

Abstract
Motivation
Time-series nuclear magnetic resonance (NMR) has advanced our knowledge about metabolic dynamics. Before analyzing compounds through modeling or statistical methods, chemical features need to be tracked and quantified. However, because of peak overlap and peak shifting, the available protocols are time consuming at best or even impossible for some regions in NMR spectra.
Results
We introduce Ridge Tracking-based Extract (RTExtract), a computer vision-based algorithm, to quantify time-series NMR spectra. The NMR spectra of multiple time points were formulated as a 3D surface. Candidate points were first filtered using local curvature and optima, then connected into ridges by a greedy algorithm. Interactive steps were implemented to refine results. Among 173 simulated ridges, 115 can be tracked (RMSD < 0.001). For reproducing previous results, RTExtract took less than 2 h instead of ∼48 h, and two instead of seven parameters need tuning. Multiple regions with overlapping and changing chemical shifts are accurately tracked.
Availability and implementation
Source code is freely available within Metabolomics toolbox GitHub repository (https://github.com/artedison/Edison_Lab_Shared_Metabolomics_UGA/tree/master/metabolomics_toolbox/code/ridge_tracking) and is implemented in MATLAB and R.
Supplementary information
Supplementary data are available at Bioinformatics online.
1. Introduction
Experimental approaches have been developed in time-series metabolic measurements by both nuclear magnetic resonance (NMR) and mass spectrometry (Bastawrous et al., 2018; Judge et al., 2019; Koczula et al., 2016; Link et al., 2015; Montana et al., 2011; Tabatabaei Anaraki et al., 2018). These experimental methods provide opportunities to understand metabolic dynamics, including metabolic changes under variation in carbon sources or oxygen levels (Judge et al., 2019; Link et al., 2014, 2015). Among existing approaches, continuous in vivo monitoring of metabolism by NMR (CIVM-NMR) provided high time resolution, in vivo measurements of metabolites in Neurospora crassa under aerobic and anaerobic conditions (Judge et al., 2019). These measurements covered a large proportion of pathways in central metabolism, and interesting dynamics in compound concentration were observed.
NMR provides a highly reproducible way to identify and quantify compounds. In an NMR spectrum, different metabolites are represented by different peaks (features), and peak height (intensity) is proportional to compound concentration, when peak shape does not change. Peak resonance frequency is sensitive to the local electronic structure and some environmental variables. Resonance frequency is reported as chemical shift,, which is derived by dividing the frequency in Hz by the spectrometer frequency in MHz and thus has units of parts per million (ppm). The dependence on local electronic structure allows for reliable compound identification. Additionally, some metabolites are sensitive to changes in the local chemical environment (e.g. pH or metal ion concentration) and systematically change their chemical shift, providing a useful way to measure these environmental factors (Takis et al., 2017; Tredwell et al., 2016).
Metabolism yields changes in metabolite concentration and local pH, resulting in NMR peak intensity and chemical shift changes. These changes not only provide important information about metabolic dynamics but also complicate feature extraction. Moreover, peaks in NMR spectra often overlap, which affects both compound annotation and quantification. The combination of systematic chemical shift, overlap and amplitude changes makes peak tracking and quantification a difficult problem. A practical, stable computational approach is needed to track and quantify peaks over time, regardless of overlap, amplitude and chemical shift changes.
Traditional alignment-based methods are popular for processing NMR spectra from different samples and aligning shifting peaks. However, these methods often introduce artifacts and are unreliable for the regions where peaks cross (Csenki et al., 2007; Vu and Laukens, 2013). In CIVM-NMR data, the pattern of peak shifting is less noisy and more continuous than in discrete extracted samples by traditional methods. These properties provide new information for quantifying crossing peaks as discussed below.
Multiple methods have been implemented to track peaks in time-series NMR spectra. The TSATool can track a peak by peak picking and a predefined function describing shifting trajectory (Koczula et al., 2016; Ludwig and Günther, 2011). It was used to track NMR peaks in leukemia cells and interesting hypoxia metabolic response was observed. This method, though capable of tracking individual peaks, does not provide a general solution for quantifying multiple peaks efficiently. In our initial CIVM-NMR study, a better framework for multiple peak tracking was introduced. Peak tracking was achieved by a smoothing filter to reduce noise (filtering step) and hierarchical clustering (connection step) to connect candidate peaks (Judge et al., 2019). While this method tracked peaks with chemical shift variation, substantial manual effort was still needed in parameter tuning to accommodate different spectral regions. For instance, the proper scaling factor for the extent of chemical shift variation and the number of expected clusters were crucial parameters but difficult to optimize. About 48 h of work were needed for the original CIVM-NMR quantification, which can be a significant bottleneck and cost (Judge et al., 2019). Additionally, none of the aforementioned methods can deal with crossing or severely overlapped peaks.
Computer vision methods have been adapted to solve other spectroscopy (Klukowski et al., 2015, 2018) and biological object tracking problems (Steger, 1998; Tinevez et al., 2017). It can also be implemented here to promote efficiency. The steps of both filtering candidate points and their subsequent connection can be improved by treating NMR peak extraction as a ridge tracking problem. Time-series NMR data can be viewed as a 2D matrix (or a 3D surface if we treat matrix elements as height) with each row being a spectrum at one time point and each column being the intensity of a particular resonance frequency across time. As the same peaks change continuously through time, they can be conceptualized as surface ridges, for which efficient detection algorithms exist (Suk and Bhandarkar, 1992). Surface segmentation techniques have been implemented in computer vision to classify 3D surface points based on their local curvature into qualitative surface types: inter alia, ridge, peak and valley (Supplementary Fig. S1) (Besl and Jain, 1986, 1988; Suk and Bhandarkar, 1992).
In Ridge Tracking-based Extract (RTExtract), to filter candidate peak points, we combined ridge classification with other information such as local maxima. This combination of multiple filters provided cleaner results with fewer false positives, and tuning parameters were fewer and more intuitive. Candidate points were then connected by a 2-step greedy method, which is composed of simple local optimal connections without global evaluations; this is possible because of the better filter on candidate points. Additionally, manual refinement steps were introduced to expand flexibility in tracking and increase tracking accuracy.
In this article, we present our new method (RTExtract) to extract and quantify time-series NMR spectra. We simulated time-series NMR data specifically presenting the challenges that limited previous methods. We also conducted a direct comparison of our previous method and RTExtract on experimental datasets (Judge et al., 2019) and found that RTExtract was faster and easier than our previous approach. Previous tracking results were reproduced in less than 2 h instead of ∼48 h. Additionally, we were able to track complex spectral regions, such as those with high amounts of overlap, that were impossible with previous published methods. RTExtract therefore significantly expands the utility of the rich data collected in CIVM-NMR and accelerates its analysis. Furthermore, we show that it can be applied to other time series NMR methods such as pH titration analysis (Brockerman et al., 2019; Edison et al., 1999; Joshi et al., 2008; Liebeke et al., 2013; Zachariah et al., 2001).
2. Materials and methods
2.1. Ridge point classification
Local curvature was used to classify ridge points and functions as one of the filters for candidate points. Including a ridge point filter with local optima filters reduced noise levels in selecting candidate points and increased accuracy in ridge tracking. The following section describes the ridge point filter.
For each point on the 3D surface (Fig. 1), a normal vector () can be defined. All planes that contain (the normal planes) will intersect the 3D surface along a curve, and for each such curve at the point of interest, the curvature can be computed. The maximum and minimum curvature values, denoted by and , respectively, correspond to two mutually orthogonal orientations of the normal planes and are referred to as the principle curvatures of the 3D surface at that point. From and , the Gaussian curvature () and mean curvature () are defined [Equation () and () and Fig. 1] (Besl and Jain, 1986, 1988; Suk and Bhandarkar, 1992). The curvatures and can be used to classify 3D surface points locally into qualitative types, including, inter alia, peak, ridge and valley. Specifically, when and , the surface is classified as a ridge, and the central point of the surface is the candidate point (Supplementary Fig. S1).
| [1] |
| [2] |
Fig. 1.
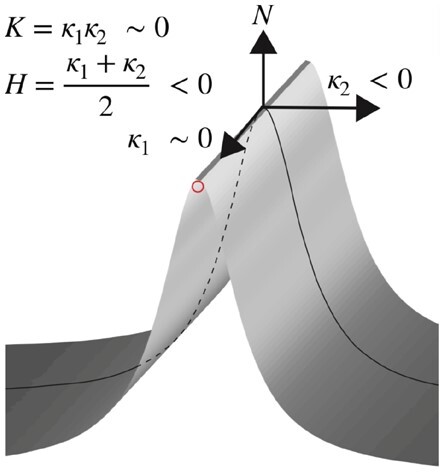
Illustration of the concept of curvatures. is the normal vector, and the curve is one of the intersecting curves. The two principal curvatures, and , correspond to the two vectors. Gaussian curvature () and mean curvature () are calculated by and . For the ridge surface shown here, and which results in and . Computation of and curvature is in Section 2.1. Other surface types are illustrated in Supplementary Figure S1
As an alternative to Equations () and (), the values of and can also be derived through the fundamental form matrices and , which provide a practical computation process (Stoker, 1969). Let be the surface and be a point on it. The first fundamental form G of the surface and the second fundamental form B of the surface can be computed from partial derivatives (e.g. ) and the unit surface normal vector () [Equations (), (), ()] (Suk and Bhandarkar, 1992).
| [3] |
| [4] |
| [5] |
We use a discrete biorthogonal second-order Chebychev polynomial with the interaction term ignored to approximate the local 3D surface within a 7 by 7 window (Besl and Jain, 1986; Haralick et al., 1983). Using biorthogonal polynomials instead of a more general fitting process increased computational speed. As the surface of interest was large (e.g. ∼50 spectra points for each spectrum in our experimental dataset) (Judge et al., 2019), this approximation was necessary to incorporate real-time analysis input within the workflow (wait time < 5 s). From the biorthogonal polynomial approximation, the first- and second-order derivatives, the fundamental forms and the curvatures ( and ) were computed in order (Suk and Bhandarkar, 1992).
Multiple surface types were generated from a second-order polynomial with the interaction term in a window (Equation [] and Supplementary Fig. S1). The parameters , and in Equation [] were varied to produce different surface types, including saddle ridge, minimal surface, saddle valley, ridge, flat surface, valley, peak and pit. The curvatures and were computed for the central point in the window to check with the expected values. The expected and curvatures were derived based on Equation [] and computed with Equations [] and [].
| [6] |
| [7] |
| [8] |
2.2. Feature quantification by ridge tracking
The entire workflow of RTExtract is presented in Figure 2. The steps include filtering candidate points, connecting candidate points into initial ridges, ridge refinement and manual ridge selection.
Fig. 2.

Illustration of the RTExtract algorithm. The algorithm is presented based on an example time-series NMR dataset, which was measured under aerobic conditions (A) (Judge et al., 2019). The stack spectral plot (A) shows changes of whole NMR spectra (X-axis chemical shift ) through time (Y-axis), and each time point is distinguished by a different line color as in all stack spectral plot in this article. Two regions (ppm [5.5, 8.5] and [1.3, 2.2]) are expanded to show spectral complexity. (B) A column slice (ppm [1.30, 1.35], indicated as orange box and arrow in (A) is chosen as show case for RTExtract (B–F). and curvatures were computed based on the 7 by 7 window (B) around each point, which was used to define ridge points (H < 0 and ). (C) To form candidate points (red points), these ridge points are intersected with local maxima and combined with the first global maximal points for each spectrum. (D) These points were connected in two steps by a greedy method to form ridges (red line). The first step was based on chemical shift distance () between points, and the second step was based on distance and angles. (E) Ridges were refined and manually selected for feature quantification. For the red ridge in (E), intensity and chemical shift () were plotted against time (F). The intensity is measured in arbitrary units (AUs), time is measured in hours, and chemical shift () is measured in ppm. Details on the RTExtract algorithm and tuning parameters can be found in Sections 2.1 and 2.2
The tested experimental datasets contain ∼50 spectra acquired at different time points (∼11 h), and each of them comprised ∼35 000 points in chemical shift resolution. The original time-series datasets were collected with finer time resolution, and the averaged (denoised) datasets were used to evaluate RTExtract, identical to our original study (Judge et al., 2019). In NMR spectra, even a small region can exhibit high complexity, and peaks of interest also differ considerably from each other in intensity (Fig. 2A, green and blue boxes). A region of interest (ROI) (ppm [1.3, 1.35] (Orange box in Fig. 2A) was selected as an example to illustrate the computational pipeline (Fig. 2B–E). The ROI is presented as a surface, in which different intensities can be visualized as different colors like a topographic map (Fig. 2B). Each row of the surface matrix is a single spectrum acquired at one time point. To filter candidate points, information from curvature, local maxima and a controlled number () of global maxima were combined (Fig. 2C). Local maxima are defined for each spectrum and several local maxima (including true peaks and noise) exist in a realistic NMR spectrum. Points on the surface were classified as ridge points (Set ) if they satisfied the curvature criteria in Equation () in the 7 by 7 window, for which no changes in the thresholds (1 and 0) were needed to accommodate different spectral regions. Besides ridge points, candidate points (global maxima) were also supplied through (the number of highest local maxima to add for each spectrum), which define the set . For each selected ROI, more local maximal points were added as candidate points for each spectrum from high to low in intensity. These candidate points were then intersected with local maxima (set ) to filter out points which did not correspond to true peaks. The combination of the three criteria () helped identify most ridges and improved accuracy.
| [9] |
A two-step greedy connection procedure was implemented to connect candidate points into ridges for quantifying individual peaks through time (Fig. 2D). This procedure assumes that chemical shift variation of peaks at nearby time points is local and continuous, which is typically the case in time-series measurements. First, points adjacent in time and with the closest chemical shift distance within (largest step size in chemical shift dimension) were connected into segments. Second, these segments were connected into ridges to cover the entire time-range for the peak. The segment connection was based on the shortest distance and a user-adjustable threshold on angle () between them. The angle threshold ensured smooth shift pattern in ridges. The order of the segment connection was ranked from high to low based on their average intensity.
In the ridge tracking process, only the parameters and required tuning. The remaining parameters in the program required no modification for the simulated and experimental datasets we tested. Choices for and values were also intuitive. In the majority of cases, we recommend the same small for most regions. The parameter can be increased when there is peak shifting and can be decreased when there are peaks that are close to each other. For , we recommend using = plus the number of ridges expected but not yet tracked. The values used in the script (, ) can be used as an initial guess for other datasets (Supplementary File S1). More details in parameter tuning for RTExtract and the previous method (Judge et al., 2019) are described in Supplementary Method S3.
2.3. Refinement of the tracking results
While most peaks can be tracked without refinement, in some cases, the tracking is imperfect, which can be solved in the refinement step (Fig. 2E). Chemical expertise adds value to this step, especially in regions with a low signal-to-noise ratio (SNR). Besides removing short ridges (default minimum ridge length is 5 time points), the refinement steps also include retracking for small regions, manual ridge selection, and removal of imperfect ridge ends (Supplementary File S1).
When multiple peaks overlap and change frequency, the greedy connection method (Section 2.2) had difficulty deciding which direction to continue through peak crossing. This was ameliorated by local retracking. In retracking, we imposed a more stringent constraint that the peaks tend to maintain their original directions when they cross. For each time point, a small search window (length 5) for connecting next candidate points was centered at the linear extrapolation of previous (last 5 time points) chemical shift values. That is, ridges are assumed to be locally linear, which is a reasonable constraint locally. In the global tracking process, however, there are indeed rapid changes in chemical shift, so in this case, the stringent constraint is not imposed. Combining different procedures for global and local tracking increases flexibility when necessary.
The automatic ridge tracking procedure often generates false positives (Supplementary File S1 and Fig2E) and these false ridges can be easily distinguished by the analyst. Hence, the interactive step (manual ridge selection) boosted performance by allowing analysts to select peaks with high confidence according to their knowledge. Moreover, compound quantification can also be improved by selecting peaks with good peak shapes, minimal overlap and high SNR. The user can also record the annotated compound name and indicate whether the tracked peaks should be used for quantification in later steps.
When the peak intensity decreases to near 0, the ridge tracking sometimes extends to noisy regions with no peak for a few time points. These imperfect ridge ends can also be removed by the ridge end removal interactive step.
The feature quantification workflow provides an interface to walk the user through ridge tracking, retracking for overlapping regions, manual ridge picking and manual end removal. The user can decide to apply certain steps depending on their needs. All information related to the tracking process, both explicit (parameter choices) and implicit (manual tuning records), is logged in the data structure for documentation and reproducibility. The manual refinement process is easy to use and no laborious peak picking is needed. Subsequently, peak intensity and chemical shift can be plotted through time (Fig. 2F). More details can be found in the MATLAB tutorial (Supplementary File S3).
2.4. Feature mapping and quantification
Tracked ridge features need to be matched to compounds and quantified. In the simulated spectra, after ridge tracking, the time-series data for each peak were automatically matched to compounds by differences in chemical shift. The difference () was evaluated using Equation (), and only the pairs with the smallest difference for all ridges and all compound peaks were selected as the final matches. In Equation (), is a set of time points overlapping between the simulated compound peaks and the tracked ridge peaks, and is the size of the set . The variables (simulated compound peak) and (tracked ridge peak) are the corresponding chemical shift vectors within . is the sum of the squared differences between extracted and simulated chemical shifts within the overlapping time range and is normalized by the range size (). Compounds were quantified by peak intensities normalized by the intensity of DSS peak (3-(Trimethylsilyl)-1-propanesulfonic acid sodium salt, a chemical shift reference and intensity internal standard). We also computed RMSD (Root Mean Square Deviation) between simulated and extracted chemical shift ( and , is the length of the ridge) for each ridge [Equation ()]. The annotation and quantification of the experimental datasets follows our published methods (Judge et al., 2019). Intensity is chosen for quantification for simplicity and peak shape does not change for both experimental and simulated spectra.
| [10] |
| [11] |
2.5. Data and software
Programs were written in MATLAB and R. They are shared through the Edison lab metabolomics toolbox GitHub repository (https://github.com/artedison/Edison_Lab_Shared_Metabolomics_UGA/tree/master/metabolomics_toolbox/code/ridge_tracking). The example experimental data can be found in Metabolomics Workbench (https://www.metabolomicsworkbench.org PR000738) and other used data can be found in Supplementary Data. We also provide a tutorial on the workflow (Supplement File S1).
The programs were extensively run and tested in MATLAB 2018b and R (RStudio Version 1.1.456 and R Version 3.5.1) on a macOS (Mojave 10.14.5) system.
3. Results
3.1. Comparison of simulated and experimental time-series NMR spectra
By their very nature, experimental datasets cannot be used to unambiguously validate the results of an algorithm such as RTExtract, especially in regions with overlap and noise. Therefore, we used simulated datasets for method evaluation. We first briefly evaluate the complexity of both dataset and how this affects ridge tracking. Complexity in time-series NMR spectra was evaluated in SNR, peak intensity, and chemical shift variation (Supplementary Table S1 and Supplementary Method S2). Besides the SNR value in the main simulation, multiple SNR levels were tested, and the workflow can still track ridges accurately in lower SNR levels (Supplementary Fig. S2A–D). The peak in was tracked automatically for (Supplementary Fig. S2A–C) and needed some manual tuning for (Supplementary Fig. S2D). In practice, most peaks in the experimental spectra possessed good enough SNR for tracking if they were visible in a stack plot. As an example, the valine multiplet centered at ppm 2.267 had low SNR, and 6 peaks of the multiplet could still be tracked (Supplementary Fig. S2E). The ridge tracking method had robust performance under a large range of different SNRs.
For the complexity metrics in variation of intensity ( and ) and chemical shift (), the values are similar between experimental and simulated datasets (Supplementary Tables S1 and S2). We note that using our metrics, the aerobic sample is more complex than the anaerobic sample, in agreement with our original qualitative conclusion (Judge et al., 2019).
3.2. Performance evaluation of ridge tracking on the simulated datasets
RTExtract was first tested on the simulated datasets (Supplementary Fig. S3 and Supplementary Method S1). Time-series NMR spectra were simulated with known concentrations and chemical shifts (simulated value), which were used to evaluate the ridge tracking result (extracted value). Extractions were evaluated by RMSD in chemical shift and most peaks were tracked accurately (low RMSD) in simulated datasets by both RTExtract and the previous method (Supplementary Table 2 and Supplementary Fig. S4).
We simulated a mixture of 15 metabolites, which yielded a possible 173 ridges with all the multiplets (Supplementary Method S1). Of these 173 potential ridges, 58 were essentially overlapped in the final simulations, allowing for 115 distinct ridges for analysis. Of these, 61 had some overlap and 54 were without overlap. We plotted extracted concentrations () against simulated concentrations () and observed that peaks without overlap were all near the diagonal (Fig. 3A). Small differences in intensity from the line broadening function created small deviations from the slope of 1. Nearly half of the ridges were not overlapped and could be accurately quantified.
Fig. 3.

Evaluation of the RTExtract algorithm on simulated datasets. (A) Compound quantification for all ridge points was plotted. For each ridge peak, extracted compound concentrations normalized to DSS (Y-axis, ) were plotted against simulated compound concentration (X-axis, ). The black diagonal represents perfect quantification and its slope is 1. Four overlapped peaks mapped to four compounds [1: alanine (blue); 2: glycerol (pink); 3: serine (green); 4: ethanol (purple)] were selected as examples (A–C). All other ridge overlapped peaks are in gray. (B) In the stack spectra, one spectrum was highlight in black, which was then decomposed into compound peaks involved in the overlap (C). In (C), the black line is the full simulated spectrum with all the compound peaks. (D) The acetate peak was simulated with chemical shift variation under different pH conditions and overlapped with another peak. (E) The extracted concentrations () were plotted against simulated concentrations (). (F) Extracted chemical shifts () were plotted against simulated chemical shift (). Performance of concentration and chemical shift estimation with more compounds can be found in Supplementary Figures S5 and S6. Compound concentration was simulated with arbitrary unit (AU) and chemical shift was evaluated in ppm
For peaks with overlap, quantification was affected. For example, the alanine Hα peak (peak 1 in Fig. 3A–C), was overestimated with a linear curve. Alanine peaks are overlapped with glycerol peaks, leading to inaccurate quantification for both metabolites (peaks 1 and 2 in Fig. 3A–C). Overlaps with glycerol also caused quantification of the ethanol peak to change in the opposite direction (peak 4 in Fig. 3A–C). In this case, the intensity variation of the small side peak from ethanol is dominated by the variation in glycerol peak.
Besides intensity estimation, overlap can affect chemical shift (e.g. peak 3 of serine in Fig. 3A–C). In this overlapping region, the uridine concentration increased through time, and serine concentration decreased. This resulted in the green line 3 in Figure 3A–B, which had a superposition of increasing and decreasing intensities. This kind of continuous shift between two features not only caused incorrect quantification but also an incorrect chemical shift variation. Even though neither uridine nor serine had clear peak shifts for the pH range considered ([4.0, 6.0]), the overlapped peak shifted smoothly. Through the changes of relative intensities of the two peaks, the chemical shift of the overlapped peak changed.
When peaks are separated enough to be distinguishable, ridge tracking is often accurate in chemical shift estimation (Fig. 3D). For the acetate peak, the concentration () was over-estimated in the overlapped region (Fig. 3E), but the chemical shift () was estimated accurately (Fig. 3F). Both peaks could be tracked for the entire range through the overlapped region, and the relative intensity between the two overlapping peaks did not affect tracking capability. Comparison of extracted and simulated values in concentration and chemical shift for more compounds can be found in Supplementary Figures S5 and S6.
3.3. Performance evaluation of ridge tracking on the experimental datasets
The ridge tracking method was next tested on experimental datasets and compared with the approach used in the initial CIVM publication (Judge et al., 2019). We first assessed the agreement on quantification of regions without much overlap (Fig. 4 and Supplementary Fig. S7). For quantification under aerobic and anaerobic conditions, the residuals between the two methods were close to zero for most compounds (19/22, Supplementary Fig. S7), and RTExtract reproduced the earlier results with much less time and manual input. The few differences were attributed to how negative values were dealt with between the two methods in computing Mean Scaled Ridge Intensity (Fig. 4 and Supplementary Fig. S7). In RTExtract, time-series data with negative intensities are shifted to positive first, which was not done in the original publication (Judge et al., 2019).
Fig. 4.
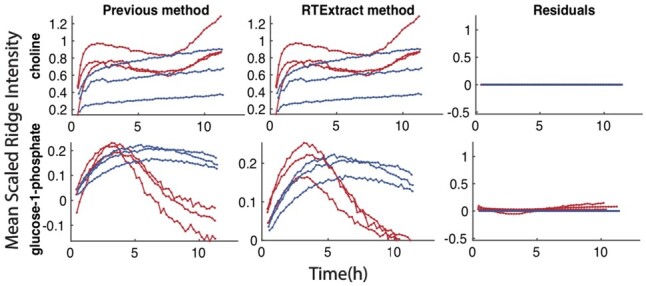
Reproduction of compound quantification results from our previous method. In each plot, the X-axes indicate time, and the Y-axes indicates Mean Scaled Ridge Intensity. Red curves are from aerobic datasets, and blue curves are from anaerobic datasets. Details in computing Mean Scaled Ridge Intensity can be found in the previous publication (Judge et al., 2019). Each row indicates quantification for one compound, including the previous, the RTExtract methods and residuals (differences between the quantifications by the two methods). Comparison for more compounds can be found in Supplementary Figure S7
RTExtract also worked for more complex regions that were difficult for the previous method (Fig. 5 and Supplementary Fig. S8) (Judge et al., 2019). The complex regions contained different degrees of overlap and/or peak shifting. When two peaks are closely overlapped with each other, the original method often produced tracking results that ‘jumped’ between the two (e.g. Fig. 5A and Supplementary Fig. S8). In RTExtract, these peaks were tracked with no jumps, resulting in fewer errors in chemical shift and intensity estimation. Parameter tuning, particularly in complex regions, was difficult in the original publication but is much easier in RTExtract. The glutamate region (ppm [2.3, 2.44], Fig. 5B) was another difficult case, in which the six peaks from glutamate shifted with pH and overlap with an unknown peak (yellow). By the previous method, only a small side peak in the multiplet was tracked for glutamate, so the quantification had a low SNR (Judge et al., 2019). By RTExtract, all the six glutamate peaks in the multiplet could be tracked with the retracking approach.
Fig. 5.
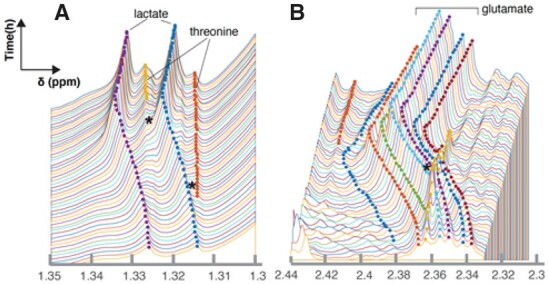
Evaluation of RTExtract on complex overlapping regions on the experimental datasets. Two ROIs (A–B) were selected as examples. Peaks in these ROIs can be precisely tracked, and the parts that are problematic in the previous method are indicated with stars. The middle six peaks in B are annotated to glutamate. Different point colors indicate different tracked peaks. Performance of the algorithm for less complex regions is in Figure 4 and Supplementary Figure S7. More example results from RTExtract and comparison with the previous method are in Supplementary Figure S8. Tracking for B is given as an example in Supplementary File S1
4. Discussion
RTExtract, a computer vision-based approach is introduced in this article to quantify time-series NMR spectra. RTExtract takes less time and exhibits better performance on complex regions than our original, less automated, approach (Judge et al., 2019). It provides a more practical way to process time-series NMR spectra and analyze in vivo metabolic dynamics of an organism.
RTExtract is an improvement from multiple perspectives. First, we reduced the number of tuning parameters from seven to two, which reduces the interactive time and is more intuitive to optimize. Second, the refinement steps allow fine-tuning of the ridge tracking process and easily remove imperfect regions. Instead of exploring a huge parameter space, the user can fix the imperfect regions through simple manual steps. With these two improvements, the published results can be replicated within 2 h by RTExtract instead of ∼48 h by the original method (Judge et al., 2019). Finally, RTExtract is also capable of dealing with more complex regions, especially with peak overlap and peak shifting (Fig. 5 and Supplementary Fig. S8). It is now possible to track most peaks in these difficult regions, without merging multiple peaks into one. Subsequently, more tracked features can be used for downstream modeling and statistical analyses. We also see that when two peaks are highly overlapped and their concentrations change in opposite way, the overlapped peak might seem to change in chemical shift (Fig. 3B peak 3). This could be mistaken for chemical shift change due to pH variation and seems to also occur on the glucose peaks (ppm region [5.2, 5.26]) under the aerobic condition in experimental datasets (Judge et al., 2019).
We still offer the option of manual interaction in the workflow, which helps produce accurate results but still requires expertise, time and manual effort. Future versions of the workflow will incorporate statistical filters accompanied by higher degrees of automation. A clustering-based method can be implemented to remove artifact ridges, which are characterized by random changes in intensity and chemical shift. Implementing this step might fully remove the manual procedure and make the full process much faster.
The RTExtract can also combine with spectral deconvolution for overlapping feature quantification. From RTExtract, chemical shift and intensity of individual overlapping peaks can be obtained and subsequently fed into the deconvolution methods. Based on the information of intensity and chemical shift, a Bayesian-based deconvolution approach can compute the underlying peak intensity (Hao et al., 2014; Krishnamurthy, 2013).
In principle, as long as peaks are changing in a continuous manner, they can be tracked by RTExtract. The experimental data tested in this article is from the CIVM method (Judge et al., 2019), and provides dense, continuous, time-series measurements. Other time-series NMR methods, such as flow NMR and in vitro sampling from NMR can also provide proper candidate measurements (Foley et al., 2014). Possible applications go beyond time-series measurements as long as the continuity constraint is met between neighboring spectra. For example, we used RTExtract to track peaks in a citrate pH titration experiment, fit the Henderson–Hasselbalch equation for the chemical shift changes under different pH, and estimate (Supplementary Fig. S9) (Edison et al., 1999; Szakacs et al., 2004; Tredwell et al., 2016; Zachariah et al., 2001). Likely, similar peaks could be tracked in pH or ligand-binding titrations of proteins (Brockerman et al., 2019; Joshi et al., 2008). A preprocessing by chemical shift sorting can even make independent samples of urine data accessible to RTExtract (Liebeke et al., 2013).
5. Conclusion
RTExtract is introduced in this article to quantify dense time-series NMR spectra by ridge tracking. It is faster, easier to use, and can deal with more complex regions than previously published methods. The extraction is accurate even in complex overlapping regions. As the ridge tracking method relies on the continuity of peaks at neighboring spectra, it can be further applied to other suitable data types.
Supplementary Material
Acknowledgements
The authors thank Heinz-Bernd Schuttler, Leidong Mao and Juan B. Gutierrez for helpful discussions on the computational method, Peter Kner for discussion on image processing, and John N. Glushka for discussion on NMR spectral processing. Goncalo Gouveia, Amanda Shaver, Mario Uchimiya and Nicole Holderman have provided helpful suggestions on figures. Laura Morris has provided technical support on computers. Rahil Taujale, Mario Uchimiya, Maxwell Colonna and Sicong Zhang have provided helpful feedback on using the RTExtract tutorial.
Funding
This work was primarily supported by National Science Foundation [NSF 1713746]. Arthur S. Edison was also supported by the Georgia Research Alliance.
Conflict of Interest: none declared.
Contributor Information
Yue Wu, Institute of Bioinformatics.
Michael T Judge, Department of Genetics.
Jonathan Arnold, Institute of Bioinformatics; Department of Genetics.
Suchendra M Bhandarkar, Department of Computer Science.
Arthur S Edison, Institute of Bioinformatics; Department of Genetics; Complex Carbohydrate Research Center; Department of Biochemistry and Molecular Biology, University of Georgia, Athens, GA 30602, USA.
References
- Bastawrous M. et al. (2018) In-vivo NMR spectroscopy: a powerful and complimentary tool for understanding environmental toxicity. Metabolites, 8, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besl P.J., Jain R.C. (1986) Invariant surface characteristics for 3D object recognition in range images. Comput. Vis. Graph. Image Process., 33, 33–80. [Google Scholar]
- Besl P.J., Jain R.C. (1988) Segmentation through variable-order surface fitting. IEEE Trans. Pattern Anal. Mach. Intell., 10, 167–192. [Google Scholar]
- Brockerman J.A. et al. (2019) The pKa values of the catalytic residues in the retaining glycoside hydrolase T26H mutant of T4 lysozyme. Protein Sci., 28, 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csenki L. et al. (2007) Proof of principle of a generalized fuzzy Hough transform approach to peak alignment of one-dimensional 1H NMR data. Anal. Bioanal. Chem., 389, 875–885. [DOI] [PubMed] [Google Scholar]
- Edison A.S. et al. (1999) Conformational ensembles: the role of neuropeptide structures in receptor binding. J. Neurosci., 19, 6318–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley D.A. et al. (2014) NMR flow tube for online NMR reaction monitoring. Anal. Chem., 86, 12008–12013. [DOI] [PubMed] [Google Scholar]
- Hao J. et al. (2014) Bayesian deconvolution and quantification of metabolites in complex 1D NMR spectra using BATMAN. Nat. Protoc., 9, 1416–1427. [DOI] [PubMed] [Google Scholar]
- Haralick R.M. et al. (1983) The topographic primal sketch. Int. J. Robot. Res., 2, 50–72. [Google Scholar]
- Joshi M.D. et al. (2008) Complete measurement of the pKa values of the carboxyl and imidazole groups in Bacillus circulans xylanase. Protein Sci., 6, 2667–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge M.T. et al. (2019) Continuous in vivo metabolism by NMR. Front. Mol. Biosci., 6, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klukowski P. et al. (2015) Computer vision-based automated peak picking applied to protein NMR spectra. Bioinformatics, 31, 2981–2988. [DOI] [PubMed] [Google Scholar]
- Klukowski P. et al. (2018) NMRNet: a deep learning approach to automated peak picking of protein NMR spectra. Bioinformatics, 34, 2590–2597. [DOI] [PubMed] [Google Scholar]
- Koczula K.M. et al. (2016) Metabolic plasticity in CLL: adaptation to the hypoxic niche. Leukemia, 30, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy K. (2013) CRAFT (complete reduction to amplitude frequency table) – robust and time-efficient Bayesian approach for quantitative mixture analysis by NMR. Magn. Reson. Chem., 51, 821–829. [DOI] [PubMed] [Google Scholar]
- Liebeke M. et al. (2013) Combining spectral ordering with peak fitting for one-dimensional NMR quantitative metabolomics. Anal. Chem., 85, 4605–4612. [DOI] [PubMed] [Google Scholar]
- Link H. et al. (2014) Advancing metabolic models with kinetic information. Curr. Opin. Biotechnol., 29, 8–14. [DOI] [PubMed] [Google Scholar]
- Link H. et al. (2015) Real-time metabolome profiling of the metabolic switch between starvation and growth. Nat. Methods, 12, 1091–1097. [DOI] [PubMed] [Google Scholar]
- Ludwig C., Günther U.L. (2011) MetaboLab—advanced NMR data processing and analysis for metabolomics. BMC Bioinformatics, 12, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana G. et al. (2011) Modelling short time series in metabolomics: a functional data analysis approach. Adv. Exp. Med. Biol., 696, 307–315. [DOI] [PubMed] [Google Scholar]
- Steger C. (1998) An unbiased detector of curvilinear structures. IEEE Trans. Pattern Anal. Mach. Intell., 20, 113–125. [Google Scholar]
- Stoker J.J. (1969) Differential Geometry. Wiley-Interscience, New York. [Google Scholar]
- Suk M., Bhandarkar S.M. (1992) Three-Dimensional Object Recognition from Range Images Springer-Verlag, New York;Tokyo, p. 308. [Google Scholar]
- Szakacs Z. et al. (2004) H-1/P-31 NMR pH indicator series to eliminate the glass electrode in NMR spectroscopic pK(a) determinations. Anal. Chim. Acta, 522, 247–258. [Google Scholar]
- Tabatabaei Anaraki M. et al. (2018) Reducing impacts of organism variability in metabolomics via time trajectory in vivo NMR. Magn. Reson. Chem., 56, 1117–1123. [DOI] [PubMed] [Google Scholar]
- Takis P.G. et al. (2017) Deconvoluting interrelationships between concentrations and chemical shifts in urine provides a powerful analysis tool. Nat. Commun., 8, 1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez J.Y. et al. (2017) TrackMate: an open and extensible platform for single-particle tracking. Methods, 115, 80–90. [DOI] [PubMed] [Google Scholar]
- Tredwell G.D. et al. (2016) Modelling the acid/base (1)H NMR chemical shift limits of metabolites in human urine. Metabolomics, 12, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu T.N., Laukens K. (2013) Getting your peaks in line: a review of alignment methods for NMR spectral data. Metabolites, 3, 259–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariah C. et al. (2001) Structural studies of a neuropeptide precursor protein with an RGD proteolytic site. Biochemistry, 40, 8790–8799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.