) in acetic acid (20 mL), and the solution was stirred at room temperature for 10 min until all the solids dissolved. The reaction mixture was heated to 100 °C for 18 h, after which LC/MS indicated the reaction to be complete. The solution was concentrated under vacuum and purified by silica gel chromatography (eluent DCM/EtOAc 0–40%). Concentration of pure fractions afforded 1.15 g (3.12 mmol, 80% yield) of dibromomaleimide derivative, 6-(3,4-dibromo-2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanoic acid. 1H NMR (400 MHz, CDCl3): δ 3.62 (t, J = 7.2 Hz, 2H), 2.36 (t, J = 7.6 Hz, 2H), 1.68–1.62 (m, 4H) 1.41–1.30 (m, 2H). LC/MS: retention time 3.172 min; acetonitrile:water gradient 5–95% acetonitrile over 5 min at 0.8 mL/min. m/z: 391.9, 389.9, 393.9 [M + Na]+.
Synthesis of DBM-MMAF Payload.
DIPC (34 mg, 0.271 mmol) and DIPEA (35 mg, 0.271 mmol) were added to a solution of the DBM linker from above (250 mg, 0.677 mmol) in DCM (5 mL), and the resulting solution was stirred for 1 h at room temperature. MMAF (208 mg, 0.271 mmol) was added in 50 mg portions over a 4 h period, and the resulting solution was stirred for a further 16 h. The DCM was removed under vacuum, and the residue was purified by preparative HPLC on an Agilent 250 mm × 30 mm C18 column. The product was eluted with a gradient of 50–90% CH3CN in water over 40 min at a flow rate of 40 mL/min. Lyophilization of the combined pure fractions afforded 170 mg of DBM-MMAF (2, Figure 1) in 58% yield. 1H NMR (500 MHz, CDCl3): δ 7.15–7.26 (m, 5H), 4.60–4.92 (m, 4H), 3.70–4.20 (m, 4H), 3.59–3.63 (m, 2H), 3.39–3.42 (m, 1H), 3.26–3.35 (m, 6H), 2.93–3.09 (m, 6H), 2.20–2.60 (m, 6H), 1.70–2.15 (m, 4H), 1.61–1.69 (m, 8H), 1.25–1.37 (m, 3H), 1.15 (dd, J = 18.5, 7.5 Hz, 2H), 0.81–1.05 (m, 20H). HPLC: 4.297 min (5–95% acetonitrile in water over 5 min). m/z: calcd 1082.38 (100.0%), 1083.38 (53.0%), 1080.38 (51.4%), 1084.37 (48.6%), 1085.38 (25.8%), 1081.38 (16.7%), 1084.38 (11.1%); obsd [M + H] 1082.1 (15%), 1083.1 (8%), 1084.1 (52%), 1085.0 (25%), 1086.0 (26%), 1087.0 (17%) [M + H].
Figure 1.
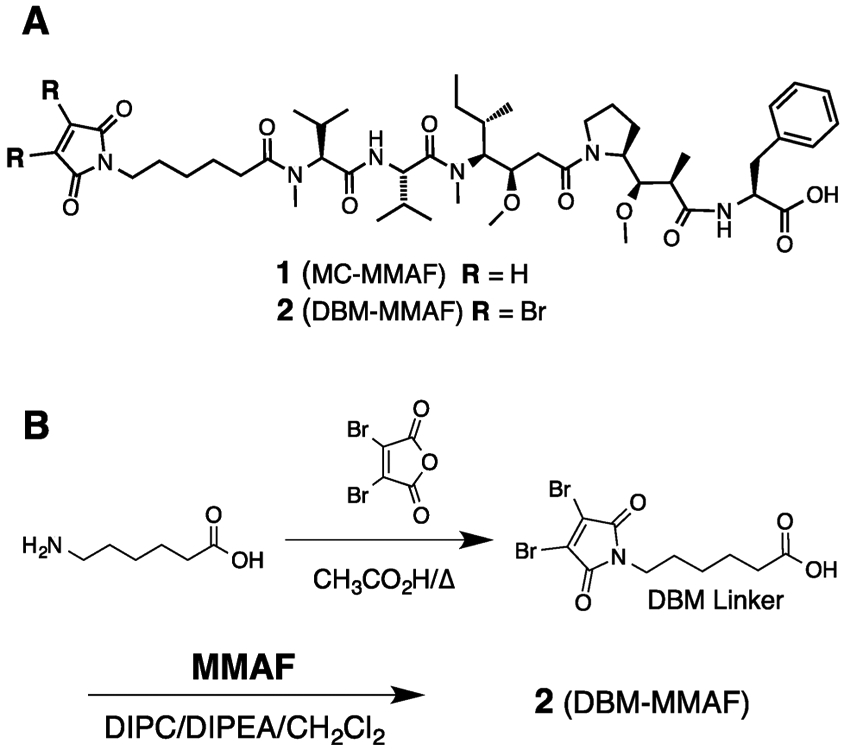
(A) Chemical structures of MMAF payloads containing conventional MC (1) or bifunctional DBM (2) linkers. (B) Synthesis of DBM-MMAF payload (2) used for synthesis of homogeneous ADCs.
Synthesis of ADCs.
The conventional maleimide ADCs 3 and 4 (Figure 2) were synthesized using protocols similar to methods described previously.24 Briefly, the purified antibody (trastuzumab or IGNX) was buffer exchanged into PBS, pH 7.4. The antibody was diluted to a final concentration of 5 mg/mL in PBS and warmed to 37 °C in a heat block. A stock solution of TCEP (50 mM in water) was freshly prepared in water, and 2.5 molar equiv (relative to the antibody concentration) was added. After 2 h the partially reduced antibody was removed from the heat block and cooled to room temperature. A stock solution of the MC-MMAF payload (2 mM in DMSO) was freshly prepared, and 5 molar equiv was added to the antibody. After 1 h the reaction mixture was buffer exchanged into PBS using PD10 spin columns to remove small MW reagents and stored at 4 °C until needed.
Figure 2.

Synthesis of ADCs via conjugation of trastuzumab or IGNX with payloads containing conventional MC (1) or bifunctional DBM (2) linkers. The conjugation protocols are identical except that excess TCEP is used to ensure full reduction of interchain disulfide bonds prior to conjugation with DBM-MMAF (2).
The DBM ADCs 5 and 6 were synthesized as follows. 60 mg of purified antibody (trastuzumab or IGNX) was buffer exchanged into PBS, pH 7.4. The antibody was diluted to a final concentration of 5 mg/mL in PBS and warmed to 37 °C in a heat block. A stock solution of TCEP (50 mM in water) was freshly prepared, and 8 molar equiv (relative to antibody concentration) was added to the antibody. The reaction mixture was incubated at 37 °C in a sealed 15 mL conical tube. After 2 h the reaction was removed from the heat block and allowed to cool to room temperature. Five molar equivalents of the DBM-MMAF derivative 2 (Figure 1) was added from a freshly prepared 2 mM stock solution in DMSO. The reaction was mixed gently at room temperature for 1 h. The crude ADC was buffer exchanged into PBS at pH 7.4 to remove excess small MW reagents and stored at 4 °C.
SEC Analysis of ADCs.
Size exclusion chromatography was performed on all ADCs to determine monomeric purity on an Acquity H class UPLC (Waters) with a BEH200 SEC 1.7 mM, 4.6 × 150 mm column with a BEH SEC Guard column 4.6 × 30 mM (Waters). The mobile phase consisted of 100 mM sodium phosphate, 150 mM sodium chloride, pH 6.8. 5 μg of sample was injected and run at a flow rate of 0.3 mL/min for 10 min at room temperature. Samples were analyzed by Empower software (Waters). The SEC results are available in the Supporting Information (see Table 1).
Table 1.
Affinity of ADCs 3–6 for Their Purified Antigens, ErbB2 and CD98, Determined via SPR
| antibody or ADC | antigen |
ka (M−1 s−1) |
kd (s−1) |
KD (nM) |
|---|---|---|---|---|
| trastuzumab | ErbB2 | 6.1 × 105 | 1.4 × 10−4 | 0.23 |
| 5 (TRA–DBM-MMAF) | ErbB2 | 5.9 × 105 | 1.4 × 10−4 | 0.24 |
| 3 (TRA–MC-MMAF) | ErbB2 | 5.7 × 105 | 1.4 × 10−4 | 0.25 |
| IGNX | CD98 | 1.6 × 105 | 2.1 × 10−4 | 0.14 |
| 6 (IGNX–DBM-MMAF) | CD98 | 1.4 × 105 | 2.6 × 10−4 | 0.18 |
| 4 (IGNX–MC-MMAF) | CD98 | 1.5 × 105 | 2.4 × 10−4 | 0.16 |
Hydrophobic Interaction Chromatography (HIC) Analysis of ADCs.
HIC analysis was performed on a Waters Acquity UPLC system on a Tosoh TSKgel Butyl-NPR, 4.6 mm × 10 cm column, 2.5 μm particle size; Tosoh (Cat. No. 42168). The mobile phase consisted of buffer A (1.5 M ammonium sulfate in 25 mM sodium phosphate, pH7) and buffer B (25 mM sodium phosphate pH 7, 25% isopropanol). A gradient of 25–100% buffer B was run over at 0.5 mL/min over 16 min.
Native LC/MS Analysis of ADCs.
Native LC/MS analysis was performed on a Waters Acquity UPLC with TUV detector (at 280 nM) and G2-S Q-ToF mass spectrometer (m/z range of 1000–8000). A 4.6 × 300 mm BEH 200 column (1.7 μm pore size) was used at a flow rate of 0.3 mL/min. 120 μg of sample was deglycosylated for 5 h at 37 °C with PNGase F prior to analysis.
Denaturing LC/MS of Analysis of ADCs.
Denaturing LC/MS analysis was performed on a Waters Acquity UPLC with TUV detector (at 280 nm) and G2-S Q-ToF mass spectrometer (scan m/z range of 1000–4000). The column used was a 2.1 × 100 mm BEH 130 (Waters, C18, 1.7 μM pore size) with a mobile phase gradient of water/acetonitrile + 0.5% formic acid.
Denaturing SEC Analysis of ADCs.
To resolve noncovalently bound antibody chains, a denaturing SEC method was developed. The method consisted of a Zenix-C SEC-300 column (4.6 × 300 mm, 3 μm particle size, 300 Å pore size) from Sepax Technologies with an isocratic mobile phase of 35% acetonitrile + 0.1% trifluoroacetic acid + 0.1% formic acid. The flow rate was 0.2 mL/min, and the run time was 25 min. 10 μg of antibody or ADC was injected as a 2 mg/mL aqueous solution. To confirm the identity of different isoforms present in trastuzumab–DBM-MMAF, the isoforms were separated using a semipreparative method similar to the analytical method described above except that the column size used was 10 × 300 mm, the flow rate was 0.87 mL/min, and 65 μL of 12 mg/mL ADC was injected. The relevant fractions were then frozen at −80 °C until denaturing LC/MS analysis was performed as described above.
Antigen Binding via Surface Plasmon Resonance (SPR).
ADC binding to purified antigens, Her2 and CD98, was performed via surface plasmon resonance analysis conducted on a Biacore 1000 instrument. Each ADC sample was diluted to a concentration of 100 nM and captured onto a goat anti-human Fc surface (Invitrogen). The running buffer included 10 mM HEPES pH 7.4, 150 mM NaCl, 0.005% Tween-20, and 0.1 mg/mL BSA. All data were collected at 25 °C. Each mAb was captured 5 times and placed within each of the 5 different flow cells. Capture levels were between 100 and 200 RU and averaged ~150 RU. Surfaces were regenerated with 2 × 18 s pulses of 1/50 dilution of phosphoric acid. Recombinant human ErbB2 (Sino Cat 10004-H08H, lot LC06AP0603) or CD98 prepared at 50, 16.6, 5.55, 1.85, 0.61 nM and a buffer blank were injected at 100 μL/min over the captured mAb surfaces. The dissociation phase was monitored for 1800 s. Data were processed and fit in Scrubber-Pro6 (Biological Software Pty Ltd.). Responses were referenced using the reference channel as well as the buffer blank injection. Data were fit to a 1:1 interaction model, and a summary of the binding constants is shown in Table 1.
Antigen Binding via Cell-Based ELISA.
ADC binding to antigen expressing cells was measured using a cell based ELISA. CD98 transfected F279 sarcoma cells or BT474 cells expressing Her2 were plated at 5000 cells per well in a 384-well plate. The following day, antibodies were serially diluted in a separate plate and transferred to the plate containing the cells. After a 2 h incubation at room temperature, the plate was washed with buffer (DPBS pH 7.4 with 0.1% BSA), and 25 μL of goat anti-mouse HRP-labeled secondary antibody (Pierce Cat. No. 37069) diluted in cell culture medium was added and incubated for 30 min at room temperature. The plate was washed, 15 μL of a goat anti-mouse chemiluminescent substrate (Pierce Cat. No. 37069) was added, and the plate was read in a plate based luminescence reader (Spectramax).
In Vitro Cytotoxicity Assays.
Tumor cell lines were routinely passaged in RPMI medium (LifeTech) supplemented with 10% fetal calf serum (LifeTech). To assay toxicity, cells were plated in 384-well plates, black well/clear bottom (Greiner), at 5000 cells per well in 40 μL of medium. ADCs or antibodies were serially diluted (5×) from 100 nM in RPMI, and 10 μL of 5× solution in complete medium was added to appropriate wells in duplicate using an iPipette liquid handler (Apricot Designs). Cell plates were then incubated for 3 days, followed by lysis in 30 μL of Cell-Titer Glo assay reagent (Promega). Luminescence was then quantified on a Synergy HT plate reader (BioTek) and graphed. IC50 values were calculated by fitting to a four-parameter sigmoidal fit using GraphPad Prism software.
Efficacy in Xenograft Tumor Models.
All procedures in animals described were in compliance with the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Office of Laboratory Animal Welfare. Protocols were reviewed by the Institutional Animal Care and Use Committees of either Igenica Biotherapeutics (Burlingame, CA) or WIL Research (Ashland, OH), respectively. Methods: Immunocompromised female NOD/SCID mice were used for the H446 tumor model (Charles River, Wilmington, MA) and female NOG mice for the SKOV3 tumor model (Taconic, Hudson, NY), respectively. H446 (HTB-171) and SKOV3 (HTB-77) were obtained from ATCC. Mice were subcutaneously injected on the right flank with at least 1 × 107 viable cells. When the tumor reached a size between 65 and 300 mm3, mice were randomized to treatment. Antibodies were administered weekly and tumors measured twice weekly. Tumor volume was calculated using the modified ellipsoid formula (π/6)(length × width2). All experiments were performed on groups of 8 animals per group. Animal experiments were performed in accordance with protocols approved by the Igenica Biotherapeutics Institutional Review Board—Animal Care and Use Committee. Data are expressed as the mean ± standard error of the mean (SEM). Group means were compared using Student’s 2-tailed, unpaired t test. Probability (p) values of <0.05 were interpreted as significantly different. All statistical analyses were performed using Microsoft EXCEL (Microsoft, Redmond, WA) and GraphPad Prism v.5.0f (GraphPad Software, Inc., La Jolla, CA).
Total Antibody and ADC ELISA.
The anti-MMAF (ADC) ELISA was designed to detect all ADC species with DARs > 0 via capture with an anti-MMAF monoclonal antibody prepared at GenScript (New Jersey) using a KLH–MMAF conjugate for immunization. The MMAF was conjugated to KLH through the N-terminus using a C6 linker identical to those used for ADC synthesis. Eight different hybridomas were screened for binding vs two heterogeneous MC-MMAF ADCs, one with low drug loading (DAR < 2) and one with high drug loading (DAR > 4). A single hybridoma with equal affinity for both ADCs was selected to use as a capture antibody for the ADC PK analysis. A mouse anti-human IgG (Fc) CH2 domain-peroxidase-conjugate was utilized for detection (Southern Biotech). The total antibody (total Ab) ELISA was designed to measure all antibody components including unconjugated and conjugated ADC in plasma. For trastuzumab and ADCs, an anti-human herceptin antibody was used as the capture reagent and a mouse anti-human IgG (Fc) CH2 domain-peroxidase-conjugate was utilized for detection (Southern Biotech). The assay range for each analyte was 39.1 to 2500 ng/mL in plasma. The lower limit of assay quantitation (LLOQ) was 39.1 ng/mL in rat plasma. Briefly, anti-MMAF or anti-herceptin antibody (Serotech) was diluted in coating buffer (PBS, pH 7.4) and immobilized onto a 96-well microtiter sample plate. The plate was washed, and all unabsorbed sites were blocked with the addition of Block Buffer (PBS with 0.05% Tween-20, pH 7.4; 1% BSA) for no less than 1 h and no more than 3 h. After washing the plate, analytes were diluted 1:1000 with Block Buffer, dispensed onto the sample plate, and then incubated for approximately 1 h. After the final wash step, a tetramethylbenzidine (TMB) peroxidase substrate solution was added and incubated for approximately 13–15 min with nonvigorous shaking. The reaction was stopped with a phosphoric acid stop solution (1 M). Color developed in proportion to the amount of herceptin and herceptin conjugate, respectively, present in the sample. Plates were read on a plate reader (Bio-Tek Power Wave HT) at two wavelengths (450 nm for detection and 620 nm for background). Analyte concentrations were determined on a standard curve obtained by plotting optical density (OD) versus concentration using a four parameter logistic curve-fitting program (Gen5 Secure Software ver. 1.08 or higher).
Pharmacokinetic Analysis of ADCs.
This was a non-clinical laboratory, open-label, randomized, single-dose study of trastuzumab antibody–drug conjugates in adult Sprague–Dawley (SD) rats (ICON Laboratory Services, LLC, Whitesboro, NY). Briefly, after a seven-day acclimation period, 12 male rats were assigned to one of three treatment groups (4 per group). Animals in each group received a single intravenous bolus administration of the test article (trastuzumab, conventional ADC 3, or DBM ADC 5) on day 0. Blood samples were collected 0.083 h, 1 h, 4 h, 8 h, 24 h, 48 h, 72 h, 120 h, 168 h, 240 h, 336 h, 408 h, 504 h, 576 h, and 672 h post administration of the test articles. The PK parameters were calculated in Phoenix WinNonlin (version 6.3, Pharsight Corp., St. Louis, MO) from the concentration time data obtained from the ELISA protocols described above using a noncompartmental analysis (NCA) method with intravenous bolus input. Half-life (t1/2) was estimated by log–linear regression of the terminal phase of the concentration versus time profiles. At least 3 points clearly visible in the terminal phase, an r2 value of at least 0.8, and time interval of at least 2 half-lives were required to characterize half-life. Detailed protocols and results for PK analysis are available in the Supporting Information.
Toxicology Study of ADCs in rats.
The relative toxicity and TK profiles of trastuzumab ADCs 3 and 5 were evaluated in a repeat-dose study in male Sprague–Dawley (SD) rats (ICON Laboratory Services, LLC, Whitesboro, NY). Four rats were assigned per dose group for PK analysis. Briefly, rats were dosed intravenously (iv) weekly for a total of three doses on days 0, 7, and 14. Doses ranged from 14.4 to 14.9, 21.5 to 22.3, and 28.7 to 29.8 mg/kg of ADCs 5 and 3 respectively in order to compensate for slight variations in ADC drug loading. Body weights were recorded twice weekly, and food consumption was recorded weekly. Blood samples were taken on day 3, and a necropsy was conducted on day 17 (3 days after the third dose). Dose-proportionality of the test articles was assessed, and time to reach steady state and effective half-life were estimated. The plasma accumulation of total antibody and ADCs 3 and 5 was evaluated following repeat iv injections. The parameters with notable differences measured between the ADCs are discussed in Results, and complete results with all measured parameters for the toxicology study are available online as Supporting Information.
RESULTS
Synthesis of DBM-MMAF Payload 2.
In order to evaluate the pharmacological properties of ADCs synthesized via interchain cysteine cross-linking, we selected a clinically validated auristatin payload (MMAF) containing a conventional maleimide (MC) linker as a benchmark (1, Figure 1A).25 An analogous auristatin derivative containing a bifunctional DBM linker, 2 (Figure 1A), was synthesized in two steps from commercially available reagents as shown in Figure 1B. The DBM-MMAF derivative 2 is designed to react with two adjacent interchain cysteines to form a dithiomaleimide linkage with the antibodies. Derivatives 1 and 2 both contain stable six carbon spacers to minimize potential bystander effects sometimes observed with protease cleavable linkers and differ only in their cross-linking capabilities.26 As a result, subtle differences in activity between homogeneous and heterogeneous ADCs can be accurately measured.
Synthesis of ADCs.
Two different antibodies were selected to serve as models for ADC synthesis via conjugation with MMAF payloads 1 and 2. Trastuzumab, an anti-Her2 antibody approved for treatment of breast cancer in 2011, was selected based on its known pharmacological properties and its frequent use as a benchmark antibody for evaluating new ADC linker/payload technologies.27 A second antibody, IGNX, was selected from our in-house library of tumor specific antibodies in order to demonstrate the versatility of DBM linkers for constructing homogeneous ADCs from native antibodies. IGNX binds specifically to CD98, a proprietary tumor associated antigen expressed on a number of hematological tumor types.28 The conjugation protocols used for synthesis of ADCs with DBM derivative 2 are comparable to previously reported methods for making conventional ADCs, except that excess reducing agent (TCEP) is used to ensure complete reduction of antibody interchain disulfide bonds prior to conjugation (Figure 2).29 The reduction/conjugation process is carried out in PBS at room temperature, and purification of the reduced antibody is not required prior to conjugation. A slight excess (5 equiv) of DBM-MMAF derivative 2 was required to obtain DARs of four drugs/antibody. The resulting ADCs, 3–6 (Figure 2), were purified via buffer exchange to remove any remaining low MW reagents. The overall yield for the two-step process typically ranges from 70 to 90% based on recovered protein. Size exclusion chromatography (SEC) analysis indicated that ADCs 3–6 were >98% monomer, suggesting that minimal inter-molecular cross-linking of different antibody molecules occurred during the conjugation process (see Supporting Information Table 1).
Homogeneity and DAR Analysis of ADCs.
The relative homogeneity of ADCs 3–6 were determined by hydrophobic interaction chromatography (HIC) and native LC/MS analysis.30 ADC fractions containing different DARs are resolved by HIC based on differences in hydrophobicity, with higher DAR species eluting progressively later than the unconjugated parent antibodies. The conventional methods for conjugation with maleimide linkers typically involve partial reduction of antibody interchain disulfides and yield ADCs containing predominantly even numbered DARs. ADCs 3 and 4 were synthesized using conventional conjugation protocols optimized to obtain DARs of exactly four drugs/antibody in order to minimize variability and maintain consistency with previously reported results.19 HIC analysis of the conventional ADCs afforded typical heterogeneous profiles with even numbered DARs ranging from 0 to 8 drugs/antibody. Average DARs of 3.9 and 4.0 were calculated for ADCs 3 and 4 respectively based on peak areas shown in Figure 3. In contrast, the DBM cross-linked ADCs 5 and 6 eluted as single peaks that had retention times consistent with DARs equal to 4 drugs/antibody. The results demonstrate that conjugation via cysteine cross-linking with DBM linkers yields ADCs with significantly improved homogeneity compared to analogous ADCs synthesized from conventional maleimide linkers.
Figure 3.

Hydrophobic interaction chromatography (HIC) analysis of ADCs 3–6. Cross-linked (DBM) ADCs are shown in blue, and conventional (MC) ADCs are shown in red. Average DAR values were calculated based on peak areas. (A) HIC overlays comparing trastuzumab ADCs 3 and 5. (B) HIC overlays comparing IGNX ADCs 4 and 6.
LC/MS Analysis of ADCs.
Native LC/MS analysis of ADCs 3–6 confirmed the HIC results and provided accurate MWs for the DAR components present in the ADCs (Figure 4).31 The DAR compositions determined by native LC/MS analysis are comparable to the DARs estimated by HIC, and the observed MWs are consistent with calculated MWs (±5 AMUs) for the cross-linked ADCs. Minor quantities (<5% each) of DAR3 and/or DAR5 fractions were observed by LC/MS that were not resolved via HIC analysis due to the superior sensitivity and accuracy of native LC/MS analysis. Nonetheless, the results indicate that the cross-linked DBM ADCs 5 and 6 have average DARs of 4.0 and 4.1 drugs/antibody and have significantly improved homogeneity (>85% DAR4) over the analogous conventional ADCs. No other higher MW adducts were observed in ADCs 5 and 6. However, fragments with MWs equal to one-half the calculated MW of the cross-linked ADCs were detected when denaturing LC/MS conditions were employed. Similar fragments were observed in the conventional ADCs 3 and 4, in addition to other heavy and light chain fragments commonly observed in conventional ADCs.30
Figure 4.
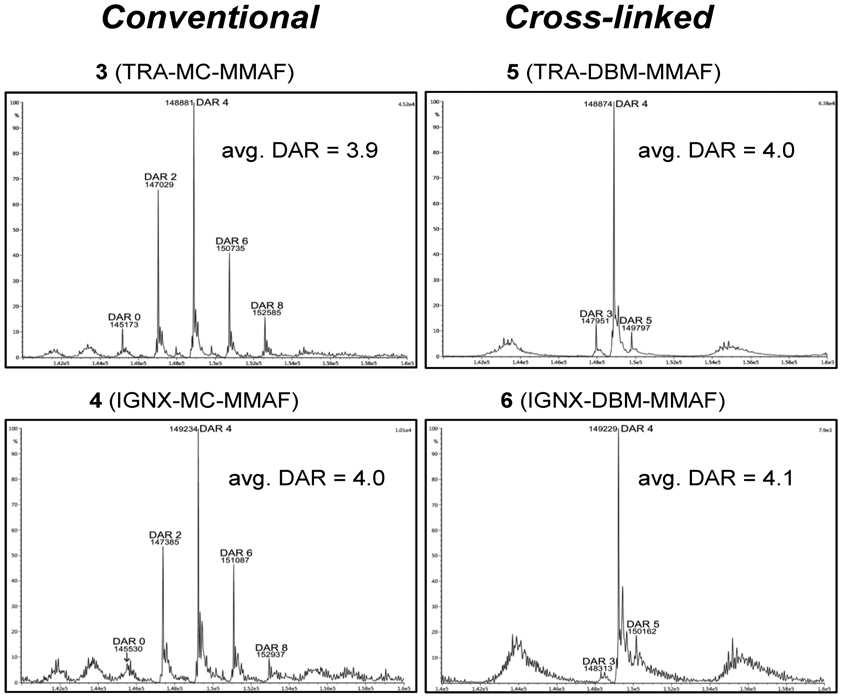
Native LC/MS analysis comparing DAR compositions of conventional ADCs, 3 and 4, with cross-linked DBM ADCs, 5 and 6. The data shown are the deconvoluted MS profiles, and observed MWs are consistent with calculated MWs within ±2 AMUs. Average DAR values shown for each ADC were calculated based on peak intensities.
Denaturing SEC Analysis of ADCs.
Denaturing LC/MS analysis of the DBM-MMAF ADCs 5 and 6 indicated the presence of an ADC fragment with half the expected mass of the fully cross-linked ADCs (data not shown). We hypothesized that this half ADC fragment potentially results from intrachain cross-linking of two hinge cysteines on the same heavy chain rather than interchain cross-linking of cysteines on two different heavy chains (see Figure 5E). Since antibody hinge region cysteines are in close proximity to each other (separated by two prolines in IgG1), the possibility of intrachain cross-linking could not be ruled out. In fact, Schumacher et al. recently reported formation of similar fragments based on SDS–PAGE analysis of ADCs synthesized using substituted maleimide linkers.26 Both inter- and intrachain cross-linking afford in ADCs with DARs of 4 drugs/antibody, but only the interchain cross-linked isoform contains covalent bonds between the two heavy chains. Intrachain cross-linking of two hinge cysteines on the same heavy chain would result in an ADC fragment that lacks covalent bonds between heavy chains with a MW equal to one-half of an ADC with DAR = 4 drugs/antibody. To further characterize and quantify the relative amounts of inter- vs intrachain cross-linking in ADCs 5 and 6, we developed a denaturing size exclusion chromatography (dSEC) method that enables separation of noncovalently linked antibody chains or fragments. In contrast to the previously reported SDS–PAGE methods for analyzing ADC fragments, the dSEC method described here does not require the use of reducing agents or high temperatures known to cause degradation of maleimide linkers.20,22
Figure 5.

Denaturing SEC analysis of ADC 5 (TRA–DBM-MMAF) showing the presence of two ADC isoforms resulting from inter- or intrachain cross-linking of hinge cysteines on the heavy chains: (A) native trastuzumab with intact interchain disulfide bonds; (B) ADC 5 (TRA–DBM-MMAF); (C) trastuzumab C225A/C228A double mutant which lacks hinge disulfide bonds; (D) fully reduced trastuzumab lacking interchain disulfides; (E) proposed structures of interand intrachain ADC isoforms. LC/MS analysis of the separated isoforms is consistent with the proposed structures (see Supporting Information).
The results shown in Figure 5 demonstrate that ADC 5 (TRA–DBM-MMAF) is composed of two different isoforms in a 7:3 ratio (Figure 5B). The major isoform elutes earlier and accounts for ~70% of the total ADC based on peak areas. Moreover, the retention time is similar to that of intact trastuzumab containing 2 heavy and 2 light chains (Figure 5A) which is consistent with interchain cysteine cross-linking (Figure 5E). The smaller, later eluting peak represents ~30% of the total ADC and has a retention time consistent with a fragment containing single heavy and light chains.
To support this observation, a trastuzumab double mutant (C225A/C228A) was prepared as a standard for comparison. The double mutant lacks both hinge region disulfide bonds between the two heavy chains and therefore elutes as a half antibody (HL) fragment under denaturing conditions (Figure 5C). To provide further confirmation for the proposed structural isoforms (Figure 5E), ADC 5 was purified via preparative dSEC and the individual isoforms were isolated and analyzed by LC/MS (see Supporting Information Figures 2 and 3). The observed MWs for the isolated isoforms are consistent with calculated MWs for the structures depicted in Figure 5E. Similar results were obtained with ADC 6 (see Supporting Information Figure 1), which indicates that isoform formation is not antibody dependent. The relative contributions of the two isoforms to ADC activity remain uncertain, but recent studies by Lyon et al. suggest that the absence of covalent bonds between ADC heavy chains has little or no impact on the pharmacological properties ADCs in vivo.32
Antigen Binding Affinity of ADCs.
The affinity and specificity of ADCs 3–6 for their respective antigens were determined using surface plasmon resonance (SPR) and cell based ELISA methods. The parent antibodies were used as benchmarks for comparison with the ADCs. The binding kinetics of the ADCs to immobilized purified antigens, Her2 or CD98, was determined on a Biacore 1000 SPR instrument. The results shown in Table 1 indicate that ADCs 3–6 have comparable affinities to their respective parent antibodies; and the observed KDs for trastuzumab ADCs 3 and 5 are consistent with previously reported values for trastuzumab.
Binding of ADCs 3–6 to antigen expressing cell lines was determined using a cell based ELISA. A murine sarcoma cell line, F279, which lacks expression of both human target antigens, was transfected with human CD98 and used for testing the IGNX ADCs, 4 and 6. Human breast carcinoma (BT474) cells that express high levels of Her2 were used for testing the trastuzumab ADCs 3 and 5. Cells were incubated with serial dilutions of the ADCs, and the relative fraction of bound antigen was measured via luminescence after addition of an HRP labeled secondary antibody. The data shown (Figure 6) indicates that the DBM ADCs 5 and 6 bind antigen-expressing cells with low nM affinities comparable to those of the parent antibodies, but they do not bind to cells lacking expression of their relevant target antigens. The results indicate that interchain cross-linking with bifunctional DBM linkers does not compromise antigen binding affinity or specificity of the ADCs, and suggests that ADCs containing bifunctional DBM linkers maintain the structural integrity and functionality of their parent antibodies.
Figure 6.

Binding affinity for antigen expressing cells: (A) binding of trastuzumab ADCs to BT474 (human breast carcinoma) cells that express Her2 but not CD98; (B) binding of IGNX ADCs to murine sarcoma cells transfected with human CD98, but lacking Her2 expression.
Potency of ADCs in Vitro.
Although the antigen binding properties of ADCs 3–6 remained comparable to those of the parent antibodies, changes in ADC stability resulting from conjugation with DBM linkers could potentially impact their potency against antigen expressing cells in vitro. To address this possibility, the relative potencies of ADCs 3–6 against tumor cell lines with and without antigen expression were determined. Three cell lines were selected based on their antigen expression profiles. The human breast carcinoma cell line, BT474, expresses high levels of Her2 but does not express CD98. The human lung carcinoma cell line, H446, expresses CD98, but not Her2. A third cell line, SKOV3 (human ovarian tumor) was selected because it expresses both antigens. Two control ADCs (7 and 8) were synthesized via conjugation of MMAF derivatives 1 and 2 (see Figure 1) using methods previously described for ADCs 3–6 with an anti-IgE isotype control antibody (C1.18) that does not bind either of the antigen targets. Cells were incubated with serial dilutions of the ADCs for 3 days, and cell growth was determined using a standard luminescence assay for measuring ATP concentrations. The results shown (Figure 7) indicate that DBM ADCs 5 and 6 inhibit growth of antigen expressing cells with comparable potency to the conventional ADCs 3 and 4. Moreover, ADCs 5 and 6 do not inhibit growth of cells lacking antigen expression and no tumor growth inhibition was observed for the control ADCs 7 and 8 against any of the cell lines, indicating that cytotoxicity is antigen dependent. Overall, the results demonstrate that DBM ADCs 5 and 6 are highly potent and selective against antigen expressing cells in vitro (Figure 7).
Figure 7.

Potency and selectivity of ADCs 3–6 in vitro: (A) growth inhibition of BT474 (human breast carcinoma) cells that express Her2, but not CD98; (B) growth inhibition of H446 (human lung carcinoma) cells that express CD98, but not Her2; (C) growth inhibition of SKOV3 (ovarian tumor cells that express both Her2 and CD98; (D) IC50s of ADCs 3–6 against three different cell lines.
Efficacy of ADCs in Xenograft Tumor Models.
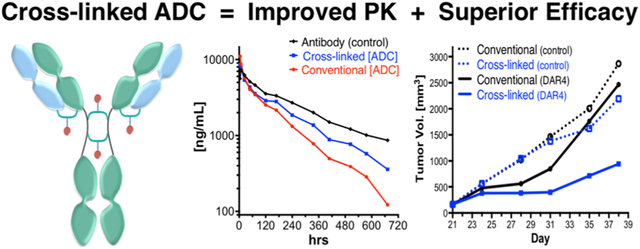
The efficacy of the ADCs was evaluated in two different xenograft tumor models and compared with that of the unconjugated parent antibodies. SKOV3 ovarian tumors express high levels of Her2 and are sensitive to treatment with trastuzumab (Figure 8A), while H446 lung carcinoma tumors express the CD98 antigen at high levels (Figure 8B). The control ADCs, 7 and 8, were included in the study as negative isotype controls to evaluate potential target independent activity. Tumor cells were implanted in mice, and when the tumors reached an appropriate size (200–300 mm3), mice were randomized into different treatment groups (10 per group) and treated twice intravenously with the test ADCs (dosed at 3 mg/kg) on days 1 and 8. Tumor growth and weight gain for each mouse were monitored at 3-day intervals for the duration of the study. Both DBM ADCs, 5 and 6, demonstrated tumor growth inhibition superior to that of the parent antibodies and resulted in complete tumor remissions in both tumor models (Figures 8A and 8B) demonstrating that cross-linked DBM ADCs are highly efficacious in vivo. A third xenograft study was performed in order to compare the relative activities of the cross-linked trastuzumab ADC 5 with the conventional ADC 3. NOG mice (which are insensitive to trastuzumab in this model) were selected for this study to remove contributions of the parent antibody to the observed efficacy. Any observed differences in tumor growth inhibition could be attributed to the ADC linkers with greater confidence by removing all other potential variables. NOG mice (10 per group) were implanted with SKOV3 tumors and treated twice with conventional ADC 3 or DBM ADC 5 at 3 mg/kg. SKOV3 tumor growth was significantly delayed in mice treated with ADC 5, while treatment with the conventional ADC 3 did not significantly inhibit tumor growth relative to the control ADCs (Figure 8C). The results demonstrate that DBM ADC 5 is more efficacious than the conventional ADC 3 at equivalent doses and suggests that improved ADC homogeneity results in superior tumor growth inhibition in vivo.
Figure 8.

(A) Tumor growth inhibition in Her2 positive SKOV3 xenograft mice treated twice (days 62 and 69) at 3 mg/kg. (B) Tumor growth inhibition in CD98 positive H446 (lung carcinoma) xenograft mice treated twice (days 41 and 48) at 3 mg/kg. (C) SKOV3 xenograft tumor model in NOG mice (insensitive to treatment with trastuzumab) treated once at 3 mg/kg with homogeneous (DBM) ADC 5 or with conventional (MC) ADC 3.
Pharmacokinetics of ADCs in Rats.
A rat pharmacokinetics study was conducted to compare the relative clearance rates of ADCs 3 and 5 with unconjugated trastuzumab. Rats were selected as the species for PK evaluation so multiple samples could be taken from each animal, enabling more accurate measurement of the relative amounts of total antibody (total mAb) and conjugated antibody (ADC) throughout the duration of the study. ADCs 3 and 5 were administered intravenously (4 rats/group), and blood samples were taken at increasing time points. The concentrations of total antibody and conjugated antibody were determined for each time point using previously described sandwich ELISA assay protocols. Total antibody concentrations [total mAb] were measured using a mouse anti-human IgG1 capture antibody, while the concentration of conjugated antibody [ADC with DAR ≥ 1] was determined using a murine anti-MMAF capture antibody.
The results shown (Figure 9) represent the average of quadruple measurements; unconjugated trastuzumab was included as a control. No significant differences in total antibody half-life were observed for ADCs 3 and 5, suggesting that the linker type has minimal impact on antibody stability in vivo. The half-lives of the conjugated antibodies (ADCs with DAR ≥ 1) however, were significantly different for the conventional heterogeneous ADC 3 (t1/2 = 130 h) compared to the DBM cross-linked ADC 5 (t1/2 = 184 h), suggesting that the MMAF payload is released more rapidly from the ADC containing a conventional maleimide linker. The increased half-life and exposure observed for DBM ADC 5 over the conventional ADC 3 likely contribute to the enhanced efficacy of the DBM ADC 5 that was observed in the SKOV3 xenograft tumor model (Figure 8C).
Figure 9.

Pharmacokinetics of trastuzumab ADCs 3 and 5 in rats after single iv injection at 1 mg/kg. Total Ab was determined using rat anti-mouse Fc capture antibody. Total ADC (DAR > 0) was determined using a murine anti-MMAF capture antibody. Detailed PK results are available in the Supporting Information.
Toxicity of ADCs in Rats.
The relative toxicity profiles of trastuzumab ADCs 3 and 5 were compared in rats. Equivalent doses of both ADCs were administered weekly on days 1, 8, and 15 via intravenous injection. Statistically significant differences were observed for the parameters shown in Figure 10. Decreases in absolute reticulocytes were observed in animals treated with conventional ADC 3, but not in groups treated with ADC 5 on study day 3 (Figure 10). On day 17 (3 days after the final dose) decreases in absolute reticulocytes were observed in all ADC treatment groups, but to a lesser extent in rats treated with the cross-linked ADC 5 (Figure 10A). Similarly, mid and high doses of the conventional ADC 3 induced a statistically significant increase in hepatic parameters, ALT and AST, on study days 3 and 17 (Figures 10B and 10C). In contrast, ALT and AST values were only elevated in the high dose group treated with ADC 5 on day 3, and in the mid and high groups on day 17. There were no test article-related effects on survival, clinical observations, or food consumption. However, gross macroscopic examinations of the testes revealed that the ADC 3 treatment groups had abnormally small (2/10-low, 3/10-mid, and 4/10-high) and/or soft (2/10-low, 2/10-mid, 6/10-high) testes, suggesting reproductive organ toxicity; while no unusual findings were observed in the ADC 5 treatment groups. Overall, the results suggest that ADC 5 has a superior safety profile compared to the conventional ADC (3). Detailed toxicology results are available in the Supporting Information, Table 6.
Figure 10.

ADC toxicity in rats treated with increasing doses of ADC 3 (red) or ADC 5 (blue) measured on days 3 and 17. Group averages (10 rats per group) are shown as a green bar. (A) Dot plot showing absolute reticulocyte counts on days 3 and 17 in individual rats. (B) Dot plot showing alanine aminotransferase (ALT) levels on days 3 and 17 in individual rats. (C) Dot plot showing aspartate aminotransferase (AST) levels on days 3 and 17 in individual rats.
Versatility and Scalability of ADCs.
In order to determine the versatility of DBM linkers for constructing ADCs from different antibodies, we selected 3 commercially available antibodies (Erbitux, Avastin, and Rituxan) and conjugated them with 2 (DBM-MMAF) using previously described protocols for ADCs 5 and 6. HIC analysis of the resulting ADCs afforded single peaks with retention times consistent with DARs of 4 drugs/antibody (Figure 11A), and LC/MS data confirms the HIC results (see Supporting Information). The results demonstrate that DBM linkers can be applied to a variety of different antibodies and consistently afford ADCs with DARs of 4 drugs/antibody. The scalability of the conjugation reaction between DBM-MMAF 2 and IGNX was investigated at three different reaction scales (1 mg, 25 mg, and 1000 mg). The HIC profiles of the resulting ADCs (Figure 11B) indicate that there was no change in ADC homogeneity up to a 1 g scale. Multiple conjugations were performed at the 1 and 25 mg scales (10 each) to demonstrate the reproducibility of the conjugation reactions. Less than 10% variation was observed in relative homogeneity, demonstrating that the conjugation reaction is highly reproducible.
Figure 11.

Versatility and scalability of DBM linkers. (A) HIC traces of ADCs derived from conjugation of 2 (DBM-MMAF) with 3 different commercially available antibodies: Avastin, Rituxan, and Erbitux. (B) HIC traces of ADCs resulting from conjugation of 2 with IGNX at 3 different reaction scales (1 mg, 25 mg, and 1000 mg).
DISCUSSION
ADCs represent a rapidly growing new class of targeted chemotherapeutic agents for treatment of cancer. The recent approvals of two new ADCs, Adcetris in 2011 and Kadcyla in 2013, has led to a marked increase in the number of ADCs entering clinical trials. There are currently at least 40 different ADCs in various phases of clinical evaluation, and the number is increasing continuously. Although ADCs have demonstrated promising antitumor activity in preclinical studies, the broad range of pharmacological properties exhibited by conventional heterogeneous ADCs suggests that they have not yet reached their optimal therapeutic potential. The absence of efficient methods for producing homogeneous ADCs, however, has compelled pharmaceutical companies to develop heterogeneous ADCs despite their known limitations.
Numerous recombinant approaches for improving ADC homogeneity have been developed to overcome the limitations of heterogeneous ADCs, and significant progress has been made toward the synthesis of ADCs with defined DARs and conjugation sites. These recombinant approaches, however, often require extensive antibody engineering to identify the optimal conjugation sites where unique side chains can be introduced for conjugation with payloads modified with appropriate linkers. As a result, they are not suitable for converting existing “off-the-shelf’ antibodies directly into ADCs. Many of these methods require complex linker and payload modifications that have not been clinically validated and therefore present a greater risk for development into therapeutic agents.
With the limitations of current recombinant methods in mind, nonrecombinant methods have been developed for synthesizing more homogeneous ADCs that do not require re-engineering of the antibody. Disubstituted maleimide linkers have been used successfully for synthesizing homogeneous ADCs via interchain cysteine cross-linking, but their impact on the pharmacological properties of the resulting ADCs relative to conventional heterogeneous ADCs was unknown.
The results presented here have demonstrated that conventional maleimide linkers can be easily modified into bifunctional DBM linkers to enable the synthesis of DAR 4 ADCs via interchain cysteine cross-linking. The DBM linkers are compatible with clinically validated ADC payloads, and they can be applied to a broad range of antibodies to consistently yield ADCs with improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. No antibody engineering or conjugation site optimization is required because the conjugation sites are conserved for most antibodies. Moreover, antigen affinity and specificity are maintained during the conjugation process, and the resulting ADCs are highly potent against antigen positive cells in vitro. In summary, these results demonstrate that interchain cysteine cross-linking with DBM linkers yields ADCs with improved pharmacokinetics, superior efficacy, and reduced toxicity in vivo over conventional ADCs. These improvements should ultimately result in a superior therapeutic window and lead to more effective therapeutic agents in the future.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported in part by the National Cancer Institute of the National Institutes of Health under Award No. R43CA171492. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS USED
- ADC
antibody–drug conjugate
- DBM
dibromomaleimide
- MC
maleimidocaproyl
- MMAF
monomethyl auristatin Phe
- PK
pharmacokinetics
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.5b00432.
SEC results, dSEC purification and analysis, LC/MS data, and PK and toxicology protocols and results (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Beck A; Reichert JM Antibody-drug conjugates. mAbs 2014, 6, 15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sievers EL; Senter PD Antibody-drug conjugates in cancer therapy. Annu. Rev. Med 2013, 64, 15–29. [DOI] [PubMed] [Google Scholar]
- (3).Hamblett KJ; Senter PD; Chace DF; Sun MMC; Lenox J; Cerveny CG; Kissler KM; Bernhardt SX; Kopcha AK; Zabinski RF; Meyer DL; Francisco JA Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res 2004, 10 (20), 7063–70. [DOI] [PubMed] [Google Scholar]
- (4).Boswell CA; Mundo EE; Zhang C; Bumbaca D; Valle NR; Kozak KR; Fourie A; Chuh J; Koppada N; Saad O; Gill H; Shen B-Q; Rubinfeld B; Tibbitts J; Kaur S; Theil F-P; Fielder PJ; Khawli LA; Lin K Impact of Drug Conjugation on Pharmacokinetics and Tissue Distribution of Anti-STEAP1 Antibody-Drug Conjugates in Rats. Bioconjugate Chem. 2011, 22 (10), 1994–2004. [DOI] [PubMed] [Google Scholar]
- (5).Wakankar AA; Feeney MB; Rivera J; Chen Y; Kim M; Sharma VK; Wang YJ Physicochemical Stability of the Antibody-Drug Conjugate Trastuzumab-DM1: Changes due to Modification and Conjugation Processes. Bioconjugate Chem. 2010, 21 (9), 1588–1595. [DOI] [PubMed] [Google Scholar]
- (6).Mullard A Maturing antibody-drug conjugate pipeline hits 30. Nat. Rev. Drug Discovery 2013, 12 (6), 483. [DOI] [PubMed] [Google Scholar]
- (7).Sassoon I; Blanc V Antibody-drug conjugate (ADC) clinical pipeline: a review. Methods Mol. Biol. (N. Y., NY, U. S.) 2013, 1045, 1–27. [DOI] [PubMed] [Google Scholar]
- (8).Francisco JA; Cerveny CG; Meyer DL; Mixan BJ; Klussman K; Chace DF; Rejniak SX; Gordon KA; DeBlanc R; Toki BE; Law C-L; Doronina SO; Siegall CB; Senter PD; Wahl AF cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102 (4), 1458–65. [DOI] [PubMed] [Google Scholar]
- (9).Sanderson RJ; Hering MA; James SF; Sun MMC; Doronina SO; Siadak AW; Senter PD; Wahl AF In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immuno-conjugate. Clin. Cancer Res. 2005, 11 (2 Part 1), 843–52. [PubMed] [Google Scholar]
- (10).Tumey LN; Charati M; He T; Sousa E; Ma D; Han X; Clark T; Casavant J; Loganzo F; Barletta F; Lucas J; Graziani EI Mild Method for Succinimide Hydrolysis on ADCs: Impact on ADC Potency, Stability, Exposure, and Efficacy. Bioconjugate Chem. 2014, 25 (10), 1871–1880. [DOI] [PubMed] [Google Scholar]
- (11).Kline T; Steiner AR; Penta K; Sato AK; Hallam TJ; Yin G Methods to Make Homogenous Antibody Drug Conjugates. Pharm. Res. 2014, DOI: 10.1007/s11095-014-1596-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Junutula JR; Raab H; Clark S; Bhakta S; Leipold DD; Weir S; Chen Y; Simpson M; Tsai SP; Dennis MS; Lu Y; Meng YG; Ng C; Yang J; Lee CC; Duenas E; Gorrell J; Katta V; Kim A; McDorman K; Flagella K; Venook R; Ross S; Spencer SD; Wong WL; Lowman HB; Vandlen R; Sliwkowski MX; Scheller RH; Polakis P; Mallet W Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol 2008, 26 (8), 925–32. [DOI] [PubMed] [Google Scholar]
- (13).Junutula JR; Flagella KM; Graham RA; Parsons KL; Ha E; Raab H; Bhakta S; Nguyen T; Dugger DL; Li G; Mai E; Lewis Phillips GD; Hiraragi H; Fuji RN; Tibbitts J; Vandlen R; Spencer SD; Scheller RH; Polakis P; Sliwkowski MX Engineered Thio-Trastuzumab-DM1 Conjugate with an Improved Therapeutic Index to Target Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer. Clin. Cancer Res. 2010, 16 (19), 4769–4778. [DOI] [PubMed] [Google Scholar]
- (14).Shen B-Q; Xu K; Liu L; Raab H; Bhakta S; Kenrick M; Parsons-Reponte KL; Tien J; Yu S-F; Mai E; Li D; Tibbitts J; Baudys J; Saad OM; Scales SJ; McDonald PJ; Hass PE; Eigenbrot C; Nguyen T; Solis WA; Fuji RN; Flagella KM; Patel D; Spencer SD; Khawli LA; Ebens A; Wong WL; Vandlen R; Kaur S; Sliwkowski MX; Scheller RH; Polakis P; Junutula JR Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol 2012, 30 (2), 184–189. [DOI] [PubMed] [Google Scholar]
- (15).Tian F; Lu Y; Manibusan A; Sellers A; Tran H; Sun Y; Phuong T; Barnett R; Hehli B; Song F; De Guzman MJ; Ensari S; Pinkstaff JK; Sullivan LM; Biroc SL; Cho H; Schultz PG; Di Joseph J; Dougher M; Ma D; Dushin R; Leal M; Tchistiakova L; Feyfant E; Gerber H-P; Sapra P A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. U. S. A 2014, 111 (5), 1766–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zimmerman ES; Heibeck TH; Gill A; Li X; Murray CJ; Madlansacay MR; Tran C; Uter NT; Yin G; Rivers PJ; Yam AY; Wang WD; Steiner AR; Bajad SU; Penta K; Yang W; Hallam TJ; Thanos CD; Sato AK Production of Site-Specific Antibody-Drug Conjugates Using Optimized Non-Natural Amino Acids in a Cell-Free Expression System. Bioconjugate Chem. 2014, 25 (2), 351–361. [DOI] [PubMed] [Google Scholar]
- (17).Strop P; Liu S-H; Dorywalska M; Delaria K; Dushin RG; Tran T-T; Ho W-H; Farias S; Casas MG; Abdiche Y; Zhou D; Chandrasekaran R; Samain C; Loo C; Rossi A; Rickert M; Krimm S; Wong T; Chin SM; Yu J; Dilley J; Chaparro-Riggers J; Filzen GF; O’Donnell CJ; Wang F; Myers JS; Pons J; Shelton DL; Rajpal A Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol. (Oxford, U. K.) 2013, 20 (2), 161–167. [DOI] [PubMed] [Google Scholar]
- (18).Lhospice F; Bregeon D; Belmant C; Dennler P; Chiotellis A; Fischer E; Gauthier L; Boedec A; Rispaud H; Savard-Chambard S; Represa A; Schneider N; Paturel C; Sapet M; Delcambre C; Ingoure S; Viaud N; Bonnafous C; Schibli R; Romagne F Site-Specific Conjugation of Monomethyl Auristatin E to Anti-CD30 Antibodies Improves Their Pharmacokinetics and Therapeutic Index in Rodent Models. Mol. Pharmaceutics 2015, 12 (6), 1863–1871. [DOI] [PubMed] [Google Scholar]
- (19).Drake PM; Albers AE; Baker J; Banas S; Barfield RM; Bhat AS; de Hart GW; Garofalo AW; Holder P; Jones LC; Kudirka R; McFarland J; Zmolek W; Rabuka D Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct in Vivo Efficacy and PK Outcomes. Bioconjugate Chem. 2014, 25 (7), 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Badescu G; Bryant P; Bird M; Henseleit K; Swierkosz J; Parekh V; Tommasi R; Pawlisz E; Jurlewicz K; Farys M; Camper N; Sheng X; Fisher M; Grygorash R; Kyle A; Abhilash A; Frigerio M; Edwards J; Godwin A Bridging Disulfides for Stable and Defined Antibody Drug Conjugates. Bioconjugate Chem. 2014, 25 (6), 1124–1136. [DOI] [PubMed] [Google Scholar]
- (21).Bryant P; Pabst M; Badescu G; Bird M; McDowell W; Jamieson E; Swierkosz J; Jurlewicz K; Tommasi R; Henseleit K; Sheng X; Camper N; Manin A; Kozakowska K; Peciak K; Laurine E; Grygorash R; Kyle A; Morris D; Parekh V; Abhilash A; Choi J.-w.; Edwards J; Frigerio M; Baker MP; Godwin A In Vitro and In Vivo Evaluation of Cysteine Rebridged Trastuzumab-MMAE Antibody Drug Conjugates with Defined Drug-to-Antibody Ratios. Mol. Pharmaceutics 2015, 12 (6), 1872–1879. [DOI] [PubMed] [Google Scholar]
- (22).Schumacher FF; Nunes JPM; Maruani A; Chudasama V; Smith MEB; Chester KA; Baker JR; Caddick S Next generation maleimides enable the controlled assembly of antibody-drug conjugates via native disulfide bond bridging. Org. Biomol. Chem 2014, 12 (37), 7261–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Maruani A; Smith MEB; Miranda E; Chester KA; Chudasama V; Caddick S A plug-and-play approach to antibody-based therapeutics via a chemoselective dual click strategy. Nat. Commun 2015, 6, 6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Doronina SO; Bovee TD; Meyer DW; Miyamoto JB; Anderson ME; Morris-Tilden CA; Senter PD Novel Peptide Linkers for Highly Potent Antibody-Auristatin Conjugate. Bioconjugate Chem. 2008, 19 (10), 1960–1963. [DOI] [PubMed] [Google Scholar]
- (25).Doronina SO; Mendelsohn BA; Bovee TD; Cerveny CG; Alley SC; Meyer DL; Oflazoglu E; Toki BE; Sanderson RJ; Zabinski RF; Wahl AF; Senter PD Enhanced Activity of Monomethylauristatin F through Monoclonal Antibody Delivery: Effects of Linker Technology on Efficacy and Toxicity. Bioconjugate Chem. 2006, 17 (1), 114–124. [DOI] [PubMed] [Google Scholar]
- (26).Polakis P Arming antibodies for cancer therapy. Curr. Opin. Pharmacol 2005, 5 (4), 382–387. [DOI] [PubMed] [Google Scholar]
- (27).Pillow TH; Tien J; Parsons-Reponte KL; Bhakta S; Li H; Staben LR; Li G; Chuh J; Fourie-O’Donohue A; Darwish M; Yip V; Liu L; Leipold DD; Su D; Wu E; Spencer SD; Shen B-Q; Xu K; Kozak KR; Raab H; Vandlen R; Lewis Phillips GD; Scheller RH; Polakis P; Sliwkowski MX; Flygare JA; Junutula JR Site-Specific Trastuzumab Maytansinoid Antibody-Drug Conjugates with Improved Therapeutic Activity through Linker and Antibody Engineering. J. Med. Chem 2014, 57 (19), 7890–7899. [DOI] [PubMed] [Google Scholar]
- (28).Hayes GM; Chinn L; Cantor JM; Cairns B; Levashova Z; Tran H; Velilla T; Duey D; Lippincott J; Zachwieja J; Ginsberg MH; van der Horst EH Antitumor activity of an anti-CD98 antibody. Int. J. Cancer 2015, 137 (3), 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Doronina SO; Toki BE; Torgov MY; Mendelsohn BA; Cerveny CG; Chace DF; DeBlanc RL; Gearing RP; Bovee TD; Siegall CB; Francisco JA; Wahl AF; Meyer DL; Senter PD Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol 2003, 21 (7), 778–84. [DOI] [PubMed] [Google Scholar]
- (30).Wakankar A; Chen Y; Gokarn Y; Jacobson FS Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs 2011, 3 (2), 161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Valliere-Douglass JF; McFee WA; Salas-Solano O Native Intact Mass Determination of Antibodies Conjugated with Monomethyl Auristatin E and F at Interchain Cysteine Residues. Anal. Chem. (Washington, DC, U. S.) 2012, 84 (6), 2843–2849. [DOI] [PubMed] [Google Scholar]
- (32).Lyon RP; Bovee TD; Doronina SO; Burke PJ; Hunter JH; Neff-LaFord HD; Jonas M; Anderson ME; Setter JR; Senter PD Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol 2015, 33 (7), 733–735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.