

Abstract
Background & Aims.
IL-33/IL-1F11 is an important mediator for the development of Th2-driven inflammatory disorders and has also been implicated in the pathogenesis of GI-related cancers, including gastric carcinoma. We therefore sought to mechanistically determine IL-33’s potential role as a critical factor linking chronic inflammation and gastric carcinogenesis using gastritis-prone SAMP1/YitFc (SAMP) mice.
Methods.
SAMP and (parental control) AKR mice were assessed for baseline gastritis and progression to metaplasia. Expression/localization of IL-33 and its receptor, ST2/IL-1R4, were characterized in corpus tissues, and both activation and neutralization studies were performed targeting the IL-33/ST2 axis. Dissection of immune pathways leading to metaplasia were evaluated, including eosinophil depletion studies using anti-IL-5/anti-CCR3 treatment.
Results.
Progressive gastritis and ultimately, intestinalized spasmolytic polypeptide-expressing metaplasia (SPEM) was detected in SAMP stomachs, which was absent in AKR, but could be moderately-induced with exogenous, recombinant (r)IL-33. Robust peripheral (bone-marrow, BM) expansion of eosinophils and local recruitment of both eosinophils and IL-33-expressing M2 macrophages into corpus tissues were evident in SAMP. Interestingly, IL-33 blockade did not affect BM-derived expansion and local infiltration of eosinophils, but markedly decreased M2 macrophages and SPEM features, while eosinophil depletion caused a significant reduction in both local IL-33-producing M2 macrophages and SPEM in SAMP.
Conclusions.
IL-33 promotes metaplasia and the sequelae of eosinophil-dependent downstream infiltration of IL-33-producing M2 macrophages leading to intestinalized SPEM in SAMP, suggesting that IL-33 represents a critical link between chronic gastritis and intestinalizing metaplasia that may serve as a potential therapeutic target for pre-neoplastic conditions of the GI tract.
Keywords: IL-33/ST2 axis, M2 macrophages, SPEM/intestinalized SPEM, gastric cancer
Lay Summary
IL-33-activated eosinophils are important in the early cascade of events leading to intestinalized metaplasia in gastritis-prone mice, and represents a potential mechanism that promotes the inflammation-metaplasia-dysplasia-carcinoma sequelae.
Graphical Abstract
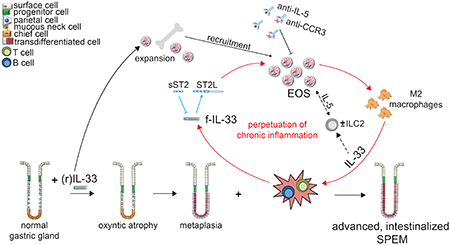
Introduction
The link between chronic inflammation and cancer is well established, with inflammation-associated cancers among the most highly represented and frequently-occurring neoplasias worldwide1,2. Investigation over the last several years has focused on determining critical pathways involved in this process, with a number of candidate molecules identified, including members of the interleukin-1 (IL-1) family that are particularly important in GI-related cancers3,4. Of these, IL-33 plays a prominent role in intestinal tumorigenesis, both in mouse models of adenomatous polyposis (i.e., ApcMin strain) and inflammation-associated colorectal cancer, as well as in patients with colorectal adenocarcinoma5,6,7. Less is known regarding IL-33’s contribution to the development of upper GI cancers, including gastric adenomas, which generally occur as a consequence of chronic gastritis after prolonged Helicobacter (H.) pylori infection or as a result of autoimmune gastritis8. As such, while IL-33 clearly plays a paramount role in both inflammation and cancer, a definitive link between IL-33-dependent inflammation and carcinogenesis in the GI tract has yet to be determined.
IL-33/IL-1F11 is widely distributed throughout various organ systems, primarily in non-hematopoietic cells, but also in cells of hematopoietic origin, particularly in restricted populations of professional antigen presenting cells, such as macrophages9,10. IL-33 was initially associated with Th2 immunity, based on expression of its cell-bound receptor, ST2L/IL-1R4, on polarized Th2 lymphocytes9 and more recently, on group 2 innate lymphoid cells (ILC2s)11, and its ability to effectively induce M1, but more commonly M2, macrophage differentiation12. Importantly, IL-33 also potently activates and induces eosinophil infiltration into mucosal organs that interface with the external environment, such as the GI and respiratory tracts9,13,14.
Relevant to the present study, prior reports associate IL-33 with poor prognosis in gastric cancer patients15, and in vitro experiments suggest that IL-33 confers chemoresistance16, and increased invasiveness17 of gastric cancer cells. Recently, IL-33-dependent activation of mast cells was shown to promote tumor angiogenesis and growth that, in combination with tumor-associated macrophages, positively correlates with decreased survival of gastric cancer patients18. As such, although evolving, the precise role of IL-33 in gastric cancer has not yet been fully elucidated; specifically, how IL-33 mechanistically exerts the aforementioned effects, and whether IL-33 contributes to the development of pre-neoplastic states ultimately leading to gastric cancer.
In fact, chronic inflammatory conditions of the stomach often result in reactive modifications of the mucosa, including atrophy of mature acid-secreting cells as new metaplastic lineages arise. Such metaplastic changes in epithelial cells increase the risk of developing dysplasia and gastric cancer19–21. The initial metaplasia that arises, concomitant with acid-secreting parietal cell atrophy, is defined by the existence of antral-like mucous cells within the body (corpus) of the stomach and is known as spasmolytic polypeptide expressing metaplasia (SPEM), or pseudopyloric metaplasia. SPEM22 is thought to arise from reprogramming of mature chief cells after parietal cell loss23–25 and is thus named by the induced expression of spasmolytic polypeptide, i.e., trefoil factor 2 (TFF2), in metaplastic cells23,26–28. In addition to SPEM, the stomach can undergo intestinal metaplasia19, which is confirmed by the presence of intestinal lineages, such as mucus-secreting goblet cells. Chronic inflammation can promote the progression of metaplastic mucosa to a more proliferative phenotype that is at risk for progression to dysplasia and cancer29. Experimentally, SPEM can be chemically-induced by L635, a parietal cell-specific protonophore, that generates a cascade of events leading to oxyntic atrophy, infiltration of activated M2-polarized macrophages, and SPEM that can develop intestinal characteristics in mice30. Using this model, IL-33 promotes IL-13-dependent M2 polarization of recruited macrophages and the development of SPEM31.
In the present study, we evaluate a murine model that spontaneously develops chronic gastric inflammation, i.e., SAMP1/YitFc (SAMP) strain32, to uncover potential IL-33-dependent mechanism(s) leading to, and downstream of, parietal cell loss and chief cell transdifferentiation, that can result in SPEM, and an even more proliferative, intestinalized SPEM. We show that: a) gastritis-prone SAMP progressively develop metaplasia leading to advanced SPEM-like features, b) exogenous rIL-33 administration induces moderate metaplasia and M2 macrophage infiltration in stomachs of healthy, (AKR) mice, and under chronic inflammatory conditions, exacerbates and advances metaplasia-acquiring intestinal characteristics in SAMP, c) neutralization of IL-33 in SAMP with established disease dampens gastritis and progression of metaplasia/SPEM, and d) IL-33 induces potent peripheral eosinophil expansion and subsequent recruitment into stomachs of treated mice, while e) eosinophil depletion markedly diminishes M2 macrophage infiltration and SPEM-like features in SAMP. Together, our data suggest that IL-33 plays a central role in the early events leading to SPEM and that during chronic inflammation, and/or perhaps under other predisposing conditions that promote lack of immune tolerance, IL-33-dependent eosinophil activation and migration are essential for this process to occur. As such, targeting the IL-33/ST2 axis for therapeutic purposes may be beneficial for the treatment and/or prevention of pre-neoplastic states leading to gastric cancer.
Materials and Methods
Mice.
Mice were provided through core services supported by the Animal and Mouse Models Cores of NIH P01 DK091222 and P30 DK097948, respectively. SAMPxIl33−/− mice were developed by backcrossing male C57BL/6J (B6) mice homozygous for a null allele of Il33 (Il33−/−)5,33 with female SAMP for 10 generations, and validated by microsatellite analysis comparing SAMP- vs. B6-specific markers. Breeding of F1N10 heterozygous offspring generated SAMP homozygous Il33−/−, heterozygous, and WT controls, identified by PCR5,33. Mice were maintained as previously described34, with all procedures approved by CWRU’s IACUC.
Tissue harvest and histologic/SPEM assessment.
Mice were euthanized and whole stomachs removed and opened along the greater curvature. Forestomach and antrum were excised, and corpus dissected along the longitudinal plane with one strip each placed in RNAlater® (Ambion®, ThermoFisher Scientific, Waltham, MA) or RIPA buffer (Pierce Biotechnology, Rockford, IL), and maintained at −20°C until later RNA and protein extraction, respectively. Remaining tissue strip was submerged in either Bouin’s (Ricca Chemical Company, Arlington, TX), 4% PFA (ChemCruz, Biotechnology, Inc., Dallas, TX) or methacarn. Bouin’s/PFA- and methacarn-fixed tissues were rinsed in 70% ethanol after 24h or 20 min, respectively, processed, paraffin-embedded, and sectioned at 3-4μm. Specimens were stained with either H&E or Alcian Blue/PAS with optional hematoxylin 7221 (Thermo Scientific™ Richard-Allan Scientific™, Kalamazoo, MI). Samples were evaluated by trained GI pathologists (WX, AO) in a blinded-fashion for gastritis/epithelial alterations and SPEM/intestinalized SPEM as previously described30,32. SPEM was scored by calculating percentage cross-sectional involvement, defined by oxyntic atrophy with loss of parietal cells and replacement of gastric glands with mucous cell lineages staining positive for PAS/Alcian blue.
In vivo studies.
Experiments were conducted in a blinded manner, with mice randomized to different interventions using progressive numeric labeling, the code only known to animal caretakers and revealed at end of each experiment. Scientific rigor, data reproducibility and biological variables were followed, based on recently published guidelines35. SAMP and gender/age-matched AKR were evaluated for baseline gastritis and progression to SPEM/intestinalized SPEM30,32. For exogenous IL-33 experiments, 8- to-12-wk-old mice were i.p.-injected with either rIL-33 (33μg/kg) (Enzo Life Sciences, Farmingdale, NY) or HBSS (vehicle), daily for 1-wk. IL-33 neutralization was achieved using a fusion protein (5 mg/kg, i.p. 2X/wk for 4-wks) consisting of the extracellular domain of mouse soluble (s)ST2 fused to IgG1-Fc (sSt2-Fc); controls were similarly administered IgG1-Fc (Dirk E. Smith, Amgen, Inc., Seattle, WA). Eosinophil depletion was performed using monoclonal antibodies against IL-5 and CCR3, and controls treated with an isotype IgG34.
qRT-PCR and Western blots.
Total RNA was isolated, reverse-transcribed, and qPCR performed as previously described34, using specific target gene primers (Supplemental Table 1), normalized to β-actin or 36B4, and reported as relative fold-difference among groups, with baseline/controls set arbitrarily at 1. Total protein extracts from corpus were prepared and Western blotting performed10,33.
BrdU staining and Immunohistochemistry (IHC).
Mice were injected with BrdU labeling agent (Invitrogen, Carlsbad, CA) 2h before euthanization, gastric tissues harvested, fixed in 4% PFA, and stained following manufacturer’s instructions (BrdU Staining Kit; Invitrogen). IHC was performed using either a polyclonal goat anti-mouse IL-33 IgG (R&D Systems, Minneapolis, MN), biotinylated Dolichos biflorus agglutinin (DBA, Vector Laboratories, Burlingame, CA), or a monoclonal rat anti-mouse MBP IgG (clone MT-14.7) (James J. Lee, Mayo Clinic, Scottsdale, AZ), with negative controls prepared under identical conditions in the absence of respective primary antibodies34. Parietal cell/eosinophil counts were calculated by average intact, nucleated cells positive for DBA/MBP in 10 randomly-selected HPFs34. Immunofluorescence for confocal imaging was performed to detect GSII, CD44v, Clu, CD163, IL-33, GIF, and imaged/analyzed by the Vanderbilt Digital Histology Shared Resource31.
Flow Cytometry.
BM from femurs/tibias were harvested and processed as previously described34. Corpus tissues were processed into single-cell suspensions31. 2 x 106 cells were stained with LIVE/DEAD Fixable Near-IR Dead Cell Stain (ThermoFisher Scientific), and specific antibodies to detect eosinophils and M2 macrophages (Supplemental Table 2). Samples were acquired with a BD LSR II flow cytometer and data analyzed with FlowJo Software (both, Becton Dickinson)31,34.
Statistical analyses.
Data were analyzed using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Selection of appropriate statistical tests was based on variance and underlying distribution of data. Global effects between groups were assessed using one-way ANOVA with Bonferroni correction for multiple comparisons. Differences between individual groups were directly compared using two-sample unpaired Student’s t-test and results expressed as mean±SEM, unless otherwise indicated, with P<0.05 considered significant.
Results
Gastritis-prone SAMP exhibit morphologic features and a molecular profile consistent with intestinalized SPEM
SAMP mice develop chronic, immune-mediated gastritis with increased severity over time32. Stomachs from 20-wk-old SAMP show remarkable and progressive parietal cell loss compared to age-matched AKR controls, which possess abundant parietal cells (Fig. S1A). The emergence of prominent Alcian blue/PAS-stained hyperplastic mucous neck cells in SAMP highlight the presence of acidic mucins, commonly found in SPEM and adenocarcinoma36, but are absent in AKR (Fig. S1A). Older SAMP display intense staining for the mucous marker, Griffonia simplicifolia II (GSII) lectin, while co-localization with either Cd44v or clusterin (Clu), indicative of SPEM, shows more intense Clu expression along the gastric glands compared to AKRs; the presence, albeit subtle, of Cd44v at the base of gastric glands in 20-wk-old SAMP is also detected, but virtually absent in the other experimental groups (Fig. S1B).
Molecular profiling of full-thickness corpus shows a striking decrease in expression of genes associated with oxyntic atrophy and foveolar hyperplasia, including gastric intrinsic factor (Gif), ATPase H+/K+ transporting alpha subunit (Atp4a), and trefoil factor 1 (Tff1), comparing SAMP vs. AKR, particularly in older mice (Fig. S2A). During acute oxyntic atrophy, as parietal cells die, chief cells show a rapid decrease in the zymogen granule maturation transcription factor, Mist1, and an increase in Tff226,37. While no differences in Tff2 are detected among experimental groups, Mist1 is markedly decreased in SAMP vs. AKR (Fig. S2B). Furthermore, the SPEM markers, human epididymus protein 4 (He4), Clu, lysozyme (Lyz), and glutathione peroxidase 2 (Gpx2)29,37, are all dramatically increased in SAMP with established disease vs. AKR (Fig. S2C). Importantly, markers of more advanced, proliferative lesions (intestinalized SPEM)29,30, including cystic fibrosis transmembrane conductance regulator (Cftr) and deleted in malignant brain tumors protein 1 (Dmbt1), are strongly upregulated in 20-wk-old SAMP vs. AKR, while the early tumor shrinkage variant 5 (Etv5) transcription factor is unchanged (Fig. S2D).
Increased circulating IL-33, with its most bioactive form localizing to gastric M2 macrophages in SAMP with advanced SPEM
To test the hypothesis that increased and persistent exposure to IL-33 may be needed for intestinalized SPEM to occur, we initially measured systemic/circulating IL-33, which is substantially increased in SAMP, even prior to histologically-evident gastritis (at 4-wks) and dramatically rises as disease becomes more severe, compared to IL-33 in AKR that remains relatively stable (Fig. S3). Locally, within non-diseased stomachs of 4-wk-olds, IL-33 is primarily found in foveolar epithelium, almost exclusively localized to nuclei (Fig. 1A). In older mice, AKR retain some epithelial-specific nuclear expression with scant IL-33+ cells within the lamina propria (LP), while abundant and diffuse IL-33 immunoreactivity is observed in SAMP, also within the LP, as well as at the base of the mucosa (Fig. 1A). Evaluation of IL-33 isoforms reveals less abundant IL-33 in 4-wk-old SAMP compared to AKR; however, in older SAMP, only full-length IL-33 (fIL-33), representing its most bioactive 30kD form38, is prominently expressed, while other, less bioactive forms39,40 are present in AKR (Fig. 1B). In fact, SAMP express mainly fIL-33, whereas the 20-22 kD cleaved form (cIL-33) is virtually undetectable, but clearly present in AKR (Fig. 1B), suggesting increased release of bioactive fIL-33 in SAMP with established disease. Surprisingly, Il33 in SAMP stomachs is decreased compared to AKR (Fig. 1C), which may be due to high circulating IL-33 levels (Fig. S3) and potential negative feedback mechanism(s) downregulating IL-33’s bioactivity. Finally, to identify the cellular source of the considerable, yet diffuse, IL-33 staining within the gastric submucosa of older SAMP (Fig. 1A), immunofluorescent co-localization studies were performed, revealing abundant, infiltrating IL-33-expressing CD163+ M2 macrophages that are virtually absent in non-inflamed AKR, but can be elicited with rIL-33 (Fig. 1D). Phenotypic characterization of SAMP gastric mucosal cells confirm the presence of IL-33-producing M2 macrophages (Figs. S4 and 1E–F), with an overall dominant M2 vs. M1 profile vs. AKR (Fig. S5).
Figure 1. Differential IL-33 localization/expression in corpus of gastritis-prone SAMP vs. control AKR.
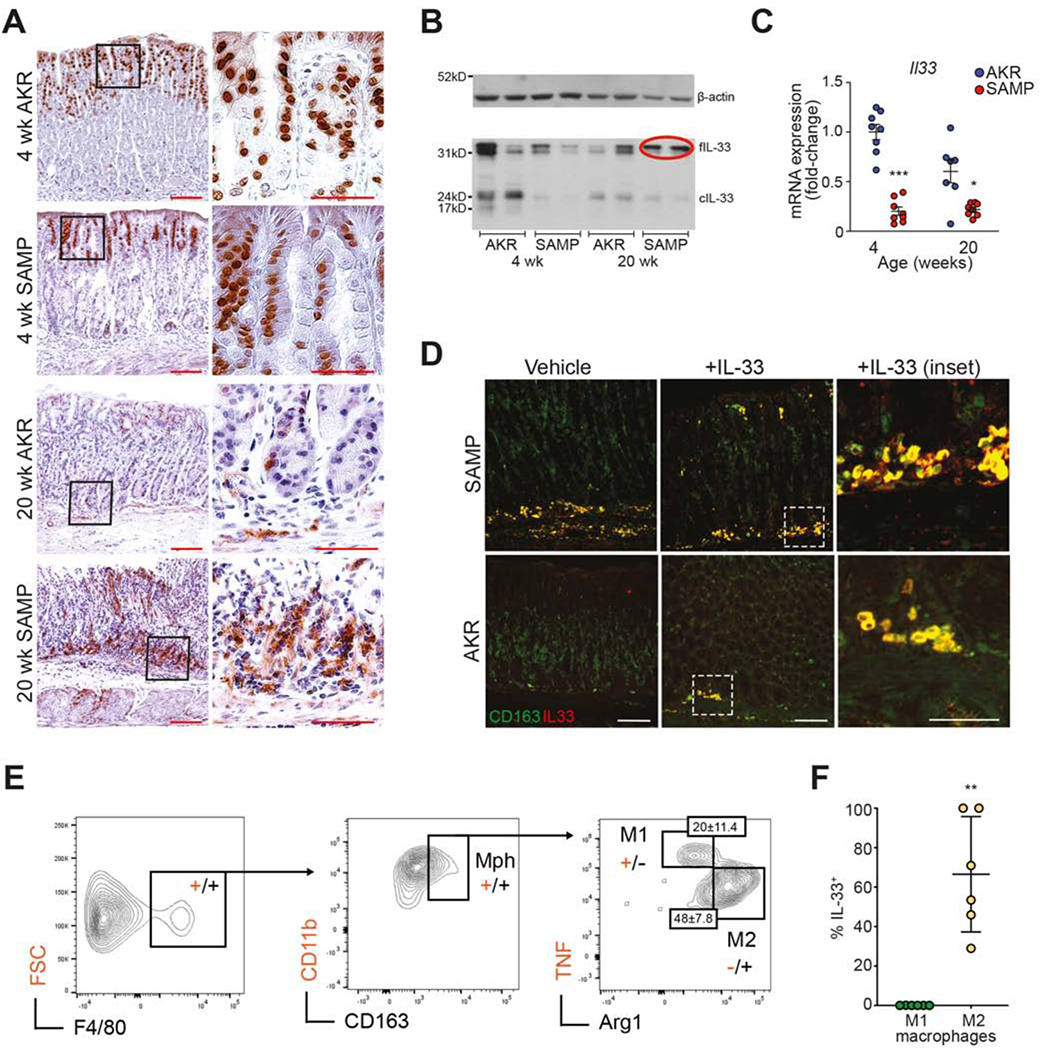
(A) Representative IHC images localizing IL-33 prior to inflammation (4-wk-old) and during established disease (20-wk-old) in SAMP (N=8). Original magnification: X10+2.0, X40+2.0 (insets); scale bars: 100μm, 50μm (insets). (B) Representative Western blot differentiating bioactive full-length (f)- from cleaved (c)-IL-33 (N=4). (C) Il33 expressed as fold-change vs. age-matched AKR, *P<0.05, ***P<0.001 (N=7-8). (D) IF co-localization of IL-33 (red) and CD163 (green) show abundance of IL-33-producing M2 macrophages (yellow), further augmented after rIL-33 in AKR (N=4), and (E) confirmed by FACS, *P<0.01 (N=6). Original magnification: X20; scale bars: 10μm, 5μm (insets).
Exogenous administration of IL-33 induces SPEM in normal (AKR) mice
One of the original observations regarding IL-33’s bioactivity was its ability to promote epithelial hyperplasia, mainly in goblet cells, within GI and airway mucosae9. These findings were confirmed in stomachs of IL-33-treated B6 mice, which results in a Th2/STAT3-driven gastric pathology41. Aside from inducing infiltration of IL-33-expressing CD163+ M2 macrophages (Fig. 1D), acute systemic exposure of IL-33 AKR consistently promotes the gross anatomical appearance of thickened gastric mucosal folds that are absent in vehicle-treated mice (Fig. 2A). Histologic evaluation of these folds reveal striking alterations, including overall hypertrophy of the gastric mucosa, epithelial hyperplasia, as well as oxyntic atrophy, while parietal and chief cells remain relatively intact in vehicle-treated AKR (Fig. 2A). Interestingly, although exogenous IL-33 results in only 25% of AKR displaying significant gastritis, increased epithelial hyperplasia suggests its contribution to inducing mucous neck cell hyperplasia and metaplasia (Fig. 2B). The presence of metaplasia is further characterized by prominent proliferation of cells within the gastric glands, demonstrated by a greater number of BrdU+ cells, along the neck and at the base of gastric glands (Fig. 2A, right panels), and Alcian blue/PAS staining, highlighting the appearance of hyperplastic acidic mucin-producing neck cells, comparing IL-33- vs. vehicle-treated AKRs (Fig. 2C). Finally, accumulation of GSII lectin (a surrogate for MUC6 expression) co-localizing with Cd44v at the base of glands in treated AKR (Fig. 2C), indicating chief cells transdifferentiation42, also supports the development of IL-33-dependent SPEM (Fig. 2D), but for the most part, in the absence of inflammation.
Figure 2. IL-33 administration to healthy AKR induces striking gastric mucosal alterations.
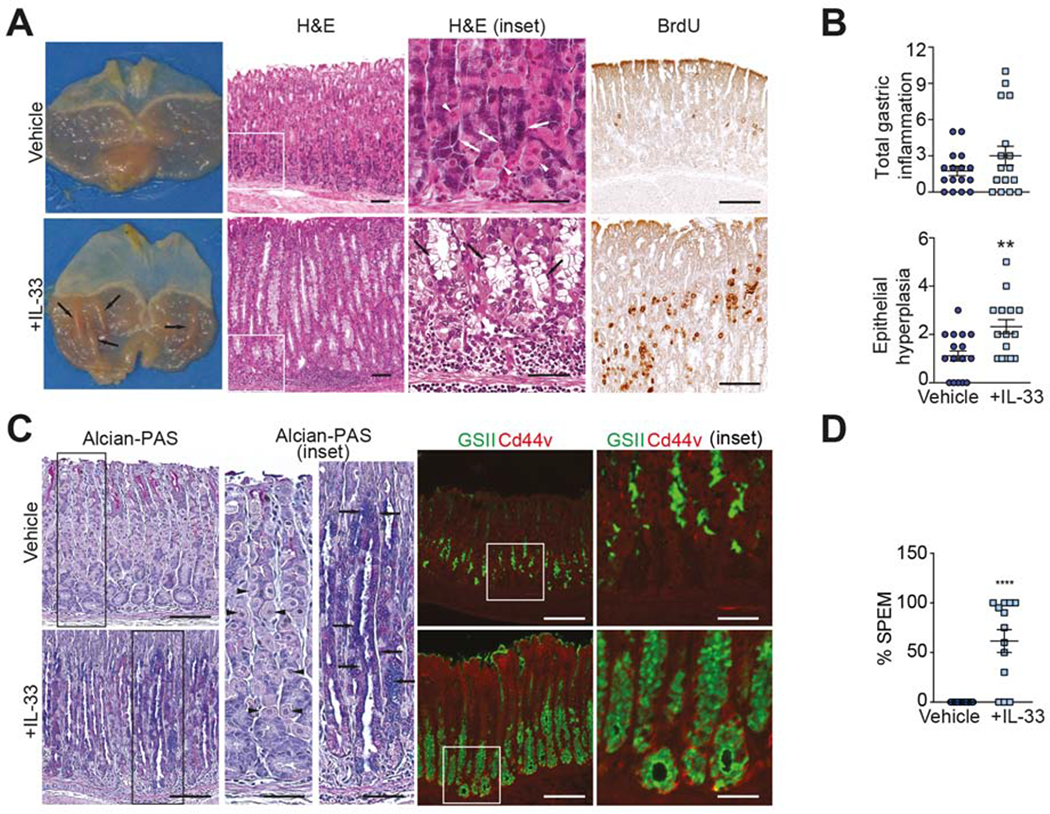
(A) Representative photos of bisected stomachs, highlighting presence of gastric mucosal folds (black arrows), and photomicrographs of corpus, displaying parietal cell (white arrowheads) atrophy, loss of chief cell (white arrows) differentiation, hyperproliferation of mucus-producing cells (black arrows), and active proliferation of BrdU+ mucous neck cells after rIL-33; original magnification: X10, X40 (insets); scale bars: 100μm, 50μm (insets). (B) Gastric inflammation, epithelial hyperplasia, and (C) representative photomicrographs showing appearance of acidic mucins by Alcian blue/PAS-staining (arrows; parietal cells indicated by arrowheads), and increased presence/intensity of GSII lectin (green) and Cd44v (red) after rIL-33; original magnification (left-to-right): X20+1.25, X40+1.25, X20, X40 (insets); scale bars: 100μm, 50μm (insets). (D) Semi-quantitative assessment of SPEM. **P<0.01, ****P<0.0001 (N=13-16).
Consistent with oxyntic atrophy, molecular profiling shows that Gif and Atp4a are downregulated in corpus tissues from IL-33-treated AKR (Fig 3A), and while Tff1 shows a trend towards decreased expression, Tff2 is strongly upregulated following short-term IL33 exposure (Fig. 3A), further indicating the occurrence of IL-33-induced chief cell transdifferentiation leading to SPEM. Additionally, He4 and Clu are consistently upregulated after rIL-33 (Fig. 3B); however, only Cftr is considerably increased for the intestinalized SPEM markers assayed (Fig. 3C), suggesting that IL-33 has a prominent effect on the development of SPEM but, in order to progress to a more advanced intestinalized phenotype, a longer, more chronic exposure to IL-33 may be needed. Interestingly, exogenous IL-33 does not affect Il1rl1 (ST2L), encoding the signaling receptor for IL-33, but Il1rl1 (sST2), encoding soluble ST2, an IL-33-specific decoy receptor43, is markedly increased and Il33 is significantly decreased (Fig. 3D), which is similar to what is observed in SAMP with established disease, and may represent a negative feedback response after rIL-33.
Figure 3. Molecular profiling indicates overt metaplasia in AKR corpus after acute exposure to IL-33.
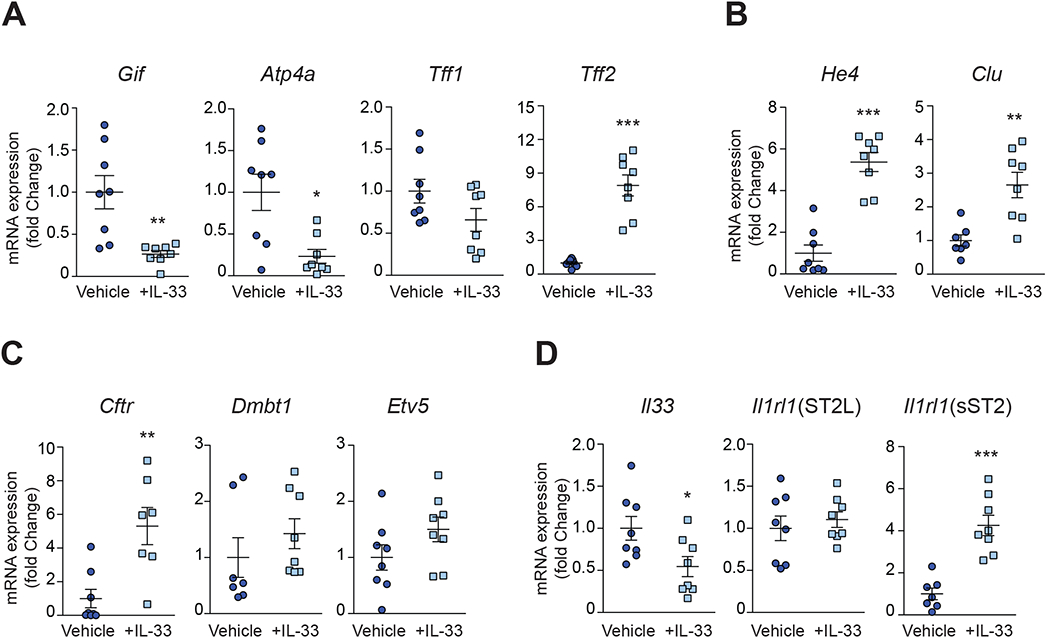
Relative transcript levels of (A) Gif, Atp4a, Tff1, Tff2, (B) He4, Clu, (C) Cftr, Dmbt1, Etv5, (D) Il33, and Il1rl1 variant 1 (ST2L) and 2 (sST2) after rIL-33. Data expressed as fold-change vs. vehicle controls; *P<0.05, **P<0.01, ***P<0.001 (N=8).
Blockade of IL-33 signaling dampens SPEM progression in SAMP mice
To test whether IL-33 neutralization is effective in dampening and/or reversing intestinalized SPEM, we administered sST2-Fc to SAMP with established disease. Treatment with sST2-Fc reduces expression of Cd44v, and does not appear to change GIF compared to vehicle controls, but has a significant impact on diminishing the number of IL-33-producing CD163+ M2 macrophages, as well as GSII expression (Fig. 4A, right panels). Histologically, global improvement is evident in regards to epithelial architecture and gland structure, with decreased Alcian blue/PAS-stained cells, indicating diminished production of acidic mucins. A substantially increased census of cells positive for the parietal cell-specific marker, DBA, is also observed, consistent with morphology of parietal cell precursors (Fig. 4A–B), which dramatically arise during restitution from SPEM44. Histologic evaluation shows a 57% decrease in epithelial hyperplasia following IL-33 blockade (Fig. 4C), suggesting restorative epithelial processes towards normal homeostasis.
Figure 4. Neutralization of IL-33 attenuates chronic gastritis and intestinalized SPEM in SAMP.
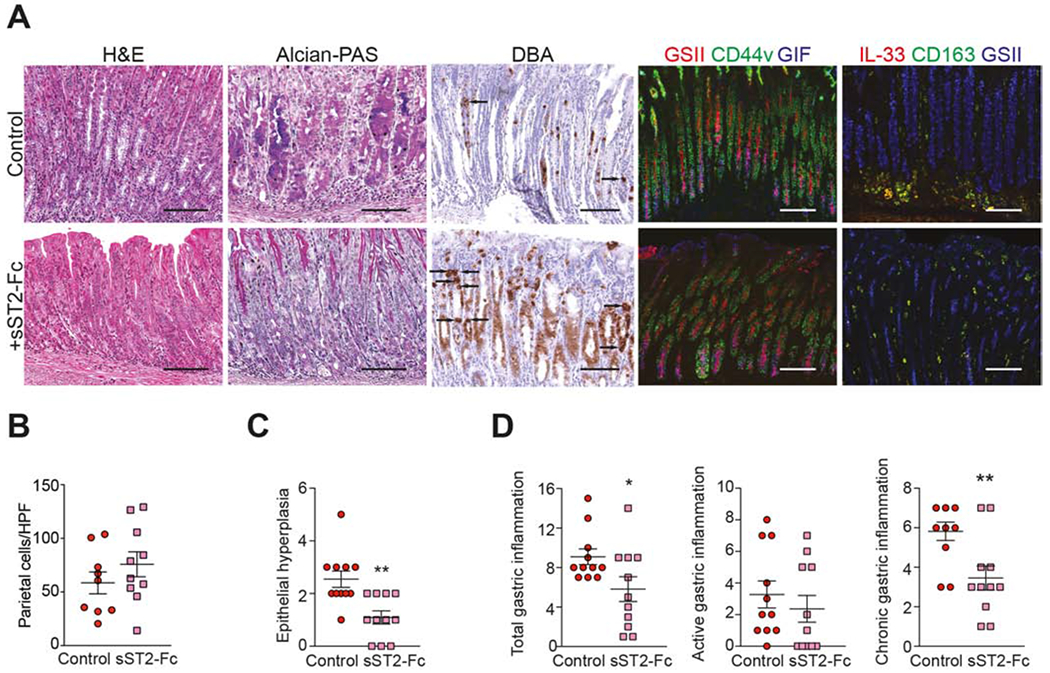
(A) Representative photomicrographs of H&E-, Alcian blue/PAS-, and DBA-stained corpus after sST2-Fc (left panels). While IF for GIF (blue) remains relatively unchanged, CD44v (green) and GSII-lectin (red/blue) are decreased, and IL-33 (red)/CD163 (green) show marked reduction of IL-33-producing M2 macrophages (yellow) after sST2-Fc (right panels). Original magnification: X20+1.25. (B) Parietal cell numbers (arrows, panel A), (C) epithelial hyperplasia, and (D) gastric inflammation after sST2-Fc; *P<0.05, **P<0.01 (N=9-11).
Histology images also show that overall gastric inflammation is visibly decreased after IL-33 blockade vs. control, confirmed by reduction in total inflammatory scores (Fig. 4D). Of note, the primary component driving total inflammation downward in sST2-Fc-treated SAMP is chronic inflammation, which decreases by 41%, and not acute inflammation, which remains relatively unchanged between experimental groups (Fig. 4D).
Peripheral expansion and local recruitment of eosinophils are dramatically increased in SAMP with advanced SPEM and rely on IL-33
We previously reported that eosinophils play a central role in mediating chronic, Th2-driven ileitis that is dependent on intestinal IL-33 induced by the gut microbiome34. In the present study, we found that expansion of peripheral BM-derived eosinophils, defined as CD11b+Siglec-F+ cells (Fig. S6), is already present in 4-wk-old SAMP, prior to the appearance of gastritis, compared to age-matched AKR (Fig. 5A). This trend is consistent with what was previously observed in older SAMP34, when intestinal-like metaplasia is fully established, and is increased vs. 4-wk-olds. Locally, baseline presence of eosinophils identified by MBP, one of the most prevalent proteins specifically-produced by eosinophils45,46, is detected in young SAMP, and also found in equal numbers in age-matched AKR (Fig. 5B). As disease progresses, the number of infiltrating MBP+ eosinophils greatly increases, first appearing at the margination between submucosa and muscularis mucosa, and then collecting at the base of the gastric glands with infiltrates percolating upwards throughout the LP vs. AKRs, wherein eosinophils are scarce and sparsely detected (Fig. 5B). Quantitatively, MBP+ eosinophils are clearly evident in SAMP corpus as intestinalizing SPEM progresses, showing a 4-fold increase by 20-weeks compared to AKR (Fig. 5B). These results indicate that systemic expansion of eosinophils and their recruitment into gastric tissues occurs early and are sustained in SAMP with advanced, intestinalized SPEM. Significant abrogation of BM-derived eosinophils (Fig. 5A) and subsequent decreased recruitment into the gastric mucosa of SAMP lacking IL-33 (SAMPxIl33−/−) compared to WT littermates (SAMPxIl33+/+) (Fig. 5C) confirm the early reliance on IL-33 for these effects.
Figure 5. Early IL-33-dependent peripheral expansion and increased eosinophil recruitment to SAMP stomachs.
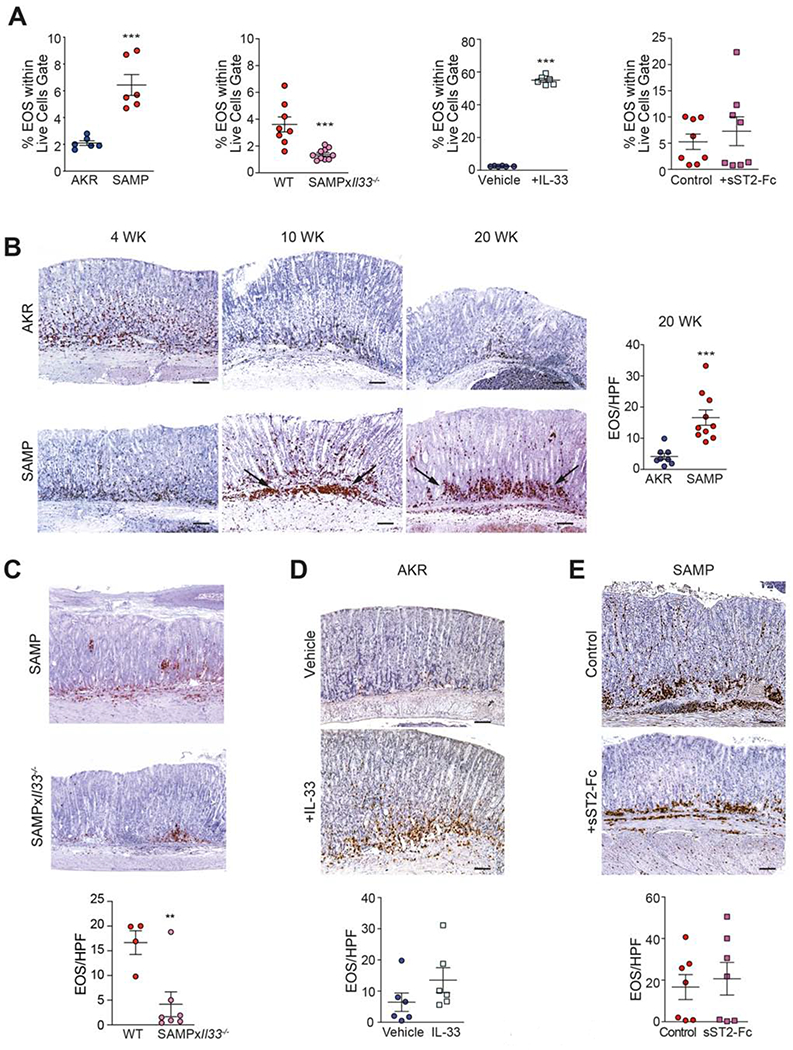
(A) Percentages of BM-derived eosinophils (EOS), and representative IHC photomicrographs of corpus from (B) SAMP vs. AKR, (C) SAMPxIl33−/− vs. WT littermates, (D) IL-33- vs. vehicle-treated AKR, and (E) sST2-Fc- vs. IgG-treated SAMP, stained for MBP and eosinophil counts. Original magnification: X10+1.25; scale bars: 100μm. **P<0.01, ***P<0.001 (N=4-10).
Eosinophil expansion, in fact, can be dramatically elicited, particularly in the periphery, in healthy AKR after acute (one-wk) IL-33 administration. Likewise, aside from the striking mucosal epithelial alterations described above (Fig. 2), IL-33 also promotes a trend of eosinophil recruitment into the corpus of treated AKR (Fig. 5D). These results indicate that, in the absence of inflammation, exogenous IL-33 induces robust systemic expansion, and to a lesser extent, local recruitment, of eosinophils into the gastric mucosa. Interestingly, IL-33 blockade in SAMP with established disease does not affect expansion of BM-derived eosinophils (Fig. 5A) or local recruitment of MBP+ eosinophils (Fig. 5E), indicating that once these processes are induced, particularly in mice exposed to chronic inflammatory conditions, they cannot be reversed.
Eosinophil depletion effectively reduces gastritis, infiltration of M2 macrophages, and intestinalized SPEM
Since IL-33 blockade is effective in dampening both infiltration of CD163+ M2 macrophages, as well as intestinal-like metaplasia, but not recruitment of eosinophils in SAMP, we tested whether earlier intervention of eosinophil depletion has downstream effects on advanced, intestinalized SPEM. As such, 14-wk-old SAMP were treated with antibodies against CCR3 and IL-5, alone and in combination, previously shown to effectively deplete eosinophils34,47,48. Eosinophil depletion was confirmed in both peripheral BM and corpus tissues (Figs. S7A and 6A, left panels), with histologic evaluation showing global improvement in restoring normal epithelial architecture, reducing the presence of cellular immune infiltrates, and decreasing overall inflammation within the LP and hyperplasia of muscle wall layers in mice treated with anti-IL-5 and anti-CCR3, alone and in combination, vs. IgG-treated controls (Figs. 6A, 6C). Eosinophil depletion is also able to effectively decrease the presence of infiltrating IL-33-expressing M2 macrophages (Figs. 6A, 6D and S7B) and potently reduces gene expression of M2-associated molecules (Fig. S7C), but does not affect epithelial-derived IL-33 (Fig. S7D). Importantly, CD44v co-labeling with GSII and GIF reveals that, while no dramatic differences are observed in GSII among experimental groups, increased intensity of GIF is noted, particularly in SAMP treated with either anti-IL-5 or combination anti-IL-5/anti-CCR3 vs. controls, indicating at least partial reversal of oxyntic atrophy (Fig. 6A, right panels). Interestingly, the decreased Mist1 seen during SPEM is dramatically reversed following eosinophil depletion (Fig. 6E), suggesting eosinophil-dependent chief cell transdifferentiation during SPEM. Taken together, these results indicate an essential role for eosinophils in the development of intestinalized SPEM.
Figure 6. Eosinophil depletion reduces gastritis, M2 macrophage recruitment and SPEM in SAMP.
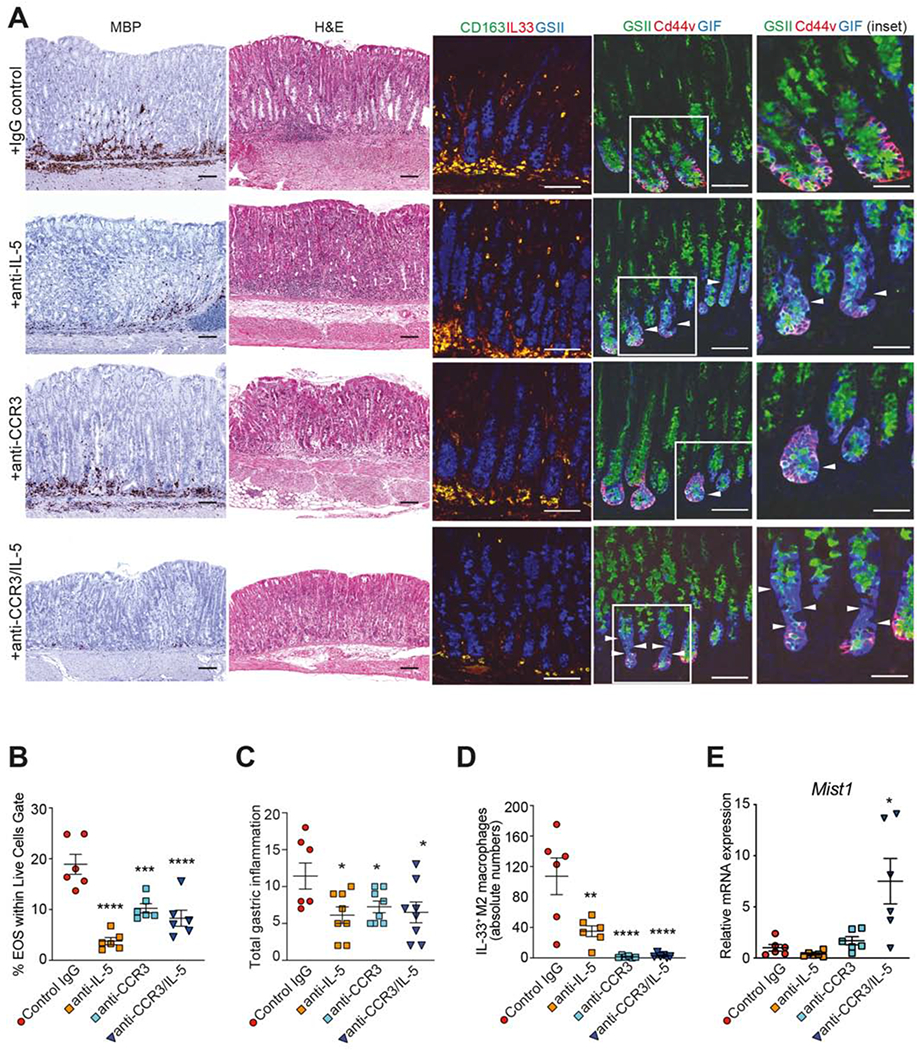
(A) Representative photomicrographs of corpus stained (left-to right): for MBP (eosinophils), with H&E, and by IF for IL-33- (red) expressing CD163+ (green) M2 macrophages (yellow), showing reduction after anti-IL-5, anti-CCR3 or combination anti-IL-5/CCR3, and for GSII (green), CD44v (red) and GIF (blue) showing specific increase in GIF (arrowheads), suggesting protection from oxyntic atrophy. Original magnification: X20, X40 (insets); scale bars: 100μm, 50μm (insets). Quantitation of gastric (B) eosinophils, (C) inflammation, (D) IL-33-expressing M2 macrophages, and (E) Mist1 after eosinophil depletion; *P<0.05. Data representative of 3 experiments (N=7-8/expt).
Discussion
Increasing evidence confirms the importance of the IL-33/ST2 axis in development of both gastritis and gastric cancer; in this context, IL-33 is an ideal candidate to link these two processes. However, while several studies report associative findings in patients, correlating increased IL-33 with intestinal-type gastric cancer18 and poor prognostic factors15,49, less is known regarding specific IL-33-dependent mechanisms leading to these pathologies. Based on the published literature and data from the present study, we propose a working hypothesis regarding the mechanistic role of IL-33 in promoting SPEM progression and the gastritis-metaplasia-dysplasia-carcinoma sequelae, in which eosinophils play a central role.
SPEM/intestinalized SPEM is considered by some to be a mandatory step for dysplasia, and eventually carcinoma, to proceed50. Once established, intestinalized SPEM and/or intestinal metaplasia may not be reversible in the presence of inciting factors, such as chronic inflammation51. In fact, along with distinct gastric epithelial changes, an essential component to attain intestinal-like characteristics is non-resolving, chronic inflammation, often thought to be dependent on H. pylori status51,52. In this setting, IL-33 represents a critical mediator for SPEM/intestinalized SPEM to manifest, considering its ability to both potently stimulate epithelial proliferation and metaplasia, as well as induce and sustain chronic, Th2-driven inflammation.
Our data show that the most prevalent form of IL-33 in SAMP with established disease is bioactive fIL-33, which is released during chronic inflammation, with minimal presence of the less bioactive cleaved form (cIL-33)38–40 that is more highly expressed in uninflamed AKR stomachs. In uninflamed young mice (AKR/SAMP) and older AKR, nuclear sequestration occurs within gastric epithelial cells, serving as reservoirs for IL-33 that is readily available for immediate release upon exposure to appropriate stimuli18. In fact, under homeostatic conditions, IL-33’s primary role in the GI tract is believed to be epithelial repair and restitution, and to promote overall mucosal wound healing33,53. Interestingly, we found that, at the transcript level, Il33 is significantly decreased in SAMP compared to AKR, which likely reflects a negative feedback mechanism in response to high systemic IL-33. In line with this finding, after exogenous IL-33 administration, Il33 is also decreased, while the Il1rl1 variant coding for sST2 (soluble decoy receptor) is greatly increased, which together serves to downregulate overall IL-33 bioactivity.
The present study also introduces the SAMP mouse strain as a model of intestinalized SPEM that develops spontaneously and progresses in severity, without chemical, genetic or immunologic manipulation. These mice provide an ideal tool to investigate the natural course of disease and a convenient system to test potential pre-clinical therapies for pre-neoplastic conditions that may lead to gastric cancer, in which both preventive and therapeutic strategies can be effectively implemented. Using a therapeutic approach, we found that IL-33 neutralization by sST2-Fc administration is effective at reversing intestinalized SPEM, primarily by targeting the epithelial compartment and reducing IL-33-dependent epithelial alterations, which are hallmark features of metaplasia. In addition, IL-33 blockade decreases severity of chronic gastritis and the presence of M2 macrophages, which are also characteristic of metaplasia and progression to intestinalized SPEM31. Importantly, using SAMP, we not only demonstrate the essential role for IL-33 in the development of intestinal-like metaplasia, but that the IL-33/ST2 axis is important for several steps throughout this process. For example, IL-33 clearly has direct effects on epithelial cells. Indeed, epithelial alterations within the GI tract were among the initial aberrancies described after healthy mice were exposed to high levels of IL-339. We and others41 confirmed these observations in the stomach, showing that rIL-33 induces severe oxyntic atrophy and upregulates specific markers of SPEM. Alternatively, IL-33 also has profound effects on the gastric immune compartment, promoting, for example, vigorous infiltration of eosinophils and M2 macrophages31, further perpetuating a chronic inflammatory state. Importantly, prolonged and elevated levels of IL-33 has the ability to sustain chronic inflammation in the stomach, which is essential for intestinal metaplasia to advance to gastric cancer.
The results of our study also underscore the importance of eosinophils in the development of intestinal-like metaplasia, and that IL-33 plays a critical role in the systemic expansion and early recruitment of eosinophils into the gastric mucosa of SAMP, as well as AKRs treated with rIL-33. Previously, eosinophils in gastric metaplasia and cancer development have been proposed to have both pathogenic as well as protective functions. A prior study investigating early gastric cancers demonstrated the presence of tumor stromal eosinophils with morphologic evidence of activation, as well as tumor cells in intimate contact with activated eosinophils with focal cytopathic changes54. Gastric carcinomas have also been reported to express eosinophil-associated chemotactic cytokines, including IL-2, IL-5 and GM–CSF, with expression of GM–CSF appearing to be specific for signet ring carcinoma cells55. On the other hand, in support of a protective function, high pre-operative eosinophil counts were found to correlate with prolonged survival after surgery56, and the −28G polymorphism in the promoter region of the eosinophil chemoattractant, RANTES, is associated with a reduced risk of developing severe intestinal metaplasia57.
Nonetheless, while our results indicate that eosinophils play an important role during early events leading to gastritis/metaplasia, one can certainly speculate that other IL-33-responsive innate immune cell populations may also contribute. Our data show that IL-33 can promote local recruitment of M2 macrophages that themselves, are potent producers of IL-33. These findings are in line with previous reports demonstrating the ability of IL-33 to increase overall macrophage numbers in the stomach and to polarize them to an alternatively-activated M2 phenotype, albeit in the periphery (peritoneum)41, and that M2 macrophages are dramatically reduced in IL-33-deficient mice in which advanced SPEM has been elicited, but that can also be rescued by administering rIL-1331. Our findings in SAMP, however, indicate that M2 macrophage infiltration is likely downstream of eosinophil activation and recruitment since eosinophil depletion, similar to systemic IL-33 blockade, has a significant impact on reducing the infiltration of gastric M2 macrophages that also happen to produce IL-33. Interestingly, however, eosinophil depletion does not appear to have an effect on IL-33 expression in the gastric epithelium of SAMP. In contrast, L635-induced SPEM does not appear to require eosinophils31, which may simply reflect differences in inciting factors between the two models, i.e., chemical obliteration of parietal cells vs. chronic, non-resolving inflammation. An alternative hypothesis is that ILC2s are also an IL-33-responsive innate immune cell population that can affect SPEM. ILC2s have not been detected and/or isolated from gastric tissues until recently, when Li et al. reported the prevalence of GATA-3+Lin− cells in tumors of patients with H. pylori-associated gastric cancer58. Of note, and counter-intuitive to this finding, Buzzelli et al., showed that IL-33 is rapidly induced after acute H. pylori infection, but suppressed in chronic infection41. However, in the former study, it is unclear what the IL-33 levels were, if they correlated with GATA-3+Lin− ILC2 frequency, and if IL-33 exhibited temporal changes during early vs. late stages in different types of gastric cancers. In the latter study, acute vs. chronic H. pylori infection is defined as 1 and 7 days vs. 2 months, which may not reflect chronic infection, especially in gastric cancer-susceptible individuals. Although indirect evidence suggests emergence of gastric ILC2s in IL-33-treated mice41, we recently reported increased ILC2s after L635-dependent acute gastric injury59. Similarly, in SAMP mice, preliminary data also show an increased frequency, albeit quite low, of ILC2s vs. AKR (Fig. S8); however, whether ILC2s contribute to the SAMP gastric phenotype is unknown. Based on studies characterizing SAMP lacking T/B cells, but with intact ILC function (i.e., SAMPxRag2−/− strain), ILCs likely play a negligible role in SPEM development since these mice, without the ability to mount adaptive immune responses and sustain a chronic inflammatory state, have virtually normal stomachs (Fig. S8). Finally, yet another IL-33-responsive innate immune cell population (i.e., mast cells) has been recently implicated in the development of gastric cancer in gp130F/F mice, specifically through mobilization of macrophages18. While similar to early, IL-33-dependent activation of eosinophils upstream of macrophage recruitment, it is unclear as to the precise temporal involvement of mast cells, and their interaction(s) with other mucosal populations to promote events leading to intestinal metaplasia and gastric cancer.
Taken together, the present study underscores a critical role for IL-33 in the development of chronic inflammation and metaplasia in the stomach, and implicates eosinophils as an early, IL-33-responsive innate immune cell population that participates in the complex orchestration leading to SPEM/intestinalized SPEM. As such, targeting the IL-33/ST2 axis for future therapeutic modalities to interrupt the early, initial events leading to gastritis and metaplasia, and potentially reversing intestinalizing SPEM, may prove to be beneficial for the prevention and/or treatment of patients susceptible to gastric cancer.
Supplementary Material
“What you Need to Know”.
Background and Context:
The link between chronic, non-resolving inflammation and cancer is well established, with inflammation-associated cancers among the most highly represented and frequently-occurring neoplasias worldwide. Investigation over the last several years has focused on determining critical pathways involved in this process, with a number of candidate molecules identified, including members of the interleukin-1 (IL-1) family that are particularly important in cancers of the GI tract.
New Findings:
IL-33 (or IL-1F11), a member of the IL-1 family of cytokines, serves as an important mediator linking chronic inflammation and metaplasia by inducing the expansion and recruitment of activated eosinophils leading to advanced, intestinalized SPEM in gastritis-prone SAMP1/YitFc (SAMP) mice.
Limitations:
Further studies are warranted to determine the precise inciting factors of increased IL-33 leading to intestinalized SPEM in SAMP mice, as well as in patients with gastric cancer.
Impact:
The present manuscript contributes to a better understanding of potential mechanism(s) that promote the inflammation-metaplasia-dysplasia-carcinoma sequelae that can apply to several GI-related cancers.
Acknowledgements
Personnel: We thank the following individuals for their important contributions to the manuscript – Xiao-Ming Wang, Danian Che, Hannah L. Havran, and Brian Marks for their technical support, as well as James J. Lee and Dirk E. Smith for provision of important reagents.
Grant support: This work was supported by grants from the NIH: DK056762, DK091222, DK042191, CA150964 Pilot & Feasibility Award (TTP), DK071590 (JRG), DK105129 (JCM), an NRSA F31 Predoctoral Fellowship (DK104600 to CPP), an Institutional T32 Fellowship from DK083251 (SL), as well as Cores from the Cleveland Silvio O. Conte Digestive Diseases Research Core Center (NIH DK097948). Other funding sources include the DeGregorio Family Foundation (TTP), the Department of Veterans Affairs: I01BX000930 (JRG), the Crohn’s & Colitis Foundation: RFA326877, CDA581292 (CDS), RFA410354 (KAB), SRFA592800 (HRF), the American Gastroenterological Association: Eli & Edythe Broad Student Research Fellowship Award (HRF, JD), and the Italian Society of Gastroenterology (LP).
Abbreviations used in this paper:
- Apc
adenomatous polyposis coli
- BM
bone marrow
- CCR3
C-C motif chemokine receptor 3
- CD44
cluster differentiation 44
- CD163
cluster differentiation 163
- Clu
clusterin
- Fc
fragment crystallizable
- GIF
gastric intrinsic factor
- GSII
Griffonia simplicifolia lectin II
- H. pylori
Helicobacter pylori
- IgG
immunoglobulin
- IHC
immunohistochemistry
- ILC2
type 2 innate lymphoid cells
- IL
interleukin
- Il1rl1
interleukin-1 receptor-like 1
- LP
lamina propria
- MBP
major basic protein
- PAS
periodic acid Schiff
- PFA
paraformaldehyde
- qPCR
quantitative polymerase chain reaction
- SAMP
SAMP1/YitFc
- SPEM
spasmolytic polypeptide-expressing metaplasia
- TFF1/2
trefoil factor 1/2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors have no conflict(s) of interest to disclose
References
- 1.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet 2001;357:539–45. [DOI] [PubMed] [Google Scholar]
- 2.Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol 2015;12:584–96. [DOI] [PubMed] [Google Scholar]
- 3.Mantovani A, Barajon I, Garlanda C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol Rev 2018;281:57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopetuso LR, Chowdhry S, Pizarro TT. Opposing functions of classic and novel IL-1 family members in gut health and disease. Front Immunol 2013;4:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maywald RL, Doerner SK, Pastorelli L, et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci USA 2015;112:E2487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malik A, Sharma D, Zhu Q, et al. IL-33 regulates the IgA-microbiota axis to restrain IL-1alpha-dependent colitis and tumorigenesis. J Clin Invest 2016;126:4469–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He Z, Chen L, Souto FO, et al. Epithelial-derived IL-33 promotes intestinal tumorigenesis in Apc(Min/+) mice. Sci Rep 2017;7:5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venerito M, Nardone G, Selgrad M, et al. Gastric cancer-epidemiologic and clinical aspects. Helicobacter 2014;19 Suppl 1:32–7. [DOI] [PubMed] [Google Scholar]
- 9.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005;23:479–90. [DOI] [PubMed] [Google Scholar]
- 10.Pastorelli L, Garg RR, Hoang SB, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci USA 2010;107:8017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010;464:1367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joshi AD, Oak SR, Hartigan AJ, et al. Interleukin-33 contributes to both M1 and M2 chemokine marker expression in human macrophages. BMC Immunology 2010;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stolarski B, Kurowska-Stolarska M, Kewin P, et al. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol 2010;185:3472–80. [DOI] [PubMed] [Google Scholar]
- 14.Hung LY, Lewkowich IP, Dawson LA, et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc Natl Acad Sci USA 2013;110:282–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun P, Ben Q, Tu S, et al. Serum interleukin-33 levels in patients with gastric cancer. Dig Dis Sci 2011;56:3596–601. [DOI] [PubMed] [Google Scholar]
- 16.Ye XL, Zhao YR, Weng GB, et al. IL-33-induced JNK pathway activation confers gastric cancer chemotherapy resistance. Oncol Rep 2015;33:2746–52. [DOI] [PubMed] [Google Scholar]
- 17.Yu XX, Hu Z, Shen X, et al. IL-33 promotes gastric cancer cell invasion and migration via ST2-ERK1/2 pathway. Dig Dis Sci 2015;60:1265–72. [DOI] [PubMed] [Google Scholar]
- 18.Eissmann MF, Dijkstra C, Jarnicki A, et al. IL-33-mediated mast cell activation promotes gastric cancer through macrophage mobilization. Nat Commun 2019;10:2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Correa P A human model of gastric carcinogenesis. Cancer Res 1988;48:3554–60. [PubMed] [Google Scholar]
- 20.Saenz JB, Mills JC. Acid and the basis for cellular plasticity and reprogramming in gastric repair and cancer. Nat Rev Gastroenterol Hepatol 2018;15:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldenring JR. Pyloric metaplasia, pseudopyloric metaplasia, ulcer-associated cell lineage and spasmolytic polypeptide-expressing metaplasia: reparative lineages in the gastrointestinal mucosa. J Pathol 2018;245:132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt PH, Lee JR, Joshi V, et al. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab Invest 1999;79:639–46. [PubMed] [Google Scholar]
- 23.Nam KT, Lee HJ, Sousa JF, et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 2010;139:2028–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills JC, Sansom OJ. Reserve stem cells: Differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal 2015;8:re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radyk MD, Burclaff J, Willet SG, et al. Metaplastic cells in the stomach arise, independently of stem cells, via dedifferentiation or transdifferentiation of chief cells. Gastroenterology 2018;154:839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lennerz JK, Kim SH, Oates EL, et al. The transcription factor MIST1 is a novel human gastric chief cell marker whose expression is lost in metaplasia, dysplasia, and carcinoma. Am J Pathol 2010;177:1514–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burclaff J, Osaki LH, Liu D, et al. Targeted apoptosis of parietal cells is insufficient to induce metaplasia in stomach. Gastroenterology 2017;152:762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willet SG, Lewis MA, Miao ZF, et al. Regenerative proliferation of differentiated cells by mTORC1-dependent paligenosis. EMBO J 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weis VG, Sousa JF, LaFleur BJ, et al. Heterogeneity in mouse spasmolytic polypeptide-expressing metaplasia lineages identifies markers of metaplastic progression. Gut 2013;62:1270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen CP, Weis VG, Nam KT, et al. Macrophages promote progression of spasmolytic polypeptide-expressing metaplasia after acute loss of parietal cells. Gastroenterology 2014;146:1727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petersen CP, Meyer AR, De Salvo C, et al. A signalling cascade of IL-33 to IL-13 regulates metaplasia in the mouse stomach. Gut 2018;67:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reuter BK, Pastorelli L, Brogi M, et al. Spontaneous, immune-mediated gastric inflammation in SAMP1/YitFc mice, a model of Crohn’s-like gastritis. Gastroenterology 2011;141:1709–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopetuso LR, De Salvo C, Pastorelli L, et al. IL-33 promotes recovery from acute colitis by inducing miR-320 to stimulate epithelial restitution and repair. Proc Natl Acad Sci USA 2018;115:E9362–E9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Salvo C, Wang XM, Pastorelli L, et al. IL-33 drives eosinophil infiltration and pathogenic type 2 helper T-cell immune responses leading to chronic experimental ileitis. Am J Pathol 2016;186:885–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arseneau KKO, Cominelli F. Improving the reproducibility and quality of reporting for animal studies in inflammatory bowel disease. Inflamm Bowel Dis 2017;23:2069–2071. [DOI] [PubMed] [Google Scholar]
- 36.Turani H, Lurie B, Chaimoff C, et al. The diagnostic significance of sulfated acid mucin content in gastric intestinal metaplasia with early gastric cancer. Am J Gastroenterol 1986;81:343–5. [PubMed] [Google Scholar]
- 37.Nozaki K, Ogawa M, Williams JA, et al. A molecular signature of gastric metaplasia arising in response to acute parietal cell loss. Gastroenterology 2008;134:511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Talabot-Ayer D, Lamacchia C, Gabay C, et al. Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem 2009;284:19420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA 2009;106:9021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luthi AU, Cullen SP, McNeela EA, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009;31:84–98. [DOI] [PubMed] [Google Scholar]
- 41.Buzzelli JN, Chalinor HV, Pavlic DI, et al. IL33 Is a stomach alarmin that initiates a skewed Th2 response to injury and infection. Cell Mol Gastroenterol Hepatol 2015;1:203–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wada T, Ishimoto T, Seishima R, et al. Functional role of CD44v-xCT system in the development of spasmolytic polypeptide-expressing metaplasia. Cancer Sci 2013;104:1323–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fagundes CT, Amaral FA, Souza AL, et al. ST2, an IL-1R family member, attenuates inflammation and lethality after intestinal ischemia and reperfusion. J Leukoc Biol 2007;81:492–9. [DOI] [PubMed] [Google Scholar]
- 44.Miao ZF, Adkins-Threats M, Burclaff JR, et al. A metformin-responsive metabolic pathway controls distinct steps in gastric progenitor fate decisions and maturation. Cell Stem Cell 2020;26:910–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mishra A, Hogan SP, Lee JJ, et al. Fundamental signals that regulate eosinophil homing to the gastrointestinal tract. J Clin Invest 1999;103:1719–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hogan SP, Rosenberg HF, Moqbel R, et al. Eosinophils: biological properties and role in health and disease. Clin Exp Allergy 2008;38:709–50. [DOI] [PubMed] [Google Scholar]
- 47.Masterson JC, McNamee EN, Jedlicka P, et al. CCR3 blockade attenuates eosinophilic ileitis and associated remodeling. Am J Pathol 2011;179:2302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song DJ, Shim MH, Lee N, et al. CCR3 monoclonal antibody inhibits eosinophilic inflammation and mucosal injury in a mouse model of eosinophilic gastroenteritis. Allergy Asthma Immunol Res 2017;9:360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu W, Li X, Li Q, et al. Interleukin-33 expression does not correlate with survival of gastric cancer patients. Pathol Oncol Res 2017;23:615–619. [DOI] [PubMed] [Google Scholar]
- 50.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest 2007;117:60–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Correa P, Piazuelo MB. The gastric precancerous cascade. J Dig Dis 2012;13:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petersen CP, Mills JC, Goldenring JR. Murine models of gastric corpus preneoplasia. Cell Mol Gastroenterol Hepatol 2017;3:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monticelli LA, Osborne LC, Noti M, et al. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci USA 2015;112:10762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caruso RA, Giuffre G, Inferrera C. Minute and small early gastric carcinoma with special reference to eosinophil infiltration. Histol Histopathol 1993;8:155–66. [PubMed] [Google Scholar]
- 55.Hong SW, Cho MY, Park C. Expression of eosinophil chemotactic factors in stomach cancer. Yonsei Med J 1999;40:131–6. [DOI] [PubMed] [Google Scholar]
- 56.Iwasaki K, Torisu M, Fujimura T. Malignant tumor and eosinophils. I. Prognostic significance in gastric cancer. Cancer 1986;58:1321–7. [DOI] [PubMed] [Google Scholar]
- 57.Tahara T, Arisawa T, Shibata T, et al. Effect of RANTES promoter genotype on the severity of intestinal metaplasia in Helicobacter pylori-infected Japanese subjects. Dig Dis Sci 2009;54:1247–52. [DOI] [PubMed] [Google Scholar]
- 58.Li R, Jiang XX, Zhang LF, et al. Group 2 innate lymphoid cells are involved in skewed type 2 immunity of gastric diseases induced by Helicobacter pylori infection. Mediators Inflamm 2017;2017:4927964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meyer AR, Engevik AC Madorsky T, et al. Group 2 innate lymphoid cells coordinate damage response in the stomach. Gastroenterology 2020;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.