

Abstract
Gut injury continues to be the devastating and unpredictable critical illness associated with increased cell death of intestinal epithelial cells (IECs). The IECs, immune system and microbiome are the interrelated entities to maintain normal intestinal homeostasis and barrier integrity. In response to microbial invasion, IEC cell death occurs to maintain intestinal epithelium function and retain the continuous renewal and tissue homeostasis. But the imbalance of IEC cell death results in increased intestinal permeability and barrier dysfunction that leads to several acute and chronic intestinal diseases, such as intestinal ischemia/reperfusion (I/R), sepsis, inflammatory bowel diseases (IBD), necrotizing enterocolitis (NEC), etc. During the pathophysiological state, the excessive IEC apoptotic cell death leads to a chronic inflammatory condition, later switches to necroptotic cell death mechanism that induces more pathological features than apoptosis and may also induce other lytic cell death mechanisms like pyroptosis and ferroptosis to increase the pathogenesis of the intestinal diseases. But still, there remains gaps in the fundamental knowledge about the IEC cell death mechanisms in chronic intestinal diseases. Together, a deep understanding of the specific cell death mechanisms underlying chronic intestinal diseases, including sepsis, IBD, NEC, and intestinal I/R, is desperately needed to develop emerging novel promising therapeutic strategies. This review aims to show how the acute and critical illness in the gut are driven by IEC cell death mechanism, such as apoptosis, necrosis, necroptosis, pyroptosis, and ferroptosis.
Keywords: Intestinal epithelial cells, apoptosis, necroptosis, pyroptosis, ferroptosis, intestinal diseases
1. Introduction:
The intestinal epithelium consists of a single layer of tightly linked columnar epithelial cells, providing the intestinal mucosa’s first-line defense. It is organized in crypt-villus units encompassing the luminal surface of the intestinal mucosa and is continuously replaced every 4–5 days. The intestinal epithelium has various critical physiological functions beyond absorption and digestion, and it mainly provides a critical barrier to prevent the translocation of destructive intraluminal substances including foreign antigens, and microorganisms, and their toxins [1]. In the event of a sensation of danger molecules, enterocytes transmit the signals to underlying cells/tissues and to the body to initiate innate and adaptive immune defense mechanisms together with specialized immune cells [2]. Therefore, the epithelium, the immune system, and the microbiome are the three closely interrelated entities to maintain the balanced homeostasis in the gut [3]. The alteration in these entities, including the dysregulation of the immune system, the perturbation of intestinal epithelial homeostasis, uncontrolled bacterial colonization, and the epithelial barrier dysfunction contribute to the onset of the gut injury [4]. Consequently, the disruption of intestinal membrane permeability by altering cell-cell junctional proteins such as occludin, E-cadherin, and ZO-1, is a pathogenic key factor and an early marker in the development of systemic inflammatory response syndrome and multiple organ failure (MOF).
Now the key question is, how disruption of intestinal epithelium homeostasis arises and progresses into the acute and chronic gut injury? What is the central risk factor for the enhanced intestinal permeability? In this regard, the growing evidence shows that increased intestinal permeability is highly associated with dysregulated intestinal epithelial cells (IECs) death [3]. The inappropriate IECs apoptosis promotes gut injury, intestinal barrier dysfunction, and translocation of bacteria, that results in chronic gastrointestinal (GI) disorders. Apoptosis is the important event to maintain the function of the intestinal epithelium at normal state, but the excessive cell death leads to the chronic inflammatory condition during the pathophysiological state, as found in the patients with critical GI symptoms [3]. Beyond apoptosis, the other programmed and non-programmed cell death mechanisms including necrosis, necroptosis, pyroptosis, and ferroptosis also play a key role in the pathogenesis of acute and chronic gut injury. We postulate that IECs cell death mechanism plays a key role in the intestinal hyperpermeability seen in the chronic intestinal diseases. This review highlights mechanisms of how intestinal epithelial cell death results in acute and chronic GI disorders, as well as preventive approaches to IECs death mechanisms for restoring and maintaining the gut barrier.
2. Intestinal epithelial cell death
The persistent IECs renewal is essential for maintaining tissue homeostasis. As we mentioned earlier, the excessive IECs cell death disrupts intestinal barrier integrity and permits the invasion of luminal antigens into the lamina propria (LP), thereby leading to a chronic inflammatory condition in the LP [5]. At present IECs undergoes several cell death pathways including apoptosis, necrosis, necroptosis, pyroptosis, and ferroptosis, depending on their stress, inflammatory, and microbial dysbiosis state. Therefore, IECs cell death is a hallmark of intestinal inflammation. To understand the pathophysiology and pathogenesis of acute and chronic GI disease, as well as the development of therapeutic approaches for GI illnesses, it is therefore essential to focus on the cellular and molecular mechanisms of IECs cell death.
2.1. Apoptosis
Apoptosis is a programmed cell death, characterized by the cell rounding, nuclear fragmentation, and blebbing of the plasma membrane [6]. It occurs spontaneously in IECs as the end-phase of migration and differentiation along the crypt-villus axis to maintain the regular gut morphology and function, including the intestinal homeostatic balance between epithelial cells proliferation and apoptosis [7]. The imbalance of this event turns out to the excessive loss of villi IECs beyond the frequency of crypt IECs regeneration, which leads to pathological IEC shedding [8]. In the early era of pathogenic microbial translocation, the dysregulated apoptosis is initially mediated by pattern recognition receptors (PRRs) including toll-like receptors (TLRs) and nucleotide-binding domain leucine-rich repeat containing receptors (NLRs) of IECs and other inflammatory cells; These receptors recognize the ligands such as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) of the pathogenic microbes [9]. After receptor-ligand binding occurs, the IECs quickly upregulate the expression levels of pro-inflammatory cytokines and mediators including tumor necrosis factor (TNF)-α and nitric oxide (NO) and pro-inflammatory chemokines to attract the inflammatory cells. Moreover, this chronic inflammatory environment induces the concomitant expression of death receptors such as Fas and tumor necrosis factor receptor-1 (TNFR1) and their ligands such as Fasl, TNF-α, and TNF-related apoptosis-inducing ligand (TRAIL), by the same or adjacent cells to induce the extrinsic cell death mechanism of enterocytes [10]. The binding of ligands to their respective death receptors recruits TNF receptor type 1-associated death domain (TRADD) protein and receptor-interacting protein kinase 1 (RIPK1) to TNFR1 to form a complex I [11]. Subsequently, RIPK1 dissociates from TNFR1 and recruits adaptor proteins such as Fas-associated death domain (FADD) to initiate the caspases cascade including Caspase-8 to form the death-induced signaling complex (DISC) [12]. Finally, the activation of Caspase-8 can either directly activates the downstream Caspases like Caspase-3, -6, and -7 or cleaves pro-Bid to form Bid which is translocated to mitochondria (intrinsic pathway) and induces cytochrome c along with the activating factor 1 (Apaf-1) to activate Caspase-3 and -9 [12], thereby results in apoptosis. Although the Caspase-mediated apoptosis is essential for IECs turnover and gastrointestinal tract morphology, growing evidence has shown the pathogenic function of Caspase-mediated IECs apoptosis in the chronic GI disease, such as IBDs, Crohn’s disease (CD) and ulcerative colitis (UC) [13] (Fig. 1).
Fig. 1.
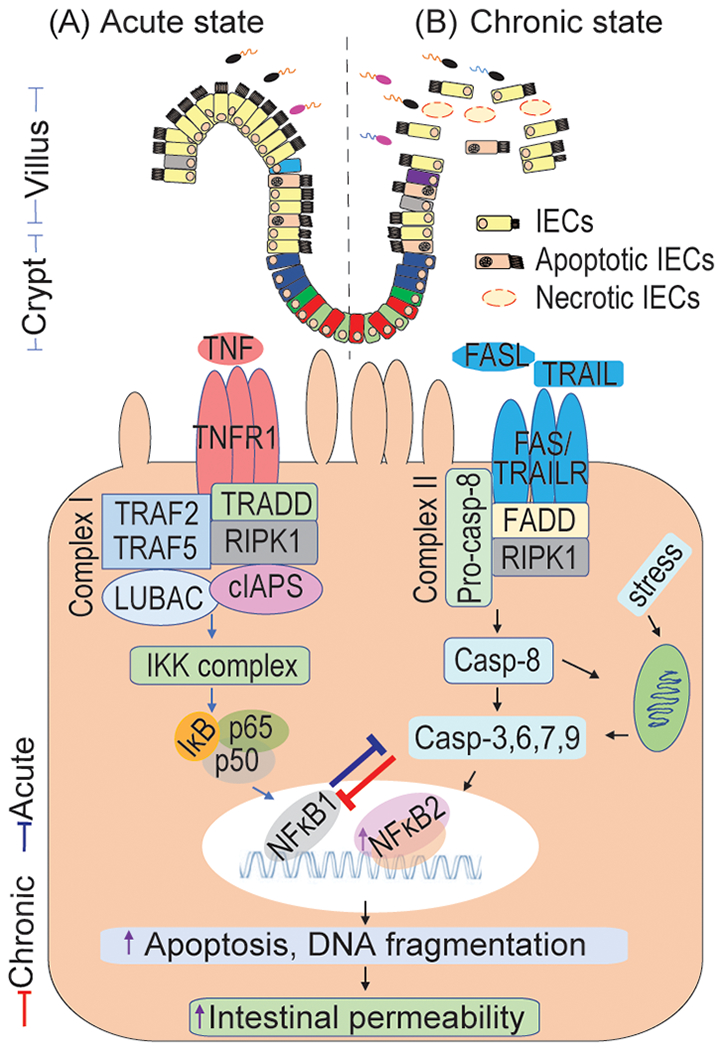
Intestinal epithelial cells (IECs) apoptosis in an acute and chronic state of intestinal diseases. Apoptosis is a crucial factor for preserving gut homeostasis. The apoptotic pathway is mediated by the stimulation of TNFR1 and FAS receptor with receptive ligands TNF and FASL or TRAIL that leading to the activation of NFκB, which turns in the regulation of cell survival, apoptosis, and pro-inflammatory genes expression. A: During the acute intestinal disease state, there is an increase in the activation of NFκB1 survival pathways and it’s mediated pro-inflammatory mediators, which plays a host defensive mechanism in regulating the caspase activity and apoptotic program to remove infected and damaged IECs. B: In meanwhile, during the chronic intestinal disease state, there is an increase in the activation of NFκB2 pro-apoptotic pathways and Caspase-3, -8 and -9 that inhibit the epithelial survival NF-kB1 pathways, provoke strikingly rapid epithelial apoptosis, reduce epithelial proliferation and result in the pathological IECs shedding. This event results in increased intestinal barrier dysfunction and intestine permeability, leading to several acute and chronic intestinal diseases, including sepsis, inflammatory bowel diseases, necrotizing enterocolitis, etc. TNF, tumor necrosis factor; FASL, Fas ligand; TNFR1, tumor necrosis factor receptor 1; TRAIL, TNF-related apoptosis-inducing ligand; TRAILR, TRAIL receptor; RIPK1, receptor-interacting serine/threonine-protein kinase 1; FADD, Fas-associated death domain; TRADD, TNFR1-associated death domain protein; cIAPs, cellular inhibitor of apoptosis proteins; LUBAC, linear ubiquitin chain assembly complex; IKK, the inhibitor of I-κB kinase; NFκB, nuclear factor kappa B; DISC, Death-induced signaling complex; Apaf-1, activating factor 1; Casp, Caspase.
2.2. Necrosis
Unlike apoptosis, necrosis is an unprogrammed or uncontrolled and accidental form of cell death, characterized by cell and organelles swelling, moderate chromatin condensation, rupture of the plasma membrane, and extensive cell lysis [6]. During the presence of various pathological stimuli, the inappropriate release of pro-inflammatory cytokines, including TNF-α, not only mediates IEC apoptotic cell death, but also induces necrotic cell death [14]. Whereas necrosis is highly associated with increase caspase-independent inflammation and reactive oxygen species (ROS) levels [15]. Once the intestinal integrity disturbance happens, intestinal mucosal T lymphocytes induce the expression levels of Th1 cytokines, such as TNF-α and interleukin (IL)-1, and stimulate IECs to generate ROS that acts as secondary messengers to regulate inflammation and its mediated signaling pathways [16]. As well the phagocytic leukocyte-derived ROS also maintains the chronic inflammatory state in the intestine and worsens the infectious GI diseases [17]. While mitochondrial generation and removal of ROS are dynamically balanced and useful for cells without causing damage, over-generating of ROS in IECs is dangerous [16] and induce intestinal injury (Fig. 2A).
Fig. 2.
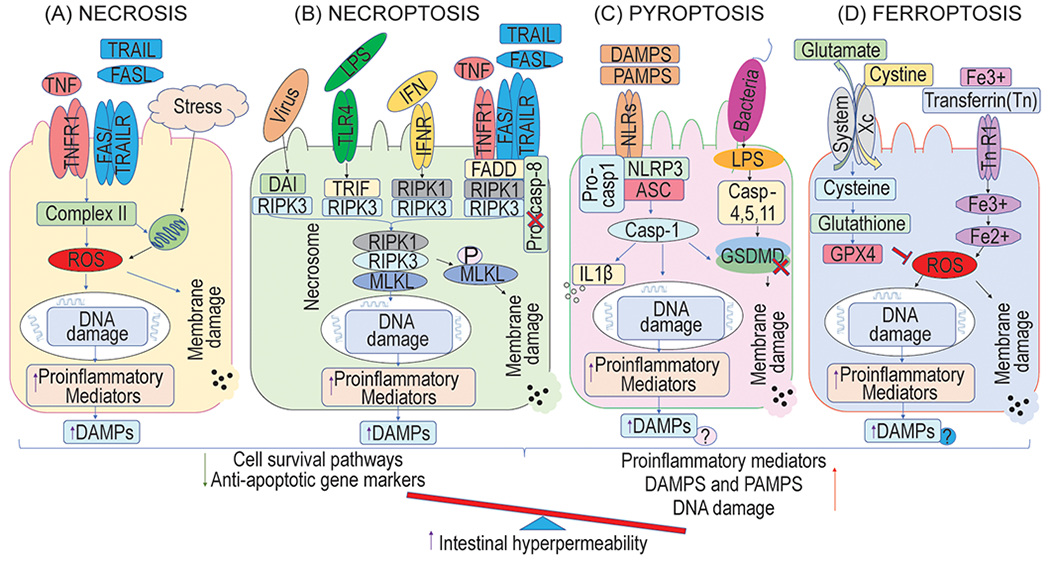
Lytic cell death pathways of intestinal epithelial cells (IECs) during the intestinal diseases. A: The inadequate release of TNF-α not only mediates IECs apoptosis, but also triggers necrotic cell death by inducing ROS, which acts as secondary messengers to regulate inflammation and its mediated signaling pathways during the presence of multiple pathological stimuli and stress condition. B: The binding of death ligands with their corresponding receptors (as shown in the figure) together with adapter proteins, but under caspase-8 or cIAP depletion, promotes the cell death pathways of necroptosis through the formation of necrosome RIPK1/RIPK3/MLKL, which leads to the phosphorylation of MLKL (p-MLKL). This event reduces the cell survival and anti-apoptotic pathways and increases the pro-inflammatory mediators along with intestinal barrier dysfunction and intestine permeability. C: Sensing of DAMPs or PAMPs by NLRs promotes the formation of inflammasome complex that involves NLRP3, ASC, and caspase-1 in the canonical pathway. In the non-canonical pathway, LPS leads to the activation of caspase-11 and results in cytoplasmic swelling and cytosolic content leakage along with DAMPs. Although pyroptosis occurs in inflammatory cells, it seems that pyroptosis in IECs plays an important role in the pathogenesis of chronic intestinal diseases, but several scientific pieces of evidences are needed to fill this gap. D: The increase in iron-dependent lipid peroxidation and lipid ROS accumulation and decrease in GPX4 activation through system Xc− inhibition lead to the destruction of IECs and intestinal hyperpermeability. However, the evidence for supporting ferroptosis in IECs is very less. DAMPS, Damage-associated molecular patterns; PAMPs, Pathogen-associated molecular pattern molecules; DAI, DNA-dependent activator of IFN-regulatory factors; MLKL, Mixed lineage kinase domain-like pseudokinase; NLRs, Nucleotide-binding oligomerization domain-like receptors; ASC, an apoptosis-associated speck-like protein containing a CARD domain; NLRP3, Nucleotide-binding oligomerization domain-, leucine-rich repeat-, and pyrin domain-containing protein 3; GPX4, glutathione peroxidase 4; LPS, lipo-polysaccharides; TLR4, Toll-like receptor 4; IL-1β, interleukin-1β; ROS, reactive oxygen species; GSDMD, gasdermin-D.
2.3. Necroptosis
Necroptosis, a novel manner of cell death modality, is an inflammatory form of programmed cell death mechanism with a necrosis phenotype characteristic, but not of apoptosis [18]. In detail, necroptosis is highly regulated by an intracellular protein platform, including the combination of death ligands and receptors along with adapter proteins, but under the inhibition of caspase activation [19]. TNFR1 and Fas are often used as a prevalent upstream signal by both necroptosis and apoptosis, but during necroptosis, suppression of caspase-8 system [13, 20] and recruitment of receptor interacting protein kinase (RPIK)-3 through the RIPK homotypic interaction motif (RHIM) domain to RIPK1 occur to form a necrosis-inducing complex [11, 21]. The interaction of RIPK1 and RIPK3 kinases through RHIM results in their auto- and trans-phosphorylation, and RIPK3-mediated phosphorylation of the downstream pseudo-kinase mixed lineage kinase domain-like (MLKL) [22, 23]. The phosphorylation of MLKL in IECs ultimately increases cytokine/alarmin expression such as interleukin 8 (IL-8), IL-1β, IL-33, and high mobility group box 1 (HMGB1), nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB)-p65 translocation and NACHT, LRR and PYD domains-containing protein 3 (NALP3) inflammasome assembly [24]. Thereby the phosphorylation of RIP3 and MLKL is essential for necrosis execution [25, 26] (Fig. 2B). Taken together, the central function of caspase-8 [13] and inhibition of RIP3 [27] and MLKL [11] in IECs preserve epithelial barrier integrity, maintains homeostasis and prevents chronic intestinal inflammation by protecting IECs from necroptotic cell death. However, the functions and detailed mechanisms of necroptosis in IECs-mediated chronic GI tract diseases remain largely unknown.
2.4. Pyroptosis
Pyroptosis is also a programmed and inflammatory form of cell death but caspase cascade dependent mechanism, described morphologically as cell rupture, followed by membrane “re-sealing,” and cell swelling with nuclear condensation [6]. In response to microbial infection, the subset of NLRs is triggered by detecting a variety of DAMPS and PAMPs, promoting to form a multimeric protein complex known as the inflammasome through oligomerize with an adaptor protein known as ASC (an apoptosis-associated speck-like protein containing a CARD domain), and a proenzyme, caspase-1 [28]. The inflammasome formation stimulates the protease activity of caspase 1 (canonical pathway) that cleaves the pore-forming effector protein, gasdermin-D, and results in the release of IL-1β and IL-18 to a form of pyroptosis-mediated inflammatory cell death [28]. The influence of lipo-polysaccharides (LPS) from gram-negative bacterial components leads to the activation of caspase-11 and pyroptosis of enterocytes [29] (Fig. 2C). Recent studies show that pyroptosis acts as a central role in intestinal immune defense and pathology by regulating microbial infections and secretion of IL-18, ROS production, or lysosomal damage [15, 28]. However, luminal content occasionally carries pathogenic microbes or toxic elements proficient of producing mucosal damage, pyroptosis-mediated NLRP3 inflammasome [30], caspase-1 [31] and cytokines, such as IL-1 and IL-18 [32], results to the chronic inflammatory state in GI tract diseases. Thus, it is important to understand the debate in the protective and destructive function of pyroptosis mechanism in IECs during the acute and chronic GI diseases.
2.5. Ferroptosis
Ferroptosis is a new form of iron-dependent, caspase-independent, lipid oxidation-mediated programmed cell death, differing from traditional apoptosis, necroptosis, and classic necrosis [33]. It is characterized by morphologically shrinkage of mitochondria and increases in mitochondrial membrane density, biochemically accumulation of iron and lipid ROS (L-ROS), and genetically involvement of a unique set of genes [34]. The increased levels of lipid peroxidation (LPO) by deficiency of glutathione peroxidase 4 (GPx4) activation through system Xc− inhibition, and oxidation of arachidonic acid (AA) and its esterifiable phosphatidylethanolamine (PE) production [34, 35], subsequently leading to the destruction of IECs and intestinal mechanical barrier [36] (Fig. 2D). Recently, the finding of reduced activity of GPx4 in the lesioned areas of the gut of the CD patients in IECs suggests that reduction of GPx4 activity in IECs induces ferroptosis [37]. It appears that dietary derived compounds, pathogenic microbial mediated metabolites, such as fatty acids, could trigger ferroptosis that contributes to the pathogenesis of the GI diseases. However, the evidence for supporting ferroptosis in IECs is very less. Thus, this review brings up an interesting research topic related to IECs ferroptosis mechanisms in chronic GI disease.
3. Cell death in IECs-mediated acute and chronic GI injury
Apoptosis is a crucial factor for usual intestinal mucosal turnover. The balance between proliferation and apoptosis of IECs is partially dependent on the microenvironment, host state, and stress categories in the intestine; the defect in balance is strongly connected with several intestinal diseases and GI injuries [38]. It is essential to characterize the duration of intestinal diseases as an acute injury that lasts only for a few (7–14) days, and chronic GI injuries that persist for months or longer [4]. Several studies have been performed to evaluate the pathological features of IEC apoptosis during the pathogenesis of acute and chronic GI injury. The clinical patients with acute and chronic intestinal diseases, in vitro IEC models, and in vivo animal models are mostly accessible to study the IEC cell death mechanism (Table 1). Here we target the pathogenic impact of cell death in IEC to the acute and chronic diseases, as well as the available therapeutic approach to prevent IEC cell death to regulate intestinal diseases.
Table1.
Intestinal epithelial cell death in the acute and chronic intestinal diseases
| Model | Subjects | Cell death | Findings |
|---|---|---|---|
| A) Sepsis | |||
| Clinical | Patients with sepsis | Apoptosis | Increase in active caspase 3 [39] |
| Clinical | Patients with trauma injuries | Apoptosis | Increase in cytokeratin 18 and active caspase 3 [40] |
| Clinical | Patients with sepsis | Apoptosis | Increase in I-FABP [41] |
| In vivo CLP induced sepsis | Transgenic mice that overexpress Bcl-2 (Fabpl-Bcl-2) | Apoptosis | Decrease in apoptosis and active caspase 3 [44] |
| In vivo pneumonia-induced sepsis | Fabpl-Bcl-2 mice | Apoptosis | Decrease in apoptosis and active caspase 3 [42] |
| In vivo pneumonia-induced sepsis | Fabpl-Bcl-2 mice | Apoptosis | Decrease in apoptosis and active caspase 3 associate with increase in S-phase cells proliferation [43] |
| In vivo MRSA pneumonia-induced sepsis model | Wild-type FVB/N mice | Apoptosis | Increase in Bid and Bax and Bcl-xL in the mitochondrial pathway [45] |
| In vivo MRSA pneumonia-induced sepsis model |
Bid−/− mice Fabpl-Bcl-2 mice |
Apoptosis | Regulate the mitochondrial apoptotic pathway [45] |
| In vivo CLP induced sepsis model | Lacking functional NF-kB in IECs (Vil-Cre/Ikkβf/Δ) | Apoptosis | Increase in mortality, apoptosis with pro-inflammatory cytokines [46] |
| In vivo CLP induced sepsis model | STING-KO mice | Apoptosis | Decrease in apoptosis, inflammation, intestinal permeability and bacterial translocation [47] |
| In vivo LPS induced sepsis model | Tnfr1−/−, Tnfr2−/−, Nfκb1−/−, Nfκb2−/−, mice | Apoptosis | Dependent on NFκB signaling, via NFκB1 favoring cell survival or via NFκB2 favoring apoptosis [8] |
| In vivo LPS induced sepsis model | Co-expressed both Bcl-2 and TAg to Fabpl | Apoptosis | Bi-transgenic animals had reduced crypt apoptosis but had a paradoxical increase in the markers of apoptosis such as caspase 3, BAX and cytochrome c in villus [38] |
| B) Intestinal ischemia/reperfuson (I/R) | |||
| Clinical | Jejunum from patients undergoing pancreaticoduodenectomy | Apoptosis | Increase in apoptosis and I-FABP during ischemia and gradually decrease during reperfusion [51] |
| Clinical | Jejunum from patients undergoing pancreaticoduodenectomy | Apoptosis | Increase in apoptosis and I-FABP associate with inflammatory markers such as C3c complement activation, IL-6, IL-8, and TNFα [52] |
| In vivo I/R rat model | Ischaemia (15–90 min) and ischaemia/reperfusion (15 minutes ischaemia followed by 15–75 min of reperfusion) | Apoptosis, Necrosis | Death cells exhibit apoptosis (80%) and necrosis (20%) characteristics; increase in DNA fragmentation [53] |
| In vivo I/R rat model | Ischemia clamping the SMA (30 or 60 min), after reperfusion various time points up to 4 days. | Apoptosis | Increase in apoptosis and decrease in intestinal ALP and lactase after ischemia, and returned normal with reperfusion [54] |
| In vitro model of ischemia | 2-deoxyglucose and oligomycin-A treated HT-29 and Caco-2 cells | Apoptosis | Greater apoptotic in differentiated cells than undifferentiated cells [54] |
| In vivo I/R rat model | Underwent occlusion of both SMA and PV for 20 minutes followed by 48h of reperfusion | Apoptosis | Increase in apoptosis along with inflammatory markers upregulation of TLR-4, MyD88, and TRAF6 [49] |
| In vivo I/R rat model | Underwent occlusion of both SMA and PV for 20 minutes followed by 24h or 48h of reperfusion | Apoptosis | Increase in apoptosis inversely associate with SHh signaling pathways [50] |
| In vivo I/R rat model | 1hr of ischemia followed by reperfusion | Necroptosis, Necrosis | Increase in necroptotic markers such as RIP-1, -3 and MLKL [19] |
| In vitro model of ischemia | Oxygen and glucose deprivation model in IEC-6 | Necroptosis, Necrosis | Increase in RIP-1, -3 and MLKL together with HMGB1 - TLR4/RAGE signaling [19] |
| In vivo I/R rat model | SMA occlusion (1.5h) of ischemia and 6 h of reperfusion | Necroptosis | RIP1/3 mediated necrosome formation [55] |
| In vivo I/R murine model | IkbkbF/ΔVil-Cre; SMA occlusion for 30 mins followed by reper 】 fusion | Apoptosis | Increase in apoptosis and pro-inflammatory markers such as TNF, IL-1, IL-6 and ICAM. Probably dual function of NFκB signaling [56] |
| In vivo I/R murine model | Fabpl-Bcl-2 mice; SMAO for 20 mins followed by reperfusion | Apoptosis | Decrease in p53-dependent death [57] |
| C) Inflammatory bowel diseases (IBD) | |||
| Clinical | patients with UC | Apoptosis | Increase in apoptosis, active caspase 3 and PUMA expression [59, 62] |
| Clinical; In vivo TNBS induced colitis murine model |
Patients with CD and UC; Wild type balb/c mice | Apoptosis | up-regulation of TRAIL in IEC [60] |
| In vitro model | TRAIL, TNF-α and IFN-γ treatment in HIEC, HT-29 or Caco-2 cells | Apoptosis | NFκB-dependent (TNF-α) or NFκB-independent (IFN-γ) pathway to induce TRAIL mediated apoptosis [60] |
| In vivo DSS or TNBS induced colitis murine model | Wildtype, PUMA−/−, Bid−/−, p53−/− | Apoptosis | PUMA inhibition can provide an efficient way of protecting IEC apoptosis and serve as a new anti-IBD approach [59] |
| In vivo model | TAK1IE-KO mice | Apoptosis | Enhance in cleaved caspase-3 and reduction in claudin-3 and antioxidant- genes and transcription factor Nrf2, and ROS accumulation, like the IBD pathology [61] |
| In vivo anti-CD3 or DSS induced colitis murine model | wild-type, p53−/−, Bid−/−, Bim−/−, Bax3−/−, Bak−/−, PUMA−/−, and Noxa−/− mice | Apoptosis | p53-dependent and - independent mechanisms; PUMA mediated intrinsic apoptosis pathway [62] |
| Clinical; In vivo TNF induced apoptosis model |
Patients with CD and UC; transgenic mice that overexpress A20 in IECs A20-Tg mice | Apoptosis | RIPK1-Dependent IEC Death [63] |
| In vivo DSS induced colitis murine model | Villin kO mice | Apoptosis | Anti-apoptotic function of villin is regulated by PI3-kinase and Akt [64] |
| In vivo DSS induced colitis murine model | TLR4−/−mice | Apoptosis | Increase in apoptosis with reduced Cox-2 and PGE-2 levels [65] |
| In vivo LPS induced injury model | Epithelial cell-specific deletion of Casp8ΔIEC mice TLR stimulation | Necrosis, Necroptosis | Rip3-dependent epithelial necroptosis [66] |
| In vivo spontaneous model | Epithelial cell-specific deletion of FADDΔIEC | Necrosis, Necroptosis | Rip3-dependent epithelial necroptosis [27] |
|
In vivo TNBS induced colitis murine model; In vitro necroptosis model |
Wildtype mice; TNF-α and Z-VAD-fmk induced Caco-2 cells | Necrosis, Necroptosis | Increase in TUNEL-positive, caspase-3 negative cells along with p-RIPK3 [11] |
| Clinical; In vivo model; In vitro model |
Patients with CD; caspase-1/IL-10 double knockout; T84 monolayers | Pyroptosis | Increase in the activated caspase-1[67] |
| Clinical | Patients with CD | Ferroptosis | Reduction in GPx4 levels [37] |
| D) Necrotizing enterocolitis (NEC) | |||
| Clinical | Infants with NEC | Apoptosis | Increase in NO and apoptosis through peroxynitrite formation [70] |
| In vivo NEC model | formula feeding, and cold/asphyxia stress induced neonatal rat | Apoptosis | Increase in caspase 3 and DNA fragmentation [71] |
| In vitro NEC model | H2O2 induced rat IECs (RIE)-1 | Apoptosis | Increase in intracellular ROS generation activates PI3-k pathway [72] |
|
In vivo; In vitro NEC model |
formula feeding/hypoxia followed by Enterobacter sakazakii (ES) mediated NEC; ES administration to IEC-6 in vitro | Apoptosis | Increase in active caspase-3 and pro-inflammatory cytokines such as IL-6 [73] |
|
In vivo; In vitro NEC model |
formula feeding/hypoxia followed by Cronobacter sakazakii (CS) mediated NEC; CS administration to HT-29 in vitro | Pyroptosis, Apoptosis | Increase in NLRP3 inflammasome, caspase-3 and caspase-1 levels [74] |
|
In vivo; In vitro NEC model |
Rat pups collected by caesarian section, followed by hand fed; TNF-α and IFN-γ induced IEC-6 cells | Apoptosis | Increase in Bax/Bcl-w ratio, cleaved caspase-3 and COX-2 levels; these events were reverted by Bifidobacterium bifidum [75] |
| In vivo NEC model | NEC induced by asphyxia and cold stress, and followed by hand fed milk | Apoptosis | Increase in pro-apoptotic Bax, cleaved caspase-3, and decrease in anti-apoptotic Bcl-2; this effect was attenuated by EGF administration [76] |
| In vivo NEC model | NEC induced by hypoxia, hypothermia, hypertonic formula feeding plus enteral administration of LPS | Apoptosis | Increase in TUNEL and active caspase 3 levels; these changes were inhibited by HB-EGF [77] |
I-FABP, Intestinal fatty acid-binding protein; CLP, Cecal ligation and puncture; MRSA, Methicillin-resistant Staphylococcus aureus; Bcl-2, B-cell lymphoma 2; Bid, BH3 Interacting Domain Death Agonist; Bax, BCL2 Associated X, Apoptosis Regulator; Bcl-xL, B-cell lymphoma-extra-large; IKK, the inhibitor of I-κB kinase; NFκB, nuclear factor kappa B; STING, Stimulator of interferon genes; LPS, lipo-polysaccharides; TAg, viral protein large T-antigen; TNF, tumor necrosis factor; IL, interleukin; IR, ischaemia/reperfusion; SMA, superior mesenteric artery; PV, portal vein; SHh, sonic hedgehog; TLR, Toll-like receptor; TRAF6, Tumor necrosis factor receptor (TNFR)-associated factor 6; MyD88, Myeloid differentiation factor 88; RIPK, receptor-interacting serine/threonine-protein kinase; MLKL, Mixed lineage kinase domain-like pseudokinase; RAGE, Receptor for advanced glycosylation end product; ICAM, Intercellular Adhesion Molecule; UC, Ulcerative colitis; CD, Crohn’s disease; PUMA, p53 upregulated modulator of apoptosis; TRAIL, TNF-related apoptosis-inducing ligand; TNBS, 2,4,6-trinitrobenzene sulfonic acid; IFN, Interferon; HIEC, human intestinal epithelial cells; IBD, Inflammatory bowel disease; DSS, Dextran sodium sulfate; TAG1, TGF-β activated kinase 1; COX-2, cyclooxygenase-2; PGE2, prostaglandin E2; FADD, Fas-associated death domain; TUNEL, Terminal deoxynucleotidyl transferase dUTP nick end labeling; NEC, Necrotizing enterocolitis; ROS, reactive oxygen species; NO, Nitric oxide; NLRs, Nucleotide-binding oligomerization domain-like receptors; NLRP3, NLR Family Pyrin Domain Containing 3; EGF, epidermal growth factor; HB-EGF, Heparin Binding EGF Like Growth Factor.
3.1. IEC cell death in Sepsis
Sepsis is a serious and life-threatening dysfunction of the organs, and it is secondary to a dysregulated host response to infection, affecting the intestine intensely [38]. There is increased apoptosis in the colon and ileum of septic patients in whom focal regions of columnar epithelial apoptosis occurred in crypt or villus [39]. In another clinical study, trauma patients who are much more prone to sepsis disclosed the increased severity of crypt epithelial apoptosis in the colon specimens, compared to control patients. Simultaneously, these trauma patients experienced a statistically significant increase in the number of cytokeratin-18 positive IECs [40]. In patients with septic shock, norepinephrine uses to maintain adequate blood pressure at ICU admission is associated with more enterocyte damage and higher intestinal fatty acid-binding protein (I-FABP), a marker for the early diagnosis of intestinal damage [41]. Thereby, the greater degree of apoptosis in the intestinal villi and crypts of septic patients suggested the enhanced IEC apoptosis in sepsis [39].
Several preclinical models of sepsis also suggested the key role of IEC apoptosis in the pathophysiology of sepsis. Mostly, cecal ligation and puncture (CLP) and pneumonia induced sepsis murine model have been used to evaluate the role of IEC apoptosis. There is a decrease in intestinal proliferation and an increase in gut epithelial cells apoptosis observed in both murine models of pneumonia-induced sepsis and CLP induced sepsis [42–44]. Whereas, methicillin-resistant Staphylococcus aureus (MRSA) pneumonia-induced sepsis showed intestinal apoptosis that highly associated with increased proapoptotic Bid and Bax proteins and the antiapoptotic Bcl-xL protein in the mitochondrial pathway, and FasL in the receptor-mediated pathway. This study shows that MRSA pneumonia-induced sepsis alters intestinal integrity by increasing IEC apoptosis and decreasing crypt proliferation and villus length, through apoptosis mechanism mediated by the mitochondrial pathway [45]. At the same time, mice lacking intestinal epithelium functional NF-kB (Vil-Cre/Ikkβf/Δ) showed an increase in IEC apoptosis and intestinal permeability along with decreased villus length and atrophy in CLP induced sepsis animals, compared to septic in wild-type mice. During ongoing sepsis, Vil-Cre/Ikkβf/Δ mice showed increased serum levels of pro-inflammatory, including TNF and monocyte chemoattractant protein-1 (MCP-1) and anti-inflammatory cytokines, such as IL-10, compared to septic wild-type mice. This result indicates that inhibition of IEC specific NF-κB worsens the sepsis-induced intestinal injury and aggravates mortality in CLP mice [46]. TNFR1 is crucial for LPS-induced IEC apoptosis and shedding, and the destiny of IECs is also dependent on NFκB signaling, via NFκB1 favoring cell survival or via NFκB2 favoring apoptosis [8]. Neutralizing TNF activity with anti-TNF antibody in CLP induced septic Vil-Cre/Ikkβf/Δ and septic wild-type mice, prevented intestinal hyperpermeability by increasing claudin-2 gene expression. This outcome suggests that TNF is the main mediator of the dysfunction of the intestinal barrier in sepsis, where anti-TNF antibody significantly decreases sepsis-induced IECs apoptosis and hyperpermeability [46].
In order to enhance our understanding to highlight the link of IECs apoptotic cell death in sepsis, Hu et al., showed that increased STING signaling that promotes the phosphorylation of STAT3, STAT6, IRF3, and NFκB, is highly associated with intestinal inflammation and induction of IEC apoptosis in patients with sepsis, as well as in CLP induced septic mice [47]. The mice with STING genetic depletion showed a reduced inflammatory response, intestinal permeability, and bacterial translocation. The treatment of DNase I protects the intestinal injury by decreasing mtDNA levels in CLP-induced septic animals. These findings indicate us that mtDNA-STING pathway regulation can be a promising therapeutic approach to improve mucosal healing in patients with sepsis and protect the intestinal barrier [47]. In a very current study, both genetic (hepcidin-1 knockout [HKO]) iron overload and iatrogenic (intravenous) iron overload mice developed sepsis after administration of clinical isolates E. coli within 24 h and associated with high bacterial multiplication and dissemination [48]. Hence host and pathogenic microbes are using iron as an essential micronutrient, it seems that iron-dependent ferroptosis cell death mechanism in IECs may have an impact on the severity of the pathogenesis of sepsis during the pathogenic microbial infection. But still, scientific evidence is needed to fill this gap.
Inhibition of cell death mechanism by blocking apoptotic markers or over-expressing anti-apoptotic markers prevents the pathogenesis of sepsis and enhances survival rate. Transgenic mice Fabpl-Bcl-2 (intestine-specific overexpression of Bcl-2, linked to Fabpl) showed decreased gut epithelial apoptosis in both sepsis model by decreasing the levels of active caspase 3. Therefore, preventing IEC apoptosis by overexpression of Bcl-2 was related to a survival advantage in sepsis [42, 44]. Bid−/− mice and Fabpl-Bcl-2 mice had decreased intestinal apoptosis, thereby, inhibited MRSA pneumonia-induced sepsis, compared to wild type animals. It is now highlighted that the potential to genetically manipulate the mitochondrial pathway could have theoretically therapeutic benefit in more lethal sepsis models whose high apoptosis of the gut is associated with increased mortality [45]. In the continuation of the previous study, Lyons et al. co-expressed both the genes such as Bcl-2 and TAg (large T-antigen, is limited to villus enterocytes) to Fabp that resulted in the expression of both genes in their villus enterocytes, but Bcl-2 alone in the crypt. As anticipated, bi-transgenic sepsis animals had reduced crypt apoptosis, but had a paradoxical increase in the markers of apoptosis such as caspase 3, BAX and cytochrome c in villus, compared with septic fabpi-Tag (TAg alone) mice, associated with decreased proliferation in both compartments [38]. Other than programmed cell death, the non-programmed cell death mechanism also plays a significant role in the pathogenesis of sepsis. But still, scientific evidence is needed to fill this knowledge gap.
3.2. IEC cell death in intestine ischemia/reperfusion (I/R)
Intestinal I/R injury is a complex and multifactorial pathophysiological process triggered by ROS formation and lipid mediator synthesis alteration [49]. The nonspecific damage caused by ischemia and injury increases the levels of several inflammatory cytokines and attracts the inflammatory cells, such as polymorphonuclear leukocytes and mast cells, into the intestinal wall, leading to intestinal epithelial apoptosis, intestinal hyperpermeability and intestinal barrier dysfunction which could result in MOF and death [50]. Thus, dysregulated apoptosis and inflammation are the main mediators in the pathogenesis of I/R-induced intestinal injury. The clinically relevant intestinal I/R models have resulted in a deeper understanding of the pathophysiology of human small intestinal and colon I/R over the past years. It has been shown that isolated jejunum from the patients undergoing pancreaticoduodenectomy was subjected to ischemia followed by reperfusion [51]. After ischemia, there is a significant increase in I-FABP across the jejunum, revealing the progression of epithelial cell damage. But at the same time, a decrease in I-FABP staining after reperfusion reveals the endogenous clearing mechanism for damaged enterocytes that results in normal epithelial lining [51]. The causative agents of the human intestinal I/R-induced inflammation were characterized by complement activation, cytokines production, and release into the systemic circulation, endothelial activation, and neutrophil influx into intestinal I/R-damaged tissue [52].
In preclinical studies, briefly, Ikeda et al. [53] exhibited that ischemia and I/R injury leads to apoptosis, which becomes the main mediator of cell death to intestinal epithelium after IR. Rats subjected to ischemia and I/R showed increased mucosal injury along with detached epithelial cells that exhibited majority with characteristic morphological features of apoptosis and rest of the cells with necrosis features [53]. It is evident that villus tip epithelial cells are more susceptible to ischemia and loss of intestinal alkaline phosphatase and lactase instantly following ischemia and returned with reperfusion, confirming that differentiated cells are particularly sensitive to ischemic injury [54]. I/R rats underwent laparotomy and vascular occlusion of both superior mesenteric artery (SMA) and portal vein (PV) for 20 min followed by reperfusion showed a significant decrease in the enterocyte proliferation index in both jejunum and ileum [49, 50]; this reduction in cell turnover is highly associated with SHh signaling pathway inhibition [50].
Few studies have investigated the efficacy of regulated IEC necrosis after intestinal I/R. I/R group rats received 1hr of ischemia followed by reperfusion showed an increase in the levels of necroptosis mediated markers RIP1/3 and MLKL, and these levels are inhibited by RIP1 kinase inhibitor necrostatin-1. In meanwhile, blocking both apoptotic and necroptotic cell death mechanism, using the pan-caspase inhibitor Z-VAD and necrostatin-1 respectively, confers better protection against intestinal I/R injury. But at the same time, in the presence of either only one of the inhibitor, these two pathways can be converted to one another. In in vitro oxygen and glucose deprivation-induced IR (IEC-6) model, necrostatin-1 decreases cell death and pro-inflammatory cytokine gene expression and confers IEC-6 protection via inhibiting HMGB1-TLR4/RAGE signaling activation [19]. Rats subjected to SMA occlusion consisting of 1.5 h of ischemia and 6 h of reperfusion, showed the activation of poly (adenosine diphosphate-ribose) polymerase 1 (PARP-1) and RIP1/3 mediated necrosome formation. The pretreatment of PARP-1-specific inhibitor PJ34 and the RIP1-specific inhibitor Necrostatin-1 resulted in a decrease in intestinal epithelial cell death and optimal protection of the intestine. Thus, in the development of I/R-induced intestinal injury, PARP-1 could function as a RIP1 downstream signaling molecule, and the RIP1/PARP-1-dependent cell death signaling pathway functioned independently of caspase 3 inhibition [55]. IEC specific ablation of IkB kinase (IKK)-β resulted in the prevention of the systemic inflammatory response triggered by IR and in severe apoptotic damage to the re-perfused intestinal mucosa, suggesting that NFκB system has a dual role for both tissue safety and systemic inflammation which could be inhibited by using NFκB and IKK inhibitors [56]. The forced overexpression of Bcl-2 inhibits I/R-induced p53-dependent apoptosis pathways in the intestinal epithelium of transgenic mice Fabpl-Bcl-2 [57]. Overall, targeting the IEC cell death mechanisms could be beneficial to alleviate intestinal IR tissue injury.
3.3. IEC cell death in inflammatory bowel diseases
Inflammatory bowel diseases (IBD), which involves CD and ulcerative colitis (UC), is the result of the breakdown of the symbiotic relationship between the commensal microbial/host intestinal immunity [58]. IEC death is a prevalent pathological characteristic of IBD causing inflammation by altering the integrity of the intestinal barrier. Despite this, in response to intestinal inflammation, little is known about the molecular mechanisms of IEC apoptosis. Increased apoptosis of IECs was identified in patients with UC and CD at the involved inflammatory sites/tissues as well as in colitis animal models with the disruption of intestinal mucosal integrity and barrier function [59]. Stimulation of TNF-α, TLR and platelet-activating factor (PAF) can lead to a stable or transient rise in IECs shedding with loss of epithelial barrier function. TRAIL is significantly up-regulated during intestinal inflammation in IECs and LP lymphocytes to detect molecular events causing IEC destruction during inflammatory processes such as IBD. The increase in TRAIL-induced IEC apoptosis is mediated via increasing the levels of proinflammatory cytokines such as TNF-α and interferon (IFN)-γ, the expression of the pro-apoptotic receptor TRAIL-R2 and the functional levels of caspase-3 [60]. Intestinal epithelial-specific deletion of transforming growth factor (TGF)-b-activated kinase 1 (TAK1) leads to enhanced apoptosis (cleaved caspase-3) and disturbance of tight junctions (claudin-3) and reduced antioxidant-responsive genes through transcription factor Nrf2, resulting in ROS accumulation. These observed pathological scenarios are very similar to the IBD pathology. Targeting TAK1-Nrf2 pathway could, therefore, control the ROS levels and enhance the survival and integrity of enterocytes [61].
A key apoptotic molecule p53-upregulated modulator of apoptosis (PUMA) is significantly increased in colonic epithelial cells in colitis induced by either dextran sulfate sodium salt (DSS; 5%) or 2,4,6-trinitrobenzene sulfonic acid (TNBS; 100 mg/kg). At the same time, PUMA KO relieved DSS- and TNBS-induced colitis and inhibited IEC apoptosis in mice. These findings indicate that by promoting IEC apoptosis, PUMA induction contributes to colitis pathogenesis [59]. In both mice and human, colon inflammation induces IEC apoptosis through p53-dependent and -independent mechanisms and PUMA mediated intrinsic apoptosis pathway [62]. A latest study demonstrates that enhanced expression of the TNFAIP3 gene encoding A20 is expressed in IECs from patients with IBD in the areas of apoptosis. TNF-induced cell death is extremely prone in transgenic mice that overexpress A20 in IECs. In these mice, by activation of Ripoptosome/RIPK1, A20 potentiates TNF-induced mucosal erosion and IEC apoptosis. Whereas, RIPK1 inhibitors can prevent A20-enhanced IEC damage and intestinal inflammation, suggesting a new strategy for IBD therapy [63]. Wang et al., [64] showed that villin, an actin regulatory protein, acts as an anti-apoptotic function. The overexpression of villin in the Madin-Darby canine kidney Tet-Off epithelial cell line protects the cells from apoptosis by inhibiting the activation of caspase-9 and -3 and activating the pro-survival proteins such as phosphatidylinositol 3-kinase and phosphorylated Akt, thereby maintaining IEC mitochondrial integrity. Increased apoptosis in DSS induced villin KO mice, suggesting the possible anti-apoptotic role in the development and progression of IBD [64].
As stated previously, TNF and TLRs can trigger caspase-dependent apoptosis through the FADD and procaspase 8. TLR4-deficient mice exhibit significantly lower IEC proliferation and increase apoptosis with reduced Cox-2 and PGE-2 levels in DSS-induced injury. Although short term TLR4 signaling is useful, persistent TLR4 signaling may lead to colitis-associated cancers [65]. TLR’s stimulation in IECs are highly associated with activated caspase-8 and increased shedding of IECs. Epithelial cell-specific deletion of caspase-8 triggered Rip3-dependent epithelial necroptosis that resulted in serious tissue damage and death instead of apoptosis [66]. Furthermore, this study emphasized that the release of TNF-α from non-epithelial cells is responsible for TLR4-mediated epithelial necroptosis [66]. In another study, IEC-specific FADD knockout spontaneously developed epithelial cell necrosis, loss of Paneth cells, enteritis and severe erosive colitis. Prevention of these changes by RIP3 inhibitor suggests that intestinal epithelial permeability and inflammation is caused by RIP3-dependent death of FADD-deficient IECs [27]. Recently the increase in TUNEL-positive, caspase-3 negative cells along with p-RIPK3 is found in TNBS-induced colitis mice. At the same time, the increasing levels of p-RIPK3 and p-MLKL on IECs Caco-2 cells under the stimulation of TNF-α and Z-VAD-fmk, a novel in vitro necroptosis model that mimics IBD, confirmed the regulated necroptosis cell death; these effects are reversed by necroptosis inhibitor necrosulfonamide [11]. Therefore, RIP3-mediated IECs are critical for maintaining intestinal homeostasis and indicate that programmed IECs necrosis may be involved in the pathogenesis of IBD.
In a recent study demonstrates that IEC pyroptosis is crucial to the development of mucosal barrier dysfunction and intestinal inflammation using cell culture, animal model (IL10 and casp-1 KO mice) and patients with IBD. Specific caspase-1 inhibitor YVAD and IBD therapeutic agents, such as mesalamine and dexamethasone, considerably inhibit IEC pyroptosis cell pathway, the pyroptosis cell mechanism in IECs [67]. To fully investigate the pathogenic impact of IECs pyroptosis in IBD, more studies are warranted. Recently, the reduced levels of GPx4, a key mediator of ferroptosis, were showed in the colon tissue of patients with CD, suggesting the role of IEC ferroptosis in the pathogenesis of IBD [37]. Still, it is essential to bring more scientific evidence to confirm the ferroptosis cell death in IBD. It is urgent to develop the pharmacological target for apoptosis as a therapy due to increasing rate of apoptosis in intestinal pathologies like IBD. Mostly, in patients with IBD, anti-TNF therapy have been found to inhibit IEC apoptosis. Treating mice with infliximab suppressed DSS- and TNBS-induced colitis and IEC apoptosis via suppressing PUMA expression [59]. Overall, a better understanding of the role of cell death machinery in the epithelial cell might aid the design of better therapeutic or preventive strategies for IBDs.
3.4. IEC cell death in necrotizing enterocolitis (NEC)
NEC is a devastating and life-threatening inflammatory GI disease in 2%–5% of all premature infants, characterized by intestinal inflammation, ischemia, apoptosis and necrosis [68]. Although evidence shows that various risk factors have been involved in the pathogenesis of NEC, including prematurity, hypoxemia, formula feeding, bacterial exposure, and intestinal ischemia, the provocative events leading to NEC remains unclear [69].
IEC apoptosis is considered as one of the prominent pathological features in NEC. It has been shown that elevated levels of NO by enterocytes of infants with NEC, leading to apoptosis in IECs through peroxynitrite formation at apical villi [70]. Indeed, it remains unclear whether the observation of epithelial apoptosis is due primarily to gross tissue necrosis or corresponds only with extensive tissue destruction in NEC. IEC apoptosis and tissue morphology were assessed to test the hypothesis that enhanced epithelial apoptosis is a preliminary event that underlies the gross histologic modifications in formula feeding and cold/asphyxia stress (FFCAS) induced neonatal rat model of NEC. In this model, the increased coincidence of morphologic damage and apoptosis in the respective tissue sections along with caspase-3 and DNA fragmentation levels in FFCAS compared to mother-fed (MF), suggested that IEC apoptosis preceded gross morphologic changes for subsequent gross tissue necrosis [71]. ROS-mediated IECs apoptosis plays a significant role in the pathogenesis of NEC in premature infants. Induction of H2O2 in rat IECs (RIE)-1 led in enhanced IEC apoptosis with intracellular ROS generation and depolarization of the mitochondrial membrane [72]. As we discussed earlier, the pathogenic microbial invasion is also one of the risk factors in the pathogenesis of NEC. Enterobacter sakazakii (ES), a prevalent contaminant in milk-based powdered infant formula, was found to bind to enterocytes in rat pups at the tips of villi, and exposure to ES resulted in apoptosis and enhanced IL-6 levels in IEC-6 cells and the animal model [73]. Cronobacter sakazakii (CS), a major pathogen, relates to NEC, induce dual pyroptosis and apoptosis cell death mechanism including NLRP3 inflammasome, caspase-3 and -1 levels in HT-29 IECs and neonatal rat model, resulting in increased intestinal permeability [74].
Interestingly, the findings of this study [74] have shown the probiotic, Bacteroides fragilis ZY-312f suppresses CS-induced NEC by modulating apoptosis and pyroptosis dual cell death. Bifidobacterium bifidum can reduce apoptosis in both in vivo and in vitro (IEC-6) NEC models by a COX-2-dependent matter, which suggests a molecular mechanism by which this probiotic preserve intestinal integrity [75]. Whereas, EGF reduces the incidence of NEC in a formula milk fed-neonatal rat model by controlling the presence of caspase-3-positive epithelial cells and altering the balance between pro-apoptotic BAX and anti-apoptotic Bcl-2 proteins in the site of NEC injury to maintain intestinal integrity and protects intestinal epithelium [76]. Insulin-like growth factor (IGF)-1 activates PI3-K pathway to promote IECs survival in H2O2 induced in vitro NEC model [72]. In meanwhile, with the administration of heparin-binding EGF (HB-EGF), the median TUNEL and active caspase 3 scores are significantly decreased in the incidence of NEC in the group of hypoxia, hypothermia, hypertonic formula feeding plus enteral administration of LPS [77]. Erythropoietin (Epo), a breast milk component is shown to reduce the incidence of NEC and maintain the function of intestinal barriers. Yu et al., [78] demonstrated that Epo protects the intestinal epithelium from excessive apoptosis by decreasing the number of total cleaved caspase-3 positive ileal epithelial cells and upregulating Bcl-2 expression through MAPK/ERK pathway in both in vitro TNF-α-induced IEC-6 cells and in vivo traditional rat neonatal NEC model. Lactoferrin, a milk supplement, administration modulates intestinal injury by reducing inflammatory cytokines such as IL-6 secretion and upregulating cell proliferation through the Wnt/β-catenin pathway in H2O2-induced IEC-18 and Caco-2 IECs NEC in vitro model [79]. In this regard, focusing on both programmed and non-programmed cell death pathways of IECs may shed light on novel therapeutic approach for the NEC. Overall, these findings highlight the pathological features of IECs cell death and pharmacological intervention for the prevention and recovery of IECs injury in NEC.
4. Future Directions and concluding remarks
Here remain several questions regarding how pathological IECs cell death mechanism interconnects and induces intestinal permeability? Is targeting one of the cell death mechanisms as a therapeutic approach provides promising treatment to patients with chronic intestinal diseases? Thus, understanding the mechanism of IECs cell death brings new insights to explore a novel therapeutic approach for the disease. In this review, we highlight the vital role of IECs death mechanisms in the acute and chronic intestinal disorders such as sepsis, I/R, NEC, IBD, and RIGS, and address the potential therapeutic approach to such intestinal diseases by preventing the IECs cell death mechanism.
Several studies have revealed that the extended IEC apoptosis progresses intestinal injury and permeability via caspase mediated inhibition of the epithelial survival NF-κB pathways, provoke epithelial apoptosis and reduce proliferation, which results in pathological IEC shedding and chronic GI disorders. Simultaneously, deficient in caspase-8 or FADD leads to Ripk1-Ripk3-MLKL mediated IEC necroptosis cell death [27, 66] which has more pathological features than apoptosis. Few studies showed that the pyroptosis cell death mechanism is highly associated with mucosal barrier dysfunction and intestinal inflammation in intestinal diseases such as NEC and IBD [67, 74]. But still, the IECs pyroptosis cell death mechanism to induce intestinal permeability remains unclear. Another rapidly expanding cell-death mechanism in IECs is ferroptosis. The findings of the reduction of GPx4 activity in CD patients [37], iron supplementation influenced bacterial dysbiosis to NEC [80], and iron overload worsened the sepsis pathogenesis [48], suggest us the contribution of the IECs ferroptosis cell-death mechanism in the development and progression of chronic intestinal diseases. But more scientific evidence is warranted to fill this gap.
A recent study shows that blocking both apoptotic and necroptotic mechanism using Z-VAD and necrostatin-1 deliver better protection than any one of these inhibitors [19]. This result suggesting that pathogenic events may switch over the cell-death mechanism and escape from the therapeutic inhibitors in the chronic state of the intestinal diseases. Therefore, it is important to understand what the exact cell-death mechanisms are underlying chronic intestinal diseases to develop emerging novel promising therapeutic approaches. This review enlightens the need for new therapeutic activators or inhibitors in a disease-specific and IECs cell-death mechanism-specific manner. However, further investigations are wanted to explore a novel therapeutic approach for the development and clinical testing in patients with intestinal diseases.
Acknowledgments
This work was supported by grants including R01GM117628, R01GM122406, R01DK123826 from National Institutes of Health and Merit Review Award (I01BX001690) from US Department of Veterans Affairs.
REFERENCES
- 1.Geng H, Bu HF, Liu F, Wu L, Pfeifer K, Chou PM, Wang X, Sun J, Lu L, Pandey A, Bartolomei MS, De Plaen IG, Wang P, Yu J, Qian J, Tan XD. In Inflamed Intestinal Tissues and Epithelial Cells, Interleukin 22 Signaling Increases Expression of H19 Long Noncoding RNA, Which Promotes Mucosal Regeneration. Gastroenterology 2018; 155: 144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hulst M, van der Weide R, Hoekman A, van Krimpen M. Transcriptional response of cultured porcine intestinal epithelial cells to micro algae extracts in the presence and absence of enterotoxigenic Escherichia coli. Genes Nutr 2019; 14: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armacki M, Trugenberger AK, Ellwanger AK, Eiseler T, Schwerdt C, Bettac L, Langgartner D, Azoitei N, Halbgebauer R, Gross R, Barth T, Lechel A, Walter BM, Kraus JM, Wiegreffe C, Grimm J, Scheffold A, Schneider MR, Peuker K, Zeissig S, Britsch S, Rose-John S, Vettorazzi S, Wolf E, Tannapfel A, Steinestel K, Reber SO, Walther P, Kestler HA, Radermacher P, Barth TF, Huber-Lang M, Kleger A, Seufferlein T. Thirty-eight-negative kinase 1 mediates trauma-induced intestinal injury and multi-organ failure. J Clin Invest 2018; 128: 5056–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiminez JA, Uwiera TC, Douglas Inglis G, Uwiera RR. Animal models to study acute and chronic intestinal inflammation in mammals. Gut Pathog 2015; 7: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jozawa H, Inoue-Yamauchi A, Arimura S, Yamanashi Y. Loss of C/EBPdelta enhances apoptosis of intestinal epithelial cells and exacerbates experimental colitis in mice. Genes Cells 2019; 24: 619–26. [DOI] [PubMed] [Google Scholar]
- 6.Thomas AJ, Pulsipher A, Davis BM, Alt JA. LL-37 causes cell death of human nasal epithelial cells, which is inhibited with a synthetic glycosaminoglycan. PLoS One 2017; 12: e0183542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruemmele FM, Seidman EG, Lentze MJ. Regulation of intestinal epithelial cell apoptosis and the pathogenesis of inflammatory bowel disorders. J Pediatr Gastroenterol Nutr 2002; 34: 254–60. [DOI] [PubMed] [Google Scholar]
- 8.Williams JM, Duckworth CA, Watson AJ, Frey MR, Miguel JC, Burkitt MD, Sutton R, Hughes KR, Hall LJ, Caamano JH, Campbell BJ, Pritchard DM. A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Dis Model Mech 2013; 6: 1388–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmadi Badi S, Khatami SH, Irani SH, Siadat SD. Induction Effects of Bacteroides fragilis Derived Outer Membrane Vesicles on Toll Like Receptor 2, Toll Like Receptor 4 Genes Expression and Cytokines Concentration in Human Intestinal Epithelial Cells. Cell J 2019; 21: 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denning TL, Takaishi H, Crowe SE, Boldogh I, Jevnikar A, Ernst PB. Oxidative stress induces the expression of Fas and Fas ligand and apoptosis in murine intestinal epithelial cells. Free Radic Biol Med 2002; 33: 1641–50. [DOI] [PubMed] [Google Scholar]
- 11.Dong W, Zhang M, Zhu Y, Chen Y, Zhao X, Li R, Zhang L, Ye Z, Liang X. Protective effect of NSA on intestinal epithelial cells in a necroptosis model. Oncotarget 2017; 8: 86726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou X, Hong T, Yu Q, Nie S, Gong D, Xiong T, Xie M. Exopolysaccharides from Lactobacillus plantarum NCU116 induce c-Jun dependent Fas/Fasl-mediated apoptosis via TLR2 in mouse intestinal epithelial cancer cells. Sci Rep 2017; 7: 14247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 2011; 477: 335–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nunes T, Bernardazzi C, de Souza HS. Cell death and inflammatory bowel diseases: apoptosis, necrosis, and autophagy in the intestinal epithelium. Biomed Res Int 2014; 2014: 218493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SJ, Jung YH, Song EJ, Jang KK, Choi SH, Han HJ. Vibrio vulnificus VvpE Stimulates IL-1beta Production by the Hypomethylation of the IL-1beta Promoter and NF-kappaB Activation via Lipid Raft-Dependent ANXA2 Recruitment and Reactive Oxygen Species Signaling in Intestinal Epithelial Cells. J Immunol 2015; 195: 2282–93. [DOI] [PubMed] [Google Scholar]
- 16.Zhao M, Tang S, Xin J, Wei Y, Liu D. Reactive oxygen species induce injury of the intestinal epithelium during hyperoxia. Int J Mol Med 2018; 41: 322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong HG, Satchell KJ. Additive function of Vibrio vulnificus MARTX(Vv) and VvhA cytolysins promotes rapid growth and epithelial tissue necrosis during intestinal infection. PLoS Pathog 2012; 8: e1002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu F, Luo M, He L, Cao Y, Li W, Ying S, Chen Z, Shen H. Necroptosis Contributes to Urban Particulate Matter-Induced Airway Epithelial Injury. Cell Physiol Biochem 2018; 46: 699–712. [DOI] [PubMed] [Google Scholar]
- 19.Wen S, Ling Y, Yang W, Shen J, Li C, Deng W, Liu W, Liu K. Necroptosis is a key mediator of enterocytes loss in intestinal ischaemia/reperfusion injury. J Cell Mol Med 2017; 21: 432–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hefele M, Stolzer I, Ruder B, He GW, Mahapatro M, Wirtz S, Neurath MF, Gunther C. Intestinal epithelial Caspase-8 signaling is essential to prevent necroptosis during Salmonella Typhimurium induced enteritis. Mucosal Immunol 2018; 11: 1191–202. [DOI] [PubMed] [Google Scholar]
- 21.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–11. [DOI] [PubMed] [Google Scholar]
- 22.Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight 2019; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zelic M, Roderick JE, O’Donnell JA, Lehman J, Lim SE, Janardhan HP, Trivedi CM, Pasparakis M, Kelliher MA. RIP kinase 1-dependent endothelial necroptosis underlies systemic inflammatory response syndrome. J Clin Invest 2018; 128: 2064–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Negroni A, Colantoni E, Pierdomenico M, Palone F, Costanzo M, Oliva S, Tiberti A, Cucchiara S, Stronati L. RIP3 AND pMLKL promote necroptosis-induced inflammation and alter membrane permeability in intestinal epithelial cells. Dig Liver Dis 2017; 49: 1201–10. [DOI] [PubMed] [Google Scholar]
- 25.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148: 213–27. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014; 54: 133–46. [DOI] [PubMed] [Google Scholar]
- 27.Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 2011; 477: 330–4. [DOI] [PubMed] [Google Scholar]
- 28.Song-Zhao GX, Srinivasan N, Pott J, Baban D, Frankel G, Maloy KJ. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol 2014; 7: 763–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang XY, Chen X, Zhang HF, Guan S, Wen SH, Huang WQ, Liu ZM. Propofol Does Not Reduce Pyroptosis of Enterocytes and Intestinal Epithelial Injury After Lipopolysaccharide Challenge. Dig Dis Sci 2018; 63: 81–91. [DOI] [PubMed] [Google Scholar]
- 30.Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, Tschopp J, Endres S, Latz E, Schnurr M. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010; 59: 1192–9. [DOI] [PubMed] [Google Scholar]
- 31.Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A 2001; 98: 13249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sivakumar PV, Westrich GM, Kanaly S, Garka K, Born TL, Derry JM, Viney JL. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut 2002; 50: 812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinman A, Holst CR, Latham JC, Bruegger JJ, Ulas G, McCusker KP, Amagata A, Davis D, Hoff KG, Kahn-Kirby AH, Kim V, Kosaka Y, Lee E, Malone SA, Mei JJ, Richards SJ, Rivera V, Miller G, Trimmer JK, Shrader WD. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS One 2018; 13: e0201369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, Liu D, Zhang F, Ning S, Yao J, Tian X. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ 2019; 26: 2284–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Nino MD, Ruiz Ortega M, Egido J, Linkermann A, Ortiz A, Sanz AB. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J Am Soc Nephrol 2017; 28: 218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qi X, Zhang Y, Guo H, Hai Y, Luo Y, Yue T. Mechanism and intervention measures of iron side effects on the intestine. Crit Rev Food Sci Nutr 2019; 1–13. [DOI] [PubMed] [Google Scholar]
- 37.Grabherr FML, Schwärzler J, Reitmeier I, Gehmacher T, Niederreiter L, Koch R, Zoller H, Effenberger M, Moschen A, Kaser A, Tilg H, Adolph T. Western-diet-derived arachidonic acid induces epithelial ferroptosis which is a feature of Crohn’s disease. Z Gastroenterol 2019; 57: e131. [Google Scholar]
- 38.Lyons JD, Klingensmith NJ, Otani S, Mittal R, Liang Z, Ford ML, Coopersmith CM. Sepsis reveals compartment-specific responses in intestinal proliferation and apoptosis in transgenic mice whose enterocytes re-enter the cell cycle. FASEB J 2017; 31: 5507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 1999; 27: 1230–51. [DOI] [PubMed] [Google Scholar]
- 40.Hotchkiss RS, Schmieg RE Jr., Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Karl IE, Buchman TG. Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit Care Med 2000; 28: 3207–17. [DOI] [PubMed] [Google Scholar]
- 41.Habes QLM, van Ede L, Gerretsen J, Kox M, Pickkers P. Norepinephrine Contributes to Enterocyte Damage in Septic Shock Patients: A Prospective Cohort Study. Shock 2018; 49: 137–43. [DOI] [PubMed] [Google Scholar]
- 42.Coopersmith CM, Stromberg PE, Dunne WM, Davis CG, Amiot DM 2nd, Buchman TG, Karl IE, Hotchkiss RS. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA 2002; 287: 1716–21. [DOI] [PubMed] [Google Scholar]
- 43.Coopersmith CM, Stromberg PE, Davis CG, Dunne WM, Amiot DM 2nd, Karl IE, Hotchkiss RS, Buchman TG. Sepsis from Pseudomonas aeruginosa pneumonia decreases intestinal proliferation and induces gut epithelial cell cycle arrest. Crit Care Med 2003; 31: 1630–7. [DOI] [PubMed] [Google Scholar]
- 44.Coopersmith CM, Chang KC, Swanson PE, Tinsley KW, Stromberg PE, Buchman TG, Karl IE, Hotchkiss RS. Overexpression of Bcl-2 in the intestinal epithelium improves survival in septic mice. Crit Care Med 2002; 30: 195–201. [DOI] [PubMed] [Google Scholar]
- 45.Perrone EE, Jung E, Breed E, Dominguez JA, Liang Z, Clark AT, Dunne WM, Burd EM, Coopersmith CM. Mechanisms of methicillin-resistant Staphylococcus aureus pneumonia-induced intestinal epithelial apoptosis. Shock 2012; 38: 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dominguez JA, Samocha AJ, Liang Z, Burd EM, Farris AB, Coopersmith CM. Inhibition of IKKbeta in enterocytes exacerbates sepsis-induced intestinal injury and worsens mortality. Crit Care Med 2013; 41: e275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu Q, Ren H, Li G, Wang D, Zhou Q, Wu J, Zheng J, Huang J, Slade DA, Wu X, Ren J. STING-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine 2019; 41: 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stefanova D, Raychev A, Deville J, Humphries R, Campeau S, Ruchala P, Nemeth E, Ganz T, Bulut Y. Hepcidin Protects against Lethal Escherichia coli Sepsis in Mice Inoculated with Isolates from Septic Patients. Infect Immun 2018; 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sukhotnik I, Ben Shahar Y, Halabi S, Bitterman N, Dorfman T, Pollak Y, Coran A, Bitterman A. Effect of N-Acetylserotonin on TLR-4 and MyD88 Expression during Intestinal Ischemia-Reperfusion in a Rat Model. Eur J Pediatr Surg 2019; 29: 188–95. [DOI] [PubMed] [Google Scholar]
- 50.Ben-Shahar Y, Pollak Y, Bitterman A, Coran AG, Bejar IN, Sukhotnik I. Sonic hedgehog signaling controls gut epithelium homeostasis following intestinal ischemia-reperfusion in a rat. Pediatr Surg Int 2019; 35: 255–61. [DOI] [PubMed] [Google Scholar]
- 51.Derikx JP, Matthijsen RA, de Bruine AP, van Bijnen AA, Heineman E, van Dam RM, Dejong CH, Buurman WA. Rapid reversal of human intestinal ischemia-reperfusion induced damage by shedding of injured enterocytes and reepithelialisation. PLoS One 2008; 3: e3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grootjans J, Lenaerts K, Derikx JP, Matthijsen RA, de Bruine AP, van Bijnen AA, van Dam RM, Dejong CH, Buurman WA. Human intestinal ischemia-reperfusion-induced inflammation characterized: experiences from a new translational model. Am J Pathol 2010; 176: 2283–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ikeda H, Suzuki Y, Suzuki M, Koike M, Tamura J, Tong J, Nomura M, Itoh G. Apoptosis is a major mode of cell death caused by ischaemia and ischaemia/reperfusion injury to the rat intestinal epithelium. Gut 1998; 42: 530–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinnebusch BF, Ma Q, Henderson JW, Siddique A, Archer SY, Hodin RA. Enterocyte response to ischemia is dependent on differentiation state. J Gastrointest Surg 2002; 6: 403–9. [DOI] [PubMed] [Google Scholar]
- 55.Li X, Ling Y, Cao Z, Shen J, Chen S, Liu W, Yuan B, Wen S. Targeting intestinal epithelial cell-programmed necrosis alleviates tissue injury after intestinal ischemia/reperfusion in rats. J Surg Res 2018; 225: 108–17. [DOI] [PubMed] [Google Scholar]
- 56.Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med 2003; 9: 575–81. [DOI] [PubMed] [Google Scholar]
- 57.Coopersmith CM, O’Donnell D, Gordon JI. Bcl-2 inhibits ischemia-reperfusion-induced apoptosis in the intestinal epithelium of transgenic mice. Am J Physiol 1999; 276: G677–86. [DOI] [PubMed] [Google Scholar]
- 58.Parikh K, Antanaviciute A, Fawkner-Corbett D, Jagielowicz M, Aulicino A, Lagerholm C, Davis S, Kinchen J, Chen HH, Alham NK, Ashley N, Johnson E, Hublitz P, Bao L, Lukomska J, Andev RS, Bjorklund E, Kessler BM, Fischer R, Goldin R, Koohy H, Simmons A. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 2019; 567: 49–55. [DOI] [PubMed] [Google Scholar]
- 59.Qiu W, Wu B, Wang X, Buchanan ME, Regueiro MD, Hartman DJ, Schoen RE, Yu J, Zhang L. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J Clin Invest 2011; 121: 1722–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Begue B, Wajant H, Bambou JC, Dubuquoy L, Siegmund D, Beaulieu JF, Canioni D, Berrebi D, Brousse N, Desreumaux P, Schmitz J, Lentze MJ, Goulet O, Cerf-Bensussan N, Ruemmele FM. Implication of TNF-related apoptosis-inducing ligand in inflammatory intestinal epithelial lesions. Gastroenterology 2006; 130: 1962–74. [DOI] [PubMed] [Google Scholar]
- 61.Kajino-Sakamoto R, Omori E, Nighot PK, Blikslager AT, Matsumoto K, Ninomiya-Tsuji J. TGF-beta-activated kinase 1 signaling maintains intestinal integrity by preventing accumulation of reactive oxygen species in the intestinal epithelium. J Immunol 2010; 185: 4729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dirisina R, Katzman RB, Goretsky T, Managlia E, Mittal N, Williams DB, Qiu W, Yu J, Chandel NS, Zhang L, Barrett TA. p53 and PUMA independently regulate apoptosis of intestinal epithelial cells in patients and mice with colitis. Gastroenterology 2011; 141: 1036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garcia-Carbonell R, Wong J, Kim JY, Close LA, Boland BS, Wong TL, Harris PA, Ho SB, Das S, Ernst PB, Sasik R, Sandborn WJ, Bertin J, Gough PJ, Chang JT, Kelliher M, Boone D, Guma M, Karin M. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc Natl Acad Sci U S A 2018; 115: E9192–E200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y, Srinivasan K, Siddiqui MR, George SP, Tomar A, Khurana S. A novel role for villin in intestinal epithelial cell survival and homeostasis. J Biol Chem 2008; 283: 9454–64. [DOI] [PubMed] [Google Scholar]
- 65.Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, Xu R, Inoue H, Arditi M, Dannenberg AJ, Abreu MT. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology 2006; 131: 862–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gunther C, Buchen B, He GW, Hornef M, Torow N, Neumann H, Wittkopf N, Martini E, Basic M, Bleich A, Watson AJ, Neurath MF, Becker C. Caspase-8 controls the gut response to microbial challenges by Tnf-alpha-dependent and independent pathways. Gut 2015; 64: 601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davis EMKY, Goyne H, Wang Y, Chen T, Theus S, Lai K, Glover SC, Claggett BL, Jobin C, Liu JJ. Pyroptosis of Intestinal Epithelial Cells is Crucial to the Development of Mucosal Barrier Dysfunction and Intestinal Inflammation. Gastroenterology 2017; 152: S967. [Google Scholar]
- 68.Managlia E, Liu SXL, Yan X, Tan XD, Chou PM, Barrett TA, De Plaen IG. Blocking NF-kappaB Activation in Ly6c(+) Monocytes Attenuates Necrotizing Enterocolitis. Am J Pathol 2019; 189: 604–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chokshi NK, Guner YS, Hunter CJ, Upperman JS, Grishin A, Ford HR. The role of nitric oxide in intestinal epithelial injury and restitution in neonatal necrotizing enterocolitis. Semin Perinatol 2008; 32: 92–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ford H, Watkins S, Reblock K, Rowe M. The role of inflammatory cytokines and nitric oxide in the pathogenesis of necrotizing enterocolitis. J Pediatr Surg 1997; 32: 275–82. [DOI] [PubMed] [Google Scholar]
- 71.Jilling T, Lu J, Jackson M, Caplan MS. Intestinal epithelial apoptosis initiates gross bowel necrosis in an experimental rat model of neonatal necrotizing enterocolitis. Pediatr Res 2004; 55: 622–9. [DOI] [PubMed] [Google Scholar]
- 72.Baregamian N, Song J, Jeschke MG, Evers BM, Chung DH. IGF-1 protects intestinal epithelial cells from oxidative stress-induced apoptosis. J Surg Res 2006; 136: 31–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hunter CJ, Singamsetty VK, Chokshi NK, Boyle P, Camerini V, Grishin AV, Upperman JS, Ford HR, Prasadarao NV. Enterobacter sakazakii enhances epithelial cell injury by inducing apoptosis in a rat model of necrotizing enterocolitis. J Infect Dis 2008; 198: 586–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fan HLR, Chen Z, Leng X, Wu X, Yiduo Z, Zhu B, Zhang Q, Bai Y, Zhi F. Bacteroides fragilis defense against Cronobacter sakazakii-induced pathogenicity by regulating the intestinal epithelial barrier function and attenuating both apoptotic and pyroptotic cell death. bioRxiv 2018; In press. [Google Scholar]
- 75.Khailova L, Mount Patrick SK, Arganbright KM, Halpern MD, Kinouchi T, Dvorak B. Bifidobacterium bifidum reduces apoptosis in the intestinal epithelium in necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 2010; 299: G1118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clark JA, Lane RH, Maclennan NK, Holubec H, Dvorakova K, Halpern MD, Williams CS, Payne CM, Dvorak B. Epidermal growth factor reduces intestinal apoptosis in an experimental model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 2005; 288: G755–62. [DOI] [PubMed] [Google Scholar]
- 77.Feng J, El-Assal ON, Besner GE. Heparin-binding epidermal growth factor-like growth factor reduces intestinal apoptosis in neonatal rats with necrotizing enterocolitis. J Pediatr Surg 2006; 41: 742–7; discussion -7. [DOI] [PubMed] [Google Scholar]
- 78.Yu Y, Shiou SR, Guo Y, Lu L, Westerhoff M, Sun J, Petrof EO, Claud EC. Erythropoietin protects epithelial cells from excessive autophagy and apoptosis in experimental neonatal necrotizing enterocolitis. PLoS One 2013; 8: e69620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu JLB, Lee C, Zhu H, Zheng S, Pierro A. Protective effects of Lactoferrin on injured intestinal epithelial cells. Journal of Pediatric Surgery 2019; In press. [DOI] [PubMed] [Google Scholar]
- 80.Gopel W, Drese J, Rausch TK, Twisselmann N, Bohnhorst B, Muller A, Franz A, Ziegler A, Hartel C, Herting E. Necrotizing enterocolitis and high intestinal iron uptake due to genetic variants. Pediatr Res 2018; 83: 57–62. [DOI] [PubMed] [Google Scholar]