

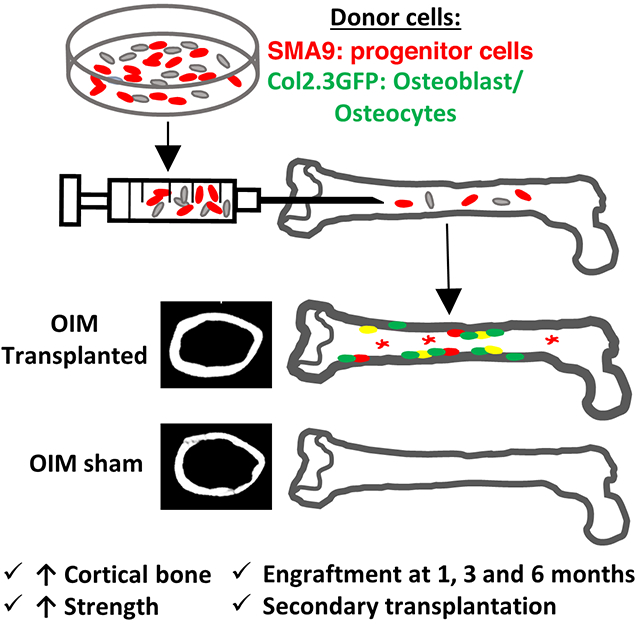
Abstract
Osteogenesis imperfecta (OI) is a genetic disorder most commonly caused by mutations associated with type I collagen, resulting in a defective collagen bone matrix. Current treatments for OI focus on pharmaceutical strategies to increase the amount of defective bone matrix, but do not address the underlying collagen defect. Introducing healthy donor stem cells that differentiate into osteoblasts producing normal collagen in OI patients has the potential to increase bone mass and correct the mutant collagen matrix. In this study, donor bone marrow stromal cells (BMSCs, also known as bone marrow mesenchymal stem cells) expressing both αSMACreERT2/Ai9 progenitor reporter and osteoblast reporter Col2.3GFP were locally transplanted into the femur of OIM mice. One month post-transplantation, 18% of the endosteal surface was lined by donor Col2.3GFP expressing osteoblasts indicating robust engraftment. Long-term engraftment in the marrow was observed 3 and 6 months post-transplantation. The presence of Col1a2-expressing donor cell derived cortical bone matrix was detected in transplanted OIM femurs. Local transplantation of BMSCs increased cortical thickness (+12%), the polar moment of inertia (+14%), bone strength (+30%) and stiffness (+30%) 3 months post-transplantation. Engrafted cells expressed progenitor markers CD51 and Sca-1 up to 3 months post-transplantation. Most importantly, 3 months post-transplantation donor cells maintained the ability to differentiate into Col2.3GFP+ osteoblasts in vitro, and in vivo following secondary transplantation into OIM animals. Locally transplanted BMSCs can improve cortical structure and strength, and persist as continued source of osteoblast progenitors in the OIM mouse for at least 6 months.
Keywords: Osteogenesis imperfecta, transplantation, stem cells, differentiation
Graphical Abstract
INTRODUCTION
Osteogenesis imperfecta (OI) is a genetic disorder commonly characterized by a variety of mutations associated with Type I collagen [1]. Patients produce defective bone matrix, and as a result have a high risk of fracture. Presently the standard of care for osteogenesis imperfecta patients centers on pharmaceutical treatments such as anti-resorptive bisphosphonates [2]. More, recently, additional treatments such as teriparatide, sclerostin- and RANKL- inhibiting antibodies, and TGF-beta inhibition have been studied in both pre-clinical and clinical trials [3–7]. A common aspect of pharmaceutical treatment options for OI is reduction of fracture risk by increasing the amount of defective bone matrix, without addressing the underlying OI collagen defect. As a result, there is great interest in developing treatments for OI which can replace mutant collagen that is the root cause of the weak and brittle bones.
Cell therapy represents an attractive therapeutic option with the potential to improve OI bone strength by transplanting osteoprogenitor cells which can subsequently differentiate into osteoblasts and form a healthy and strong collagen bone matrix. Previously utilized strategies for cell therapy center around the systemic introduction of healthy/wild-type (WT) cells into OI mice [8–15], or patients [16–19]. In OI patients, transient benefits such as increased growth velocity have been reported with cell transplantation but overall engraftment rates are low and proof of differentiation into the osteoblast lineage is lacking [17–19]. Studies in murine models have evaluated both intrauterine and adult cell transplantation strategies. Despite very low to absent differentiation of donor cells into osteoblasts and osteocytes, murine OI cell transplantation studies have reported bone lining cells of donor origin without clear histological confirmation of an osteoblast phenotype. Interestingly, it was recently reported that donor bone marrow cells systemically injected into OI mice do not become osteoblasts, but rather myeloid lineage cells such as osteoclasts and osteal macrophages lining endosteal and trabecular surfaces [10, 20, 21]. Therefore, a fundamental limitation of many OI cell therapy studies is the generally low cell engraftment and lack of conclusive detection of donor cell differentiation into the osteoblast lineage.
Local transplantation of cells directly into the bone marrow represents a different strategy for cell delivery with the potential to improve engraftment [22]. Although we have demonstrated the engraftment potential of local transplantation in proof-of-concept studies, this work did not investigate if there was an improvement in bone morphology, structure and bone strength or the phenotype of the engrafted donor cells [23]. To directly address the issues surrounding the accurate determination of donor cell differentiation into the osteoblast lineage, we employed donor cells with two fluorescent reporters. Specifically, we used donor cells where ɑSMA-expressing bone marrow stromal stem and progenitor cells (BMSCs) and their progeny are labeled with an Ai9 reporter [24, 25], and tracked their differentiation into osteoblasts and osteocytes with a Col2.3GFP reporter [26].
Therefore, in this study fluorescently labeled donor cells were used to identify short-term and long-term engraftment after local transplantation into an OI mouse model. We demonstrate their continued ability to give rise to osteoblasts, and show that local transplantation improves bone morphology and mechanical properties.
MATERIALS AND METHODS
Animals
All animal procedures were approved by an institutional animal care committee. To model osteogenesis imperfecta, oim on a C57Bl/6 background (obtained from Dr. Charlotte Philips, Univ. of Missouri) that lack functional Col1a2 were utilized [27]. OIM homozygous mice were used for all experiments. OIM mice were genotyped as previously described [28]. Donor cells were sourced from mice that are a combination of the three previously described lines: αSMACreERT2 [24], Col2.3GFP [26] and Ai9 reporter mice (stock # 007909, Jackson Laboratory) [29]. To generate αSMACreERT2/Ai9/Col2.3GFP mice, αSMACreERT2/Col2.3GFP were bred with Ai9, and termed SMA9/Col2.3GFP. To assess recombination efficiency αSMAGFP mice were crossed with αSMACreERT2/Ai9 [30].
Bone marrow stromal cell cultures
Donor BMSCs for transplantation were prepared from 6–10 week old SMA9/Col2.3GFP transgenic mice. Bone marrow was flushed from both femurs and tibiae, clumps broken up by passing through an 18G needle, then strained through a 70μm filter to achieve a single cell suspension. Cells were seeded at ~4×105/cm2, and expanded in 5% oxygen to promote CFU-F formation in alpha minimum essential medium (ɑMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals) and 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were moved to a normoxic incubator (5% CO2) on day 4, and half the medium was changed. To activate Cre recombination and Tomato expression, cells were treated with 1μM 4-hydroxytamoxifen (Sigma) on days 4 and 6. At day 7, adherent cells were harvested with Accutase cell detachment solution (Innovative Cell Technologies, San Diego, CA) and resuspended in serum free DMEM for transplantation. Cultures at day 7 were used for transplantation, a time point when the ɑSMA progenitor population is expanded, and yet not differentiated into Col2.3GFP+ osteoblasts [23, 24, 26]. All donor cells were sex matched to the recipient mice.
To assess transplanted cell engraftment and osteogenic ability, whole bone marrow from the femurs of mice that received transplantation 3 months earlier was flushed and seeded into a single well of a 6-well plate in ɑMEM 10% FBS. After 7 days, osteogenic media (50 μg/ml ascorbic acid and 8 mM β-glycerophosphate) was added to cultures and media changed every 2 days until day 14 or 21 [26]. The presence of SMA9+ and Col2.3GFP+ cells was evaluated with an Observer Zeiss fluorescence microscope. A subset of plates was stained for Alkaline Phosphatase (ALP, 86R-1 Kit, Sigma) and Von Kossa and imaged on a scanner [26].
For secondary transplantation, cultures were established from femurs of mice 3 months after transplantation in a similar manner to the initial transplantation (except 4-hyrdoxytamoxifen treatment was omitted) and expanded for 7 days. Once expanded, these cells were locally transplanted again into OIM mice. Secondary transplantation was performed in male OIM mice (n=3 secondary transplantation recipients).
To assess SMA9 recombination efficiency, BMSC cultures were established from SMA9/αSMAGFP mice [30]. Cultures were treated with 4-hydroxytamoxifen on days 4 and 6, and the percent of GFP cells that were labeled by SMA9 was assessed by flow cytometry.
Local Transplantation
One day prior to local BMSC transplantation, 8–10wk old homozygous oim mice were exposed to 900 cGy or 500 cGy of total body gamma irradiation using a 137Cs source. Except in the study in which we tested effects of sublethal irradiation, mice were always exposed to 900 cGy of total body gamma irradiation. Several hours after irradiation, mice irradiated with 900 cGy were briefly anesthetized with isoflurane and given 1×107 wild-type C57Bl/6 whole bone marrow cells by retro orbital injection to enable bone marrow reconstitution. Before local transplantation, recipient oim mice were anesthetized with ketamine (140 mg/kg) and xylazine (11 mg/kg). The knee was flexed between 90–120° and a 26G needle was inserted into the joint surface from the anterior side, and into the medullary space through the growth plate. Immediately after, a 25G needle was inserted into this hole to aspirate marrow. Finally, 106 donor SMA9/Col2.3GFP cells in 20μL of serum free DMEM were delivered with a different 25G needle into the marrow space. The cells were injected into the femur as the needle was withdrawn to distribute them evenly. Mice were euthanized 1, 3, or 6 months post-transplantation. Mice used for microCT and mechanical testing received contralateral control “sham” injections. Contralateral injections were performed identically to the local transplantation of cells including needle insertion, marrow aspiration, and injection of the same volume of vehicle, without cells.
Histology
Bones were fixed for 3 days in 10% formalin, decalcified in 14% EDTA, incubated in 30% sucrose overnight and embedded in cryomatrix (Shandon). Soft tissue was similarly processed, without EDTA decalcification. A subset of mice were injected with demeclocycline two days before they were euthanized (37.5 mg/kg, i.p.) and bones collected and sectioned undecalcified. Cryosections (7μm) were obtained using a Leica cryostat (Wetzler, Germany) and the tape transfer system (Section-lab, Hiroshima, Japan) as previously described [31]. Fluorescent images were obtained with an Axioscan slide scanner.
To quantify engrafted cells, images were first exported at consistent brightness and contrast. For analysis, a 6 mm long region spanning the entire cortical width and centered between the growth plate and third trochanter was analyzed. Endosteal surface, cortical bone area, and marrow area were quantified in ImageJ. Cell counting was performed and SMA9+, Col2.3GFP+, and dual positive SMA9+/Col2.3GFP+ cells were quantified in each of the three compartments (cortical, endosteal surface, marrow).
To detect WT matrix in an OIM mouse, bone sections from OIM mice 3 months post-transplantation were immunostained for the presence of collagen I α2 using an antibody kindly provided by Dr. Charlotte Phillips, University of Missouri–Columbia [23]. After permeabilization in PBS 0.1% triton X, and blocking with Powerblock (Biogenex, San Ramon, CA), primary rabbit antibody was diluted 1:400 and applied at 4°C overnight, followed by incubation with donkey secondary antibody conjugated to Alexa Fluor 647.
Flow Cytometry
Flow cytometry was performed on an LSR II system (BD Bioscience, San Jose, CA). Voltages and gates were set based on unstained samples and single stain controls. Antibodies were purchased from eBioscience unless otherwise stated and included Sca1 (1:100, Alexa Fluor 700, D7 Clone), CD51 (1:100, biotin, RMV-7 clone) with Streptavidin-APCeFluor780 (1:400), CD90.2 (1:100, BV605, 53–2.1 clone, BD Bioscience), CD11b (1:800, APC, M1/70 clone), CD45 (1:200, eFluor450, 30-F11), CD31 (1:400, eFluor450, clone 390), Ter119 (1:200, eFluor450), and DAPI was added immediately before analysis to exclude dead cells. Bone marrow analysis was performed by flushing marrow, lysing red blood cells with ACK (Ammonium-Chloride-Potassium), and staining 1 million cells. Endosteal cells were collected following flushing of bone marrow, cutting up the bone tissue, and digesting with collagenase P (0.5 mg/mL in PBS). Cell culture plates were analyzed after removal with accutase and 1 million cells were stained. FACSDIVA8 software (BD Bioscience) was used for data analysis.
MicroCT
Femurs were harvested from transplanted female oim mice and frozen in PBS soaked gauze at −20°C (for later mechanical testing), or fixed in 10% formalin for subsequent histology. Femurs were imaged by microcomputed tomography (μCT40, Scanco Medical AG) at 55 kV at a 16 μm voxel size. Cortical morphometry near the mid-diaphysis was measured in a 600 μm span referenced 5.1 mm from the growth plate. Segmentation of bone was performed in conjunction with a constrained Gaussian filter to reduce noise, applying a calibrated hydroxyapatite-equivalent density threshold of 740 mg/cm3 for cortical bone. Femurs with fractures were excluded from the microCT analysis (n=9 sham, n=10 transplanted intact femurs were analyzed). One sample was excluded due to poor injection noted during transplantation.
Mechanical Testing
After microCT imaging, samples were soaked in PBS and stored at −20°C until torsion testing was performed as previously reported [32]. Briefly, samples were potted in methyl methacrylate (Orthodontic Resin, Dentisply Caulk Inc. Milford, DE, USA) and tested at 1 degree/s of rotation until failure. Bone strength (maximum torque) and bone stiffness were assessed. Femora with fractures were excluded from the mechanical testing analysis, as well as two intact sham femurs that broke during potting, resulting in n=7 sham, and n=10 transplanted femurs analyzed for mechanical testing.
Statistical Analysis
Comparison between sham and transplanted mice were made with a two-tailed Student’s t-test in Excel, with p<0.05 considered significant.
RESULTS
Undifferentiated Donor Cells Express Progenitor Markers and Differentiate to Form Col2.3GFP+ Mineralized Colonies In Vitro
BMSC cultures were established from SMA9/Col2.3GFP transgenic mice allowing labeling and lineage tracing of cells with Col2.3GFP marking mature osteoblasts and osteocytes (Fig 1A). At Day 7 of culture, the time point at which BMSCs were used for transplantation, they were undifferentiated as evidenced by the absence of Col2.3GFP expression (Fig. 1B). By D14, and D21, activation of Col2.3GFP is observed that co-localizes with areas of mineral deposition visible on the brightfield image. In contrast, at all time points a large number of αSMA-labeled cells are observed. At D14 and D21, the osteoprogenitor potential of the αSMA+ cells is confirmed by the presence of dual positive (yellow) cells indicating differentiation of SMA9+ progenitor cells into Col2.3GFP+ mature osteoblasts. To further validate the osteoprogenitor potential of SMA9+ cells, donor derived cell cultures (day 7) were analyzed for cell surface markers by flow cytometry (Fig 1C). At Day 7, 17.2% of cells were SMA9+/(CD31/CD45/TER119)− (Fig 1C), and 0.6% of all cells were SMA9+ /(CD31/CD45/TER119)+. SMA9+/(CD31/CD45/TER119)− donor derived progenitor cells were mostly Sca-1+/CD51+ (37.8%) stem/progenitor cells, or Sca-1−CD51+ (55.4%) thought to represent more committed osteoprogenitors, suggesting their potential to give rise to osteoblasts once transplanted in vivo.
Figure 1: Donor SMA9 cells express mesenchymal progenitor markers and can differentiate into Col2.3GFP+ osteoblasts.

A) SMA9 mice, labeling BMSCs, were crossed with Col2.3GFP mice, labeling mature osteoblasts and osteocytes, to generate SMA9/Col2.3GFP mice. n=3 male for all of Figure 1.
B) Bone marrow stromal cells from these mice were cultured, and after Day 7, osteogenic differentiation was induced. Col2.3GFP+ cells are largely absent at Day 7, begin to be present at Day 14 with differentiation, and are strongly present at Day 21. Note the co-localization of Col2.3GFP cells to mineralized regions in the brightfield (BF) images. Merged images at day 14 and day 21 contain dual positive yellow cells, indicating that αSMA labeled progenitors differentiated into Col2.3GFP expressing osteoblasts. αSMA+ cells were labeled by treating the culture with 4-OH tamoxifen on Days 4 and 6.
C) As Day 7 cells were used for transplantation into OIM mice, these cells were profiled for progenitor markers by flow cytometry. Within the (CD45/CD31/Ter119)− SMA9+ population, approximately 37.8% of cells were dual positive for mesenchymal and osteoprogenitor markers Sca-1 and CD51, while majority of cells were CD51+ (>90%). Representative dot plot is shown.
Robust Engraftment One month Post-Transplantation
One month after local transplantation, robust engraftment of SMA9/Col2.3GFP donor cells was observed in the marrow, along the endosteal surface, and in the cortical bone matrix of transplanted OIM mice (Fig. 2A, B). To quantify the engraftment of donor osteoblasts, the percent of the endosteal cortical surface covered by donor Col2.3GFP+ or dual Col2.3GFP+/SMA9+ cells was quantified. The average donor osteoblast surface per bone surface was 18.0±9.0% (mean±SD) (Fig. 2C). On average, donor osteocytes comprised 1.3% of all osteocytes. We quantified the cell populations (SMA9, Col2.3GFP, or double positive) in each of three bone compartments in a mid-diaphyseal region: the marrow (stromal progenitors), the endosteal surface (osteoblasts) and embedded within the cortical bone matrix (osteocytes) (Fig. 2D). We found that Col2.3GFP+ cells were absent in marrow as expected, confirming the specificity of the reporter. Donor cells localized on the endosteal surface and within the bone matrix were nearly all Col2.3GFP+, and a portion of these cells were dual positive for αSMA and Col2.3GFP.
Figure 2: Transplanted BMSCs differentiate into osteoblasts and engraft into the bone matrix as osteocytes.

A) Summary of experimental design. Cells were injected into the bone marrow of 8–10wk old OIM mice. Mice were evaluated on Day 30 for histologic outcomes. n=9/group, female.
B) Representative images highlight the presence of SMA9 cells within the marrow and along the endosteal surface. Scale bar is 500 μm on low magnification images, 100 μm on high-magnification merged image.
C) The presence of donor Col2.3GFP+ cells along the endosteal surfaces was quantified along a 6 mm ROI centered about the mid-diaphysis.
D) Labeled cells in various bone compartments (marrow, endosteal, osteocyte/matrix) were quantified over a 6 mm region about the mid-diaphysis. As expected, the marrow was devoid of Col2.3GFP+ cells, and dual positive cells were observed on the endosteal surface and in the bone matrix as osteocytes.
Similar to studies in non-OI mice [33], we observed that irradiation was required to achieve successful cell engraftment 1 month post-transplantation (Supplemental Figure 1). Without irradiation, engraftment was low with 5 of 11 female animals showing no engraftment of SMA9+ or Col2.3GFP+ donor endosteal osteoblasts. We detected engrafted osteocytes in only 1 of 11 mice. SMA9+ donor cells were present within bone marrow of non-irradiated animals, but numbers were greatly reduced. Quantification of the engrafted cells showed an 8x-400x increase (depending on skeletal compartment) in engraftment with irradiation. The irradiation effect was confirmed in a small subset of male mice (n=4 irradiated, n=3 non-irradiated, data not shown) which similarly showed few engrafted cells without irradiation, but strong engraftment at 900 cGy. In addition, we note that a lower sub-lethal (500cGy) level of irradiation showed significantly reduced engraftment of marrow cells, endosteal osteoblasts, and osteocytes compared to 900 cGy (Supplemental Figure 2). Specifically, at the lower irradiation level of 500 cGy, engraftment of donor cells as osteocytes was 84.8% less than at 900 cGy, engraftment of osteoblasts was 64.7% less, and engraftment of marrow cells was 83.9% less at 500 cGy than 900 cGy. Because we observed that engraftment was very low without irradiation, all subsequent transplantation experiments were performed in mice exposed to 900 cGy irradiation.
Improved Cortical Structure and Mechanical Properties 3 months Post-Transplantation
Given the observed engraftment of donor cells, we determined if local transplantation resulted in improved bone morphology by microCT. Femurs transplanted with donor progenitor cells (3 months following transplantation) had significantly increased cortical area (+12%, p=0.019) and cortical thickness (+13%, p=0.013) at the mid-diaphysis (Fig 3A). In addition, the polar moment of inertia was also increased (+16%, p=0.029) compared to contralateral sham controls. Representative microCT images highlight the changes to cortical structure (Fig. 3B).
Figure 3: Local transplantation of skeletal progenitors increases cortical structure and bone strength.

To test if local transplantation impacted bone structure and bone strength, microCT and mechanical testing were performed. A) MicroCT analysis of mid-diaphyseal cortical bone showed that transplanted bones had a greater cortical area and polar moment of inertia (MOI) compared to sham contralateral controls. B) Representative images from bones with near average values highlight local cell transplantation effects on bone structure. n=9 sham, n=10 transplanted, female.
C) Femora were tested to failure in torsion. Transplanted femurs had significantly higher maximum torque and stiffness compared to contralateral sham controls. D) Representative torque vs rotation curves from samples with near average maximum torque values highlight the differences. n=7 sham, n=10 transplanted, female.
While local transplantation increased bone volume and showed engrafted donor cells, we performed mechanical testing to determine if the structural increases resulted in increased bone strength. In comparison to sham contralateral control femurs, local transplantation significantly improved maximum torque (+30%, p=0.031) and torsional stiffness (+30%, p<0.001) (Fig. 3C). Representative torque vs displacement plots highlight the increased mechanical properties with local transplantation, confirming that transplanted bones require more force to break than controls (Fig 3D). All bones failed in a mechanically brittle manner, without detectable post-yield displacement, indicating that local transplantation did not rescue OI bone brittleness as assessed by torsion testing. However, different mechanical tests may be more sensitive for detection of any improvement in brittleness in transplanted OI mice.
Long-Term Engraftment of SMA9 Donor Cells 3 and 6 months Post-Transplantation
Given that the average life span of an osteoblast is estimated to be somewhere between 14–60 days [34, 35], we analyzed bones 3 and 6 months post-transplantation to determine whether GFP+ osteoblasts were still present, suggesting their continued supply by the locally transplanted donor cells (Fig. 4A). Additionally, we wanted to determine the long-term residence of αSMA progenitor cells in the bone marrow and their ability to produce normal collagen. Three months post-transplantation, OIM mice were assessed by staining for Col1a2 (Supplemental Figure 3). Within the bone matrix of OIM mice, Col1a2 expression is absent (negative control). In cortical bone matrix from a positive control WT mouse, we see abundant staining for Col1a2 through the entire cortex as expected. In the OIM transplanted bone, we see a layer of strong Col1a2 staining near the endosteal surface similar to that observed in the WT matrix. We note that this area of Col1a2+ matrix in the transplanted OIM mouse co-localizes with a region with donor osteoblasts on the endosteal bone surface, as well as several donor osteocytes.
Figure 4: Long-term transplantation shows continued presence of osteoblasts and osteocytes up to 6 months post-transplantation.

A) Experimental design for long-term transplantation.
B) At 3 months (90 days) post-transplantation, strong engraftment of αSMA-labeled progenitors in the marrow was observed. In addition, there were regions with donor osteoblasts lining the bone surface but relatively few osteocytes (top set of high magnification images), potentially indicating a newly formed group of osteoblasts. In addition, there are also regions of the cortex (bottom set of high-mag images) with strong osteocyte presence within the cortical bone matrix. n=6, female.
C) At 6 months (180 days) post-transplantation, engraftment of αSMA-labeled progenitors in the marrow was observed. In addition, there were regions with donor osteoblasts lining the bone surface but relatively few osteocytes (top set of high magnification images), potentially indicating a newly formed group of osteoblasts. There were also regions of cortical bone (bottom set of high-mag images) where most of the cortical width contains donor osteocytes, indicating strong engraftment in the matrix. n=3, female. Scale bars are 500μm in low-mag images, and 100μm in high-mag images.
At 3 and 6 months post-transplantation, Col2.3GFP+ cells were observed along the endosteal surface, and in the bone matrix (Fig 4B,C). SMA9+ cells were observed on the endosteal surface, embedded in the bone as osteocytes, and throughout the marrow. We note that total donor SMA9+ marrow cells in the 6 mm long centrally located region analyzed were 641±308 cells 1 month post-transplant, 651±178 cells 3 months post-transplant, and 1738±398 cells 6 months post-transplant. Similarly, donor endosteal osteoblast surface was 18.1±9.0% 1 month post-transplant, 11.7±7.2% 3 months post-transplant, and 21.9±7.8% 6 months post-transplant. Finally, total osteocyte number was 40±35 cells 1 month post-transplant, 43±26 cells 3 months post-transplant, and 108±88 cells 6 months post-transplant. While these values over time are useful to rule-out a major reduction in donor cells and confirm cell persistence, we caution against over-interpretation of time-dependent effects given the variability of transplantation and unique sets of donor cells and recipient mice for each time point. While engraftment was primarily assessed in females, long-term engraftment was confirmed in male mice (n=2) at 6 months (data not quantified).
Engrafted cells express stem/progenitor markers and show stem cell potential
To determine if SMA9 donor cells existing in host OI mice expressed cell surface markers for osteoprogenitors, flow cytometry was performed on marrow cells derived from host femurs that had been transplanted 3 months prior and expanded for 5 days in vitro (Fig 5A). Expanded SMA9+ cells that were non-hematopoietic (CD45/CD31/Ter119)− expressed osteoprogenitor markers including CD51 (47%) and a subset were dual positive for Sca-1/CD51 (Fig 5B).
Figure 5: Engrafted SMA9 cells maintain the expression of progenitor markers three months post-transplantation and the ability to differentiate into osteoblasts in vitro.

A) Transplanted mice were collected 3 months post-transplantation and engrafted marrow cells were assessed for mesenchymal progenitor markers by flow cytometry and for their continued ability to differentiate into Col2.3GFP+ osteoblasts in vitro. B) BMSCs from transplanted femurs were expanded for 5 days in culture, and assessed by flow cytometry. Non-hematopoietic (CD45-/CD31-/Ter119-) SMA+ cells expressed mesenchymal progenitor markers such as CD51 and Sca-1, n=3, male. C) From a separate cohort of transplanted mice (n=5, male), marrow was flushed, and cultured under differentiating conditions. On Day 21, mineralization and robust ALP staining are evident. Some mineralized colonies co-localized with Col2.3GFP+ cells that derived from a cell initially labeled as SMA9+ before it was transplanted into the OIM animal.
We also verified if engrafted cells maintained the functional ability to differentiate into osteoblasts in vitro and in vivo. Marrow cells cultured from transplanted femurs formed mineralized colonies, some of which co-localized with dual positive αSMA and Col2.3GFP cells (Fig 5C). As Col2.3GFP+ cells were absent from cultures at D7, this indicates the continued progenitor ability of αSMA cells that have been engrafted for 3 months to differentiate into osteoblasts. We also observed colonies that are Col2.3GFP+ but not SMA9+, which is a finding also detected in vivo.
Finally, we evaluated the long-term osteoprogenitor potential of engrafted cells by secondary transplantation (Fig 6A). BMSCs from transplanted femurs were expanded for 7 days and then transplanted into a new cohort of OIM mice for 1 month and histologically evaluated for engraftment. We observed the presence of Col2.3GFP+ cells on the bone surface derived from an αSMA+ progenitor that was labeled before transplantation into the first OIM mouse, highlighting the long-term osteoprogenitor potential of the engrafted cells (Fig. 6B, yellow arrows). In addition, the red arrow also highlights an engrafted osteocyte within the cortical bone matrix. Demeclocycline mineral label was given to the secondary transplanted OIM mouse 2 days before they were euthanized to provide further confirmation that engrafted Col2.3GFP cells were osteoblasts as evidenced by their localization along a mineralizing surface. The average donor osteoblast surface in secondary transplantation mice was 5.8% (n=3).
Figure 6: Engrafted SMA9+ progenitors maintain the ability to differentiate into osteoblasts in vivo following secondary transplantation.

A) Experimental design for secondary transplantation.
B) Representative section of a bone that received secondary transplantation. Demeclocycline mineral label was given to the secondary recipient mouse 2 days before euthanasia to provide further confirmation that any engrafted Col2.3GFP+ cells were osteoblasts. The presence of Col2.3GFP+ cells on the bone surface highlights the long-term osteoprogenitor potential of the engrafted cells. The yellow arrows highlight dual positive (SMA9+/Col2.3GFP+) osteoblasts on the endosteal surface, and the red arrow highlights an engrafted osteocyte within the cortical bone matrix. Low-scale bar on merged imaged is 500μm, high-mag scale bars are 100μm. n=3, males: specifically, n=3 separate cultures were made, each from two mice transplanted 3 months prior, and each of these 3 cultures was injected into separate secondary transplantation host mice.
DISCUSSION
OI is a highly heterogeneous genetic disease with a very large number of mutations, many of which are dominantly inherited, causing a large spectrum of disease severity. There is accumulating evidence that the weakness and brittleness of OI bones, especially in patients with more severe forms of disease, is caused by a combination of insufficient type I collagen production, inappropriate processing of the collagen that is produced, and cellular stress caused by production of mutant collagen that affects osteoblast differentiation [36, 37]. These pathologies are challenging to target pharmacologically. Therefore, cell transplantation represents an ideal treatment strategy with the potential for engraftment of healthy cells that make a strong collagen matrix.
In OI, achieving a 100% donor cell engraftment rate is likely unnecessary to achieve the bulk of the therapeutic benefit for OI patients. Indeed, a well cited example is provided by families mosaic for OI. OI patients that have ~40% of cells producing normal collagen minimize the impact of the mutant collagen to the level of a sub-clinical phenotype [38]. However, systemic delivery of cells has proved challenging. Moreover, accurate determination of the fate of any engrafted cells has proved elusive or difficult. In this study, we used fluorescent lineage tracing of bone marrow stromal progenitor cells as well as osteoblast specific reporters to positively trace cell fate and detect differentiation. Using a local transplantation strategy, we found donor progenitor cells were able to differentiate into osteoblasts and osteocytes, improve mechanical properties, and engraft in the marrow as long-term skeletal progenitors.
In our study, we observed 18% of the cortical endosteal surface was covered by Col2.3GFP+ donor osteoblasts indicating a relatively strong level of engraftment. We note that average values for endosteal mineralizing surface in OI models have been reported to be ~25–70%, depending on age [7, 39, 40]. While the engraftment outcomes (donor cell enumeration, normal collagen gene expression) vary between studies, systemic cell delivery strategies in adult mice typically report average values in the 0.1%−2% range such as this report for “donor osteopoetic” engraftment [12]. More critically, histologic images from these studies often show little to no osteocyte engraftment, unlike the regions of strong engraftment observed in this study with local transplantation. Of note, we found osteoblast and osteocyte cell engraftment in our study was not randomly distributed but appeared to occur in concentrated regions. This may suggest that certain regions of the cortex significantly benefit from cell transplantation, while others show a more modest impact.
The engraftment of long-term progenitors observed in this study is of particular interest. It represents the potential for a durable therapeutic effect with benefits that may extend well beyond the acute post-transplantation period. In this study, we observed that osteoblasts were present up to 6 months post-transplantation; given the estimated 14–60 day osteoblast lifespan this suggests that a continued source of osteoblast progenitors has engrafted. This was further explored by flow cytometry showing donor cells expressing stem/progenitor cell markers and most importantly functional assays showing the continued ability of engrafted cells to differentiate into osteoblasts both in vitro and in vivo by secondary transplantation. The potential for engrafted cells to continue to reside in the bone marrow and differentiate into osteoblasts represents a continued source of osteoprogenitors with the potential to have a protracted and durable therapeutic effect.
We did not observe significant levels of donor cell engraftment in non-irradiated animals, while sublethal irradiation (500 cGy) has significantly lower engraftment indicating that high dose (900 cGy) irradiation was required to achieve robust donor cell engraftment as osteoblasts and osteocytes. This is consistent with reports in non-OI mouse models as well as our prior experience [10, 23, 33]. The specific reasons that irradiation is permissive for cell engraftment is of particular interest as it relates to translational optimization. It appears that putting osteoprogenitors in close contact to the bone surface by local transplantation is not sufficient in the absence of irradiation. Irradiation may permit engraftment by opening the endosteal niche and compromising the host cells. Another positive effect of irradiation may be to suppress a systemic host vs graft reaction and also, to prevent response of the re-established host immune system towards the GFP and Tomato proteins. In addition to inhibiting cells of the host environment, the response to irradiation also leads to the induction of a cascade of signaling factors such as SDF-1 that may also influence observed engraftment [33]. A better understanding of these specific mechanisms may facilitate similar engraftment rates with pharmaceutical or related targeted methods that are less burdensome than irradiation. One potential method for translating lethal irradiation into clinical practice could involve irradiation of specific bones or body regions, which will require prior evaluation using mouse models.
In this study, we observed that not all Col2.3GFP+ donor cells were derived from αSMACreERT2/Ai9+ cells indicating that they can originate from other cell progenitor types or from αSMA expressing cells in which Cre recombination did not occur. Specifically, one month after transplantation, 30–40% of Col2.3GFP+ osteoblasts and osteocytes were also SMA9+. There are several possible reasons that this number is less than 100%. First, we have shown that the efficiency SMA9 recombination in vitro is ~50% of what is labeled by αSMA-GFP (Supplemental Figure 4). Thus, even if all osteoblasts were derived from an αSMA+ cell, one would only expect half of Col2.3GFP osteoblasts to also be red as a result of cre efficiency. A second reason for this observation centers on the fact that αSMA does not label all progenitors that can differentiate into an osteoblast. There are many different markers for mesenchymal stem cells [35, 41–43], and although there is significant overlap between these markers, there are also distinct sub-populations. While the use of different mesenchymal markers, or ubiquitous expressing GFPs, could be beneficial in our study, utilizing a well characterized progenitor cell directed Cre such as SMA allows for tracking of an established sub-population that comprises a significant percentage of osteoprogenitors.
One limitation of this study is that a local treatment for a systemic disorder is not an ideal strategy, although local cell therapy has shown promising results in other diseases such as in Duchenne Muscular Dystrophy [44]. With this limitation of local transplantation in mind, several related points are worthy of consideration. First, children with OI commonly suffer multiple fractures and orthopedic procedures are routinely performed. As a result, there are ample opportunities during orthopedic procedures to perform local cell transplantation as part of an existing operation. While local transplantation has limitations, it also shows significant engraftment in a way that systemic transplantations have struggled to achieve. Once locally transplanted cells are engrafted, there may be opportunities to mobilize the engrafted progenitors to move to other tissues with less invasive pharmaceutical strategies. Contralateral sham transplantation serves as an internal control isolating the effect of locally transplanted cells. Previous studies involving systemic transplantation of expanded stromal cells or parabiosis, indicated that any donor cells present on bone surfaces were not osteoblasts, but rather hematopoietic in origin [23, 45, 46].
In summary, we have demonstrated that local transplantation of skeletal progenitor cells into an OI mouse model results in donor osteoblasts and osteocytes, improved cortical structure and strength, and the engraftment of stem/progenitor cells with the ability to continue to differentiate into osteoblasts.
Supplementary Material
Supplemental Figure 1: Minimal engraftment is observed with local transplantation in non-irradiated Mice
A cohort of mice received local transplantation without irradiation. Minimal engraftment was observed without irradiation, with 5/11 animals showing no engrafted donor osteoblasts.
A-E) Examination of non-irradiated femurs 1 month post-transplantation showed a small number of engrafted osteoblasts (representative images provided). Scale bar 500μm in A, and 100μm in B-D.
At higher magnification (B-D) some engrafted SMA9+ cells were detected in the marrow and a small number of Col2.3GFP+ osteoblasts on the bone surface but no engrafted osteocytes. However, among all 11 transplanted animals without irradiation, only a single osteocyte was detected in one femur.
F-H) To quantify the impact of irradiation on engraftment, the total amount of donor cells (SMA or Col2.3GFP) was quantified in three bone compartments (Cortical bone/Osteocytes, Endosteal surface/osteoblasts, and marrow) at a cortical region centered between the third trochanter and the growth plate. Irradiation significantly improved engraftment of donor cells in each compartment compared to non-irradiated controls. The irradiated mice used for comparison here are identical to those presented in Figure 2. n=11 Non-Irradiated, n=9 Irradiated. *p<0.05
Supplemental Figure 2: Assessing the effects of sub-lethal of irradiation.
A cohort of female OIM mice were irradiated with 900cGy, or a lower level, 500cGy, to determine the effect of a sub-lethal dose of irradiation on donor cell engraftment. The same set of donor cells were transplanted into mice of either irradiation level, and sections were analyzed for engraftment 1 month post-transplantation. Representative images highlight that 900cGy resulted in a greater level of engraftment than 500cGy. In a 6mm ROI centered about the mid-diaphysis of a femoral section, the total number of donor marrow cells, osteocytes, and endosteal cells (osteoblasts) were quantified. We found that 900cGy results in significantly more donor cell engraftment. Scale bars in low magnification image 500μm, and high magnification images are 100μm. n=5 500cGy, and n=3, 900cGy. *p<0.05.
Supplemental Figure 3: Presence of Col1α2 in transplanted mice co-localizes with area of donor cells
Three months post-transplantation, presence of Col1α2+ “WT” collagen in transplanted OIM was assessed by immunohistochemical staining for Col1α2 in the bone matrix. In OIM transplanted mice (A-B) we note the presence of Col1α2 staining near the endosteal surface. The high magnification image in panel B highlights the area of Col1α2 staining, as outlined by the red dashed lined. We note that this area of Col1α2 staining is directly adjacent to the endosteal surface of donor osteoblasts (white arrows), and surrounds donor osteocytes located inside the bone matrix (red arrows). Regions without donor osteocytes were negative for Col1α2 matrix staining, as expected. C) Staining for Col1α2 was validated in a healthy WT C57/Bl6 femur as a positive control. D) A non-transplanted OIM mouse served a negative control.
Supplemental Figure 4: Efficiency of αSMACreERT2/Ai9 (SMA9)
To assess the efficiency of the SMA9 mouse model, we crossed it with αSMAGFP reporter mice and made BMSC cultures. On Days 4 and 6 of culture, BMSCs were treated with hydroxytamoxifen to induce tdTomato expression. On Day 7, cells were analyzed by flow cytometry. Single cells that were CD45− and αSMAGFP+ were used, as shown in A-B. C) Representative histogram highlighting the percent of αSMAGFP cells that are SMA9+, and D) shows quantification of this number across three samples. Results suggest the efficiency of SMA9 recombination is ~50%. Data is n=3, male.
SIGNIFICANCE STATEMENT.
We present a novel translational approach for the treatment of osteogenesis imperfecta (OI) using a murine model. Attempts to treat OI using stem cells via systemic transplantation have not been successful. By direct intra bone transplantation of bone marrow stromal cells (also known as bone marrow-derived mesenchymal stem cells) we achieved positive long-term effects on bone microarchitecture and biomechanics. Using fluorescent genetic markers we tracked donor stem cells and their ability to differentiate into mature cells of the osteoblast lineage: osteoblasts and osteocytes. Engraftment persisted at least 6 months following transplantation, and cells show stem cell potential based on engraftment following secondary transplantation.
ACKNOWLEDGMENTS
We acknowledge Renata Rydzik for assistance with the microCT scanning. This work has been supported by NIH/NIAMS grants AR055607, AR070813 and Regenerative Medicine Research Fund (RMRF) grant 16-RMB-UCHC-10 to I.K.
Supported by: This work has been supported by NIH/NIAMS grants AR055607, AR070813 and Regenerative Medicine Research Fund (RMRF) grant 16-RMB-UCHC-10 to I.K.
Data availability and sharing:
The data that support the findings of this study are available from the corresponding author on request.
REFERENCES
- 1.Forlino A, Cabral WA, Barnes AM et al. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. 2011;7:540–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glorieux FH, Bishop NJ, Plotkin H et al. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med. 1998;339:947–952. [DOI] [PubMed] [Google Scholar]
- 3.Grafe I, Yang T, Alexander S et al. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat Med. 2014;20:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobsen CM, Barber LA, Ayturk UM et al. Targeting the LRP5 pathway improves bone properties in a mouse model of osteogenesis imperfecta. J Bone Miner Res. 2014;29:2297–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orwoll ES, Shapiro J, Veith S et al. Evaluation of teriparatide treatment in adults with osteogenesis imperfecta. J Clin Invest. 2014;124:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semler O, Netzer C, Hoyer-Kuhn H et al. First use of the RANKL antibody denosumab in osteogenesis imperfecta type VI. J Musculoskelet Neuronal Interact. 2012;12:183–188. [PubMed] [Google Scholar]
- 7.Sinder BP, Eddy MM, Ominsky MS et al. Sclerostin antibody improves skeletal parameters in a Brtl/+ mouse model of osteogenesis imperfecta. J Bone Miner Res. 2013;28:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guillot PV, Abass O, Bassett JH et al. Intrauterine transplantation of human fetal mesenchymal stem cells from first-trimester blood repairs bone and reduces fractures in osteogenesis imperfecta mice. Blood. 2008;111:1717–1725. [DOI] [PubMed] [Google Scholar]
- 9.Jones GN, Moschidou D, Lay K et al. Upregulating CXCR4 in human fetal mesenchymal stem cells enhances engraftment and bone mechanics in a mouse model of osteogenesis imperfecta. Stem Cells Transl Med. 2012;1:70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee LR, Peacock L, Ginn SL et al. Bone Marrow Transplantation for Treatment of the Col1a2(+/G610C) Osteogenesis Imperfecta Mouse Model. Calcif Tissue Int. 2019;104:426–436. [DOI] [PubMed] [Google Scholar]
- 11.Li F, Wang X, Niyibizi C. Distribution of single-cell expanded marrow derived progenitors in a developing mouse model of osteogenesis imperfecta following systemic transplantation. Stem Cells. 2007;25:3183–3193. [DOI] [PubMed] [Google Scholar]
- 12.Otsuru S, Gordon PL, Shimono K et al. Transplanted bone marrow mononuclear cells and MSCs impart clinical benefit to children with osteogenesis imperfecta through different mechanisms. Blood. 2012;120:1933–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panaroni C, Gioia R, Lupi A et al. In utero transplantation of adult bone marrow decreases perinatal lethality and rescues the bone phenotype in the knockin murine model for classical, dominant osteogenesis imperfecta. Blood. 2009;114:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pereira RF, O’Hara MD, Laptev AV et al. Marrow stromal cells as a source of progenitor cells for nonhematopoietic tissues in transgenic mice with a phenotype of osteogenesis imperfecta. Proc Natl Acad Sci U S A. 1998;95:1142–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vanleene M, Saldanha Z, Cloyd KL et al. Transplantation of human fetal blood stem cells in the osteogenesis imperfecta mouse leads to improvement in multiscale tissue properties. Blood. 2011;117:1053–1060. [DOI] [PubMed] [Google Scholar]
- 16.Gotherstrom C, Westgren M, Shaw SW et al. Pre- and postnatal transplantation of fetal mesenchymal stem cells in osteogenesis imperfecta: a two-center experience. Stem Cells Transl Med. 2014;3:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horwitz EM, Prockop DJ, Fitzpatrick LA et al. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med. 1999;5:309–313. [DOI] [PubMed] [Google Scholar]
- 18.Horwitz EM, Prockop DJ, Gordon PL et al. Clinical responses to bone marrow transplantation in children with severe osteogenesis imperfecta. Blood. 2001;97:1227–1231. [DOI] [PubMed] [Google Scholar]
- 19.Horwitz EM, Gordon PL, Koo WK et al. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone. Proc Natl Acad Sci U S A. 2002;99:8932–8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang MK, Raggatt LJ, Alexander KA et al. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol. 2008;181:1232–1244. [DOI] [PubMed] [Google Scholar]
- 21.Millard SM, Pettit AR, Ellis R et al. Intrauterine Bone Marrow Transplantation in Osteogenesis Imperfecta Mice Yields Donor Osteoclasts and Osteomacs but Not Osteoblasts. Stem Cell Reports. 2015;5:682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li F, Wang X, Niyibizi C. Bone marrow stromal cells contribute to bone formation following infusion into femoral cavities of a mouse model of osteogenesis imperfecta. Bone. 2010;47:546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pauley P, Matthews BG, Wang L et al. Local transplantation is an effective method for cell delivery in the osteogenesis imperfecta murine model. Int Orthop. 2014;38:1955–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grcevic D, Pejda S, Matthews BG et al. In vivo fate mapping identifies mesenchymal progenitor cells. Stem Cells. 2012;30:187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthews BG, Grcevic D, Wang L et al. Analysis of alphaSMA-labeled progenitor cell commitment identifies notch signaling as an important pathway in fracture healing. J Bone Miner Res. 2014;29:1283–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalajzic I, Kalajzic Z, Kaliterna M et al. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J Bone Miner Res. 2002;17:15–25. [DOI] [PubMed] [Google Scholar]
- 27.Chipman SD, Sweet HO, McBride DJ Jr. et al. Defective pro alpha 2(I) collagen synthesis in a recessive mutation in mice: a model of human osteogenesis imperfecta. Proc Natl Acad Sci U S A. 1993;90:1701–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saban J, King D. PCR genotyping of oim mutant mice. Biotechniques. 1996;21:190–192. [DOI] [PubMed] [Google Scholar]
- 29.Madisen L, Zwingman TA, Sunkin SM et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalajzic Z, Li H, Wang LP et al. Use of an alpha-smooth muscle actin GFP reporter to identify an osteoprogenitor population. Bone. 2008;43:501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dyment NA, Jiang X, Chen L et al. High-Throughput, Multi-Image Cryohistology of Mineralized Tissues. J Vis Exp. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wee NKY, Sinder BP, Novak S et al. Skeletal phenotype of the neuropeptide Y knockout mouse. Neuropeptides. 2019;73:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herberg S, Kondrikova G, Hussein KA et al. Total body irradiation is permissive for mesenchymal stem cell-mediated new bone formation following local transplantation. Tissue Eng Part A. 2014;20:3212–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40:1434–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park D, Spencer JA, Koh BI et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell. 2012;10:259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Jiang X, Delaney J et al. Immature osteoblast lineage cells increase osteoclastogenesis in osteogenesis imperfecta murine. Am J Pathol. 2010;176:2405–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mirigian LS, Makareeva E, Mertz EL et al. Osteoblast Malfunction Caused by Cell Stress Response to Procollagen Misfolding in alpha2(I)-G610C Mouse Model of Osteogenesis Imperfecta. J Bone Miner Res. 2016;31:1608–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cabral WA, Marini JC. High proportion of mutant osteoblasts is compatible with normal skeletal function in mosaic carriers of osteogenesis imperfecta. Am J Hum Genet. 2004;74:752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacobsen CM, Schwartz MA, Roberts HJ et al. Enhanced Wnt signaling improves bone mass and strength, but not brittleness, in the Col1a1(+/mov13) mouse model of type I Osteogenesis Imperfecta. Bone. 2016;90:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sinder BP, White LE, Salemi JD et al. Adult Brtl/+ mouse model of osteogenesis imperfecta demonstrates anabolic response to sclerostin antibody treatment with increased bone mass and strength. Osteoporos Int. 2014;25:2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Houlihan DD, Mabuchi Y, Morikawa S et al. Isolation of mouse mesenchymal stem cells on the basis of expression of Sca-1 and PDGFR-alpha. Nat Protoc. 2012;7:2103–2111. [DOI] [PubMed] [Google Scholar]
- 42.Shi Y, He G, Lee WC et al. Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat Commun. 2017;8:2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou BO, Yue R, Murphy MM et al. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15:154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siemionow M, Cwykiel J, Heydemann A et al. Dystrophin Expressing Chimeric (DEC) Human Cells Provide a Potential Therapy for Duchenne Muscular Dystrophy. Stem Cell Rev Rep. 2018;14:370–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boban I, Barisic-Dujmovic T, Clark SH. Parabiosis model does not show presence of circulating osteoprogenitor cells. Genesis. 2010;48:171–182. [DOI] [PubMed] [Google Scholar]
- 46.Boban I, Jacquin C, Prior K et al. The 3.6 kb DNA fragment from the rat Col1a1 gene promoter drives the expression of genes in both osteoblast and osteoclast lineage cells. Bone. 2006;39:1302–1312. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Minimal engraftment is observed with local transplantation in non-irradiated Mice
A cohort of mice received local transplantation without irradiation. Minimal engraftment was observed without irradiation, with 5/11 animals showing no engrafted donor osteoblasts.
A-E) Examination of non-irradiated femurs 1 month post-transplantation showed a small number of engrafted osteoblasts (representative images provided). Scale bar 500μm in A, and 100μm in B-D.
At higher magnification (B-D) some engrafted SMA9+ cells were detected in the marrow and a small number of Col2.3GFP+ osteoblasts on the bone surface but no engrafted osteocytes. However, among all 11 transplanted animals without irradiation, only a single osteocyte was detected in one femur.
F-H) To quantify the impact of irradiation on engraftment, the total amount of donor cells (SMA or Col2.3GFP) was quantified in three bone compartments (Cortical bone/Osteocytes, Endosteal surface/osteoblasts, and marrow) at a cortical region centered between the third trochanter and the growth plate. Irradiation significantly improved engraftment of donor cells in each compartment compared to non-irradiated controls. The irradiated mice used for comparison here are identical to those presented in Figure 2. n=11 Non-Irradiated, n=9 Irradiated. *p<0.05
Supplemental Figure 2: Assessing the effects of sub-lethal of irradiation.
A cohort of female OIM mice were irradiated with 900cGy, or a lower level, 500cGy, to determine the effect of a sub-lethal dose of irradiation on donor cell engraftment. The same set of donor cells were transplanted into mice of either irradiation level, and sections were analyzed for engraftment 1 month post-transplantation. Representative images highlight that 900cGy resulted in a greater level of engraftment than 500cGy. In a 6mm ROI centered about the mid-diaphysis of a femoral section, the total number of donor marrow cells, osteocytes, and endosteal cells (osteoblasts) were quantified. We found that 900cGy results in significantly more donor cell engraftment. Scale bars in low magnification image 500μm, and high magnification images are 100μm. n=5 500cGy, and n=3, 900cGy. *p<0.05.
Supplemental Figure 3: Presence of Col1α2 in transplanted mice co-localizes with area of donor cells
Three months post-transplantation, presence of Col1α2+ “WT” collagen in transplanted OIM was assessed by immunohistochemical staining for Col1α2 in the bone matrix. In OIM transplanted mice (A-B) we note the presence of Col1α2 staining near the endosteal surface. The high magnification image in panel B highlights the area of Col1α2 staining, as outlined by the red dashed lined. We note that this area of Col1α2 staining is directly adjacent to the endosteal surface of donor osteoblasts (white arrows), and surrounds donor osteocytes located inside the bone matrix (red arrows). Regions without donor osteocytes were negative for Col1α2 matrix staining, as expected. C) Staining for Col1α2 was validated in a healthy WT C57/Bl6 femur as a positive control. D) A non-transplanted OIM mouse served a negative control.
Supplemental Figure 4: Efficiency of αSMACreERT2/Ai9 (SMA9)
To assess the efficiency of the SMA9 mouse model, we crossed it with αSMAGFP reporter mice and made BMSC cultures. On Days 4 and 6 of culture, BMSCs were treated with hydroxytamoxifen to induce tdTomato expression. On Day 7, cells were analyzed by flow cytometry. Single cells that were CD45− and αSMAGFP+ were used, as shown in A-B. C) Representative histogram highlighting the percent of αSMAGFP cells that are SMA9+, and D) shows quantification of this number across three samples. Results suggest the efficiency of SMA9 recombination is ~50%. Data is n=3, male.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on request.