

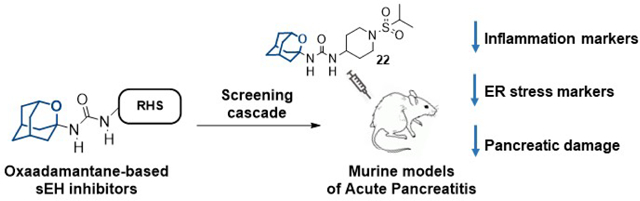
Abstract
In vivo pharmacological inhibition of soluble epoxide hydrolase (sEH) reduces inflammatory diseases, including acute pancreatitis (AP). Adamantyl ureas are very potent sEH inhibitors but the lipophilicity and metabolism of the adamantane group compromises their overall usefulness. Herein, we report that the replacement of a methylene unit of the adamantane group by an oxygen atom increases the solubility, permeability and stability of three series of urea-based sEH inhibitors. Most of these oxa-analogs are nanomolar inhibitors of both the human and murine sEH. Molecular dynamics simulations rationalize the molecular basis for their activity and suggest that the presence of the oxygen atom on the adamantane scaffold results in active site rearrangements to establish a weak hydrogen bond. The 2-oxaadamantane 22, which has a good solubility, microsomal stability and selectivity for sEH was selected for further in vitro and in vivo studies in models of cerulein-induced AP. Both in prophylactic and treatment studies, 22 diminished the overexpression of inflammatory and endoplasmic reticulum stress markers induced by cerulein and reduced the pancreatic damage.
Keywords: acute pancreatitis, inflammation, oxaadamantane, soluble epoxide hydrolase, urea
Graphical Abstract
INTRODUCTION
Arachidonic acid (AA) is a polyunsaturated fatty acid that is released from membrane phospholipids of activated cells by the action of phospholipase A2 stimulation. AA can be converted into different metabolites, which can either enhance inflammation or help in its resolution. The cyclooxygenases (COXs) and the lipoxygenases (LOXs) convert AA to pro-inflammatory and nociceptive prostaglandins and leukotrienes, respectively.1 Both pathways have been pharmaceutically targeted.2 In contrast, a third metabolic route, the cytochrome P450 pathway, has been scarcely explored. Cytochrome enzymes can convert AA to the epoxyeicosatrienoic acids (EETs), which are endowed with potent anti-inflammatory properties.3–5 However, soluble epoxide hydrolase (sEH, EPHX2, EC 3.3.2.3) rapidly hydrolyzes EETs to their corresponding dihydroxyeicosatrienoic acids (DiHETrEs), that show altered biological activity.6–7 In vivo pharmacological inhibition of sEH has been previously shown to stabilize the concentration of EETs, reducing inflammation and pain, suggesting that sEH is a potential target for the treatment of various diseases.8–9
Structural studies revealed that sEH has an L-shaped hydrophobic pocket with the catalytic residues at the corner.10–11 Ureas, amides and carbamates were shown to bind strongly to the catalytic residues of sEH. Taking into account the characteristics of the active site and the overall high hydrophobicity of the pocket, the introduction of lipophilic groups on both sides of the central urea has proven a successful strategy for the space-filling of the cavity and increasing van der Waals interactions, leading to several potent sEH inhibitors.12 Indeed, a vast number of potent sEH inhibitors incorporate an adamantane moiety, including AR9281, the first sEH inhibitor to enter clinical trials (Figure 1).13–15
Figure 1.
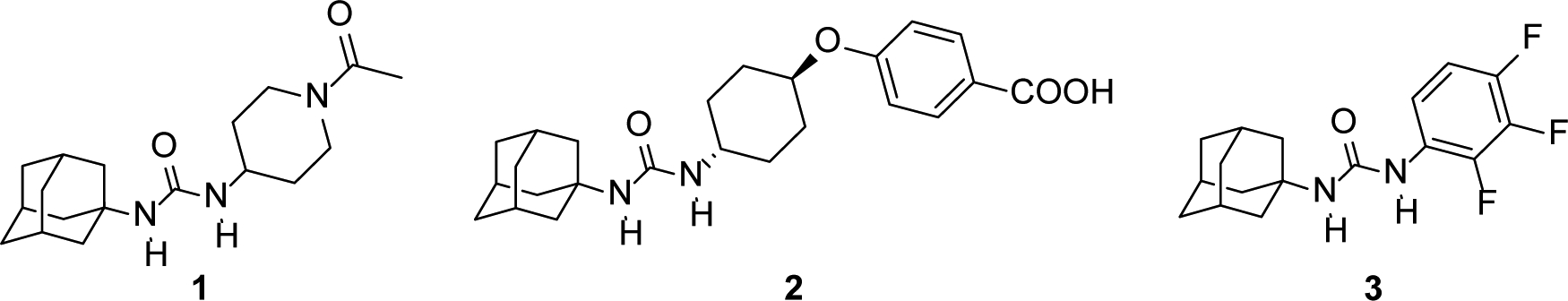
Adamantane-based sEH inhibitors 1 (AR9281, APAU, UC1153), 2 (t-AUCB),16 and 3.17
However, regardless of the efforts made to procure drug candidates with excellent inhibitory activities, the pronounced lipophilicity of the adamantane group compromises negatively the overall water solubility of the molecule, an important physicochemical parameter in the early stages of drug discovery,18–19 thus limiting the discovery of new drug-like sEH inhibitors.
To solve this problem, we hypothesized that the introduction of an oxygen atom should increase the solubility and may impact in the overall drug-like properties of the known adamantane-based sEH inhibitors without seriously compromising the inhibitory potency. Taking into account the availability of several oxapolycyclic amines,20–23 herein we have replaced the adamantane nucleus of known sEH inhibitors by selected polycyclic scaffolds bearing an oxygen atom. Encouragingly, previous research conducted around adamantane surrogates showed that the 2-oxaadamant-1-yl substituent is a suitable replacement moiety for the clinically approved antiviral drug amantadine.24
RESULTS AND DISCUSSION
Chemistry.
To test our hypothesis, the adamantane core present in inhibitor 3 was initially replaced by three known oxapolycyclic amines, 5, 6 and 8.20–23 The reaction of commercially available 2,3,4-trifluorophenylisocyanate with 5, 6 and 8 gave ureas 4, 7 and 9, respectively, in 83, 94 and 94% yield (Scheme 1).
Scheme 1.
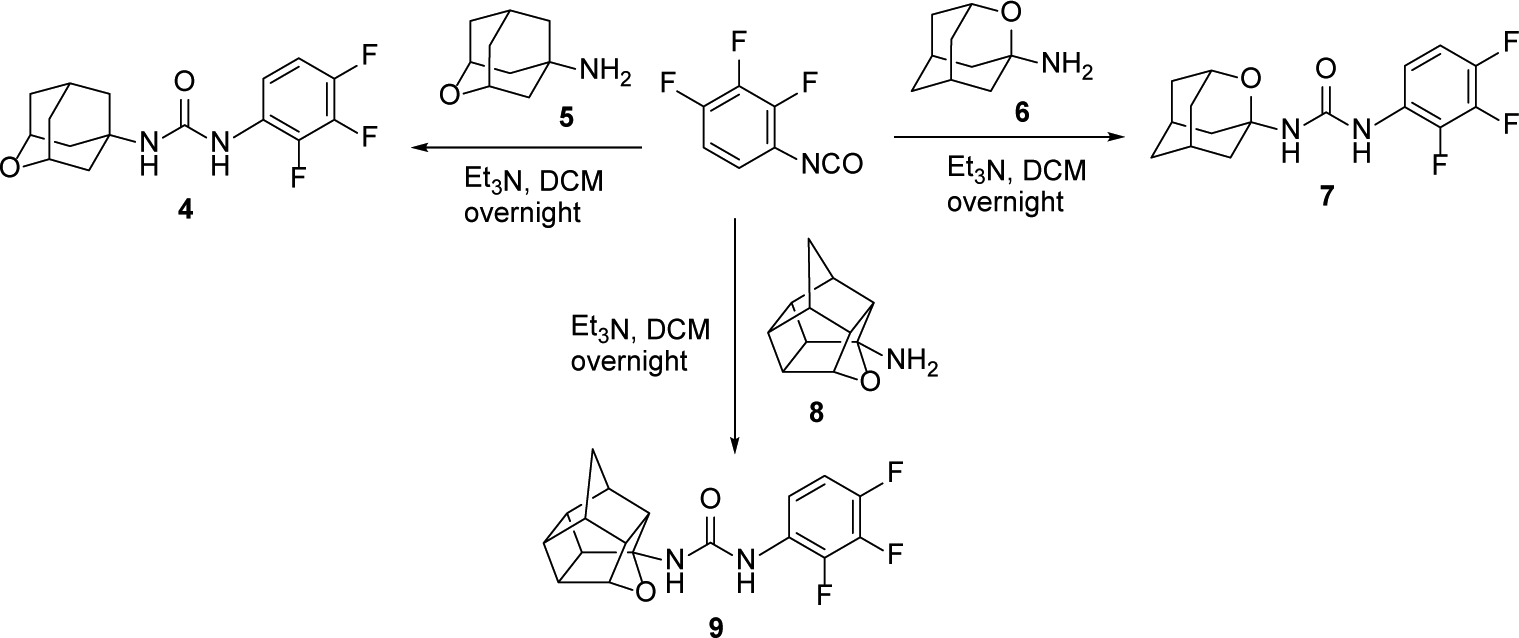
Synthesis of 2,3,4-triflurophenyl-derived ureas, 4, 7 and 9.
The three ureas were tested as human sEH inhibitors using a previously reported fluorescent-based assay.25 The three compounds displayed IC50 values in the same range of potency (47.4, 21.3 and 31.3 nM, for 4, 7, and 9, respectively), although less potent than 3 (IC50 = 7.7 nM).
Taking into account that, i) the 2-oxaadamantane-1-amine 6 was synthetically much more accessible than its isomer 2-oxaadamantane-5-amine, 5, and ii) the chiral nature of amine 8, the synthesis of the oxa-analogs of AR9281 and t-AUCB was subsequently performed only with amine 6.
For the synthesis of urea 12, amine 6 was treated with triphosgene to yield isocyanate 10, which was then reacted with the commercially available 1-acetyl-4-aminopiperidine, 11. Similarly, the reaction of 10 with 4-((trans-4-aminocyclohexyl)oxy)benzoic acid, 13, prepared as previously reported,26 furnished urea 14 (Scheme 2).
Scheme 2.
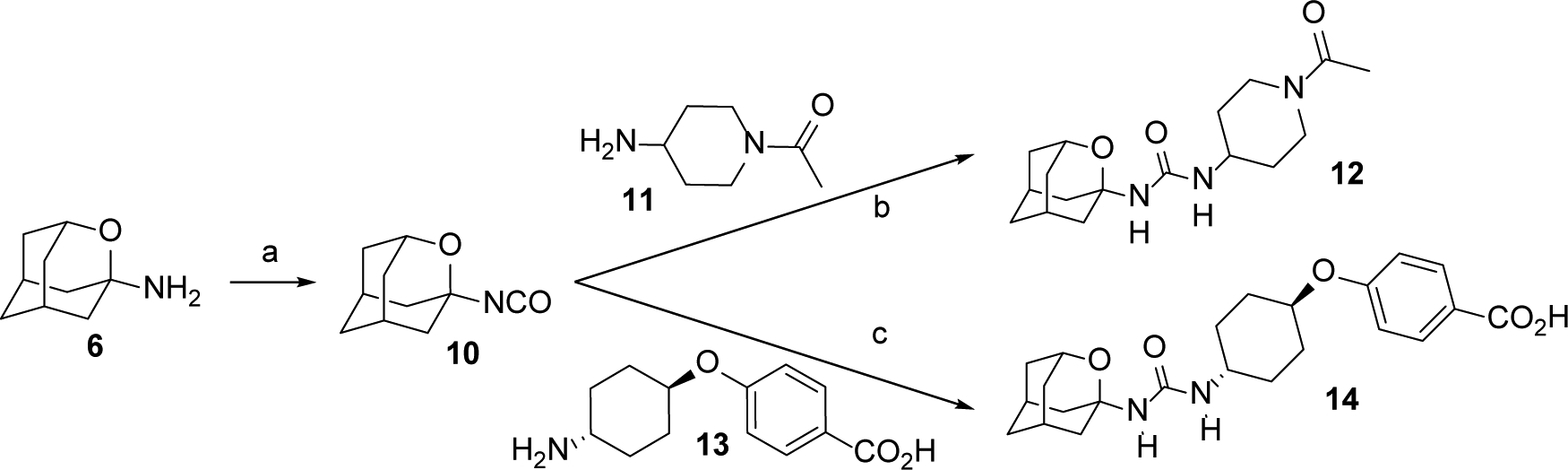
Synthesis of ureas 12 and 14.a
aReagents and conditions: a) Triphosgene, sat. NaHCO3, DCM, 30 min; b) 11, DCM, overnight, 52% overall yield; c) 13, DCM, overnight, 24% overall yield. See Experimental Section and Supporting Information for further details.
Interestingly, although less potent than their hydrocarbon counterparts, the novel oxaadamantane-derived ureas were nanomolar inhibitors of the human sEH (see Table 1). Often, a high melting point is indicative for bad physical properties such as low solubility in water and organic solvents. The adamantane-derived inhibitors have limited solubility in water and high melting points (Table 1), which likely affect their in vivo efficacy and certainly make formulation difficult. Gratifyingly, in AR9281 and compound 3 series, the oxaadamantane derivatives exhibited lower melting points than their adamantane counterparts, while t-AUCB and 14 presented approximately the same value (Table 1). Remarkably, as expected, the compounds featuring the 2-oxaadamantane moiety exhibited higher solubility values, in the three series, than the ones presenting the adamantane scaffold (compare AR9281 vs 12, t-AUCB vs 14, and 3 vs 7 in Table 1). Pleasingly, in addition to the expected improvement in the solubility, the membrane permeability improved for the three series. Thus in the AR9281 (1 and 12) and t-AUCB (2 and 14) series, a decrease in the efflux ratio was observed when the methylene unit was replaced by an oxygen atom. On the other hand, in the trifluorophenyl analogs, the efflux ratio was already low, but the substitution led to an increase in both AB and BA permeability (see Table 1).
Table 1.
Human sEH IC50 and solubility, melting point and permeability values for known adamantane inhibitors AR9281, t-AUCB and 3, and their oxygen analogs 12, 14 and 7.
| Compounds | X | h sEH IC50 (nM)[a] | Solubility (μM)[b] | Melting Point (°C) | Permeability (Caco-2) | |||
|---|---|---|---|---|---|---|---|---|
| Papp (nm/s) | ER[c] | |||||||
| A→B | B→A | |||||||
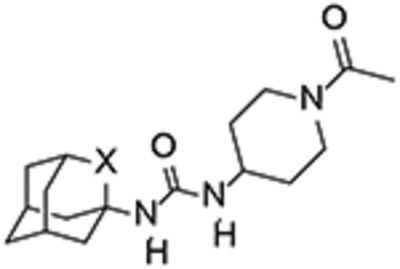 |
1, AR9281 (UC1153) | CH2 | 8.0 | 24 | 202–204[d] | 2.2 | 141.2 | 64.5 |
| 12 | O | 29.9 | 59 | 172–173 | 22.4 | 94.5 | 4.3 | |
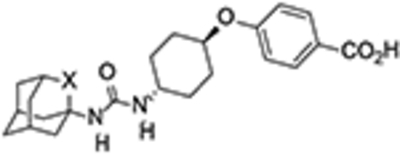 |
2, t-AUCB | CH2 | 0.5 | 25 | 250–255[e] | 1.9 | 210.3 | 111 |
| 14 | O | 9.0 | >100 | 255–257 | 1.4 | 75.5 | 55.4 | |
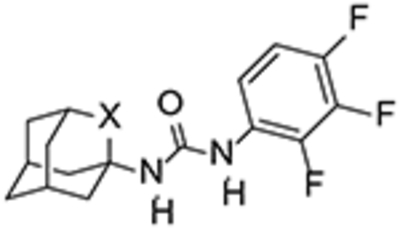 |
3 | CH2 | 7.7 | 16 | 216–219 | 6.7 | 4.6 | 0.69 |
| 7 | O | 21.3 | 27 | 196–198 | 168 | 151 | 0.9 | |
IC50 values are the average of three replicates. The fluorescent assay as performed here has a standard error between 10 and 20% suggesting that differences of two-fold or greater are significant. Because of the limitations of the assay, it is difficult to distinguish among potencies < 0.5 nM.25
Solubility in a 1% DMSO: 99% PBS buffer solution, see Experimental Section for details.
The efflux ratio was calculated as ER = (Papp B→A) / (Papp A→B). See the Experimental Section for further details.
Taken from reference 27.
Taken from reference 16.
Certainly, the introduction of the oxygen atom in the adamantane scaffold reduced the potency. Notwithstanding, designing bioactive compounds is a multifactorial process, so in view of the significant increase in aqueous solubility and permeability and the decrease of the melting point arising from the replacement of a methylene unit of the adamantane by an oxygen atom, the synthesis of two families of analogs of 7 and 12 was undertaken using diverse right-hand side (RHS) fragments selected from previous series of known sEH inhibitors.15,17,28–32 After exploring their potency as sEH inhibitors, their cytotoxicity and DMPK properties were assessed for selecting a candidate to perform an in vivo proof of concept study in a murine model of acute pancreatitis (AP), for which a sEH inhibitor showed effectiveness at reducing symptoms.33–34 Of note, an in vivo study of the third oxa-derivative, 14, was disclosed recently elsewhere.35
Two different synthetic procedures were followed to obtain the derivatives of 7 and 12 (Schemes 3 and 4). The first route involved the synthesis of isocyanate 10 from amine 6, followed by its reaction with the amines containing the RHS of the ureas, either directly (compounds 15 and 24–25), or, for the less nucleophilic amines, after deprotonation with a strong base such as n-butyllithium (compounds 26–29). The second pathway involved the reaction of amine 6 in the presence of triethylamine with the isocyanate derived from the different anilines containing the RHS of the urea (compounds 30–32).
Scheme 3.
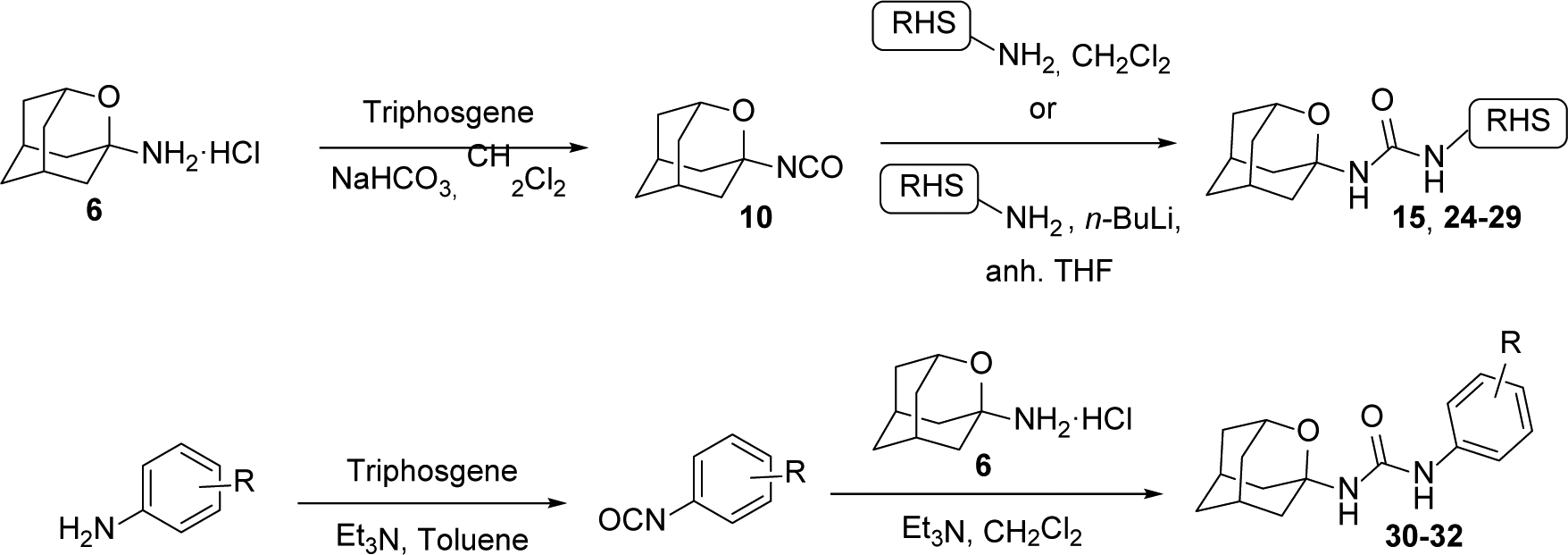
Synthetic procedures for the preparation of 2-oxaadamant-1-yl ureas.
Scheme 4.
Synthesis of new 2-oxaadamant-1-yl ureas.
aReagents and conditions: a) Triphosgene, sat. NaHCO3, DCM, 30 min; b) 1-benzylpiperidin-4-amine, DCM, overnight; c) H2, Pd/C, MeOH, conc. HCl, 5 days. d) Corresponding carboxylic acid, HOBt, EDCI·HCl, Et3N, EtOAc, 24 h; e) Corresponding sulfonyl chloride, Et3N, DCM, overnight; f) corresponding acyl chloride, Et3N, DCM, overnight; g) 2,4-dichloro-6-methyl-1,3,5-triazine, DIPEA, DCM, 30 min; h) Methylamine hydrochloride, DIPEA, DCM, 40 °C, 40 min; i) Corresponding amine, n-BuLi, anh. THF, overnight; j) Corresponding amine, DCM, overnight; k) Triphosgene, Et3N, toluene, 70 °C, 2 h; l) 6·HCl, Et3N, DCM, overnight. See Experimental Section and Supporting Information for further details.
The reaction of isocyanate 10 with commercially available 1-benzylpiperidin-4-amine furnished urea 15, which, upon debenzylation by catalytic hydrogenation in methanol, led to 16. The reaction of piperidine 16 with commercially available 2,4-dichloro-6-methyl-1,3,5-triazine furnished urea 17, which upon reaction with methylamine led to urea 18. Of note, the triazine unit is a feature of several sEH inhibitors disclosed by GSK, including its clinical candidate GSK2256294.31–32 The reaction of 16 with a series of acyl chlorides, sulfonyl chlorides or carboxylic acids provided the desired ureas 19–23 in moderate to good yields. Compound 24 was directly obtained from 10 and the commercially available 1-(4-aminopiperidin-1-yl)propan-1-one. Similarly, the reaction of 10 with 1-(4-(4-aminopiperidin-1-yl)phenyl)ethan-1-one, prepared as reported in the literature,36 furnished urea 25. When the amines were not nucleophilic enough to attack isocyanate 10, the presence of a strong base such as n-butyllithium was necessary in order to deprotonate the amine. The subsequent reaction of the anion with 10 led to ureas 26–29 in low to moderate yields (Scheme 4).
Finally, a different synthetic strategy was undertaken for the synthesis of the phenyl derivatives 30–32, which involved the treatment of 6 with the corresponding arylisocyanates in the presence of triethylamine. In turn, the intermediate isocyanates were either commercially available or synthesized from the reaction of the corresponding anilines with triphosgene, in the presence of triethylamine in hot toluene (Scheme 4).
sEH inhibition and structure-activity relationships.
As a first step for the biological characterization of the novel ureas, their potency as human and murine sEH inhibitors was tested (Tables 2 and 3). In line with the good activity of 7, compounds 30–32 were potent inhibitors of the human sEH, while the compounds featuring a heteroaromatic ring attached to the urea group, 27–29, were poor inhibitors of the human enzyme (IC50 > 500 nM). However, regardless of the presence of an aromatic or heteroaromatic ring as the RHS of the urea, all compounds were very poor inhibitors of the murine enzyme. For this reason, this set of derivatives was not further evaluated.
Table 2.
Human (hsEH) and murine (msEH) sEH IC50 of the aromatic and heteroaromatic series of 2-oxaadamant-1-yl ureas.
 | |||
|---|---|---|---|
| Compound | R | IC50 (nM)[a] | |
| hsEH | msEH | ||
| 7 |  |
21.3 | 553 |
| 27 | 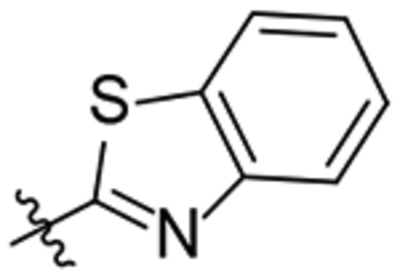 |
782 | 14721 |
| 28 | 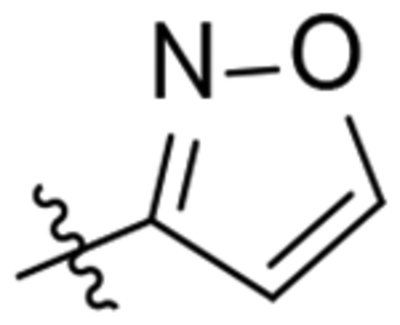 |
911 | 3853 |
| 29 | 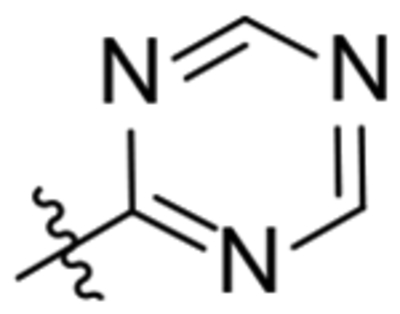 |
1448 | >100.000 |
| 30 | 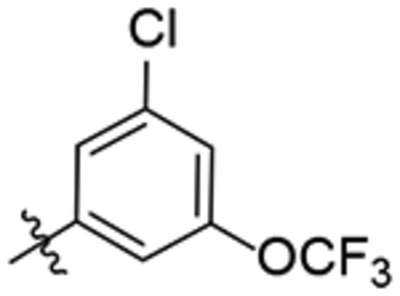 |
52.6 | 1857 |
| 31 | 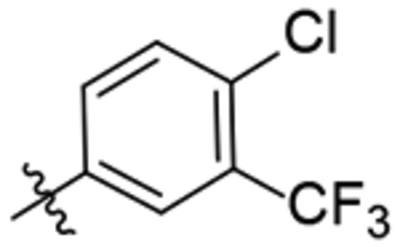 |
19.7 | 344 |
| 32 | 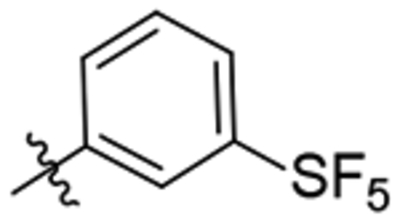 |
35.9 | 723 |
Reported IC50 values are the average of three replicates. The fluorescent assay as performed here has a standard error between 10 and 20% suggesting that differences of two-fold or greater are significant. Because of the limitations of the assay it is difficult to distinguish among potencies < 0.5 nM.25
Table 3.
Human (hsEH) and murine (msEH) sEH IC50 of the piperidine series of 2-oxaadamant-1-yl ureas.
 | |||
|---|---|---|---|
| Compound | R | IC50 (nM)[a] | |
| hsEH | msEH | ||
| 12 |  |
29.9 | 92.3 |
| 15 |  |
1093 | n.d.[b] |
| 18 |  |
496 | 365 |
| 19 | 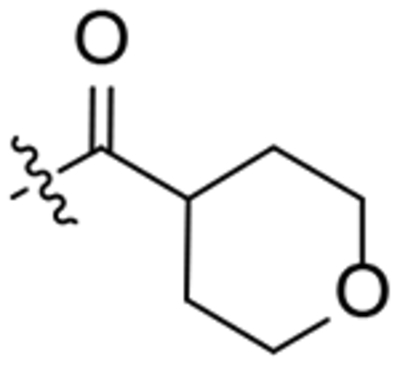 |
491 | 512 |
| 20 | 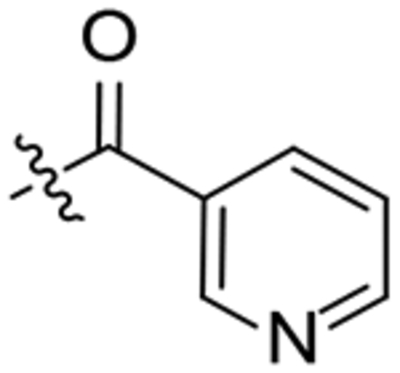 |
159 | 291 |
| 21 | 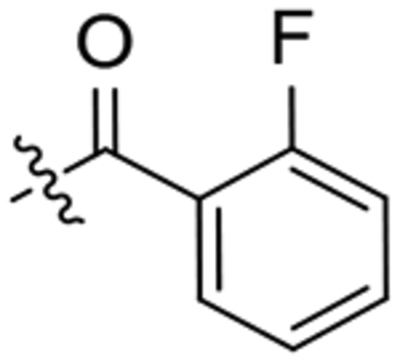 |
46 | 75.9 |
| 22 | 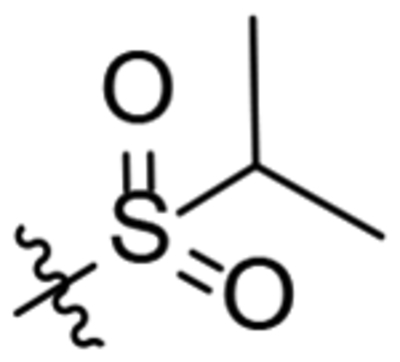 |
197 | 79.1 |
| 23 | 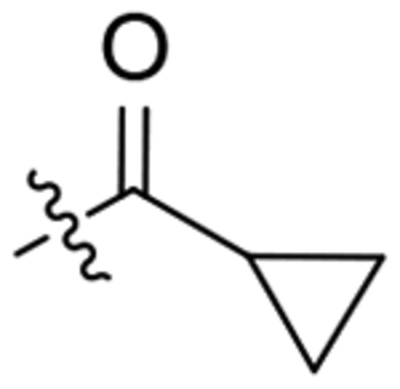 |
213 | 130 |
| 24 | 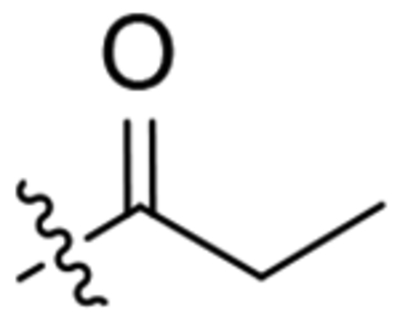 |
356 | 234 |
| 25 | 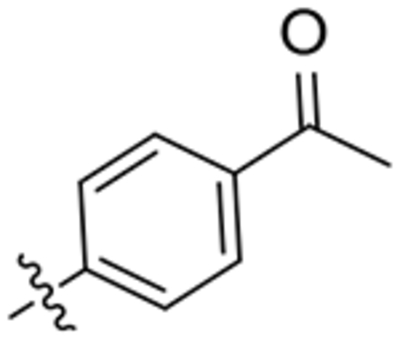 |
82.5 | 107 |
| 26 | 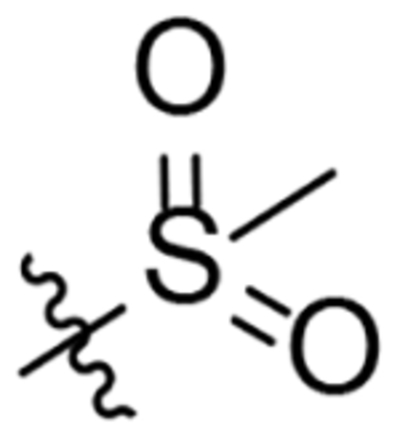 |
247 | 174 |
Reported IC50 values are the average of three replicates. The fluorescent assay as performed here has a standard error between 10 and 20% suggesting that differences of two-fold or greater are significant. Because of the limitations of the assay it is difficult to distinguish among potencies < 0.5 nM.25
n.d.: not determined.
Table 3 collects the structures and the IC50 values of ten analogs of 12. As previously seen with the analogs of 7, the introduction of the triazine group, 18, yielded a poor inhibitor of the human sEH. However, the introduction of an aryl derivative, 25, resulted into a two-digit nanomolar inhibitor. In the adamantane series of AR9281 derivatives the replacement of the methyl group of the acetyl unit in the piperidine by a broad range of alkyl and aryl substituents did not significantly impact the activity.15 By way of contrast, within our series of 2-oxaadamantane derivatives, subtle variations in the N-substituent of the piperidine, led to significant changes. Increasing the size of the alkyl group from the methyl of 12 to propyl (24), cyclopropyl (23) and tetrahydropyranyl (19) resulted in a 10–15-folds reduction of the potency. Also, while the introduction of a 2-fluorophenyl group (21) furnished a good sEH inhibitor, the 3-pyridyl unit, as in 20, caused a 3-folds drop in inhibitory activity. While the replacement of the acetyl group in AR9281 by a benzyl unit did not affect the inhibitory activity,15 the same change in the 2-oxaadamantane series (12 vs 15) led to an important reduction (almost 40-fold) of the potency (Table 3). Finally, the substitution of the acetyl group of 12 for two alkylsulfonyl groups, as in 22 and 26, yielded a six- and eight-fold reduction of the activity, respectively.
Furthermore, all the compounds were also evaluated as murine sEH inhibitors (Table 3). Although there were a few minor inconsistences, in general, the most potent compounds in the human sEH were also the most potent inhibitors of the murine sEH. Considering both the human and murine sEH inhibitory activity, compounds 12, 21, 22 and 25 were selected for further studies.
In silico study: Molecular basis of 2-oxaadamant-1-yl ureas as sEH inhibitors.
To elucidate the conformational and molecular basis of the inhibitory mechanism of 2-oxaadamant-1-yl ureas and, in particular, the molecular impact of the replacement of a methylene unit by an oxygen atom on the activity, Molecular Dynamics (MD) simulations of sEH were carried out in the presence of two selected compounds (22 and 26). The ability of sEH to recognize and properly bind both small and bulky inhibitors (as the ones depicted in Tables 1–3) will depend on the grade of plasticity of its active site. Indeed, some epoxide hydrolases present rich conformational dynamics of key structural elements surrounding the active site, which play a crucial role for recognizing bulky epoxide substrates and inhibitors of different size.37 To explore the conformational dynamics of sEH, and how compounds 22 and 26 are accommodated in the active site, three replicates of 250 ns of MD simulations were performed for four different systems: a) in the apo state; b) in the presence of compound 26 (IC50 = 247.2 nM) bound in the active site; c) with the inhibitor AR9273 (the hydrocarbon counterpart of 26, IC50 = 1.9 nM,13 PDB ID: 5ALZ38) bound; and d) with compound 22 (IC50 = 197.1 nM) present in the active site (see Figure 2). The changes on the active site volume along the MD simulations were monitored using the computational tool POcket Volume MEasurer (POVME)39 to capture the conformational plasticity of the sEH active site. In the apo state, the volume fluctuates between 70 and 500 Å3 providing an average value of 253 ± 62 Å3 (see Figure 2b). The high fluctuation of the active site volume in the apo state already suggests that inhibitors of different size can fit in the binding pocket. The visual analysis of the shape of the pocket clearly shows the presence of two hydrophobic pockets separated by catalytic Asp335 and Tyr383 residues (L-shaped hydrophobic pocket, see Figure 2c). The first hydrophobic pocket is located below the catalytic His524, which is known to accommodate the adamantane moiety, according to X-ray data (see Figure 2 and PDB ID: 5ALZ). The other pocket is located on the right-hand site of Asp335 and Tyr383 residues and encompasses also the hydrophobic entrance tunnel. The size of both pockets significantly changes along the MD simulations being the “adamantane” pocket the smallest most of the simulation time. When either compound 22 or 26 are bound in the active site, the average volume is 361 ± 43 Å3 and 324 ± 41 Å3 respectively, in line with the bulkier isopropylsulfonyl group of 22. As shown in Figure 2b, both inhibitors restrain the flexibility of the active site in comparison with the significant fluctuations observed in the apo state. MD simulations show that sEH presents a pocket with significant conformational plasticity and its breathing capability can be key to recognize and bind inhibitors of different sizes.40 Compounds 22 and 26 are both capable of restricting the inner flexibility of sEH active site residues keeping the active site blocked. The large active site volumes sampled indicate that there is room for further functionalization of both the oxaadamantyl moiety and the RHS of the urea.
Figure 2.
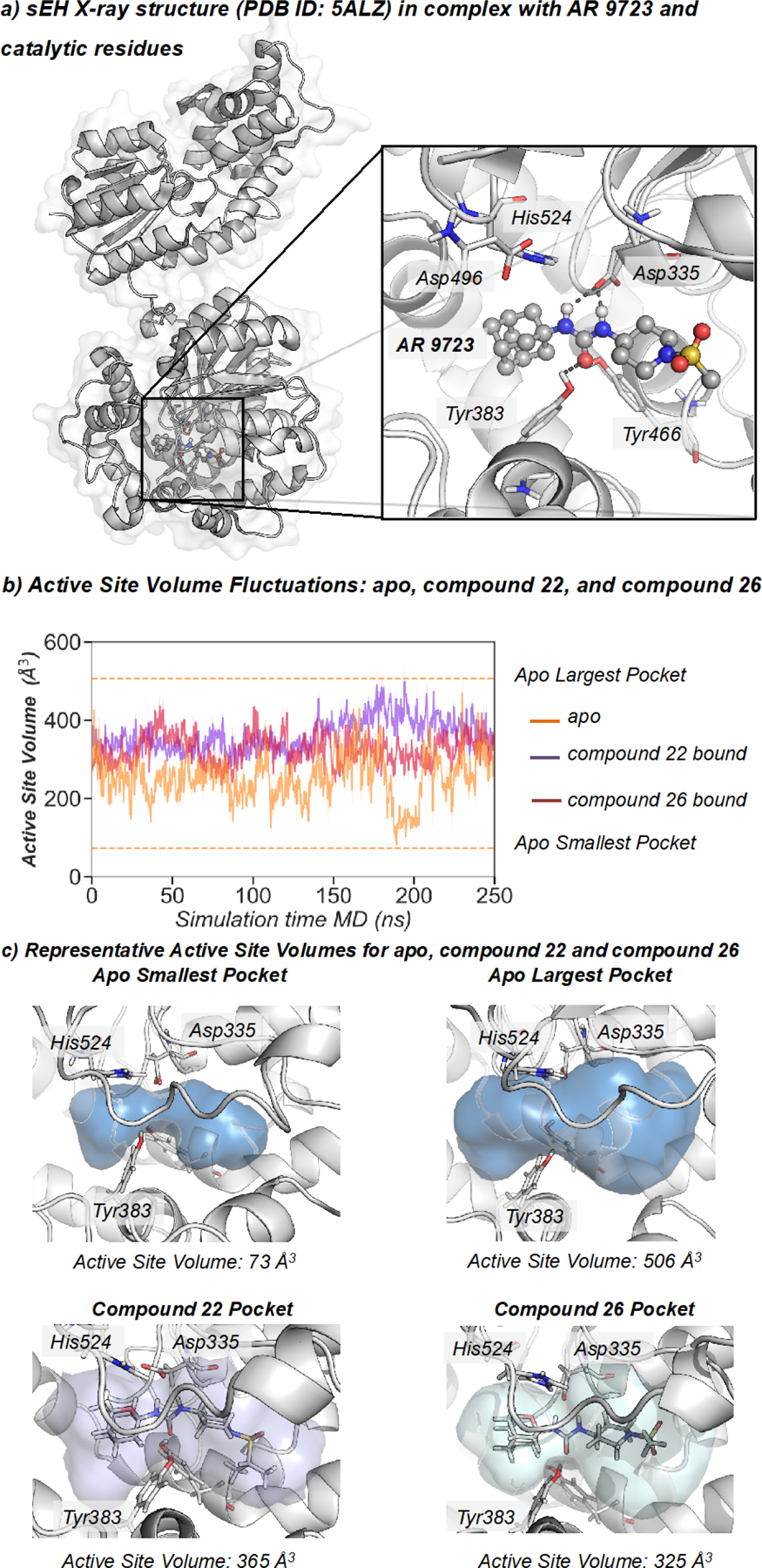
a) Representation of sEH structure (PDB: 5ALZ), active site catalytic residues (nucleophilic Asp335, Tyr383, Tyr466, and the histidine-aspartic acid pair His524-Asp496. b) Plot of the fluctuations of the active site volume for the apo state (orange line, 253 ± 62 Å3), compound 22 bound (purple line, 361 ± 43 Å3), and compound 26 bound (red line, 324 ± 41 Å3) along a representative 250 ns MD simulation trajectory. c) Representative sEH structures with the active site volume obtained from MD simulations of apo, 22, and 26.
To gain a deeper insight into the molecular basis of the inhibitory mechanism of 2-oxaadamantyl derivatives, we monitored key interactions between the three inhibitors (AR9273, 26, and 22) and relevant active site residues along the 250 ns MD simulations of sEH (see Figure 3). The three selected distances correspond to the one between the carboxylic group of the catalytic Asp335 and one of the NH groups of the inhibitor, and the distances between the carbonyl group of the urea inhibitors and the OH group of either Tyr383 or Tyr466 residues (Figure 3a). MD simulations show that the three hydrogen bonds remain significantly stable along the whole simulation time for the hydrocarbon based AR9273 (d(CγAsp335-NHAR 9273) = 2.55 ± 0.17 Å, d(OHTyr383-OAR 9273) = 1.76 ± 0.17 Å, and d(OHTyr466-OAR 9273) = 2.21 ± 0.83 Å). In the case of oxadamantane-based 22 and 26 compounds, the hydrogen bonds are less stable than for AR9273 and fluctuate along the simulation time indicating a lower affinity towards the active site of sEH (d(CγAsp335-NH26) = 2.66 ± 0.19 Å, d(OHTyr383-O26) = 2.01 ± 0.83 Å, d(OHTyr466-O26) = 2.67 ± 0.81 Å for 26 and d(CγAsp335-NH22) = 2.74 ± 0.40 Å, d(OHTyr383-O22) = 2.50 ± 1.54 Å, d(OHTyr466-O22) = 2.06 ± 0.71 Å for 22).
Figure 3.
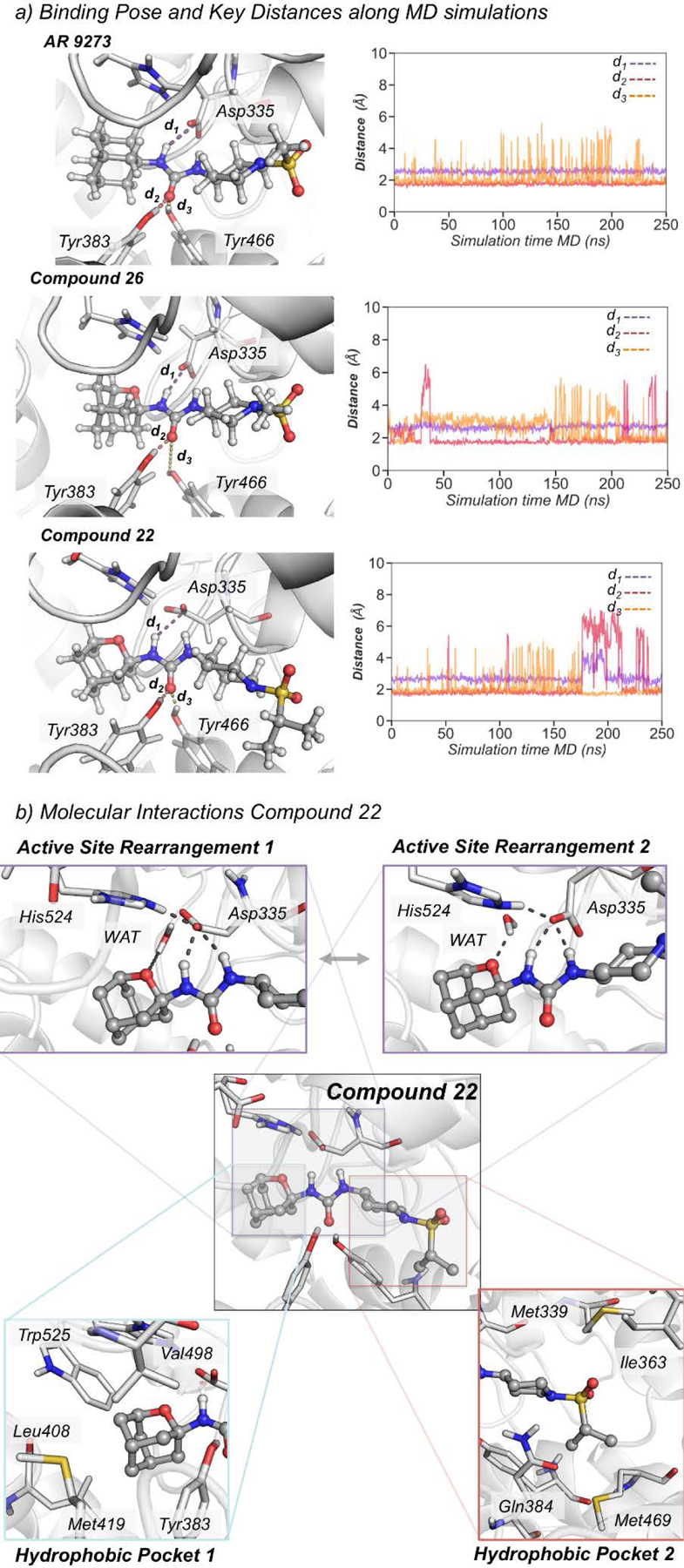
a) Representative structures of AR9273, 22, and 26 bound in the active site of sEH obtained from MD simulations. Plot of the distance between the carboxylic group of the catalytic Asp335 and the amide groups of the inhibitor (purple line, d1(CγAsp335-NHINH)), and the distances between the carbonyl group of the urea inhibitors and the OH group of either Tyr383 (red line, d2(OHTyr383-OINH)) or Tyr466 residues (orange line, d3(OHTyr466-OINH)) along the MD simulations of AR 9273 (d1(CγAsp335-NHAR 9273) = 2.55 ± 0.17 Å, d2(OHTyr383-OAR 9273) = 1.76 ± 0.17 Å, and d3(OHTyr466-OAR 9273) = 2.21 ± 0.83 Å), 22 (d1(CγAsp335-NH26) = 2.66 ± 0.19 Å, d2(OHTyr383-O26) = 2.01 ± 0.83 Å, d3(OHTyr466-O26) = 2.67 ± 0.81 Å), and 26 (d1(CγAsp335-NH22) = 2.74 ± 0.40 Å, d2(OHTyr383-O22) = 2.50 ± 1.54 Å, d3(OHTyr466-O22) = 2.06 ± 0.71 Å). b) Key molecular interactions between 22 and active site residues.
The simulations indicate that the adamantane group of AR9273 freely rotates inside the hydrophobic pocket establishing weak interactions with the surrounding Tyr383, Leu408, Met419, Val498, His524, and Trp525. This situation totally changes with the introduction of an oxygen atom into the adamantane scaffold, which restricts the rotation of the adamantane moiety inside the hydrophobic pocket introducing an entropic penalty that is partially compensated by more persistent interactions with surrounding residues (see the detailed analysis of non-covalent interactions in Figure S1). This restricted flexibility induces a dipole moment in the pocket that reshapes the interactions between catalytic residues (see Figure 3b). Interestingly, the oxaadamantane group rapidly orients to establish an interaction between the oxa group and both a water molecule and the positively charged His524 residue (see Figure 3b and Figure S1). The orientation towards the water molecule occupying the active site occurs in both 22 and 26 simulations. To unravel in more detail the network of interactions established when the oxygen group is present in the adamantane scaffold, we represented the non-covalent interactions (NCI) between compound 22 and active site residues computed with NCIplot (see Figure S1).41 The NCI analysis reveals a hydrogen bond interaction between the water molecule and the oxa group, and a wide ion dipole interaction surface between the oxygen and the protonated His524. Additionally, several hydrophobic interactions between the adamantane group and residues Phe267, Tyr383, Leu408, Leu417, Met419, and Trp525 are observed (see Figure S1). However, in the presence of AR9273, the water molecule establishes a network of hydrogen bonds with His524 and Glu269 that keeps Asp335 interacting with the NH groups of the inhibitor. In 22 and 26, this water molecule preferentially interacts with the oxaadamantane moiety rather than with His524, which establishes a hydrogen bond with Asp335 that has undergone a reorientation of its side chain (see active site rearrangements in Figure 3b). This destabilizes the interactions between Asp335 and the carbonyl group of the inhibitor, lowering the affinity of 22 and 26 for the sEH active site. To estimate the binding affinities, we computed the relative binding free energy using the thermodynamic integration (TI) method between compound 22 and its adamantane counterpart containing a methylene unit instead of an oxygen (22-CH2). Both ligands present a similar affinity towards sEH active site being the relative binding free energy of compound 22-CH2 with respect to compound 22 around −0.8 kcal/mol. These observations are in line with the obtained IC50 values that indicate slightly higher affinity for adamantane derivatives. Therefore, the introduction of an oxygen atom into the adamantane scaffold introduces a network of interactions that altogether restrict its orientation within the hydrophobic pocket having an impact on the affinity of oxaadamantane inhibitors for sEH.
Finally, we assessed the in silico lipophilicity of compounds 22 and 22-CH2 by computing the LogP from the solvation free energy differences using the M06–2X functional with SMD implicit solvation and the def2-SVP basis set.42 The computed LogP of compound 22 is 0.77 while the 22-CH2 is 2.44. In line with the above reported experimental solubilities, these results predict a significant decrease in lipophilicity when the oxygen atom is introduced in the adamantane scaffold of compound 22.
Biological profiling of the selected sEH inhibitors.
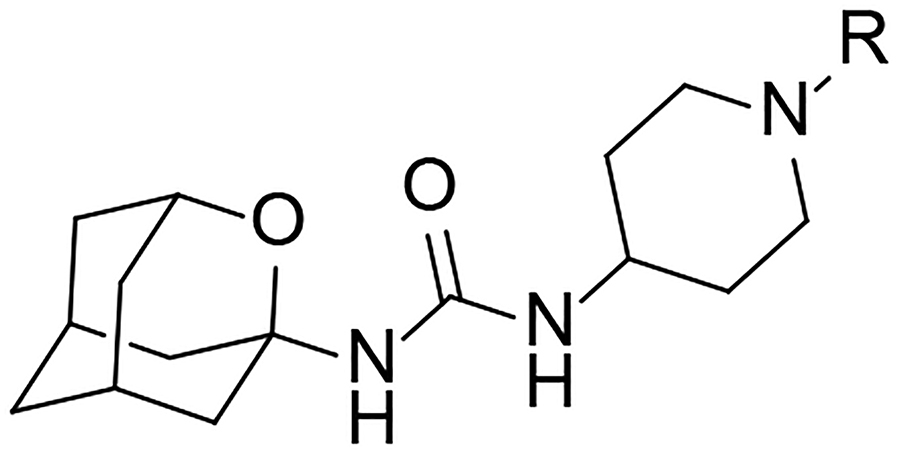
The four more potent inhibitors were characterized in terms of cytotoxicity in 2 different cell lines [Transformed Human Liver Epithelial-2 cell line (THLE-2) and Peripheral Blood Mononuclear Cells (PBMC)], solubility, microsomal stability (human, mouse and rat species), hERG (human ether-a-go-go-related gene) inhibition, cytochromes P450 (CYP) inhibition and predicted brain permeability, in order to select the best candidate to perform an in vivo study in a murine model of AP. None of the tested compounds showed to be cytotoxic for the THLE-2 at the highest concentration tested (100 μM). Additionally, compounds 12, 21 and 22 were not cytotoxic in the PBMC at the highest concentration tested (20 μM) (Table 4).
Table 4.
Cytotoxicity (in THLE-2 and PBMC cell lines), PAMPA-BBB, solubility and microsomal stability values of selected compounds.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R | Cytotoxicity LC50 (μM) | pampa-BBB | Solubility[a] (μM) | Microsomal stability[b] | |||
| PBMC | THLE-2 | Human | Mouse | Rat | ||||
| 12 |  |
>20 | >100 | CNS− | 59 | 88.8 | 80.5 | 85.1 |
| 21 | 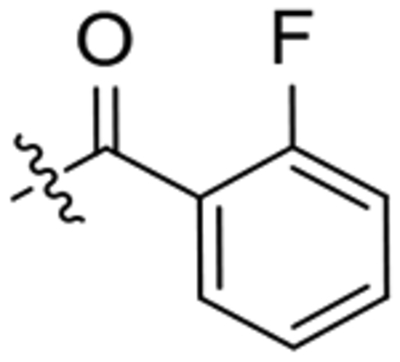 |
>20 | >100 | CNS− | >100 | 30.2 | 39.5 | 19.3 |
| 22 | 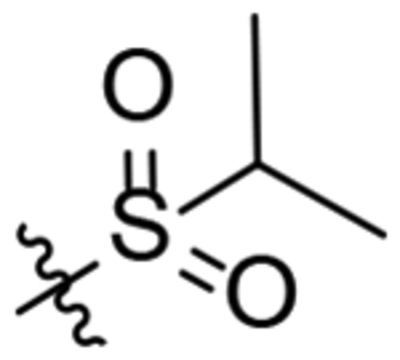 |
>20 | >100 | CNS− | 34 | 72.7 | 63.9 | 63.9 |
| 25 | 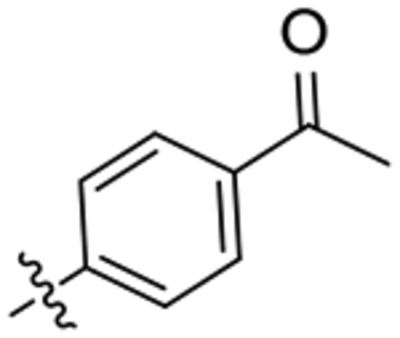 |
ND | >100 | CNS+ | 81.5 | 40 | 73 | 84 |
Solubility in a 1% DMSO : 99% PBS buffer solution.
Percentage of remaining compound after 60 min of incubation with human, mice and rat microsomes in the presence of NADPH at 37 °C. Metabolism of testosterone was used as a positive control. See Experimental Section for details.
Taking into account that AP is a peripherally restricted inflammatory disease, we wanted to prioritize compounds showing negative BBB permeation in order to avoid potential side effects resulting from central inhibition of sEH. The selected compounds were further tested for predicted brain permeation in the widely used in vitro parallel artificial membrane permeability assay-blood brain barrier (PAMPA-BBB) model.43 With the sole exception of 25, all the compounds have predicted limited BBB penetration (Table 4).
Next, the solubility was experimentally determined in a 1% DMSO: 99% PBS buffer solution. In the adamantane series, when the methyl group of the amide of AR9281 was replaced by bigger substituents, the potency was retained or even increased, but the solubility dramatically dropped.15 Pleasantly, all the oxaadamantanes presented good to excellent solubilities (see Table 4), greater than that of the adamantane-based inhibitors 1-3, as expected.
The microsomal stability of 12, 21, 22 and 25 was evaluated in human, mice and rat liver microsomes, which are widely used to determine the likely degree of primary metabolic clearance in the liver. Although compound 21 displayed unsatisfactory stabilities in the three species, and 25 presented poor stability in human microsomes, compounds 12 and 22 showed good microsomal stabilities in the three species (see Table 4). Furthermore, the study of cytochromes P450 (CYP) inhibition was performed in order to examine the inhibitory potency of 12 and 22, the compounds with better overall microsomal stabilities, against human recombinant cytochrome P450 enzymes, mainly CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3AE, through a fluorescence-detection method. These assays were of great interest, not only for the detection of possible drug-drug interactions, but also in terms of selectivity, as EETs are formed by cytochrome P450 isoforms, especially CYP2C19, followed by CYP1A2 and CYP2J2 to a lesser extent. Satisfactorily, the tested compounds showed very low potency (higher than 10 μM) inhibiting the different cytochromes tested (see Table S2 in the Supporting Information). This potency is much lower (more than 100-fold) than the potency observed either in human or murine sEH. Inhibition of the hERG channel is an important toxicology screen due to its known association with cardiotoxicity. Inhibitors 12 and 22 showed minimal inhibition on hERG at 10 μM (see Table S2 in Supporting Information). Therefore, the selected inhibitors are considered not to present a risk with CYP or hERG inhibition. Finally, three representative inhibitors were tested for selectivity against hCOX-2 and hLOX-5, two enzymes involved in the AA cascade. Neither the N-acyl piperidines 21 and 24 nor the N-sulfonyl piperidine 22 displayed significant inhibition of these enzymes (see Table S3 in Supporting Information).
In vitro proof of concept: Acute Pancreatitis.
AP is a serious and life-threatening inflammatory disease, and one of the most common gastrointestinal disorders worldwide without specific therapies available.44–46 Recently, it was shown that the EPHX2 whole-body knockout (KO) mice exhibit attenuated cerulein- and arginine-induced AP,33 and that pharmacological inhibition of sEH can modulate the severity of AP before and after induction of disease, due to the potent anti-inflammatory properties of the EETs and the reduction of the endoplasmic reticulum (ER) stress.34
In order to select a candidate for in vivo studies and verify the positive effect of the new sEH inhibitors on AP, we performed an in vitro proof of concept in the AR42J pancreatic acinar rat cell line that expresses sEH,47 a well-stablished model for evaluating the potential therapeutic activity of compounds for AP.48–49 Given that this is a rat-derived cell line and the inhibitory activity of 12 and 22 was previously evaluated for the human and murine enzymes (see above), we first confirmed that the selected inhibitors presented potency in the rat enzyme. Both compounds inhibited the rat sEH, being compound 22 slightly more potent than 12 (IC50 of 1.9 nM and 3.3 nM, respectively).
For the in vitro study of efficacy, AR42J cells were treated with cerulein (Cer), a well-known inducer of AP, and with the selected compounds, following an established procedure.49 Inflammatory (IL-6 and TNF-α) and ER stress (CHOP and TRB3) markers were measured (Figure 4). As expected, cells treated with cerulein (10 nM) showed an increase in the expression of inflammatory and ER stress markers, while co-incubation of the cells with cerulein and the sEH inhibitors 12 and 22 (60 μM) caused a significant reduction in the mRNA levels of these markers. Notably, compound 22 completely abolished the increase in the expression of IL-6, CHOP and TRB3, and strongly attenuated the increment in TNF-α caused by cerulein exposure. Consequently, compound 22 was chosen for the in vivo efficacy studies in two mice models of AP. Overall, 22 has low nanomolar inhibition of the murine sEH, does not exhibit cytotoxicity, does not inhibit cytochrome P450s nor hERG, has good microsomal stability, predicted poor brain penetration and has a reasonable solubility value.
Figure 4:
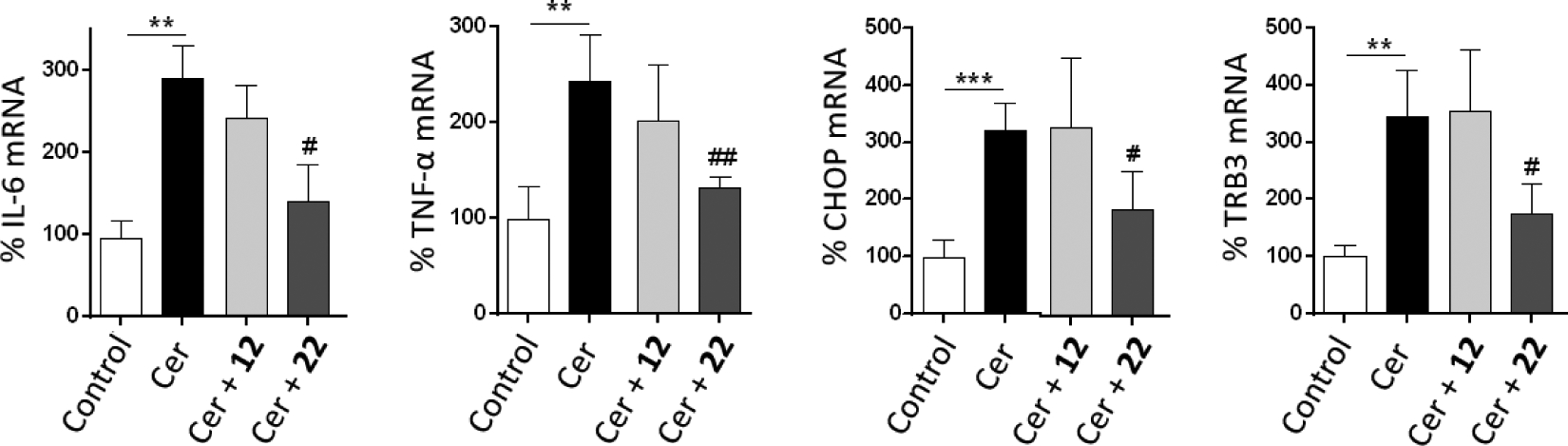
Results of the in vitro study of cerulein-induced AP in AR42J pancreatic rat cells. IL-6, TNFa, CHOP and TRB3 mRNA levels are presented as the mean ± SEM (n = 5 per group). **p<0.01, ***p<0.001 vs. control. #p<0.05, ##p<0.01 vs. Cer.
Pharmacokinetic study of 22.
To determine its pharmacokinetic profile in mouse, compound 22 was administered either by intravenous (iv) or intraperitoneal (ip) routes at a single dose of 1 mg/kg (see Figure S2 and Tables S4 and S5 in the supporting information). Pharmacokinetic parameters are shown in Table 5. For both administration routes, Tmax and Cmax values were similar. Good exposures of compound 22 were obtained, indicating bioavailability of 59%. High volumes of distribution and clearance were obtained with both administration routes, indicating that while 22 is well distributed across all tissues it is eliminated quite fast. Considering the results obtained, 22 was administered ip in the subsequent in vivo studies.
Table 5:
Pharmacokinetic parameters in mouse for ip and iv administration routes of compound 22 at 1 mg/kg.
| Route | T1/2 | Tmax | Cmax | C0 | AUClast | AUCINF_obs | Vz | Cl |
|---|---|---|---|---|---|---|---|---|
| (h) | (h) | (ng/mL) | (ng/mL) | (h·ng/mL) | (h·ng/mL) | (L/kg) | (L/h/kg) | |
| IP | 3.02 | 0.16 | 117.33 | - | 34.67 | 35.20 | 124.02 | 28.41 |
| IV | 2.80 | 0.16 | 108.33 | 236.23 | 59.1 | 59.63 | 67.73 | 16.77 |
Maximum Tolerated Dose (MTD) study of 22.
In order to determine potential acute toxicological effect, compound 22 was intraperitoneally daily administered to mice at variable doses (5, 10, 20 and 80 mg/kg) for five consecutive days. No toxic effects were observed based on body weight along the treatment, as well as the absence of apparent behavior problems and petechiae, indicating that compound 22 is well tolerated.
In vivo efficacy study.
To determine the effects of the administration of compound 22 on AP, C57BL/6J mice were treated with this sEH inhibitor before (prevention) and after (treatment) the induction of AP by cerulein, following already published protocols (see Figure S3 in the Supporting Information and the Experimental Section).34 The post-induction efficacy study was performed in order to simulate a treatment study. In this protocol, cerulein was administered several times before the administration of compound 22.
Results of the post-induction efficacy study are shown in Figure 5. In line with previous reports,34 mice treated with cerulein presented a significant increase in the mRNA abundance of inflammatory (TNF-α, IL-1β and MCP-1) and ER stress (ATF3) markers. Albeit the administration of compound 22 at 3 mg/kg did not cause a significant reduction of inflammatory and ER stress markers, administration of 30 mg/kg produced a significant reduction in the four markers studied compared to mice treated only with cerulein.
Figure 5:
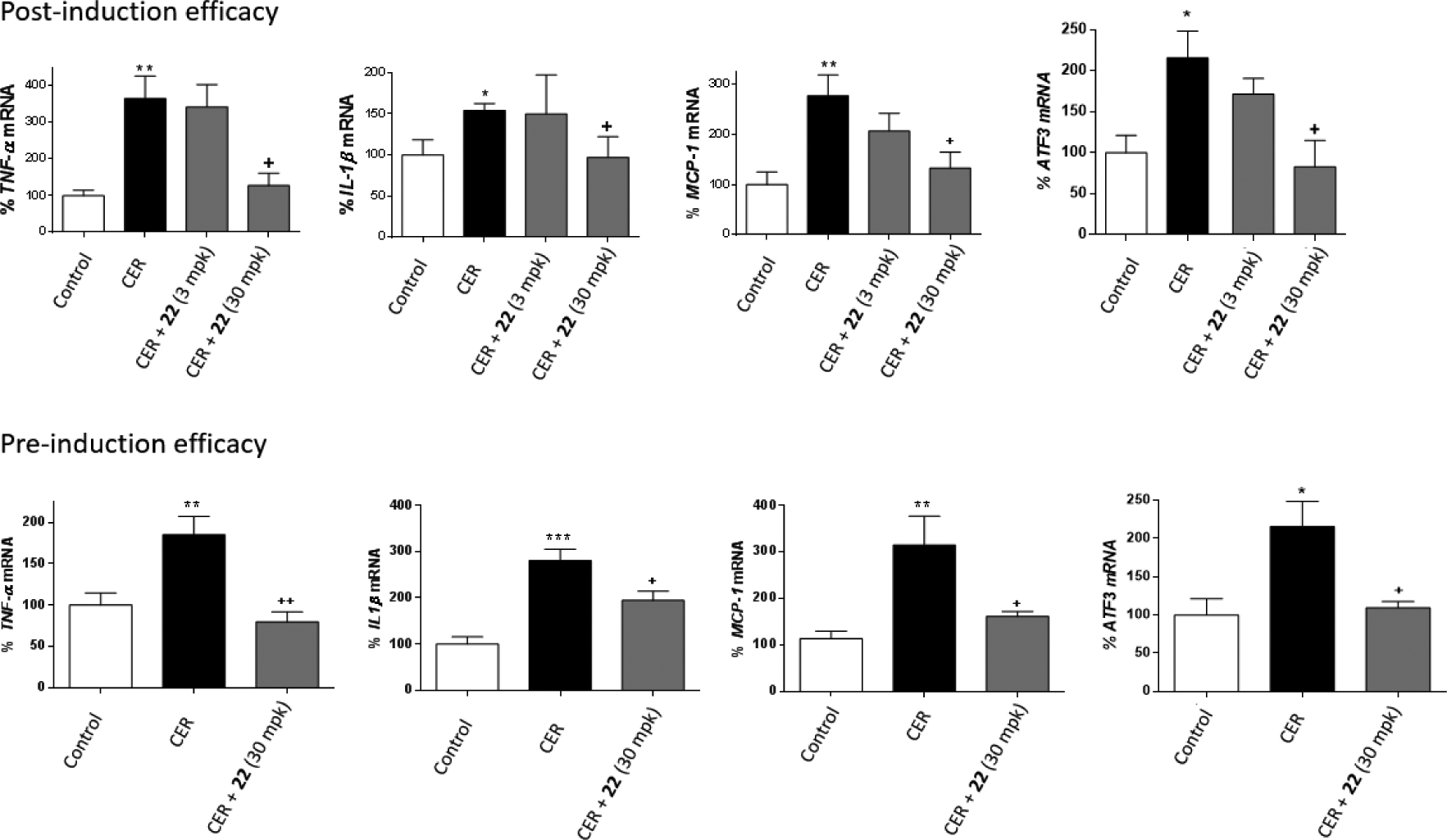
Results of the in vivo studies of cerulein-induced AP in the post- and pre-induction models. TNFa, IL-1β, MCP-1 and ATF3 mRNA levels are presented as the mean ± SEM (n = 5 per group). *p<0.05, **p<0.01, ***p<0.001 vs. control. +p<0.05, ++p<0.01 vs. cerulein (CER).
Considering that the dosage of 30 mg/kg was the one that presented significant effects on the post-induction model, the pre-induction efficacy study was performed only at 30 mg/kg. Results (Figure 5) indicate that administration of 22 significantly reduced an increase of inflammatory (TNF-α, IL1-β and MCP-1) and ER stress (ATF3) markers produced by cerulein administration. Interestingly, AP is the most common complication of the endoscopic retrograde cholangiopancreatography (ERCP), a well-established technique for the treatment of pathological conditions of the biliary tract and pancreas. ERCP-related AP occurs in up to 9% of ERCPs in unselected prospective studies. The severity of post-ERCP pancreatitis can range from mild disease with full recovery to critical illness with necrotizing pancreatitis, multiorgan failure, prolonged hospitalization, and even death.50 The efficacy of pre-induction compound 22 on AP suggests that such treatment might be of interest to patients before the ERCP procedure, in order to diminish the probabilities of suffering of AP as a complications of this classic procedure.
Histopathology.
Additionally, the severity of cerulein-induced pancreatitis in control and sEH inhibitor-treated mice was determined by histologic analyses. Pathologic changes were studied on H&E-stained pancreas sections from control, cerulein-treated and cerulein- and 22-treated mice in both the pre-induction and post-induction AP models. As anticipated, the administration of cerulein caused a significant increase in edema and inflammation. In contrast, mice treated with compound 22 after or before the induction of AP exhibited a significant decrease in cerulein-induced edema and inflammation (Figure 6).
Figure 6.
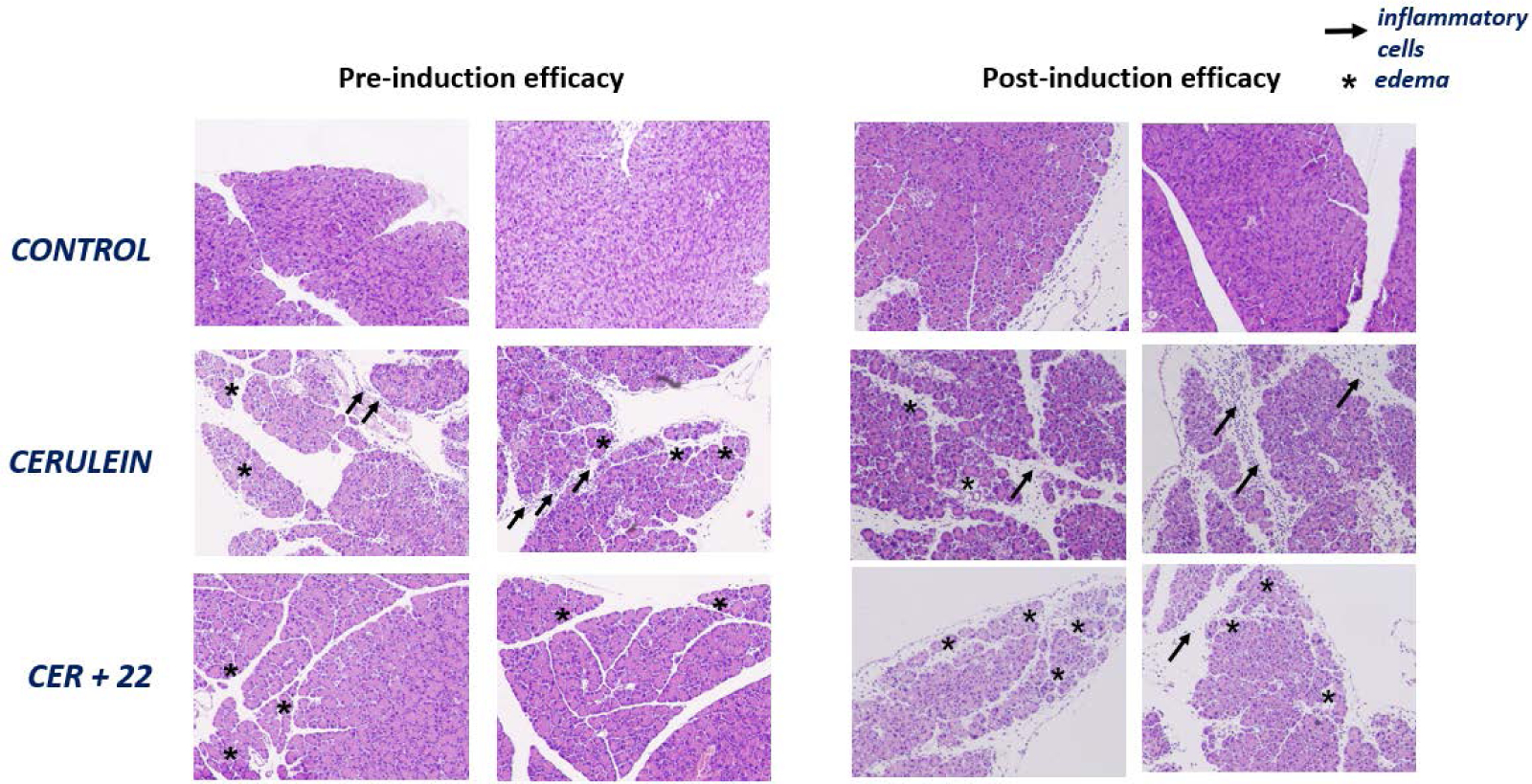
Representative H&E-stained sections of the pancreas from the pre-induction (left) and post-induction (right) efficacy studies. CER: cerulein. Arrows indicate the presence of inflammatory cells and asterisks the presence of edema.
CONCLUSIONS
Several sEH inhibitors feature an adamantane moiety in their structure, however, its lipophilicity negatively compromises the solubility and PK properties of these compounds. In this work, a new family of sEH inhibitors bearing a 2-oxaadamantane moiety in order to improve those characteristics were synthesized. Most of the compounds were nanomolar inhibitors of the human and murine sEH, although somehow less potent than their adamantane counterparts. Molecular modeling suggests that the introduction of the oxygen atom into the adamantane scaffold restricts its orientation within the hydrophobic pocket and weakes hydrogen bonds between the inhibitor and the enzyme. Biological profiling (solubility, cytotoxicity, metabolic stability, predicted BBB penetration, CYP450s and hERG inhibition) allowed us to select compound 22 for in vivo studies in a murine model of cerulein-induced AP. In both pre-induction and post-induction studies, 22 diminished the overexpression of inflammatory and ER stress markers induced by cerulein and reduced pancreatic damage. Due to the promising biological activity of 22, further optimization of new sEH inhibitors for the treatment of AP, an unmet medical need, is underway.
Finally, the adamantane unit is a typically used polycyclic scaffold in many drug discovery programs.51–52 It is already present in nine clinically approved drugs and in many clinical candidates. Nonetheless, the use of its oxapolycyclic analogs has largely been neglected in drug discovery. This work may pave the way for the evaluation of less lipophilic scaffolds in several other targets, currently being explored with adamantanes.
EXPERIMENTAL SECTION
Chemical Synthesis. General methods.
Commercially available reagents and solvents were used without further purification unless stated otherwise. Preparative normal phase chromatography was performed on a CombiFlash Rf 150 (Teledyne Isco) with pre-packed RediSep Rf silica gel cartridges. Thin-layer chromatography was performed with aluminum-backed sheets with silica gel 60 F254 (Merck, ref 1.05554), and spots were visualized with UV light and 1% aqueous solution of KMnO4. Melting points were determined in open capillary tubes with a MFB 595010M Gallenkamp. 400 MHz 1H and 100.6 MHz 13C NMR spectra were recorded on a Varian Mercury 400 or on a Bruker 400 Avance III spectrometers. 500 MHz 1H NMR spectra were recorded on a Varian Inova 500 spectrometer. The chemical shifts are reported in ppm (δ scale) relative to internal tetramethylsilane, and coupling constants are reported in Hertz (Hz). Assignments given for the NMR spectra of selected new compounds have been carried out on the basis of DEPT, COSY 1H/1H (standard procedures), and COSY 1H/13C (gHSQC and gHMBC sequences) experiments. IR spectra were run on Perkin-Elmer Spectrum RX I, Perkin-Elmer Spectrum TWO or Nicolet Avatar 320 FT-IR spectrophotometers. Absorption values are expressed as wave-numbers (cm−1); only significant absorption bands are given. High-resolution mass spectrometry (HRMS) analyses were performed with an LC/MSD TOF Agilent Technologies spectrometer. The elemental analyses were carried out in a Flash 1112 series Thermofinnigan elemental microanalyzator (A5) to determine C, H, N and S. The structure of all new compounds was confirmed by elemental analysis and/or accurate mass measurement, IR, 1H NMR and 13C NMR. The analytical samples of all the new compounds, which were subjected to pharmacological evaluation, possessed purity ≥95% as evidenced by their elemental analyses or their HPLC/MS. HPLC/MS were determined with a HPLC Agilent 1260 Infinity II LC/MSD coupled to a photodiode array and mass spectrometer. 5 μL of sample 0.5 mg/mL in methanol:acetonitrile were injected, using a Agilent Poroshell 120 EC-C18, 2.7 μm, 50 mm × 4.6 mm column at 40 °C. The mobile phase was a mixture of A = water with 0.05% formic acid and B = acetonitrile with 0.05% formic acid, with the method described as follows: flow 0.6 mL/min, 5%B-95%A 3 min, 100%B 4 min, 95%B-5%A 1 min. Purity is given as % of absorbance at 210 or 254 nm.
1-(2-oxaadamant-5-yl)-3-(2,3,4-trifluorophenyl)urea, 4.
In a round-bottom flask equipped with a stir bar under nitrogen atmosphere 1.2 equiv. of the 2-oxaadamantyl-1-amine hydrochloride was added to anh. DCM (~110 mM). To this suspension 1.0 equiv. of 2,3,4-trifluorophenylisocyanate followed by 7 equiv. of triethylamine was added. The reaction mixture was stirred at room temperature overnight. Then, the solvent was removed under vacuum and the resulting crude was purified by column chromatography (SiO2, hexane/EtOAc mixtures). Evaporation under vacuum of the appropriate fractions gave urea 4 (24 mg, 83% yield) as a white solid. Mp 211–213 °C. IR (ATR) ν: 3344, 2937, 2856, 2478, 2362, 1681, 1621, 1556, 1504, 1473, 1434, 1402, 1361, 1320, 1288, 1236, 1183, 1146, 1107, 1076, 1035, 1020, 1005, 992, 948, 921, 890, 810, 793, 775, 761, 724, 686, 656, 636 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.66 [dm, J = 12.8 Hz, 2 H, 8(10)-Ha], 1.98 [dm, J = 12.4 Hz, 2 H, 8(10)-Hb], 2.06 [d, J = 12 Hz, 2 H, 4(9)-Ha], 2.14–2.21 [complex signal, 4 H, 4(9)-Ha, 6-H2], 2.25 (m, 1 H, 7-H), 4.17 [broad s, 2 H, 1(3)-H], 7.01 (m, 1 H, 5’-H), 7.68 (m, 1 H, 6’-H). 13C-NMR (100.6 MHz, CD3OD) δ: 28.8 (CH, C7), 36.0 [CH2, C8(10)], 41.2 [CH2, C6], 41.8 [CH2, C4(9)], 50.6 (C, C5), 70.9 [CH, C1(3)], 112.2 (CH, dd, 2JC-F = 17.8 Hz, 3JC-F = 3.7 Hz, C5’), 116.8 (CH, C6’), 126.8 (C, d, 2JC-F = 6.5 Hz, Ar-C1’), 141.0 (C, d, 1JC-F = 247 Hz, Ar-C3’), 143.5 (C, d, 1JC-F ~ 245 Hz, Ar-C4’), 147.5 (C, d, 1JC-F = 242 Hz, Ar-C2’), 156.2 (C, CO). MS (DIP), m/z (%); significant ions: 173 [(C7H2F3NO)·+, 4], 149 (100), 148 (62), 147 (16), 137 [(C9H13O)+, 8], 93 (49). HRMS: Calculated for [C16H17F3N2O2-H]−: 325.1169. Found: 325.1173.
1-(2-oxaadamant-1-yl)-3-(2,3,4-trifluorophenyl)urea, 7.
In a round-bottom flask equipped with a stir bar under nitrogen atmosphere 1.2 equiv. of the 2-oxaadamantyl-1-amine hydrochloride was added to anh. DCM (~110 mM). To this suspension 1.0 equiv. of 2,3,4-trifluorophenylisocyanate followed by 7 equiv. of triethylamine was added. The reaction mixture was stirred at room temperature overnight. Then the solvent was removed under vacuum. Purification by column chromatography (SiO2, hexane/EtOAc mixtures) of the crude and evaporation under vacuum of the appropriate fractions gave urea 7 (163 mg, 94% yield) as a white solid. Mp 196–198 °C. IR (ATR) ν: 3300–2800 (3293, 3232, 3127, 2933, 2857), 1702, 1640, 1621, 1563, 1509, 1489, 1471, 1446, 1373, 1349, 1340, 1317, 1294, 1257, 1239, 1227, 1200, 1165, 1117, 1099, 1080, 1020, 996, 976, 963, 932, 912, 884, 840, 805, 788, 757, 683, 653 cm−1. 1H-NMR (500 MHz, CD3OD) δ: 1.68 [dm, J = 12.5 Hz, 2 H, 4(10)-Ha], 1.89 [complex signal, 4 H, 6-H2, 8(9)-Ha], 2.00 [dm, J = 12.5 Hz, 2 H, 4(10)-Hb], 2.26 [broad signal, 2 H, 5(7)-H], 2.32 [d, J = 12.5 Hz, 8(9)-Hb], 4.23 (broad s, 1 H, 3-H), 7.03 (m, 1 H, 5’-H), 7.75 (m, 1 H, 6’-H). 13C-NMR (125.7 MHz, CD3OD) δ: 29.6 [CH, C5(7)], 35.6 [CH2, C4(10)], 35.8 (CH2, C6), 40.9 [CH2, C8(9)], 72.5 (CH, C3), 82.2 (C, C1), 112.3 (CH, dd, 2JC-F = 18 Hz, 3JC-F = 4 Hz, C5’), 116.8 (CH, C6’), 126.5 (C, d, 2JC-F = 8.8 Hz, Ar-C1’), 141.0 (C, dt, 1JC-F = 246 Hz, 2JC-F = 15 Hz, Ar-C3’), 143.8 (C, dd, 1JC-F = 244 Hz, 2JC-F = 11 Hz, Ar-C4’), 147.6 (C, dd, 1JC-F = 244 Hz, 2JC-F = 9 Hz, Ar-C2’), 156.0 (C, CO). HRMS: Calculated for [C16H17F3N2O2+H]+: 327.1315. Found: 327.1319.
1-(4-oxahexacyclo[5.4.1.02,6.03,10.05,9.08,11]dodecan-3-yl)-3-(2,3,4-trifluorophenyl)urea, 9.
In a round-bottom flask equipped with a stir bar under nitrogen atmosphere 1.2 equiv. of the 2-oxaadamantyl-1-amine hydrochloride was added to anh. DCM (~110 mM). To this suspension 1.0 equiv. of 24-trifluorophenyl isocyanate followed by 7 equiv. of triethylamine was added. The reaction mixture was stirred at room temperature overnight. Then the solvent was removed under vacuum. Purification by column chromatography (SiO2, hexane/EtOAc mixtures) of the crude and evaporation under vacuum of the appropriate fractions gave urea 9 (374 mg, 94% yield) as a white solid. Mp 152–154 °C. IR (ATR) ν: 3329, 3192, 3118, 2975, 1701, 1641, 1621, 1559, 1509, 1470, 1342, 1321, 1287, 1253, 1205, 1183, 1166, 1127, 1071, 1020, 996, 951, 933, 911, 867, 839, 798, 757, 708, 688, 655, 635, 593 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.60 (dt, J = 10.8 Hz, J’ = 1.6 Hz, 1 H, 12-Ha), 1.95 (dt, J = 10.8 Hz, J’ = 1.6 Hz, 1 H, 12-Hb), 2.50 (broad t, J = 5.2 Hz, 1 H, 7-H), 2.57 (t, J = 4.8 Hz, 1 H, 1-H), 2.66 (m, 8 H, 2-H), 2.75–2.84 (complex signal, 3 H, 2-H, 10-H, 11-H), 2.88–3.01 (complex signal, 2 H, 6-H, 9-H), 4.83 (t, J = 5.2 Hz, 1 H, 5-H), 5.94 (broad s, 1 H, 1-NH), 6.90 (m, 1 H, 5’-H), 7.82 (m, 1 H, 6’-H), 8.06 (broad s, 1 H, 3-NH). 13C-NMR (100.6 MHz, CDCl3) δ: 41.4 (CH, C8), 41.6 (CH, C11), 43.2 (CH, C1), 43.5 (CH2, C12), 44.6 (CH, C9), 44.9 (CH, C7), 46.3 (CH, C10), 54.8 (CH, C6), 57.0 (CH, C2), 84.1 (CH, C5), 103.1 (C, C3), 111.3 (CH, dd, 2JC-F = 17.7 Hz, 3JC-F = 3.9 Hz, C5’), 115.2 (CH, m, C6’), 124.5 (C, dd, 2JC-F = 8 Hz, 3JC-F = 3.5 Hz, C1’), 139.8 (C, ddd, 1JC-F = 252 Hz, 2JC-F = 16.4 Hz, 2JC-F = 13.7 Hz, C3’), 142.3 (C, ddd, 1JC-F = 251 Hz, 2JC-F = 11.7 Hz, 3JC-F = 3.2 Hz, C4’), 146.6 (C, ddd, 1JC-F = 245.8 Hz, 2JC-F = 10 Hz, 3JC-F = 2.7 Hz, C2’), 154.7 (C, CO). MS (DIP), m/z (%); significant ions: 348 (M·+, 18), 173 [(C7H2F3NO)·+, 17], 159 [(C11H11O)+, 35], 147(100), 146(13), 131(24), 91(15). HRMS: Calculated for [C18H15F3N2O2+H]+: 349.1158. Found: 349.1160.
2-oxaadamant-1-yl isocyanate, 10.
Triphosgene (940 mg, 3.16 mmol) was added in a single portion to a solution of 2-oxaadamantyl-1-amine hydrochloride (1.20 g, 6.32 mmol) in DCM (42 mL) and saturated aqueous NaHCO3 solution (17 mL). The resulting biphasic mixture was stirred at room temperature for 30 min. Thereafter, the two phases were separated and the organic layer was washed with brine, dried over anh. Na2SO4 and filtered. Evaporation under vacuum below 30 °C provided isocyanate 10 as a yellow gum, which was used in the next step without further purification.
1-(1-acetylpiperidin-4-yl)-3-(oxaadamant-1-yl)urea, 12.
Under anhydrous conditions, a solution of isocyanate 10 (323 mg, 1.80 mmol) in anh. DCM (20 mL) was added to a solution of 1-acetyl-4-aminopiperidine (308 mg, 2.16 mmol) in anh. DCM (10 mL), followed by triethylamine (0.50 mL, 3.61 mmol). The reaction mixture was stirred at room temperature overnight. The solution was then concentrated under vacuum to give an orange gum (720 mg). Purification by column chromatography (SiO2, DCM/methanol mixtures) gave urea 12 (300 mg, 52% yield) as a white solid. The analytical sample was obtained by washing with pentane. Mp 172–173 °C. IR (ATR) ν: 3322, 2920, 2850, 2188, 2153, 2000, 1637, 1549, 1428, 1369, 1313, 1264, 1234, 1192, 1139, 1090, 1046, 995, 959, 879, 816, 773, 731 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.28–1.41 (complex signal, 2 H, 3-Hax and 5-Hax), 1.58–1.96 (complex signal, 9 H, 8’-Ha, 9’-Ha, 4’-H2, 10’-H2, 6’-H2, and 3-Heq or 5-Heq), 1.98–2.22 (complex signal, 6 H, COCH3, 8’-Hb, 9’-Hb and 5-Heq or 3-Heq), 2.27 [broad s, 2 H, 5’(7’)-H], 2.86 (dt, J = 11.2 Hz, J’ = 2.8 Hz, 1 H, 6-Hax or 2-Hax), 3.18 (dt, J = 10.8 Hz, J’ = 2.8 Hz, 1 H, 2-Hax or 6-Hax), 3.70 (dm, J = 14 Hz, 1 H, 2-Heq or 6-Heq), 3.87 (m, 1 H, 4-H), 4.27 (broad s, 1 H, 3’-H), 4.35 (broad d, J = 14.0 Hz, 1 H, 6-Heq or 2-Heq), 4.75 (s, 1 H, 3-NH), 6.06 (d, J = 8 Hz, 1-NH). 13C-NMR (100.6 MHz, CDCl3) δ: 21.4 (CH3, CH3CO), 27.9 [CH, C5’(7’)], 32.1 (CH2, C3 or C5), 33.2 (CH2, C5 or C3), 34.6 (broad CH2, C4’, C6’ and C10’), 40.0 (CH2, C8’ or C9’), 40.2 (CH2, C9’ or C8’), 40.4 (CH2, C2 or C6), 45.1 (CH2, C6 or C2), 46.5 (CH, C4), 71.0 (CH, C3’), 80.6 (C, C1’), 156.7 (C, CO), 168.9 (C, CH3CO). MS (DIP), m/z (%); significant ions: 321 (M·+, 34), 197 (32), 179 [(C10H13NO2)·+, 34], 154 (100), 153 (18), 137 [(C9H13O)+, 33], 136 (32), 126 (15), 125 (51), 122 (21), 111 (17), 96 (41), 95 (18), 94 (45), 84 (19), 83 (37), 82 (54), 79 (22), 67(20), 57 (23), 56 (32), 55 (15). HRMS: Calculated for [C17H27N3O3+H]+: 322.2125. Found: 322.2124.
Trans-4-[4-(3-oxaadamant-1-yl-ureido)-cyclohexyloxy]benzoic acid, 14.
A solution of 2-oxaadamant-1-yl isocyanate (400 mg, 2.23 mmol) in anh. DCM (25 mL) was added to a solution of t-4-[(4-aminocyclohexyl)oxy]benzoic acid26 (728 mg, 2.68 mmol) in anh. DCM (12 mL), followed by triethylamine (1.24 mL, 8.94 mmol) under nitrogen. The reaction mixture was stirred at room temperature overnight. Water (50 mL) was then added and the phases were separated. The organic layer was extracted with further water (2 × 50 mL) and the pH of the combined aqueous phases was adjusted to pH ~2 with 5N HCl solution, prior extraction with DCM (3 × 50 mL). The combined organic layers were dried over anh. Na2SO4, filtered and concentrated under vacuum yielding urea 14 (220 mg, 24% yield) as a white solid. The analytical sample was obtained by crystallization from methanol/diethyl ether. Mp 255–275 °C. IR (ATR) ν: 3364, 3267, 3198, 3061, 2922, 2559, 2348, 2187, 2068, 2011, 1977, 1672, 1601, 1552, 1443, 1369, 1347, 1320, 1231, 1196, 1172, 1110, 1091, 1049, 1027, 989, 959, 863, 828, 774, 698, 640 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.37 [m, 2 H, 3’(5’)-Hax], 1.55 [m, 2 H, 2’(6’)-Hax], 1.65 [dm, J = 12.4 Hz, 2 H, 4”(10”)-Ha], 1.78 [dm, J = 11.6 Hz, 2 H, 8”(9”)-Ha], 1.86 (complex signal, 2 H, 6”-H2), 1.94 [m, 2 H, 4”(10”)-Hb], 2.01 [m, 2 H, 3’(5’)-Heq], 2.10 [m, 2 H, 2’(6’)-Heq], 2.22 [broad s, 2 H, 5”(7”)-H], 2.28 [dm, J = 15.5 Hz, 2 H, 8”(9”)-Hb], 3.57 (m, 1 H, 4’-H), 4.18 (broad s, 1 H, 3”-H), 4.40 (m, 1 H, 1’-H), 6.92 [d, J = 8.8 Hz, 2 H, 3(5)-H], 7.93 [d, J = 8.8 Hz, 2 H, 2(6)-H]. 13C-NMR (100.6 MHz, CD3OD) δ: 29.6 [CH, C5”(7”)], 31.0 [CH2, C2’(6’)], 31.4 [CH2, C3’(5’)], 35.7 [CH2, C4”(10”)], 35.9 (CH2, C6”), 41.0 [CH2, C8”(9”)], 48.6 (CH, C4’), 72.3 (CH, C3”), 75.8 (CH, C1’), 81.9 (C, C1”), 115.9 [CH, C3(5)], 126.8 (C, C1), 132.6 [CH, C2(4)], 159.0 (C, CO), 162.4 (C, C4 and CO2H). MS (DIP), m/z (%); significant ions: 414 (M·+, 0.2), 179 [(C10H13NO2)·+, 27], 138 [(C9H14O)+, (C7H5O3)+, 100], 122 (29), 121 (39), 111 (21), 108 (10), 98 (99), 96 (30), 94 (45), 82 (18), 81 (97), 79 (41), 67 (19), 65 (15), 56 (42), 55 (16). HRMS: Calculated for [C23H30N2O5+H]+: 415.2227. Found: 415.2230.
1-(2-Oxaadamant-1-yl)-3-(1-benzylpiperidin-4-yl)urea, 15.
To a solution of 2-oxaadamant-1-yl isocyanate (1.25 g, 6.97 mmol) in DCM (10 mL) was added 1-benzylpiperidin-4-amine (1.60 g, 8.37 mmol). The reaction mixture was stirred at room temperature overnight. The solvents were evaporated under vacuum to give a yellow gum (3.06 g). Column chromatography (DCM/methanol mixtures) gave urea 15 as a yellowish solid (2.54 g, 82% yield). Mp 153–154 °C. IR (ATR): 694, 745, 768, 989, 110, 1194, 1225, 1319, 1372, 1441, 1484, 1540, 1664, 1918, 1959, 2918 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.50 [dq, J = 3.6 Hz, J’ = 10.4 Hz, 2 H, 3(5)-Ha], 1.59–1.70 [complex signal, 4 H, 8’(9’)-Ha, 4’(10’)-Ha], 1.75–1.87 [complex signal, 2 H, 6’-H2], 1.86–1.97 [complex signal, 4 H, 3(5)-Hb, 4’(10’)-Hb], 2.13–2.22 [complex signal, 4 H, 2(6)-Ha, 8’(9’)-Hb], 2.26 [broad s, 2 H, 5’(7’)-H], 2.74 [d, J = 11.2 Hz, 2 H, 2(6)-Hb], 3.49 [s, 2 H, 7-H2], 3.71 (m, 1 H, 4-H), 4.24 (t, J = 4 Hz, 1 H, 3’-H), 4.65 (s, 1 H, 1’-NH), 6.04 (d, J = 8.4 Hz, 1 H, 4-NH), 7.24 (m, 1 H, 11-H), 7.28–7.32 (complex signal, 4 H, 9-H, 10-H, 12-H and 13-H). 13C-NMR (100.5 MHz, CDCl3) δ: 27.9 [CH, C5’(7’)], 32.6 [CH2, C3(5)], 34.6 [(CH2, C4’(10’)], 34.7 (CH2, C6’), 40.1 [(CH2, C8’(9’)], 46.2 (CH, C4), 52.1 [CH2, C2(6)], 63.2 (CH2, C7), 70.9 (CH, C3’), 80.6 (C, C1’), 126.9 (CH, C11), 128.2 [CH, C10(12) or C9(13)], 129.2 [CH, C9(13) or C10(12)], 138.2 (C, C8), 156.7 (C, CO). HRMS: Calculated for [C22H31N3O2+H]+: 370.2489 Found: 370.2488.
1-(2-Oxaadamant-1-yl)-3-(piperidin-4-yl)urea, 16.
To a solution of 1-(2-oxaadamant-1-yl)-3-(1-benzylpiperidin-4-yl)urea (2.40 g, 6.50 mmol) in methanol (20 mL), palladium on carbon 10% wt. (300 mg) and HCl 37% (1 mL) were added. The reaction mixture was hydrogenated for 5 days. The palladium on carbon was filtered and the solvent was evaporated under vacuum. The crude was dissolved in DCM and washed with 2N NaOH solution (2 × 30 mL). The organic phase was dried over anh. Na2SO4 and filtered. Evaporation under vacuum of the organics gave urea 16 as a white solid (1.28 g, 70% yield). The product was used in the next step without further purification. The analytical sample was obtained by crystallization from hot DCM (825 mg). Mp 164–165 °C. 1H-NMR (400 MHz, CDCl3) δ: 1.37 [m, 2 H, 3(5)-Hax], 1.60–1.73 [complex signal, 6 H, 4’(10’)-Ha, 8’(9’)-Ha], 1.77–1.88 (m, 2 H, 6’-H2), 1.90–2.01 [complex signal, 4 H, 4’(10’)-Hb, 3(5)-Hb], 2.19 [dm, J = 12 Hz, 2 H, 8’(9’)-Hb], 2.28 [broad s, 2 H, 5’(7’)-H], 2.71 [ddd, 2 H, J = 13 Hz, J’ = 11.8 Hz, J = 2.8 Hz, 2(6)-Ha], 3.04 [dt, 2 H, J = 13 Hz, J’ = 4 Hz, 2(6)-Hb], 3.77 (m, 1 H, 4-H), 4.29 (broad s, 1 H, 3’-H), 4.56 (s, 1 H, 1’-NH), 6.06 (d, J = 8.0 Hz, 4-NH). HRMS: Calculated for [C15H25N3O2+H]+: 280.2020. Found: 280.2022.
1-(2-Oxaadamant-1-yl)-3-(1-(4-chloro-6-methyl-1,3,5-triazin-2-yl)piperidin-4-yl)urea, 17.
To a solution of 2,4-dichloro-6-methyl-1,3,5-triazine (130 mg, 0.78 mmol) in DCM (4 mL) were added 1-(2-oxaadamantan-1-yl)-3-(piperidin-4-yl)urea (220 mg, 0.78 mmol) and DIPEA (305 mg, 2.36 mmol). The reaction mixture was stirred at room temperature for 30 min. Column chromatography (DCM/methanol mixtures) gave urea 17 as a white solid (54 mg, 9% yield). Mp 196–197 °C. IR (ATR) ν: 708, 762, 845, 907, 964, 992, 1075, 1116, 1168, 1194, 1219, 1243, 1271, 1315, 1364, 1444, 1485, 1527, 1578, 1671, 1953, 1974, 1994, 2180, 2335, 2852, 2914 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.38–1.47 [complex signal, 2 H, 3(5)-Ha], 1.60–1.71 [complex signal, 4 H, 6’-H2 and 8’(9’)-Ha], 1.76–1.87 [m, 2 H, 4’(10’)-Ha], 1.87–1.95 [m, 2 H, 4’(10’)-Hb], 2.02–2.21 [complex signal, 4 H, 3(5)-Hb and 8’(9’)-Hb], 2.27 [s, 2 H, 5’(7’)-H], 2.39 (s, 3 H, CH3), 3.16–3.24 [m, 2 H, 2(6)-Ha], 3.69 (m, 1 H, 4-H), 4.26 (broad s, 1 H, 3’-H), 4.55 (d, J = 13.6 Hz, 1 H, 2-Hb or 6-Hb), 4.63 (d, J = 13.6 Hz, 1 H, 6-Hb or 2-Hb], 4.68 (s, 1 H, 1’-NH), 6.10 (d, J = 7.6 Hz, 1H, 1-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 25.4 (CH3), 27.9 [CH, C5’(7’)], 32.36 (CH2, C3 or C5), 32.39 (CH2, C5 or C3), 34.63 [CH2, C4’(10’)], 34.64 (CH2, C6’), 40.1 (CH2, C8’ or C9’), 40.2 (CH2, C9’ or C8’), 42.4 (CH2, C2 or C6), 42.5 (CH2, C6 or C2), 46.6 (CH, C4), 71.0 (CH, C3’), 80.7 (C, C1’), 156.7 (C, CO), 164.1 (C, C8), 170.2 (C, C7), 177.3 (C, C9). HRMS: Calculated for [C19H27ClN6O2+H]+: 407.1957. Found: 407.1952.
1-(2-Oxaadamant-1-yl)-3-(1-(4-methyl-6-(methylamino)-1,3,5-triazin-2-yl)piperidin-4-yl)urea, 18.
Methylamine hydrochloride (160 mg, 2.36 mmol) and DIPEA (407 mg, 3.15 mmol) were added to the solution of 1-(2-oxaadamantan-1-yl)-3-(1-(4-chloro-6-methyl-1,3,5-triazin-2-yl)piperidin-4-yl)urea in DCM (5 mL) obtained in the previous step. The reaction mixture was stirred at 40 °C for 4 hours. The solvent was evaporated under vacuum to give a yellow gum (830 mg). Column chromatography (DCM/methanol mixtures) gave urea 18 as a grey solid (27 mg, 8% yield). Mp 203–204 °C. IR (ATR) ν: 653, 803, 880, 993, 1085, 1118, 1189, 1235, 1317, 1366, 1442, 1532, 1644, 1943, 2143, 2337, 2843, 2920 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.35–1.44 [complex signal, 2 H, 3(5)-Ha)], 1.61 (d, J = 12.0 Hz, 2 H, 6’-H2), 1.69 [d, J = 12.4, 2 H, 8’(9’)-Ha], 1.77–1.86 [complex signal, 2 H, 4’(10’)-Ha], 1.91 (d, J = 10.4 Hz, 2 H, 4’(10’)-Hb], 1.99–2.04 [complex signal, 2 H, 3(5)-Hb)], 2.17 [d, J = 12.0 Hz, 2 H, 8’(9’)-Hb], 2.24–2.27 [complex signal, 5 H, 5’(7’)-H and CCH3], 2.93 (d, J = 4.8 Hz, 3 H, NCH3), 3.15 [t, J = 12.0 Hz, 2 H, 2(6)-Ha], 3.93 (m, 1 H, 4-H), 4.25 (t, J = 4.4 Hz, 1 H, 3’-H), 4.56 [broad s, 2 H, 2(6)-Hb], 4.73 (s, 1 H, 1’-NH), 6.09 (d, J = 7.6 Hz, 1 H,1-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 25.2 (CH3, C10), 27.5 (CH3, C11), 27.9 [CH, C5’(7’)], 32.5 (CH2, C3(5)], 34.63 [CH2, C4’(10’)], 34.66 (CH2, C6’), 40.1 [CH2, C9’(8’)], 41.7 [CH2, C2(6)], 46.8 (CH, C4), 70.9 (CH, C3’), 80.6 (C, C1’), 156.7 (C, CO), 164.3 (C, C8), 173.9 (C, C7), 177.3 (C, C9). HRMS: Calculated for [C20H31N7O2+H]+: 402.2612. Found: 402.2608.
1-(2-Oxaadamant-1-yl)-3-(1-(tetrahydro-2H-pyran-4-carbonyl)piperidin-4-yl)urea, 19.
To a solution of 1-(2-oxaadamant-1-yl)-3-(piperidin-4-yl)urea (150 mg, 0.53 mmol) in EtOAc (10 mL), tetrahydro-2H-pyran-4-carboxylic acid (70 mg, 0.53 mmol), HOBt (109 mg, 0.80 mmol), EDCI·HCl (125 mg, 0.80 mmol) and triethylamine (0.15 mL, 1.07 mmol) were added. The reaction mixture was stirred at room temperature for 24 hours. To the resulting suspension was added water (15 mL) and the two phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (15 mL) and brine (15 mL). The combined aqueous phases were extracted with DCM (3 × 30 mL). The combined organic phases were dried over anh. Na2SO4 and filtered. Evaporation under vacuum of the organics gave urea 19 as colorless crystals (190 mg, 90% yield). Mp 150–152 °C. IR (ATR) ν: 641, 770, 878, 990, 1085, 1121, 1194, 1240, 1318, 1367, 1442, 1550, 1633, 2010, 2067, 2341, 2919 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.30–1.39 (complex signal, 2 H, 6-Ha, 2-Ha), 1.55–1.71 [complex signal, 6 H, 4’(10’)-Ha, 8’-Ha, 9’-Ha, 9-Ha, 13-Ha], 1.75–2.00 [complex signal, 7 H, 6’-H2, 2-Hb or 6-Hb, 4’(10’)-Hb, 9-Hb, 13-Hb], 2.10 (t, J = 13.2 Hz, 2 H, 2-Hb or 6-Hb, 8’-Hb or 9’-Hb), 2.22 (d, J = 12.0 Hz, 1 H, 9’-Hb or 8’-Hb), 2.27 [broad s, 2 H, 5’(7’)-H], 2.72 (tt, J = 11.2 Hz, J’ = 3.6 Hz, 1 H, 8-H), 2.86 (t, J = 11.2 Hz, 1 H, 3-Ha or 5-Ha), 3.18 (t, J = 11.2 Hz, 1 H, 5-Ha or 3-Ha), 3.43 [td, J = 12.0 Hz, J’ = 2 Hz, 2 H, 10(12)-Ha], 3.80 (d, J = 14.0 Hz, 1 H, 5-Hb or 3-Hb), 3.88 (m, 1 H, 4-H), 4.00 [d, J = 11.2 Hz, 2 H, 10(12)-Hb], 4.27 (t, J = 3.6 Hz, 1 H, 3’-H), 4.38 (dm, J = 13.6 Hz, 1 H, 3-Hb or 5-Hb), 4.75 (s, 1 H, 1’-NH), 6.07 (d, J = 8.0 Hz, 1 H, 4-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 27.9 [CH, C5’(7’)], 29.08 (CH2, C9 or C13), 29.14 (CH2, C13 or C9), 32.2 (CH2, C2 or C6), 33.6 (CH2, C6 or C2), 34.59 (CH2, C6’), 34.61 [CH2, C4’(10’)], 37.6 (CH, C8), 40.0 (CH2, C8’ or C9’), 40.2 (CH2, C9’ or C8’), 40.7 (CH2, C3 or C5), 44.0 (CH2, C5 or C3), 46.7 (CH, C4), 67.2 [CH2, C10(12)], 71.0 (CH, C3’), 80.7 (C, C1’), 156.7 (C, CO), 172.7 (COR). HRMS: Calculated for [C21H33N3O4+H]+: 392.2544. Found: 392.2553.
1-(2-Oxaadamant-1-yl)-3-(1-nicotinoylpiperidin-4-yl)urea, 20.
To a solution of 1-(2-oxaadamant-1-yl)-3-(piperidin-4-yl)urea (150 mg, 0.53 mmol) in EtOAc (10 mL), nicotinic acid (66 mg, 0.53 mmol), HOBt (109 mg, 0.805 mmol), EDCI·HCl (125 mg, 0.80 mmol) and triethylamine (0.15 mL, 1.07 mmol) were added. The reaction mixture was stirred at room temperature for 24 hours. Water (15 mL) was added to the resulting suspension and the two phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (15 mL) and brine (15 mL). The combined aqueous phases were basified with 1N NaOH solution (30 mL) and extracted with DCM (3 × 30 mL). The combined organic phases were dried over anh. Na2SO4 and filtered. Evaporation under vacuum of the organics gave a white solid (140 mg). Column chromatography (DCM/methanol mixtures) gave pure urea 20 as a white solid (63 mg, 32% yield). Mp 187–188 °C. IR (ATR) ν: 618, 711, 736, 767, 824, 990, 1114, 1132, 1194, 1219, 1245, 1269, 1318, 1367, 1436, 1483, 1537, 1622, 1666, 2051, 2144, 2217, 2919 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.37 (m, 1 H, 5-Ha or 3-Ha), 1.51 (m, 1 H, 3-Ha or 5-Ha), 1.60–1.72 [complex signal, 4 H, 8’(9’)-Ha and 6’-H2], 1.82 [m, 2 H, 4’(10’)-Ha], 1.92 (d, J = 12.4 Hz, 2 H, 3-Hb and 5-Hb), 1.97–2.09 [complex signal, 2 H, 4’(10’)-Hb], 2.15 [broad s, 2 H, 8’(9’)-Hb], 2.27 [broad s, 2 H, 5’(7’)-H], 3.02–3.28 [complex signal, 2 H, 2(6)-Ha], 3.57–3.75 (complex signal, 1 H, 2-Hb or 6-Hb), 3.93 (m, 1 H, 4-H), 4.29 (t, J = 4.4 Hz, 1 H, 3’-H), 4.50 (m, 1 H, 6-Hb or 2-Hb), 4.80 (s, 1 H, 1’-NH), 6.12 (d, J = 7.6 Hz, 1 H, 4-NH), 7.31–7.40 (dd, J = 8.0 Hz, J’ = 4.4 Hz, 1 H, 10-H), 7.74 (d, J = 8.0 Hz, 1 H, 9-H), 8.66 (complex signal, 2 H, 11-H, 13-H).13C-NMR (100.5 MHz, CDCl3) δ: 27.9 [CH, C5’(7’)], 32.2 (CH2, C3 or C5), 33.3 (CH2, C5 or C3), 34.60 (CH2, C6’), 34.63 [CH2, C4’(10’)], 40.1 (CH2, C8’(9’)], 41.3 (CH2, C2 or C6), 46.6 (CH, C4 and CH2, C6 or C2), 71.0 (CH, C3’), 80.7 (C, C1’), 123.5 (C, C10), 131.7 (C, C8), 134.8 (C, C9), 147.7 (C, C11 or C13), 150.7 (C, C13 or C11), 156.7 (C, CO), 167.7 (C, COR). HRMS: Calculated for [C21H28N4O3+H]+: 385.2234. Found: 385.2238.
1-(2-Oxaadamant-1-yl)-3-(1-(2-fluorobenzoyl)piperidin- 4-yl)urea, 21.
To a solution of 1-(2-oxaadamant-1-yl)-3-(piperidin-4-yl)urea (120 mg, 0.43 mmol) in EtOAc (10 mL), 2-fluorobenzoic acid (61 mg, 0.43 mmol), HOBt (87mg, 0.64 mmol), EDCI·HCl (100 mg, 0.64 mmol) and triethylamine (0.12 mL, 0.86 mmol) were added. The reaction mixture was stirred at room temperature for 24 hours. Water (15 mL) and DCM (20 mL) were added to the resulting suspension and the two phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (15 mL), brine (15 mL), dried over anh. Na2SO4 and filtered. Evaporation under vacuum of the organics gave urea 21 as a white solid (131 mg, 77% yield). The analytical sample was obtained as a white solid (111 mg) by crystallization from hot EtOAc. Mp 193–194 °C. IR (ATR) ν: 630, 785, 925, 987, 1010, 1093, 1121, 1191, 1243, 1318, 1372, 1447, 1462, 1491, 1555, 1615, 1684, 1974, 2351, 2925, 3338 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.38 (broad s, 1 H, 5-Ha or 3-Ha), 1.45–1.54 (m, 1 H, 3-Ha or 5-Ha), 1.61–1.71 (complex signal, 4 H, 6’-H2, 8’-Ha, 9’-Ha), 1.82 [m, 2 H, 4’(10’)-Ha], 1.90–1.99 [complex signal, 3 H, 5-Hb or 3-Hb, 4’(10’)-Hb], 2.06 (m, 1 H, 3-Hb or 5-Hb), 2.13 (m, 1 H, 9-Hb or 8-Hb), 2.20 (dq, J = 12.4 Hz, J’ = 2.0 Hz, 1 H, 8’-Hb or 9’-Hb), 2.27 [s, 2 H, 5’(7’)-H], 3.06–3.19 (complex signal, 2 H, 2-Ha, 6-Ha), 3.5 (d, J = 13.6 Hz, 1 H, 2-Hb or 6-Hb), 3.92 (m, 1 H, 4-H), 4.29 (s, 1 H, 3’-H), 4.53 (dm, J = 13.6 Hz, 1 H, 6-Hb or 2-Hb), 4.74 (s, 1 H, 1’-NH), 6.09 (d, J = 7.6 Hz, 1 H, 4-NH), 7.08 (m, 1 H, 10-H), 7.19 (td, J = 7.6 Hz, J’ = 1.2 Hz, 1 H, 13-H), 7.34–7.41 (complex signal, 2 H, 11-H, 12-H). 13C-NMR (100.5 MHz, CDCl3) δ: 27.95 (CH, C5’or C7’), 27.97 (CH, C7’or C5’), 32.2 (CH2, C3 or C5), 33.1 (CH2, C5 or C3), 34.7 [CH2, C6’ and C4’(10’)], 40.1 (CH2, C8’ or C9’), 40.2 (CH2, C9’ or C8’), 40.8 (CH2, C6 or C2), 45.9 (CH2, C2 or C6), 46.6 (CH, C4), 71.0 (CH, C3’), 80.7 (C, C1’), 115.7 (CH, d, 2JC-F = 21.4 Hz, C10), 124.2 (C, d, 2JC-F = 17.9 Hz, C8), 124.7 (CH, d, 4JC-F = 3.5 Hz, C13), 129.0 (CH, d, 3JC-F = 3.8 Hz, C11), 131.2 (CH, d, 3JC-F = 7.9 Hz, C12), 156.7 (C, CO), 158.1 (C, d, 1JC-F = 247.3 Hz, C9), 165.2 (C, COR). HRMS: Calculated for [C22H28FN3O3+H]+: 402.2187. Found: 402.2187.
1-(2-Oxaadamant-1-yl)-3-(1-(isopropylsulfonyl)piperidin-4-yl)urea, 22.
To a solution of 1-(oxaadamant-1-yl)-3-(piperidin-4-yl)urea (250 mg, 0.895 mmol) in DCM (10 mL), triethylamine (0.15 mL, 1.07 mmol) was added. The mixture was cooled down with an ice bath (0 °C) and propane-2-sulfonyl chloride (127 mg, 0.89 mmol) was added dropwise. The reaction mixture was stirred at room temperature overnight and quenched by the addition of HCl solution 37% (2 mL). The organic phase was collected, and the aqueous layer was extracted with EtOAc (4 × 30 mL). The combined organic phases were dried over anh. Na2SO4 and filtered. Evaporation of the organics gave an oil that was then dissolved in DCM (20 mL) and washed with 2N NaOH solution (3 × 20 mL). The organic phase was dried over anh. Na2SO4 and filtered. Evaporation under vacuum of the organics gave 22 as a white solid (88 mg, 26% yield). The analytical sample was obtained by crystallization from hot DCM as a white solid (60 mg). Mp 190–191 °C. IR (ATR) ν: 618, 729, 842, 884, 935, 961, 1010, 1041, 1093, 1116, 1132, 1196, 1243, 1269, 1292, 1320, 1374, 1444, 1547, 1635, 2930, 3333 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.32 [d, J = 6.8 Hz, 6 H, 8(9)-H], 1.50 [dq, J = 4.0 Hz, J’ = 11.2 Hz, 2 H, 3(5)-Ha], 1.63 [d, J = 12.0 Hz, 2 H, 4’(10’)-Ha], 1.69 [d, J = 12.4, 2 H, 8’(9’)-Ha], 1.82 (complex signal, 2 H, 6’-H2), 1.92 [d, J = 12.0 Hz, 2 H, 4’(10’)-Hb], 2.0 [broad d, J = 13.2 Hz, 2 H, 3(5)-Hb], 2.16 [d, J = 11.2 Hz, 2 H, 8’(9’)-Hb], 2.27 [broad s, 2 H, 5’(7’)-H], 3.01 [ddd, J = 2.4 Hz, J’ = J” = 13.6 Hz, 2 H, 2(6)-Ha], 3.17 (sept, 1 H, 7-H), 3.75 [dm, J = 13.2 Hz, 2 H, 2(6)-Hb], 3.81 (m, 1 H, 4-H), 4.28 (t, J = 4.4 Hz, 1 H, 3’-H), 4.74 (s, 1 H, 1’-NH), 6.08 (d, J = 8.0 Hz, 1 H, 4-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 16.7 [CH3, C8(9)], 27.9 [CH, C5’(7’)], 33.1 [CH2, C3(5)], 34.61 [CH2, C4’(10’)], 34.63 (CH2, C6’), 40.1 [CH2, C8’(9’)], 45.4 [CH2, C2(6)], 46.3 (CH, C4), 53.4 (CH, C7), 71.0 (CH, C3’), 80.6 (C, C1’), 156.7 (C, CO). HRMS: Calculated for [C18H31N3O4S+H]+: 386.2108. Found: 386.2113.
1-(2-Oxaadamant-1-yl)-3-(1-(cyclopropanecarbonyl)-piperidin-4-yl)urea, 23.
To a solution of 1-(2-oxaadamant-1-yl)-3-(piperidin-4-yl)urea (300 mg, 1.07 mmol) in DCM (10 mL), cyclopropanecarbonyl chloride (112 mg, 1.07 mmol) and triethylamine (0.18 mL, 1.29 mmol) were added. The reaction mixture was stirred at room temperature overnight and quenched by the addition of aqueous HCl 37% solution (3 mL). The organic phase was collected, and the aqueous phase was extracted with EtOAc (4 × 10 mL). The combined organic phases were washed with NaOH 2N (2 × 30 mL), dried over anh. Na2SO4 and filtered. Evaporation under vacuum of the organics gave urea 23 as a yellow oil (382 mg, 48% yield). The analytical sample was obtained as a white solid (180 mg) by crystallization from hot EtOAc. Mp 197–198 °C. IR (ATR) ν: 612, 729, 816, 876, 922, 961, 992, 1085, 1132, 1191, 1219, 1266, 1310, 1369, 1447, 1555, 1604, 1640, 2925, 3307 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.73 [m, 2 H, 9(10)-Ha], 0.95 [m, 2 H, 9(10)-Hb], 1.32–1.46 (complex signal, 2 H, 3-Ha, 5-Ha), 1.60–1.71 (complex signal, 4 H, 6’-H2, 8’-Ha, 9’-Ha), 1.74 (m, 1 H, 8-H), 1.79–1.97 [complex signal, 5 H, 3-Hb or 5-Hb and 4’(10’)-H2], 2.05–2.21 (complex signal, 3 H, 5-Hb or 3-Hb and 9’-Hb, 8’-Hb), 2.27 [broad s, 2 H, 5’(7’)-H], 2.90 (t, J = 12.4 Hz, 1 H, 6-Ha or 2-Ha), 3.27 (t, J = 11.6 Hz, 1 H, 2-Ha or 6-Ha), 3.90 (m, 1 H, 4-H), 4.07 (m, 1 H, 2-Hb or 6-Hb), 4.27 (t, J = 4.4 Hz, 1 H, 3’-H), 4.35 (m, 1 H, 6-Hb or 2-Hb), 4.80 (broad s, 1 H, 1’-NH), 6.08 (d, J = 8.0 Hz, 1 H, 4-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 7.3 [CH2, C9(10)], 10.9 (CH, C8), 27.9 [CH, C5’(7’)], 32.2 (CH2, C3 or C5), 33.4 (CH2, C5 or C3), 34.6 [CH2, C6’ and C4’(10’)], 40.0 (CH2, C9’ or C8’), 40.2 (CH2, C8’ or C9’), 41.2 (CH2, C6 or C2), 44.3 (CH2, C2 or C6), 46.8 (CH, C4), 71.0 (CH, C3’), 80.7 (C, C1’), 156.8 (C, CO), 171.9 (C, COR). HRMS: Calculated for [C19H29N3O3+H]+: 348.2282. Found: 348.2289.
1-(2-Oxaadamantan-1-yl)-3-(1-propionylpiperidin-4-yl)urea, 24.
To a solution of 2-oxaadamant-1-yl isocyanate (2.11 mmol) in DCM (5 mL) was added 1-(4-aminopiperidin-1-yl)propan-1-one (330 mg, 2.11 mmol). The reaction mixture was stirred at room temperature overnight. The solvent was evaporated under vacuum to give a yellow gum (823 mg). Column chromatography (DCM/methanol mixtures) gave the urea 24 as a white solid (280 mg, 40% yield). Mp 165–166 °C. The analytical sample was obtained by crystallization from hot EtOAc. IR (ATR) ν: 887, 930, 965, 995, 1011, 1092, 1119, 1137, 1195, 1218, 1247, 1267, 1321, 1375, 1444, 1556, 1620, 1647, 2332, 2367, 2852, 2927, 3336 cm−1. 1H-NMR (400 MHz, CDCl3) δ 1.13 (t, J = 7.6 Hz, 3 H, CH3), 1.28–1.40 (complex signal, 2 H, 3-Ha, 5-Ha), 1.60–1.70 [complex signal, 4 H, 9’-Ha, 8’-Ha, 4’(10’)-Ha], 1.75–1.87 (m, 2 H, 6’-H2), 1.87–1.97 [complex signal, 3 H, 4’(10’)-Hb, 3-Hb or 5-Hb], 2.04 (dm, J = 12.8 Hz, 1 H, 5-Hb or 3-Hb), 2.09–2.22 (complex signal, 2 H, 9’-Hb, 8’-Hb), 2.27 [broad s, 2 H, 5’(7’)-H], 2.33 (q, J = 7.6 Hz, 2 H, CH2CH3), 2.87 (m, 1 H, 6-Ha or 2-Ha), 3.15 (m, 1 H, 2-Ha or 6-Ha), 3.74 (dm, J = 13.6 Hz, 1 H, 2-Hb or 6-Hb), 3.87 (m, 1 H, 4-H), 4.26 (t, J = 4.0 Hz, 1 H, 3’-H), 4.37 (dm, J = 13.6 Hz, 1 H, 6-Hb or 2-Hb), 4.72 (s, 1 H, 1’-NH), 6.06 (d, J = 8.0 Hz, 1 H, 4-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 9.5 (CH3, CH2CH3), 26.5 (CH2, CH2CH3), 27.9 [CH, C5’(7’)], 32.2 (CH2, C3 or C5), 33.3 (CH2, C5 or C3), 34.6 [CH2, C6’, C4’(10’)], 40.0 (CH2, C9’ or C8’), 40.2 (CH2, C8’ or C9’), 40.5 (CH2, C6 or C2), 44.1 (CH2, C2 or C6), 46.6 (CH, C4), 71.0 (CH, C3’), 80.6 (C, C1’), 156.7 (C, CO), 172.2 (C, COR). HRMS: Calculated for [C18H29N3O3+H]+: 336.2282 Found: 336.2274.
1-(1-(4-Acetylphenyl)piperidin-4-yl)-3-(2-oxaadamant-1-yl)urea, 25.
To a solution of 2-oxaadamant-1-yl isocyanate (188 mg, 1.05 mmol) in DCM (5 ml), 1-(4-(4-aminopiperidin-1-yl)phenyl)ethan-1-one36 (230 mg, 1.05 mmol) and triethylamine (0.15 mL, 1.05 mmol) were added. The reaction mixture was stirred at room temperature overnight. The solvents were evaporated under vacuum to give an orange solid (410 mg). Column chromatography (DCM/methanol mixtures) gave the urea 25 as a white solid (183 mg, 45% yield). Mp 190–191 °C. IR (ATR) ν: 674, 723, 770, 819, 866, 915, 953, 974, 995, 1111, 1134, 1194, 1222, 1279, 1315, 1330, 1475, 1537, 1597, 1653, 1992, 2160, 2341, 2930 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.53 [dq, J = 4.0 Hz, J’ = 12.8 Hz, 2 H, 3(5)-Ha], 1.59–1.64 (complex signal, 2 H, 6’-H2), 1.69 [dm, J = 12.0 Hz, 2 H, 9’(8’)-Ha], 1.77–1.94 [complex signal, 4 H, 4’(10’)-H2], 2.06 [dm, J = 12.8 Hz, 2 H, 3(5)-Hb], 2.17 [dm, J = 12.4 Hz, 2 H, 9’(8’)-Hb], 2.27 [broad s, 2 H, 5’(7’)-H], 2.50 (s, 3 H, COCH3), 3.08 [ddd, J = 2.6 Hz, J’ = 11.2 Hz, J” = 13.6 Hz, 2 H, 2(6)-Ha], 3.78 [dt, J = 13.6 Hz, J’ = 4.0 Hz, 2 H, 2(6)-Hb], 3.92 (m, 1 H, 4-H), 4.26 (broad s, 1 H, 3’-H), 4.74 (s, 1 H, 1’-NH), 6.07 (d, J = 7.6 Hz, 1 H, 4-NH), 6.86 [d, J = 9.2 Hz, 2 H, 9(11)-H], 7.85 [d, J = 9.2 Hz, 2 H, 8(12)-H]. 13C-NMR (100.5 MHz, CDCl3) δ: 26.0 (CH3, C13), 27.9 [CH, C5’(7’)], 32.0 [CH2, C3(5)], 34.6 [CH2, C6’ and C4’(10’)], 40.1 [CH2, C9’(8’)], 46.57 (CH, C4), 46.59 [CH2, C2(6)], 71.0 (CH, C3’), 80.6 (C, C1’), 113.4 [CH, C9(11)], 127.1 (C, C10), 130.5 [CH, C8(12)], 153.7 (C, C7), 156.7 (C, CO), 196.3 (C, COCH3). HRMS: Calculated for [C23H31N3O3+H]+: 398.2438. Found: 398.2448.
1-(2-Oxaadamantan-1-yl)-3-(1-(methylsulfonyl)piperidin-4-yl)urea, 26.
To a solution of 1-amino-2-oxa-adamantane hydrochloride (265 mg, 1.4 mmol) in DCM (6 mL) and saturated aqueous NaHCO3 solution (6 mL) was added triphosgene (153 mg, 0.52 mmol). The biphasic mixture was stirred at room temperature for 30 min and then the two phases were separated and the organic layer was washed with brine (5 mL), dried over anh. Na2SO4, filtered and evaporated under vacuum to obtain 1–2 mL of a solution of the isocyanate in DCM. 1-(methylsulfonyl)piperidin-4-amine was suspended in anh. THF (10 mL) under argon atmosphere and the mixture was cooled down to −78 °C. Then, n-butyllithium (2.5 M in hexanes, 0.73 mL, 1.82 mmol) was added dropwise during 20 min. Meanwhile, the isocyanate from the previous step was dissolved in anh. THF and cooled down to 0 °C. The deprotonated amine was then tempered to 0 °C and the solution of isocyanate in THF was added. The mixture was stirred at room temperature overnight. The reaction was quenched by the addition of methanol (5 mL) and the solvent was evaporated to obtain a yellow gum (707 mg). Column chromatography (DCM/methanol mixtures) gave urea 26 as a white solid (47 mg, 10% yield). Mp 213–214 °C. IR (NaCl disk) ν: 629, 667, 729, 772, 824, 849, 885, 929, 954, 960, 968, 993, 1012, 1042, 1052, 1095, 1118, 1142, 1159, 1203, 1246, 1272, 1293, 1334, 1355, 1376, 1446, 1466, 1562, 1637, 2850, 2922, 3323 cm−1. 1H-NMR (400 MHz, CDCl3), δ: 1.54 [m, 2 H, 3(5)-Ha], 1.63 [d, J = 12.4 Hz, 2 H, 4’(10’)-Ha], 1.70 [d, J = 12.0 Hz, 2 H, 9’(8’)-Ha], 1.81 (m, 2 H, 6’-H2), 1.91 [dm, J = 11.2 Hz, 2 H, 4’(10’)-Hb], 2.06 [ddm, J = 12.8 Hz, J’ = 3.6 Hz, 2 H, 3(5)-Hb], 2.15 [d, J = 11.6 Hz, 2 H, 9’(8’)-Hb], 2.27 [broad s, J = 3.2 Hz, 2 H, 5’(7’)-H], 2.77 (s, 3 H, 7-H), 2.82 [ddd, J = J’ = 12.0 Hz, J” = 2.8 Hz, 2 H, 2(6)-Ha], 3.70 [dm, J = 12.0 Hz, 2 H, 2(6)-Hb], 3.77 (m, 1 H, 4-H), 4.27 (t, J = 4.4 Hz, 1 H, 3’-H), 4.82 (s, 1 H, 1’-NH), 6.06 (d, J = 8.0 Hz, 1 H, 4-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 27.9 [CH, C5’(7’)], 32.2 (CH2, C3(5)], 34.6 [CH2, C4’(10’) and C6’], 34.9 (CH3, C7), 40.1 (CH2, C9’(8’)], 45.0 [CH2, C2(6)], 46.1 (CH, C4), 71.0 (CH, C3’), 80.7 (C, C1’), 156.7 (C, CO). HRMS: Calculated for [C16H27N3O4S+H]+: 358.1795. Found: 358.1803.
1-(−2-Oxaadamantan-1-yl)-3-(benzo[d]thiazol-2-yl)urea, 27.
2-Amino-1,3-benzothiazole (114 mg, 0.76 mmol) was dissolved in anh. THF (7 mL) under argon and cooled to −78 °C on a dry ice in acetone bath. Then, n-butyllithium (2.5 M in hexanes, 0.31 mL, 0.76 mmol) was added dropwise during 20 min. Afterwards, the reaction mixture was removed from the dry ice in acetone bath and tempered to 0 °C with an ice bath. Meanwhile, 2-oxaadamant-1-yl isocyanate (150 mg, 0.84 mmol) was dissolved in anh. THF (4 mL) under argon and was continuously added to the reaction mixture. The mixture was stirred at room temperature overnight. Methanol (3 mL) was added to quench any unreacted n-butyllithium. The precipitate formed was filtered and washed with ice-cold THF to afford urea 27 as a white solid (151 mg, 42% overall yield). Mp 240 °C (dec). IR (ATR) ν: 731, 757, 788, 822, 866, 884, 920, 964, 995, 1046, 1093, 1119, 1191, 1248, 1274, 1323, 1341, 1377, 1452, 1514, 1537, 1597, 1718, 1904, 1992, 2036, 2134, 2201, 2852, 2894, 2930, 3064, 3255, 3322 cm−1. 1H-NMR (400 MHz, DMSO) δ: 1.57 [dm, J = 12.0 Hz, 2 H, 4’(10’)-Ha], 1.73–1.98 [complex signal, 6 H, 4’(10’)-Hb, 6’-H2, 8’(9’)-Ha], 2.18 [m, 2 H, 5’(7’)-H], 2.23 [dm, J = 12.4 Hz, 2 H, 8’(9’)-Hb], 4.11 (m, 1 H, 3’-H), 7.16 (ddd, J = 8.0 Hz, J’ = 8.0 Hz, J” = 1.2 Hz, 1 H, 5-H or 6-H), 7.27 (ddd, J = 8.0 Hz, J’ = 7.2 Hz, J” = 1.2 Hz, 1 H, 6-H or 5-H), 7.58 (dm, J = 7.6 Hz, 1 H, 7-H), 7.79–7.86 (complex signal, 2 H, 4-H, 1’-NH or 2-NH). 13C-NMR (100.5 MHz, DMSO) δ: 27.7 [CH, C5’(7’)], 34.46 [CH2, C4’(10’)], 34.48 (CH2, C6’), 39.5 [CH2, C8’(9’)], 69.3 (CH, C3’), 80.5 (C, C1’), 118.7 (CH, C7), 120.9 (CH, C4), 121.3 [(CH, C5 or C6), (C, C7a)], 125.1 (CH, C6 or C5), 131.2 [(C, C2), (C, C3a)], 149.4 (C, CO). HRMS: Calculated for [C17H19N3O2S+H]+: 330.1271. Found: 330.1272.
1-(−2-Oxaadamantan-1-yl)-3-(isoxazol-3-yl)urea, 28.
3-Aminoisoxazole (103 mg, 1.22 mmol) was dissolved in anh. THF (13 mL) under argon and cooled to −78 °C on a dry ice in acetone bath. Then, n-butyllithium (2.5 M in hexanes, 0.50 mL, 1.22 mmol) was added dropwise during 20 min. Afterwards, the reaction mixture was removed from the dry ice in acetone bath and tempered to 0 °C with an ice bath. Meanwhile, 2-oxaadamant-1-yl isocyanate (258 mg, 1.34 mmol) was dissolved in anh. THF (6 mL) under argon and was continuously added to the reaction mixture. The mixture was stirred at room temperature overnight. Methanol (4.5 mL) was added to quench any unreacted n-butyllithium. The organic solvents were evaporated under vacuum to give an orange gum (371 mg). Column chromatography (hexane/EtOAc mixtures) gave urea 28 as a white solid (90 mg, 22% overall yield). Mp 193 °C. IR (ATR) ν: 768, 788, 824, 888, 929, 959, 965, 987, 1014, 1050, 1075, 1093, 1116, 1196, 1260, 1288, 1324, 1377, 1395, 1444, 1475, 1566, 1598, 1672, 1685, 1920, 2005, 2051, 2158, 2215, 2323, 2369, 2851, 2923, 3082, 3179, 3287 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.64 [dm, J = 11.8 Hz, 2 H, 4’(10’)-Ha], 1.77–1.96 [complex signal, 4 H, 6’-H2, 8’(9’)-Ha], 2.03 [dm, J = 11.8 Hz, 2 H, 4’(10’)-Hb], 2.25 [dm, J = 12.4 Hz, 2 H, 8’(9’)-Hb], 2.30 [m, 2 H, 5’(7’)-H], 4.34 (m, 1 H, 3’-H), 6.11 (broad s, 1’-NH), 6.75 (s, 1 H, 4-H), 8.19 (d, J = 1.6 Hz, 1 H, 5-H), 8.95 (broad s, 1 H, 3-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 28.0 [CH, C5’(7’)], 34.55 [CH2, C4’(10’)], 34.58 (CH2, C6’), 40.2 [CH2, C8’(9’)], 71.2 (CH, C3’), 81.2 (C, C1’), 98.7 (CH, C4), 153.6 (C, C3), 158.2 [(C, CO), (CH, C5)]. HRMS: Calculated for [C13H17N3O3+H]+: 264.1343. Found: 264.1345.
1-(−2-Oxaadamantan-1-yl)-3-(1,3,5-triazin-2-yl)urea, 29.
2-Amino-1,3,5-triazine (245 mg, 2.55 mmol) was dissolved in anh. THF (20 mL) under argon and cooled to −78 °C on a dry ice in acetone bath. Then, n-butyllithium (2.5 M in hexanes, 1.05 mL, 2.55 mmol) was added dropwise during 20 min. Afterwards, the reaction mixture was removed from the dry ice in acetone bath and tempered to 0 °C with an ice bath. Meanwhile, 2-oxaadamant-1-yl isocyanate (539 mg, 2.80 mmol) was dissolved in anh. THF (8 mL) under argon and was continuously added to the reaction mixture. The mixture was stirred at room temperature overnight. Methanol (9 mL) was added to quench any unreacted n-butyllithium. A white precipitate formed among the orange solution was filtered and washed with ice-cold THF to afford urea 29 as a white solid (340 mg, 35% overall yield). Mp 157–158 °C. IR (ATR) ν: 700, 783, 824, 887, 965, 997, 1080, 1117, 1186, 1194, 1270, 1320, 1343, 1372, 1395, 1402, 1480, 1482, 1502, 1590, 1625, 1700, 2000, 2055, 2170, 2260, 2345, 2546, 2847, 2922, 3233, 3383, 3498 cm−1. 1H-NMR (400 MHz, DMSO) δ: 1.54 [dm, J = 12.0 Hz, 2 H, 4’(10’)-Ha], 1.66–1.98 [complex signal, 6 H, 4’(10’)-Hb, 6’-H2, 8’(9’)-Ha], 2.15 [m, 2 H, 5’(7’)-H], 2.26 [dm, J = 12.0 Hz, 2 H, 8’(9’)-Hb], 4.05 (m, 1 H, 3’-H), 7.31 (broad s, 1 H, 2-NH), 8.34 (s, 2 H, 4-H, 6-H). 13C-NMR (100.5 MHz, DMSO) δ: 27.7 [CH, C5’(7’)], 34.60 [CH2, C4’(10’)], 34.63 (CH2, C6’), 39.6 (CH2, C8’(9’)], 69.1 (CH, C3’), 80.1 (C, C1’), 164.7 (C, CO), 165.2 [(CH, C4, C6), (C, C2)]. HRMS: Calculated for [C13H17N5O2+H]+: 276.1455. Found: 276.1454.
1-(2-Oxaadamant-1-yl)-3-(3-chloro-5-trifluoromethoxy)phenyl)urea, 30.
A solution of 3-chloro-5-(trifluoromethoxy)aniline (200 mg, 0.94 mmol) in toluene (3 mL) was treated with triphosgene (140 mg, 0.47 mmol). Immediately, triethylamine (0.13 mL, 0.94 mmol) was added and the reaction mixture was stirred at 70 °C for 2 hours. Afterwards, pentane (0.5 mL) was added and a white precipitate was formed. The mixture was filtered and pentane was evaporated under vacuum at room temperature to give the isocyanate in toluene solution that was used in the next step without further purification. To a solution of 3-(trifluoromethoxy)-5-chlorophenylisocyanate from the previous step were added DCM (5 mL), 2-oxaadamantan-1-amine hydrochloride (161 mg, 0.85 mmol) and triethylamine (0.24 mL, 1.71 mmol). The suspension was stirred at room temperature overnight. The mixture was evaporated under vacuum to give a residue that was then dissolved in DCM (20 mL) and washed with 2N HCl solution. The organic phase was dried over anh. Na2SO4 and filtered. Evaporation under vacuum of the organics gave urea 30 (284 mg, 89% overall yield) as an orange solid. The analytical sample was obtained as a white solid (100 mg) by crystallization from hot DCM. Mp 177–178 °C. IR (ATR) ν: 672, 747, 935, 964, 995, 1093, 1116, 1152, 1191, 1212, 1248, 1416, 1465, 1550, 1599, 1664, 2930, 3302 cm−1. 1H-NMR (400 MHz, CDCl3), δ: 1.70 [dm, J = 12.4 Hz, 2 H, 4’(10’)-Ha], 1.76–1.94 [complex signal, 4 H, 6’-H2, 8’(9’)-Ha], 2.01 [dm, J = 12.4 Hz, 2 H, 4’(10’)-Hb], 2.18 [dm, J = 12.4 Hz, 2 H, 8’(9’)-Hb], 2.32 [m, 2 H, 5’(7’)-H], 4.41 (m, 1 H, 3’-H), 5.36 (broad s, 1 H, 1’-NH), 6.87 (m, 1 H, 4-H), 7.29–7.35 (complex signal, 2 H, 2-H, 6-H), 8.47 (broad s, 1 H, 1-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 27.9 [CH, C5’(7’)], 34.4 (CH2, C6’), 34.7 [CH2, C4’(10’)], 40.5 [CH2, C8’(9’)], 71.5 (C, C3’), 81.2 (C, C1’), 110.0 (CH, C6), 115.1 (CH, C4), 117.2 (CH, C2), 120.3 (q, J = 258.3 Hz, C, CF3), 135.1 (C, C3), 141.0 (C, C1), 149.8 (m, C, C5), 154.6 (C, CO). HRMS: Calculated for [C17H18ClF3N2O3+H]+: 391.1031. Found: 391.1038.
1-(−2-Oxaadamantan-1-yl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea, 31.
To a solution of the commercial 4-chloro-3-(trifluoromethyl)phenyl isocyanate (191 mg, 0.84 mmol) in DCM were added 2-oxaadamantan-1-amine hydrochloride (145 mg, 0.76 mmol) and triethylamine (0.21 mL, 1.52 mmol). The reaction mixture was stirred at room temperature overnight. The mixture was evaporated under vacuum to give a solid that was then dissolved in EtOAc (20 mL) and washed with 2N HCl solution (10 mL). The organic phase was dried over anh. Na2SO4 and filtered. Evaporation under vacuum of the organics gave urea 31 as a white solid (238 mg, 83% yield). The analytical sample was obtained by crystallization from hot EtOAc (127 mg). Mp 196 °C. IR (ATR) ν: 661, 721, 765, 785, 824, 835, 881, 930, 961, 987, 1028, 1093, 1114, 1134, 1170, 1191, 1209, 1253, 1289, 1297, 1323, 1374, 1416, 1485, 1550, 1586, 1607, 1671, 2118, 2144, 2217, 2351, 2847, 2925, 3054, 3100, 3235, 3286 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.69 [dm, J = 12.6 Hz, 2 H, 4’(10’)-Ha], 1.77–1.93 [complex signal, 4 H, 6’-H2, 8’(9’)-Ha], 2.01 [dm, J = 12.6 Hz, 2 H, 4’(10’)-Hb], 2.19 [m, J = 12.0 Hz, 2 H, 8’(9’)-Hb], 2.32 [m, 2 H, 5’(7’)-H], 4.41 (m, 1 H, 3’-H), 5.42 (broad s, 1H, 1’-NH), 7.37 (d, J = 8.6 Hz, 1 H, 5-H), 7.56 (dd, J = 8.6 Hz, J’ = 2.6 Hz, 6-H), 7.71 (d, 1 H, J = 2.6 Hz, 2-H), 8.4 (broad s, 1 H, 1-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 27.9 [CH, C5’(7’)], 34.4 (CH2, C6’), 34.7 [CH2, C4’(10’)], 40.4 [CH2, C8’(9’)], 71.4 (CH, C3’), 81.2 (C, C1’), 118.2 (q, 3JC-F = 6.0 Hz, CH, C2), 122.7 (q, 1JC-F = 273 Hz, C, CF3), 123.2 (CH, C6), 125.1 (C, C4), 128.5 (q, 2JC-F = 31.3 Hz, C, C3), 131.8 (CH, C5), 137.7 (C, C1), 154.8 (C, CO). HRMS: Calculated for [C17H18ClF3N2O2+H]+: 375.1082. Found: 375.1090.
1-(−2-Oxaadamantan-1-yl)-3-(3-(pentafluoro-λ6-sulfanyl)phenyl)urea, 32.
A solution of 3-(pentafluoro-λ6-sulfanyl)aniline (185 mg, 0.84 mmol) in toluene (3.6 mL) was treated with triphosgene (125 mg, 0.42 mmol). Immediately, triethylamine (0.12 mL, 0.84 mmol) was added and the reaction mixture was stirred at 70 °C for 2 hours. Afterwards, pentane (0.5 mL) was added and a white precipitate was formed. The mixture was filtered and pentane was evaporated under vacuum at room temperature to give the isocyanate in toluene solution that was used in the next step without further purification. To a solution of the 3-(pentafluoro-λ6-sulfanyl)phenyl isocyanate from the previous step were added DCM (5 mL), 2-oxaadamantan-1-amine hydrochloride (145 mg, 0.76 mmol) and triethylamine (0.21 mL, 1.52 mmol). The suspension was stirred at room temperature overnight. The mixture was evaporated under vacuum to give a solid that was then dissolved in DCM (20 mL) and washed with aqueous 2N HCl solution. The organic phase was dried over anh. Na2SO4 and filtered. Evaporation under vacuum of the organics gave urea 32 (237 mg, 71% overall yield) as a pale yellow solid. The analytical sample was obtained by crystallization from hot EtOAc as a white solid (75 mg). Mp 203 °C. IR (ATR) ν: 649, 685, 734, 785, 824, 835, 863, 946, 959, 990, 1093, 1114, 1199, 1250, 1256, 1292, 1318, 1369, 1431, 1478, 1537, 1591, 1671, 1966, 2041, 2930, 3080, 3224, 3286 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.69 [dm, J = 11.8 Hz, 2 H, 4’(10’)-Ha], 1.78–1.94 [complex signal, 4 H, 6’-H2, 8’(9’)-Ha], 2.01 [dm, J = 11.8 Hz, 2 H, 4’(10’)-Hb], 2.21 [m, J = 12.4 Hz, 2 H, 8’(9’)-Hb], 2.32 [m, 2 H, 5’(7’)-H], 4.42 (m, 1 H, 3’-H), 5.43 (broad s, 1H, 1’-NH), 7.35 (d, J = 8.2 Hz, 1 H, 6-H), 7.39 (dq, J = 8.2 Hz, J’ = 1.2 Hz, 1 H, 5-H), 7.52 (d, J = 8.2 Hz, 1 H, 4-H), 7.86 (t, J = 2.0 Hz, 1 H, 2-H), 8.50 (broad s, 1 H, 1-NH). 13C-NMR (100.5 MHz, CDCl3) δ: 27.9 [CH, C5’(7’)], 34.5 (CH2, C6’), 34.7 [CH2, C4’(10’)], 40.4 [CH2, C8’(C9’)], 71.4 (CH, C3’), 81.2 (C, C1’), 116.9 (m, CH, C2), 120.1 (m, CH, C4), 122.3 (CH, C6), 128.9 (CH, C5), 139.2 (C, C1), 154.3 (C, C3), 154.9 (C, CO). HRMS: Calculated for [C16H19F5N2O2S+H]+: 399.1160. Found: 399.1172.
Determination of IC50 sEH inhibitors in human, murine and rat purified sEH.
IC50 is the concentration of a compound that reduces the sEH activity by 50%. The IC50 values reported herein were determined using a fluorescent based assay (CMNPC as substrate).53 The fluorescent assay was used with purified recombinant human, mouse or rat sEH proteins. The enzymes were incubated at 30 °C with the inhibitors ([I]final = 0.4 – 100,000 nM) for 5 min in 100 mM sodium phosphate buffer (200 μL, pH 7.4) containing 0.1 mg/mL of BSA and 1% of DMSO. The substrate (CMNPC) was then added ([S]final = 5 μM). Activity was assessed by measuring the appearance of the fluorescent 6-methoxynaphthaldehyde product (λex = 330 nm, λem = 465 nm) every 30 seconds for 10 min at 30 °C on a PsectraMax M2 (molecular devices). Results were obtained by regression analysis from a linear region of the curve.
In silico study. Molecular Dynamics Simulations Details.
The parameters for AR9273, 22, and 26, for the MD simulations were generated within the ANTECHAMBER module of AMBER 1854 using the general AMBER force field (GAFF),55 with partial charges set to fit the electrostatic potential generated at the HF/6-31G(d) level by the RESP model.56 The charges were calculated according to the Merz-Singh-Kollman scheme57–58 using Gaussian 09.59
MD simulations of sEH were carried out using PDB 5ALZ (crystallized with AR9273)38 as a reference. For the MD simulations in the apo state the AR9273 was removed from the active site. The oxaadamantyl derivatives corresponding to compounds 22 and 26 were manually prepared using AR9273 structure as starting point. From these coordinates, conventional MD simulations were used to explore the conformational plasticity of sEH in the apo state and in the presence of AR9273, 22, and 26 bound in the active site. Amino acid protonation states were predicted using the H++ server (http://biophysics.cs.vt.edu/H++). The MD simulations have been carried with the following protonation of histidine residues: HIE146, HIE239, HIP251, HID265, HIP334, HIE420, HIE506, HIE513, HIE518, and HIP524.
Each system was immersed in a pre-equilibrated truncated octahedral box of water molecules with an internal offset distance of 10 Å. All systems were neutralized with explicit counterions (Na+ or Cl−). A two-stage geometry optimization approach was performed. First, a short minimization of the positions of water molecules with positional restraints on solute by a harmonic potential with a force constant of 500 kcal mol−1 Å−2 was done. The second stage was an unrestrained minimization of all the atoms in the simulation cell. Then, the systems were gently heated in six 50 ps steps, increasing the temperature by 50 K each step (0–300 K) under constant-volume, periodic-boundary conditions and the particle-mesh Ewald approach60 to introduce long-range electrostatic effects. For these steps, a 10 Å cut-off was applied to Lennard-Jones and electrostatic interactions. Bonds involving hydrogen were constrained with the SHAKE algorithm.61 Harmonic restraints of 10 kcal mol−1 were applied to the solute, and the Langevin equilibration scheme was used to control and equalize the temperature.62 The time step was kept at 2 fs during the heating stages, allowing potential inhomogeneities to self-adjust. Each system was then equilibrated for 2 ns with a 2 fs timestep at a constant pressure of 1 atm. Finally, conventional MD trajectories at constant volume and temperature (300 K) were collected. In total, three replicas of 250 ns MD simulations for sEH in the apo state and in the presence of AR9273, 22, and 26 were run, gathering a total of 3 μs of MD simulation time. The relative binding free energy was calculated for compounds 22 and 22-CH2 using the TI method.63 The NPT ensemble was used for equilibrating the system and the production runs for the TI calculations using a timestep of 1fs and the same simulation protocols as described above. The schedule of lambda windows chosen consists of 11 λ values: 0.0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0. The initial equilibrations of 4 ns were performed using a λ of 0.5 and this configuration was used as starting point for neighbouring lambda windows. The TI runs were run for 4 ns and frames were collected every 0.5 ps for post-analysis and relative binding free energy calculations.
The octanol/water partition coefficient, LogP, of compound 22 and 22-CH2 was calculated from the solvation free energy differences using the M06–2X functional64 with SMD implicit solvation65 and the def2-SVP basis set66 as described by Guan et al.42 Solvated geometry optimizations were performed using M06-2X/def2-SVP amd SMD using water and 1-octanol as solvents. To calculate the LogP, the free energies obtained at 298.15 K using octanol and water as solvents were employed to compute the standard free energy associated with the transfer of the solute from the water to octanol phases as:
| (1) |
Solubility.
The stock solutions (10−2 M) of assayed compounds were diluted to decreased molarity, from 300 μM to 0.1 μM, in 384 well transparent plate (Greiner 781801) with 1% DMSO:99% PBS buffer. Then, they were incubated at 37 °C and the light scattering was measured after 2 hours in a NEPHELOstar Plus (BMG LABTECH). The results were adjusted to a segmented regression to obtain the maximum concentration in which compounds are soluble.
Permeability.
The Caco-2 cells were cultured to confluency, trypsinized and seeded onto a 96-filter transwell insert (Corning) at a density of ~10,000 cells/well in DMEM cell culture medium supplemented with 10% Foetal Bovine Serum, 2 mM L-Glutamine and 1% penicillin/streptomycin. Confluent Caco-2 cells were subcultured at passages 58–62 and grown in a humidified atmosphere of 5% CO2 at 37 °C. Following an overnight attachment period (24 h after seeding), the cell medium was replaced with fresh medium in both the apical and basolateral compartments every other day. The cell monolayers were used for transport studies 21 days post seeding. The monolayer integrity was checked by measuring the transepithelial electrical resistance (TEER) obtaining values ≥ 500 Ω/cm2. On the day of the study, after the TEER measurement, the medium was removed and the cells were washed twice with pre-warmed (37 °C) Hank’s Balanced Salt Solution (HBSS) buffer to remove traces of medium. Stock solutions were made in dimethyl sulfoxide (DMSO), and further diluted in HBSS (final DMSO concentration 1%). Each compound and reference compounds (Colchicine, E3S) were all tested at a final concentration of 10 μM. For A → B directional transport, the donor working solution was added to the apical (A) compartment and the transport media as receiver working solution was added to the basolateral (B) compartment. For B → A directional transport, the donor working was added to the basolateral (B) compartment and transport media as receiver working solution was added to the apical (A) compartment. The cells were incubated at 37 °C for 2 hours with gentle stirring.
At the end of the incubation, samples were taken from both donor and receiver compartments and transferred into 384-well plates and analyzed by UPLC-MS/MS. The detection was performed using an ACQUITY UPLC /Xevo TQD System. After the assay, Lucifer yellow was used to further validate the cell monolayer integrity, cells were incubated with LY 10 μM in HBSS for 1 hour at 37 °C, obtaining permeability (Papp) values for LY of ≤ 10 nm/s confirming the well-established Caco-2 monolayer.
Cytotoxicity in PBMCs cells + 1.5% PHA (Phytohaemagglutinin).
Cytotoxic effects of assayed compounds were tested using peripheral blood mononuclear cells isolated following the regular density gradient centrifugation procedure with Ficol. Cells were plated in 96-well black microplates at 270.000 cells/well density with RPMI medium (containing 10% FBS, 1% NEAA, 1% P/S and 1.5% PHA). The tested compounds were solubilized in 100% DMSO at a concentration curve way and then diluted with cell culture medium containing 2% DMSO. The final concentrations of the test compounds (0.2% DMSO) ranged from 0–20 μM in a final volume of 150 μL. Microplates were maintained at 37 °C (5% CO2, 95% humidity) for 3 days. Following this 72 hour exposure to test compounds, cell viability in each well was determined by measuring the concentration of cellular adenosine triphosphate (ATP) using the ATP1Step Kit as described by the manufacturer (Perkin-Elmer). In a typical procedure, 80 μL of cell reagent was added to all wells of each test plate followed by incubation for 10 min at room temperature on an orbital shaker. ATP concentration was determined by reading chemical luminescence using the Envision plate reader (PerkinElmer). The percentage of viable cells relative to the non-drug treated controls was determined for each well and LC50 values were calculated as concentrations projected to kill 50% of the cells following a 72 hours exposure.
Cytotoxicity in THLE-2 cells.
Cytotoxic effects of assayed compounds were tested using the immortalized human liver cell line THLE-2 (ATCC CRL-2706). Cells were cultured in BEGM medium (Clonetics #CC-4175) containing all the supplements kit except additional EGF and G418. Medium was completed by adding 0.7 μg/mL phosphoethanolamine, 0.5 ng/mL epidermal growth factor, antibiotics (penicillin and streptomycin) and 10% fetal bovine serum (FBS). Cells were plated in 96-well black microplates at 10.000 cells/well density and were incubated at 37 °C (5% CO2, 95% humidity) for 24 hours to allow the cells to adhere and form a monolayer. Test compounds were solubilized in 100% DMSO at a concentration curve way and then diluted with cell culture medium containing 10% DMSO. The final concentrations of the test compounds (1% DMSO) ranged from 0–100 μM in a final volume of 200 μL. Microplates were maintained at 37 °C (5% CO2, 95% humidity) during 3 days. Following this 72 h exposure to test compounds, cell viability in each well was determined by measuring the concentration of cellular adenosine triphosphate (ATP) using the ATP1Step Kit as described by the manufacturer (Perkin-Elmer). In a typical procedure, 50 μL of cell reagent was added to all wells of each test plate followed by incubation for 10 min at room temperature on an orbital shaker. ATP concentration was determined by reading chemical luminescence using the Envision plate reader (PerkinElmer). The percentage of viable cells relative to the non-drug treated controls was determined for each well and LC50 values were calculated as concentrations projected to kill 50% of the cells following a 72 hours exposure.
Parallel Artificial Membrane Permeation Assays - Blood-Brain Barrier (PAMPABBB).
To evaluate the brain penetration of the different compounds, a parallel artificial membrane permeation assay for blood-brain barrier was used, following the method described by Di et al.43 The in vitro permeability (Pe) of fourteen commercial drugs through lipid extract of porcine brain membrane together with the test compounds were determined. Commercial drugs and assayed compounds were tested using a mixture of PBS:EtOH (70:30). Assay validation was made by comparing the experimental permeability with the reported values of the commercial drugs by bibliography and lineal correlation between experimental and reported permeability of the fourteen commercial drugs using the parallel artificial membrane permeation assay was evaluated (y = 1.5219x − 0.9129; R2 = 0.9387). From this equation, and taking into account the limits established by Di et al.43 for BBB permeation, we established the ranges of permeability as compounds of high BBB permeation (CNS+): Pe (10-6 cms−1) > 5.149; compounds of low BBB permeation (CNS−): Pe (10-6 cms−1) < 2.131 and compounds of uncertain BBB permeation (CNS+/−): 5.149 > Pe (10-6 cms−1) > 2.131.
Cytochrome P450 inhibition assay.
The objective of this study was to screen the inhibition potential of the compounds using recombinant human cytochrome P450 enzymes CYP3A4 (BFC and DBF substrates) and probe substrates with fluorescent detection. Incubations were conducted in a 200 μL volume in 96 well microtiter plates (COSTAR 3915). Adding the mixture buffer-cofactor (KH2PO4 buffer, 1.3 mM NADP, 3.3 mM MgCl2, 0.4 U/mL Glucose-6-phosphate Dehydrogenase), Control supersomes the standard inhibitor Ketoconazole (Sigma K1003) and pre-diluted compounds. The plate was then pre-incubated at 37 °C for 5 min in 100 μL volume and reaction initiated by the addition of pre-warmed enzyme/substrate (E/S) mix. The E/S mix contained buffer (KH2PO4), enzyme (CYP), substrate 7-benzyloxytrifluoromethyl coumarin (7-BFC) and Dibenzylfluorescein (DBF) in a reaction volume of 200 μL. Reactions were terminated after various times depending on the substrate by addition of STOP solution (ACN/TrisHCl 0.5M 80:20 (BFC) or 2N NaOH for CYP3A4 (DBF). Fluorescence per well was measured using a fluorescence plate reader (Tecan M1000 pro) and percentage of inhibition was calculated.
Microsomal stability.
The human microsomes employed were purchased from Tebu-Xenotech. The compound was incubated at 37 °C with the microsomes in a 50 mM phosphate buffer (pH = 7.4) containing 3 mM MgCl2, 1 mM NADP, 10 mM glucose-6-phosphate and 1 U/mL glucose-6-phosphate-dehydrogenase. Samples (75 μL) were taken from each well at 0, 10, 20, 40 and 60 min and transferred to a plate containing 75 μL acetonitrile, and 30 μL of 0.5% formic acid in water were added for improving the chromatographic conditions. The plate was centrifuged (46000 g, 30 min) and supernatants were taken and analyzed in a UPLC-MS/MS (Xevo-TQD, Waters) by employing a BEH C18 column and an isocratic gradient of 0.1% formic acid in H2O : 0.1% formic acid acetonitrile (60:40). The metabolic stability of the compounds was calculated from the logarithm of the remaining compounds at each of the time points studied.
Inhibition of human lipoxygenase-5 (hLOX-5).
AA and 2’,7’-dichlorodihydrofluorescein diacetate (H2DCFDA) were obtained from Sigma. Human recombinant LOX-5 was purchased from Cayman Chemical. For the determination of hLOX-5 activity, the method described by Pufahl et al. was followed.67 The assay solution consisted of 50 mM Tris (pH 7.5), 2 mM EDTA, 2 mM CaCl2, 3 μM AA, 10 μM ATP, 10 μM H2DCFDA and 100 mU/well hLOX-5. For the enzyme inhibition studies the compounds to be tested were added to the assay solution prior to AA and ATP and were preincubated for a period of 10 min at room temperature, after which AA and ATP were added. The enzymatic reaction was carried out for 20 min and terminated by the addition of 40 μL of acetonitrile. The fluorescence measurement, 485 nm excitation and 520 nm emission, was performed on a FLUOstar OPTIMA (BMG LABTECH, Offenburg, Germany.). The IC50 is defined as the concentration of compound that inhibits enzymatic activity by 50% over the untreated enzyme control.
In vitro proof of concept.
Rat pancreatic AR42J acinar cells were purchased from ATCC and cultured in RPMI-1680 (Gibco) supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 μg/mL streptomycin and 100 units/ml penicillin at 37 °C in a humidified atmosphere containing 5% CO2. Medium was changed every 48 hours. AR42J cells plated into 6-well plate and incubated for 24–48 hours, (one or the other) were pre-treated with sEH inhibitors for 1 hour and, later, treated with sEH inhibitors plus cerulein 10 nM (treated group) for 8 hours. The same volume of saline serum (0.9% NaCl) was added to the control cultures (control group).
Pharmacokinetic study.
Mice used in this study were Hsd:ICR (CD-1) (females, 6–7 weeks old supplied by ENVIGO). Animals were housed under sterile conditions at a constant temperature of 20–22 °C and relative humidity (45–65%) under daily cycles of light/darkness (12 hours). Manipulation was performed in laminar flow hood and sterilized water and food were available ad libitum. The procedure involving experimental animals was approved by the “Ethical Committee of Animal Experimentation” of the animal facility plate at Science Park of Barcelona (Platform of Applied Research in Animal Laboratory). Once approved by the Institutional ethical committee, this procedure was additionally approved by the ethical committee of the Catalonian authorities according to the Catalonian and Spanish regulatory laws and guidelines governing experimental animal care. Along the procedures using experimental animals, we also established a continuous supervision and control of the animals to monitor their degree of suffering and if needed to sacrifice them according to the defined end point criteria described by the United Kingdom Coordinating Committee of Cancer Research (UKCCCR), by the Canadian Council on Animal Care (1998), by the Institute for Laboratory Research Journal (2000), by the Conference 22–25 November 1998 Zeist (The Netherlands) and by the Guidelines for the welfare and use of animals in cancer research (2010). The euthanasia method applied was by CO2 saturated atmosphere. Formulations were prepared the day of the study. Vehicle was saline solution + 5% DMSO. 21 mice were administered intravenously (iv) with a single dose of 1 mg/kg of compound 22. The volume of administration was 5 mL/kg. 21 mice were administered intraperitoneally (ip) with a single dose of 1 mg/kg of compound 22. The volume of administration was 10 mL/kg. Animals were weighed before each administration to adjust the required volume. Compound 22 was administered by ip or iv and animals were killed under a CO2 saturated atmosphere to obtain blood samples. Blood samples were collected at different times post administration: 0 h, 10’, 30’, 1 h, 2 h, 4 h, 6 h, and 24 h. Samples were taken by cardiac puncture at different times post administration (3 animals per time). Blood was collected on recipients containing 5 μL of EDTA 0.5 M. Blood was centrifuged at 5000 rpm for 10 min. 100 μL of each plasma sample (BK, calibration standards and study samples) were mixed with 300 μL of methanol and 60 μL of ISTD in a Sirocco plate. Plate was centrifuged at 2000 rpm for 10 min. After that, sample were dried at 40 °C under N2 streaming and reconstituted with 110 μL of MeOH:H2O 0.1% HCOOH 1:1. Concentrations of compound 22 were analyzed by UPLC-MS.
Maximum tolerated dose study.
Mice used in this study were Hsd:ICR (CD-1) females, 6–7 weeks old supplied by ENVIGO. Animals were housed under sterile conditions at a constant temperature of 20–22 °C and relative humidity (45–65%) under daily cycles of light/darkness (12 hours). Manipulation was performed in laminar flow hood and sterilized water and food were available ad libitum. The procedure involving experimental animals was approved by the “Ethical Committee of Animal Experimentation” of the animal facility plate at Science Park of Barcelona (Platform of Applied Research in Animal Laboratory). Once approved by the Institutional ethical committee, this procedure was additionally approved by the ethical committee of the Catalonian authorities according to the Catalonian and Spanish regulatory laws and guidelines governing experimental animal care. Along the procedures using experimental animals, we also establish a continuous supervision and control of the animals to monitor their degree of suffering and if needed to sacrifice them according to the defined end point criteria described by the United Kingdom Coordinating Committee of Cancer Research (UKCCCR), by the Canadian Council on Animal Care (1998), by the Institute for Laboratory Research Journal (2000), by the Conference 22–25 November 1998 Zeist (The Netherlands) and by the Guidelines for the welfare and use of animals in cancer research (2010). The euthanasia method applied was by CO2 saturated atmosphere. Animal weight was monitored daily. The criteria for end point before the end of the assay was that the mean of weight loss from any group reaches 15% or more; also, any individual mouse with a loss of 20% should be sacrificed. Body weight was monitored daily previous to administration. The body weight loss was calculated according to the formula: ((Weight − weight start) / weight start) × 100. Compound 22 was administered by ip for 5 consecutively days and, after then, animals’ weight was monitored for 5 days more. Then, animals were killed under a CO2 saturated atmosphere to obtain samples. Five groups were performed taking into account the administered dose. G1: Control vehicle, 5 days (n = 6). G2: Compound 22 at 80 mg/kg, 5 days (n = 6). G3: Compound 22 at 20 mg/kg, 5 days (n = 6). G4: Compound 22 at 10 mg/kg, 5 days (n = 6). G5: Compound at 5 mg/kg, 5 days (n = 6).
In vivo efficacy study.
Compound 22 was administered before (pre) and after (post) induction of acute pancreatitis with cerulein (50 μg/kg body weight, 12 times at 1-hour intervals) in 8- to 10-week-old male mice (C57BL/6J), as previously described.34 In the post-induction studies, 22 (3 and 30 mg/kg) was administered in one injection 24 hours after induction of AP. In the pre-induction studies, sEH inhibitors were administered at the same dose for 5 consecutive days 2-times/day before induction of AP. Animals were sacrificed 24 hours after the end of the treatment and blood and tissues were collected and flash-frozen at −80 °C. Mice were treated in accordance with the European Community Council Directive 86/609/EEC and the procedures established by the Department d’Agricultura, Ramaderia i Pesca of the Generalitat de Catalunya.
Histologic analyses.
A portion of the pancreas was fixed in 4% paraformaldehyde and sections were stained with H&E to observe morphologic changes. Histologic analysis was performed as previously described.34
RNA preparation and quantitative RT-PCR.
RNA was extracted from AR42J cells and pancreatic tissue using Trisure reagent (TRIsure Bio-38032, Bio-Rad) according to the manufacturer’s recommended protocol. Normally, 1 μg of total RNA was reverse-transcribed into cDNA and the relative levels of specific mRNAs were assessed by real-time PCR with MiniOpticon Real-Time PCR system (Bio-Rad). The primer sequences used are shown in Supporting Information.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by the Spanish Ministerio de Economía, Industria y Competitividad (Grants SAF2017-82771-R to S.V., SAF2015-64146-R and RTI2018-093999-B-I00 to M. V.-C., RTI2018-093955-B-C21 to M.I.R.-F., PGC2018-102192-B-I00 to S. O. and RTI2018-101032-J-I00 to F. F.), the CaixaImpulse 2015 Programme, the Spain EIT Health (Proof of concept 2016), the European Regional Development Fund (ERDF), the Xunta de Galicia (GRC2014/011 and ED431C2018-21), the Fundació Bosch i Gimpera, Universitat de Barcelona (F2I grant), the Generalitat de Catalunya (2017 SGR 106, 2017 SGR 124 and 2017 SGR 1707), the Fundación Fuentes Dutor, the European Research Council (ERC-2015- StG-679001-NetMoDEzyme to S.O.), and the European Community (MSCA-IF-2014-EF-661160-MetAccembly to F.F.). S.C. and E.P. acknowledge PhD fellowships from the Universitat de Barcelona (APIF grants). E.V. and R.L. thank the Institute of Biomedicine of the University of Barcelona (IBUB) and the Spanish Ministerio de Educación, Cultura y Deporte (FPU grant), respectively, for PhD grants. Partial support was provided by NIH-NIEHS River Award R35 ES03443, NIH-NIEHS Superfund Program P42 ES004699, NINDS R01 DK107767, and NIDDK R01 DK103616. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Daniel Closa (IIBB-CSIC, Barcelona, Spain) for advice regarding the hystologic analyses.
ABBREVIATIONS USED
- AP
Acute Pancreatitis
- ATF3
Activating Transcription Factor 3
- ATR
Attenuated Total Reflectance
- BEGM
Bronchial Epithelial Growth Medium
- BEH
Ethylene Bridged Hybrid
- BFC
Benzyloxytrifluoromethylcoumarin
- CER
cerulein
- CHOP
C/EBP homologous protein
- DBF
Dibenzylfluorescein
- DiHETrE
Dihydroxyeicosatrienoic Acids
- DIPEA
Diisopropylethyl Amine
- E/S
Enzyme/Substrate
- EDCI·HCl
1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride
- EETs
Epoxyeicosatrienoic Acids
- ER
Endoplasmic Reticulum
- ERCP
Endoscopic Retrograde Cholangiopancreatography
- EtOAc
Ethyl Acetate
- FBS
Fetal Bovine Serum
- Fs
Femtosecond
- G418
Geneticin
- H&E
Hematoxylin and Eosin
- HOBt
Hydroxybenzotriazole
- hsEH
human soluble Epoxide Hydrolase
- IL
Interleukin
- KO
Knockout
- LC50
Concentration of the compound that is lethal for 50% of exposed population
- LOX
Lipoxygenases
- MCP-1
Monocyte Chemoattractant Protein 1
- msEH
murine soluble Epoxide Hydrolase
- NCI
non-covalent interactions
- ND
Not Done
- NEAA
Non-Essential Amino Acid
- P/S
Penicillin/Streptomycin solution
- PBMC
Peripheral Blood Mononuclear Cells
- Pe
Permeabiliy
- PHA
Phytohaemagglutinin
- PHOME
(3-Phenyl-oxiranyl)-acetic acid cyano-(6-methoxy-naphthalen-2-yl)-methyl ester
- PMSF
Phenylmethylsulfonyl fluoride
- RHS
Right-Hand Side
- RPMI
Roswell Park Memorial Institute
- sEH
soluble Epoxide Hydrolase
- THLE-2
Transformed Human Liver Epithelial-2 cell line
- TI
Thermodynamic Integration
- TRB3
Tribbles homolog 3
Footnotes
Supporting Information
1H and 13C NMR spectra and elemental analysis data of the new compounds, inhibition of hLOX-5 and hCOX-2 and Pharmacokinetic data of compound 22 (PDF). Molecular formula string and data (CSV). This material is available free of charge via the Internet at http://pubs.acs.org.
S.C., E.V., R.L., M.V.-C. and S.V. are inventors of the Universitat de Barcelona patent application on sEH inhibitors WO2017/017048. C.M. and B.D.H. are inventors of the University of California patents on sEH inhibitors licensed to EicOsis. None of the other authors has any disclosures to declare.
REFERENCES
- 1.The Role of Bioactive Lipids in Cancer, Inflammation and Related Diseases in Advances in Experimental Medicine and Biology; Honn KV, Zeldin DC, Eds.; Vol 1161, Springer, Cham, 10.1007/978-3-030-21735-8_1. [DOI] [Google Scholar]
- 2.Meirer K; Steinhilber D; Proschak E Inhibitors of the arachidonic acid cascade: interfering with multiple pathways. Basic Clin. Pharmacol. Toxicol 2014, 114, 83–91. [DOI] [PubMed] [Google Scholar]
- 3.Kaspera R; Totah RA Epoxyeicosatrienoic acids: formation, metabolism and potential role in tissue physiology and pathophysiology. Expert Opin. Drug Metab. Toxicol 2009, 5, 757–771. [DOI] [PubMed] [Google Scholar]
- 4.Fleming I Cytochrome P450-dependent eicosanoid production and crosstalk. Curr. Opin. Lipidol 2011, 22, 403–409. [DOI] [PubMed] [Google Scholar]
- 5.Capdevila JH; Falck JR The arachidonic acid monooxygenase: from biochemical curiosity to physiological/pathophysiological significance. J. Lipid Res 2018, 59, 2047–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morisseau C; Hammock BD Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol 2013, 53, 37–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris TR; Hammock BD Soluble epoxide hydrolase: gene structure, expression and deletion. Gene 2013, 526, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pillarisetti S; Khanna I A multimodal disease modifying approach to treat neuropathic pain – inhibition of soluble epoxide hydrolase (sEH). Drug Discov. Today 2015, 20, 1382–1390. [DOI] [PubMed] [Google Scholar]
- 9.Wagner KM; McReynolds CB; Schmidt WK; Hammock BD Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol. Ther 2017, 180, 62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borhan B; Jones AD; Pinot F; Grant DF; Kurth MJ; Hammock BD Mechanism of soluble epoxide hydrolase. J. Biol. Chem 1995, 270, 26923–26930. [DOI] [PubMed] [Google Scholar]
- 11.Gómez GA; Morisseau C; Hammock BD; Christianson DW Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry 2004, 43, 4716–4723. [DOI] [PubMed] [Google Scholar]
- 12.Shen HC; Hammock BD Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J. Med. Chem 2012, 55, 1789–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anandan SK; Webb HK; Chen D; Wang Y-X; Aavula BR; Cases S; Cheng Y; Do ZN; Mehra U; Tran V; Vincelette J; Waszczuk J; White K; Wong KR; Zhang L-N; Jones PD; Hammock BD; Patel DV; Whitcomb R; MacIntyre DE; Sabry J; Gless R 1-(1-Acetyl-piperidin-4-yl)-3-adamantan-1-yl-urea (AR9281) as a potent, selective, and orally available soluble epoxide hydrolase inhibitor with efficacy in rodent models of hypertension and dysglycemia. Bioorg. Med. Chem. Lett 2011, 21, 983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen D; Whitcomb R; MacIntyre E; Tran V; Do NZ; Sabry J; Patel DV; Anandan SK; Gless R; Webb HK Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J. Clin. Pharmacol 2012, 52, 319–328. [DOI] [PubMed] [Google Scholar]
- 15.Jones PD; Tsai H-J; Do ZN; Morisseau C; Hammock BD Synthesis and SAR of conformationally restricted inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett 2006, 16, 5212–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hwang SH; Tsai H-J; Liu J-Y; Morisseau C; Hammock BD Orally bioavailable potent soluble epoxide hydrolase inhibitors. J. Med. Chem 2007, 50, 3825–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown JR; North EJ; Hurdle JG; Morisseau C; Scarborough JS; Sun D; Korduláková J; Scherman MS; Jones V; Grzegorzewicz A; Crew RM; Jackson M; McNeil MR; Lee RE The structure-activity relationship of urea derivatives as anti-tuberculosis agents. Bioorg. Med. Chem 2011, 19, 5585–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhattachar SN; Deschenes LA; Wesley JA Solubility: it’s not just for physical chemists. Drug Discov. Today 2006, 11, 1012–1018. [DOI] [PubMed] [Google Scholar]
- 19.Di L; Fish PV; Mano T Bridging solubility between drug discovery and development. Drug Discov. Today 2012, 17, 486–495. [DOI] [PubMed] [Google Scholar]
- 20.Gagneux AR; Meier R 1-Substituted 2-heteroadamantanes. Tetrahedron Lett 1969, 10, 1365–1368. [DOI] [PubMed] [Google Scholar]
- 21.Duque MD; Camps P; Profire L; Montaner S; Vázquez S; Sureda FX; Mallol J; López-Querol M; Naesens L; De Clercq E; Prathalingam SR; Kelly JM Synthesis and pharmacological evaluation of (2-oxaadamantan-1-yl)amines. Bioorg. Med. Chem 2009, 17, 3198–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Onajole OK; Coovadia Y; Kruger HG; Maguire GEM; Pillay M; Govender T Novel polycyclic ‘cage’−1,2-diamines as potential anti-tuberculosis agents. Eur. J. Med. Chem 2012, 54, 1–9. [DOI] [PubMed] [Google Scholar]
- 23.Leiva R; Gazzarrini S; Esplugas R; Moroni A; Naesens L; Sureda FX; Vázquez S Ritter reaction-mediated syntheses of 2-oxaadamantan-5-amine, a novel amantadine analog. Tetrahedron Lett 2015, 56, 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duque MD; Ma C; Torres E; Wang J; Naesens L; Juárez-Jiménez J; Camps P; Luque FJ; DeGrado WF; Lamb RA; Pinto LH; Vázquez S Exploring the size limit of templates for inhibitors of the M2 ion channel of influenza A virus. J. Med. Chem 2011, 54, 2646–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones PD; Wolf NM; Morisseau C; Whetstone P; Hock B; Hammock BD Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal. Biochem 2005, 343, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammock BD; Hwang SH; Wecksler AT; Morisseau C Sorafenib derivatives as soluble epoxide hydrolase inhibitors WO 2012/112570 A1, August 23, 2012.
- 27.Gless RD Processes for the preparation of piperidinyl-substituted urea compounds US 2008/0207908 A1, August 28, 2008.
- 28.Rose TE; Morisseau C; Liu J-Y; Inceoglu B; Jones PD; Sanborn JR; Hammock BD 1-Aryl-3(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure – activity relationships, pharmacokinetics, and reduction of inflammatory pain. J. Med. Chem 2010, 53, 7067–7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee KSS; Liu J-Y; Wagner KM; Pakhomova S; Dong H; Morisseau C; Fu SH; Yang J; Wang P; Ulu A; Mate CA; Nguyen LV; Hwang SH; Edin ML; Mara AA; Wulff H; Newcomer ME; Zeldin DC; Hammock BD Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. J. Med. Chem 2014, 57, 7016–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.North EJ; Scherman MS; Bruhn DF; Scarborough JS; Maddox MM; Jones V; Grzegorzewicz A; Yang L; Hess T; Morisseau C; Jackson M; McNeil MR; Lee RE Design, synthesis and anti-tuberculosis activity of 1-adamantyl-3-heteroaryl ureas with improved in vitro pharmacokinetic properties. Bioorg. Med. Chem 2013, 21, 2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thalji RK; McAtee JJ; Belyanskaya S; Brandt M; Brown GD; Costell MH; Ding Y; Dodson JW; Eisennagel SH; Fries RE; Gross JW; Harpel MR; Holt DA; Israel DI; Jolivette LJ; Krosky D; Li H; Lu Q; Mandichak T; Roethke T; Schnackenberg CG; Schwartz B; Shewchuk LM; Xie W; Behm DJ; Douglas SA; Shaw AL; Marino JP Jr. Discovery of 1-(1,3,5-triazin-2-yl)piperidine-4-carboxamides as inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett 2013, 23, 3584–3588. [DOI] [PubMed] [Google Scholar]
- 32.Lazaar AL; Yang L; Boardley RL; Goyal NS; Robertson J; Baldwin SJ; Newby DE; Wilkinson IB; Tal-Singer R; Mayer RJ; Cheriyan J Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol 2016, 81, 971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bettaieb A; Chahed S; Tabet G; Yang J; Morisseau C; Griffey S; Hammock BD; Haj FG Effects of soluble epoxide hydrolase deficiency on acute pancreatitis in mice. PLos One 2014, 9, e113019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bettaieb A; Chahed S; Bachaalany S; Griffey S; Hammock BD; Haj FG Soluble epoxide hydrolase pharmacological inhibition ameliorates experimental acute pancreatitis in mice. Mol. Pharmacol 2015, 88, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griñán-Ferré C; Codony S; Pujol E; Yang J; Leiva R; Escolano C; Puigoriol-Illamola D; Companys-Alemany J; Corpas R; Sanfeliu C; Loza MI; Brea J; Morisseau C; Hammock BD; Vázquez S; Pallàs M; Galdeano C Pharmacological inhibition of soluble epoxide hydrolase as a new therapy for Alzheimer’s disease. Neurotherapeutics, 2020, 10.1007/s13311-020-00854-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie L; Ochterski JW; Gao Y; Han B; Caldwell TM; Xu Y; Peterson JM; Ge P; Ohliger R Dipiperazinyl ketones and related analogs WO 2007/0106496 A2, February 8, 2007.
- 37.Serrano-Hervás E; Casadevall G; Garcia-Borràs M; Feixas F; Osuna S Epoxide hydrolase conformational heterogeneity for the resolution of bulky pharmacologically relevant epoxide substrates. Chem. Eur. J 2018, 24, 12254–12258. [DOI] [PubMed] [Google Scholar]
- 38.Öster L; Tapani S; Xue Y; Käck H Successful generation of structural information for fragment-based drug discovery. Drug Discovery Today 2015, 20, 1104–1111. [DOI] [PubMed] [Google Scholar]
- 39.Durrant JD; Votapka L; Sorensen J; Amaro RE POVME 2.0: An enhanced tool for determining pocket shape and volume characteristics. J. Chem. Theory Comput 2014, 10, 5047–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Codony S; Valverde E; Leiva R; Brea J; Loza MI; Morisseau C; Hammock BD; Vázquez S Exploring the size of the lipophilic unit of the soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem 2019, 27, 115078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Contreras-García J; Johnson ER; Keinan S; Chaudret R; Piquemal J-P; Beratan DN; Yang W NCIPLOT: a program for plotting noncovalent interaction regions. J. Chem. Theory Comput 2011, 7, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guan D; Matthews S LogP prediction performance with the SMD solvation model and the M06 density functional family for SAMPL6 blind prediction challenge molecules. J. Comput.-Aided Mol. Des 2020, 34, 511–522. [DOI] [PubMed] [Google Scholar]
- 43.Di L; Kerns EH; Fan K; McConnell OJ; Carter GT High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem 2003, 38, 223–232. [DOI] [PubMed] [Google Scholar]
- 44.Yadav D; Lowenfels AB The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forsmark CE; Vege SS; Wilcox CM Acute pancreatitis. N. Engl. J. Med 2016, 375, 1972–1981. [DOI] [PubMed] [Google Scholar]
- 46.Krishna SG; Kamboj AK; Hart PA; Hinton A; Conwell DL The changing epidemiology of acute pancreatitis hospitalizations: a decade of trends and the impact of chronic pancreatitis. Pancreas 2017, 46, 482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weber H; Hüns S; Jonas L; Sparmann G; Bastian M; Schuff-Werner P Hydrogen peroxide-indiced activation of defense mechanisms against oxidative stress in rat pancreatic acinar AR42J cells. Free Radic. Biol. Med 2007, 42, 830–841. [DOI] [PubMed] [Google Scholar]
- 48.González A; Santofimia-Castaño P; Salido GM (2011). Culture of pancreatic AR42J cell for use as a model for acinar cell function. Pancreapedia: Exocrine pancreas Knowledge Base https://www.pancreapedia.org/tools/methods/culture-of-pancreatic-ar42j-cell-for-use-as-model-for-acinar-cell-function (accessed December 23, 2019). [Google Scholar]
- 49.Mareninova OA; Orabi AI; Husain SZ (2015). Experimental acute pancreatitis: in vitro models. Pancreapedia: Exocrine pancreas Knowledge Base https://www.pancreapedia.org/reviews/experimental-acute-pancreatitis-in-vitro-models (accessed December 23, 2019). [Google Scholar]
- 50.Afghani E; Pandol SJ; Shimosegawa T; Sutton R; Wu BU; Vege SS; Gorelick F; Hirota M; Windsor J; Lo SK; Freeman ML; Lerch MM; Tsuji Y; Melmed GY; Wassef W; Mayerle J Acute pancreatitis-progress and challenges: a report on an international symposium. Pancreas 2015, 44, 1195–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu J; Obando D; Liao V; Lifa T; Codd R The many faces of the adamantyl group in drug design. Eur. J. Med. Chem 2011, 46, 1949–1963. [DOI] [PubMed] [Google Scholar]
- 52.Wanka L; Iqbal K; Schreiner PR The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives. Chem. Rev 2013, 113, 3516–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morisseau C; Hammock BD Measurement of soluble epoxide hydrolase (sEH) activity. Curr. Protoc. Toxicol 2007, Chapter 4, Unit 4.23. [DOI] [PubMed] [Google Scholar]
- 54.Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham TEI; Cruzeiro VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK; Gohlke H; Goetz AW; Greene D; Harris R; Homeyer N; Izadi S; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein DJ; Merz KM; Miao Y; Monard G; Nguyen C; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling CL; Smith J; Salomon-Ferrer R; Swails J; Walker RC; Wang J; Wei H; Wolf RM; Wu X; Xiao L; York DM; Kollman PA (2018), AMBER 2018, University of California, San Francisco.Ta [Google Scholar]
- 55.Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- 56.Bayly CI; Cieplak P; Cornell W; Kollman PA A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem 1993, 97, 10269–10280. [Google Scholar]
- 57.Besler BH; Merz KM Jr.; Kollman PA Atomic charges derived from semiempirical methods. J. Comput. Chem 1990, 11, 431–439. [Google Scholar]
- 58.Singh UC; Kollman PA An approach to computing electrostatic charges for molecules. J. Comput. Chem 1984, 5, 129–145. [Google Scholar]
- 59.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Gaussian 09, Revision A.02; Gaussian, Inc.: Pittsburgh, PA, 2009). [Google Scholar]
- 60.Sagui C; Darden TA Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct 1999, 28, 155–179. [DOI] [PubMed] [Google Scholar]
- 61.Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Phys 1977, 23, 327–341. [Google Scholar]
- 62.Wu X; Brooks BR Self-guided Langevin dynamics simulation method. Chem. Phys. Lett 2003, 381, 512–518. [Google Scholar]
- 63.Song LF; Lee T-S; Zhu C; York DM; Merz KM Jr. Using AMBER18 for relative free energy calculations. J. Chem. Inf. Model 2019, 59, 3128–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao Y; Truhlar DG The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 2008, 120, 215–241. [Google Scholar]
- 65.Marenich AV; Cramer CJ; Truhlar DG Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- 66.Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 67.Pufahl RA; Kasten TP; Hills R; Gierse JK; Reitz BA; Weinberg RA; Masferrer JL Development of a fluorescence-based enzyme assay of human 5-lipoxygenase. Anal. Biochem 2007, 364, 204–212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.