

Abstract
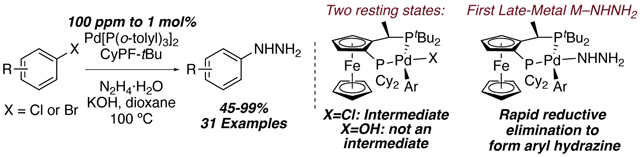
We report the Pd-catalyzed C–N coupling of hydrazine with (hetero)aryl chlorides and bromides to form aryl hydrazines with catalyst loadings as low as 100 ppm of Pd and KOH. Mechanistic studies revealed two catalyst resting states: an arylpalladium(II) hydroxide and arylpalladium(II) chloride. These compounds are present in two interconnected catalytic cycles and react with hydrazine and base or hydrazine alone to give the product. The selectivity of the hydroxide complex with hydrazine to form aryl over diaryl hydrazine was lower than that of the chloride complex, as well as the catalytic reaction. In contrast, the selectivity of the chloride complex closely matches that of the catalytic reaction, indicating that the aryl hydrazine is derived from this complex. Kinetic studies show that the coupling process occurs by rate-limiting deprotonation of a hydrazine-bound arylpalladium(II) chloride complex to give an arylpalladium(II) hydrazido complex.
Keywords: palladium, transition-metal catalysis, cross-coupling, aryl halide, hydrazine
Entry for the Table of Contents
A practical coupling of (hetero)aryl halides with hydrazine to form (hetero)aryl hydrazines with loadings down to 100 ppm of a Pd catalyst and KOH as base is reported, along with detailed mechanistic data revealing the factors that control the rate and selectivity. Two catalyst resting states are observed, but one major pathway involving rate-limiting deprotonation of hydrazine bound to an arylpalladium(II) chloride complex occurs.
Introduction
Aryl hydrazines are synthetic intermediates to a wide range of nitrogen-containing heterocycles,[1] including those in a variety of pharmaceuticals and agrochemicals.[2] Aryl hydrazines are typically prepared by diazotization of aniline, followed by reduction with tin or metabisulfite salts, a sequence that generates explosive diazonium intermediates and large amounts of salt waste.[3] Nucleophilic aromatic substitution with hydrazine also can form aryl hydrazines, but this reaction is typically limited to arenes containing highly electron-withdrawing substituents or electrophilic heterocycles.[4] Thus, a general economical synthesis of aryl hydrazines would be synthetically valuable and industrially relevant.
The palladium-catalyzed coupling of amines with aryl halides is a well-established method to prepare aryl amines.[5] However, the coupling of hydrazine with aryl halides is much less developed. [6] Pd-catalyzed couplings of hydrazine are difficult to develop because hydrazine is a strong reductant and can reduce Pd(II) species to catalytically inactive Pd black.[7] Moreover, hydrazine contains multiple N–H bonds and can undergo multiple couplings. Although prior reports by Stradiotto[6a,6b] and Buchwald[6c] showed that the hydrazine can couple with aryl halides, these reactions have required NaOtBu as base and high loadings of catalyst (1–20 mol%). This base and these loadings make this reaction impractical for production of simple aryl hydrazines.[8] Instead, for such applications, this reaction must occur with low loadings of catalyst, with hydrazine hydrate, and with a simple base, like hydroxide or carbonate. Although C−N coupling reactions with hydrazine have been reported with copper catalysts,[9] only two systems have been reported, and reactions do not occur with chloroarenes, which are less expensive and more commercially available than other haloarenes.[10]
No studies on the mechanism of the coupling of hydrazine with aryl halides have been reported that could guide the selection of catalysts and conditions.[11] It is unclear how the electronic properties and small size of hydrazine, in addition to the water in hydrazine hydrate, will affect the identity of the species in the catalytic system, the identity of the turnover-limiting step, and pathways to catalyst decomposition. Further, no late-metal, parent hydrazido complexes – the type of complex that would likely form the C–N bond – have been prepared previously. Published C–N coupling reactions with hydrazine display a high selectivity for forming monoaryl hydrazine over diaryl hydrazine, but the basis for this selectivity is not understood.
We report the synthesis of aryl hydrazines from aryl chlorides and bromides by palladium-catalyzed coupling of hydrazine hydrate with catalyst loadings down to 100 ppm and with potassium hydroxide as base, along with detailed mechanistic data that include an unusual resting state, characterization of a parent hydrazido complex, and directly observed reductive eliminations from this complex. The resting state of the catalyst comprises arylpalladium chloride and hydroxide complexes, and the rate-limiting step is the reaction of the arylpalladium chloride complex with hydrazine and hydroxide base to generate water and an arylpalladium hydrazido complex that reductively eliminates aryl hydrazine. Generation and studies of the hydrazido intermediate showed that reductive elimination is rapid.
Results and Discussion
Reaction Development
Our efforts to develop a catalyst for the monoarylation of hydrazine with high turnovers focused on the combination of a Pd precursor and the Josiphos ligand, (R)-1-[(S)-2-(dicyclohexylphosphino)ferrocenyl]ethyldi-tert-butylphosphine (CyPF-tBu). We showed that Pd complexes of this ligand catalyze the reactions of aryl halides with primary amines, ammonia, or thiols. The reactions of thiols and primary amines occurred with high turnover numbers and with catalyst loadings as low as 5 ppm,[12] and the reactions of aryl halides with ammonia and primary amines also occurred with high selectivity for the product from monoarylation.[12b,13,14]
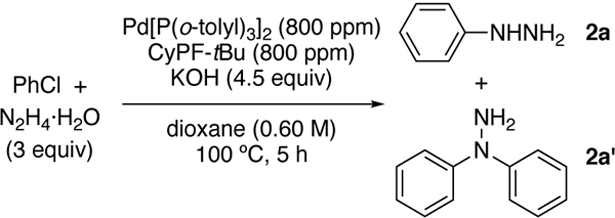
To test whether the coupling of aryl chlorides with hydrazine could occur with this type of catalyst and with high turnovers, we investigated reactions with CyPF-tBu as a ligand and Pd[P(o-tolyl)3]2 as the Pd(0) source (Table 1). The combination of 800 ppm (0.08 mol%) Pd[P(o-tolyl)3]2 and CyPF-tBu resulted in high yield of the desired hydrazine 2a and high selectivity for monoarylation (97:3) with KOH as base (Entry 1). In contrast, reactions with previously reported ligands (Entries 2–4) or catalysts from alternative palladium precursors (Entries 5–7) did not furnish the desired product at low catalyst loadings (800–1600 ppm). With loadings of Pd[P(o-tolyl)3]2 and CyPF-tBu below 800 ppm, reactions occurred with lower conversion and yield, even with longer reaction times (Entries 8–10). Although NaOtBu in place of KOH gave the highest yield and selectivity of 2a (Entry 11), we used KOH for our remaining studies because it is much less expensive than t-butoxide bases. Reactions with related NaOMe (Entry 12) and NaOH (Entry 13) occurred in similar, but slightly lower yields and selectivity for monoarylation. Reactions below 100 °C (Entries 14 and 15) or with lower loadings of base or hydrazine (Entries 16–18) also led to lower yields with poor monoarylation selectivity. The reaction with bromobenzene, instead of chlorobenzene, occurred in slightly higher yield and selectivity, despite prior low yields with bromoarenes (Entry 19).[15]
Table 1.
Survey of reaction conditions of C–N coupling with hydrazine.
 | ||||
|---|---|---|---|---|
| Entry | Variations from above | Conv. (%)[a] | Yield 2a (%)[a] | 2a : 2a’[a] |
| 1 | none | >99 | 93 | 97:3 |
| 2 | MorDalPhos (1200 ppm) | - | - | - |
| 3 | BippyPhos (1600 ppm) | - | - | - |
| 4 | BrettPhos (1200 ppm) | <1 | trace | - |
| 5 | [Pd(cinnamyl)Cl]2 (400 ppm) MorDalPhos (1200 ppm) | 1 | 1 | - |
| 6 | [Pd(cinnamyl)Cl]2 (400 ppm) BippyPhos (1600 ppm) | <1 | trace | - |
| 7 | BrettPhos-Pd-G1 (800 ppm) | 2 | 2 | - |
| 8 | [Pd] and L2 (400 ppm), 24 h | 96 | 84 | 93:7 |
| 9 | [Pd] and L2 (200 ppm), 24 h | 88 | 80 | 95:5 |
| 10 | [Pd] and L2 (100 ppm), 24 h | 77 | 73 | 97:3 |
| 11 | NaOtBu instead of KOH | >99 | 99 | >99:1 |
| 12 | NaOMe instead of KOH | >99 | 89 | 96:4 |
| 13 | NaOH instead of KOH | >99 | 91 | 96:4 |
| 14 | 80 °C, 24 h | >99 | 86 | 93:7 |
| 15 | 60 °C, 24 h | >99 | 76 | 88:12 |
| 16 | KOH (3 equiv) | >99 | 92 | 92:8 |
| 17 | N2H4·H2O (2.3 equiv), 24 h | >99 | 82 | 94:6 |
| 18 | N2H4·H2O (1.7 equiv), 24 h | >99 | 86 | 94:6 |
| 19 | PhBr instead of PhCl | >99 | 97 | 98:2 |
Conversion, yield, and selectivity were determined by 1H NMR spectroscopy.
![[a]](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/526f/7755694/ec0cf48e8667/nihms-1632403-f0019.jpg)
Reaction Scope
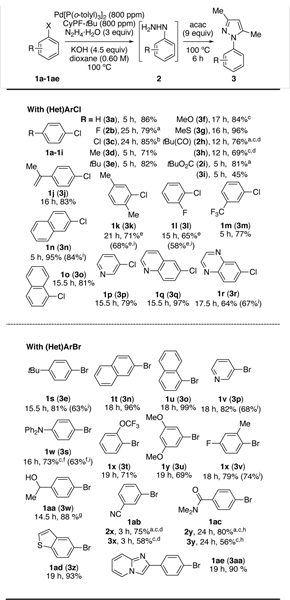
Table 2 shows the scope of the reaction of hydrazine with aryl and heteroaryl chlorides and bromides under the developed conditions. Because of the air sensitivity of aryl hydrazines, we treated the aryl hydrazines with acetylacetone (acac) in one-pot to form 3,5-dimethyl-N-arylpyrazoles, which were more amenable to isolation by column chromatography.[16]
Table 2.
Scope of C–N coupling reaction with hydrazine and (hetero)aryl chlorides or bromides.
 |
Yield of aryl hydrazine determined by 1H NMR spectroscopy.
100 ppm [Pd(o-tolyl)3]2 and CyPF-tBu.
4.5 equiv NaOtBu instead of KOH.
1 mol% [Pd(o-tolyl)3]2 and CyPF-tBu at rt.
1600 ppm [Pd(o-tolyl)3]2 and CyPF-tBu.
1200 ppm [Pd(o-tolyl)3]2 and CyPF-tBu.
Isolated as a 2:1 mixture of 3,5-dimethylpyrazole and 3w, respectively.
2400 ppm [Pd(o-tolyl)3]2 and CyPF-tBu at rt.
4.5 equiv NaOMe instead of KOH.
Most of the reactions of hydrazine with aryl and heteroaryl chlorides occurred with 800 ppm catalyst and KOH as base. However, 0.16 mol% of catalyst was required to couple hydrazine with the more hindered 2-chloro-p-xylene (1k) in good yield. Although this loading was higher than that for the unhindered chloroarenes, previously reported couplings of hydrazine with o-methyl substituted haloarenes required 6 to 30 times more Pd (1–5 mol%).[6] The reaction between hydrazine and chlorobenzene occurred in 5 h, but the those of most aryl halides required 5–24 h for high conversion. Ethers, thioethers, t-butyl esters, alcohols, amines, amides, hindered ketones, nitriles, additional halides, and terminal olefins were tolerated in the aryl halide. The reaction of 1,4-dichlorobenzene with hydrazine to form 4-chloroaryl hydrazine selectively occurred with only 100 ppm (0.01 mol%) of palladium catalyst. Heteroaryl chlorides, including those bearing pyridine, quinoline, benzothiophene, imidazo[1,2-a]pyridine, and benzopyrazine units, reacted in 64–97% yield.[17] Aryl hydrazines 2m, 2c, and 2p are precursors to the agrochemicals norflurazon,[18] pyraclostrobin,[2d] and tyclopyrazoflor,[19] respectively.
In a few cases, the reactions required NaOtBu as base for high yields. For example, the reaction of electron-rich chloroarene 1f with KOH as base gave a low yield (29%) of 4-methoxyphenyl hydrazine, but that with NaOtBu gave 84% isolated yield. In a few other cases, the pyrazoles were isolated in modest yield, but the aryl hydrazine formed in high yield. For example, pyrazoles 3h-i, 3x, and 3y were isolated in 45–69% yield, but the hydrazines formed in 75–81% yield by 1H NMR spectroscopy.[20]
The monoarylation of hydrazine also occurred on larger scales. Reactions with 3.0 mmol (0.38–0.49 g) 4-chlorotoluene (1d) and 6.0 mmol (1.0 g) of 2-chloronaphthalene (1n) gave yields and monoarylation selectivities that were nearly identical to those performed at smaller scale (0.5 mmol, Table 1).
Mechanistic Studies on C–N Cross-Coupling with Hydrazine
To reveal the mechanism of this valuable process, we analyzed the Pd complexes in the system, the relative rates of the individual steps, the effect of hydroxide base on the catalytic reaction, and the origin of the selectivity for monoarylation. We also sought to observe or independently generate arylpalladium(II) hydrazido complexes to determine their potential intermediacy, their rates and selectivity for formation, and their rates for reductive elimination to form aryl hydrazines.
The resting state of the catalytic reaction was determined by analyzing the reaction of 4-chlorotoluene at partial conversion by 31P NMR spectroscopy. At 52% conversion with 5 mol% of catalyst, two Pd species were detected in a 1:1 ratio (Scheme 1). One of these species is the arylpalladium chloride complex 4 (δ 71.6 and 19.4 ppm), while the other species was determined to be the aryl hydroxide species 5 (δ 67.1 and 20.6 ppm), which was synthesized independently (Scheme 2). This experiment implies that oxidative addition of the chloroarene to Pd(0) to form 4 is not rate-limiting. The reaction between hydrazine and one or both Pd(II) species or the reductive elimination of aryl hydrazine after reversible generation of an arylpalladium(II) hydrazido complex could be rate-limiting.
Scheme 1.

Determination of catalyst resting state by analyzing the catalytic reaction at partial conversion by 31P NMR spectroscopy. (a) 31P NMR spectrum of catalytic reaction analyzed at 52% conversion. (b) 31P NMR spectrum of (CyPF-tBu)Pd(p-tolyl)(OH) (5) in 1,4-dioxane. (c) 31P NMR spectrum of (CyPF-tBu)Pd(p-tolyl)Cl (4) in 1,4-dioxane.
Scheme 2.

Synthesis of palladium aryl hydroxide complexes 5 and 6.
The initial rates of the catalytic reactions were measured with varied concentrations of Pd catalyst, N2H4•H2O, and chloroarene. The catalytic reaction was zero-order in chloroarene and first-order in palladium catalyst and N2H4•H2O. (Figure 1). These data also indicate that the oxidative addition of the chloroarene to Pd occurs before the rate-limiting step.
Figure 1.

Kinetic studies on the monoarylation of hydrazine. (a) Dependence of initial rate on [N2H4] (1.2–3.0 M) with [ArCl] = 0.60 M and [Pd] and [CyPF-tBu] = 800 ppm. (b) Dependence of initial rate on [Pd] and [CyPF-tBu] (400–1200 ppm) with [ArCl] = 0.60 M and [N2H4] = 1.8 M. (c) Dependence of initial rate on [ArCl] (0.60–2.4 M) with [N2H4] = 1.8 M and [Pd] and [CyPF-tBu] = 800 ppm.
The rate-limiting step could be an acid-base reaction between hydrazine and an arylpalladium(II) hydroxide to generate an arylpalladium(II) hydrazido complex and water (Figure 2, Path A), an association between hydrazine and an arylpalladium(II) chloride complex to generate a five-coordinate compound that undergoes deprotonation (Figure 2, Path B), or a combination of these pathways. All scenarios lead to an arylpalladium(II) hydrazido species that could undergo reductive elimination to give aryl hydrazine.
Figure 2.

Potential mechanisms for the rate-limiting step of the catalytic reaction.
To determine whether the arylpalladium(II) hydroxide of path A can react with hydrazine, we synthesized p-tolyl and phenyl Pd(II) hydroxides 5 and 6 from CyPF-tBu and [Pd(PPh3)(μ-OH)(Ar)]2 (Ar = p-Tol or Ph) (Scheme 2). p-Tolyl complex 5 reacted with 6 equiv. of hydrazine monohydrate and PPh3 at 65 °C to generate (CyPF-tBu)Pd(PPh3) in quantitative yield, along with p-tolyl hydrazine in 85% yield after 45 min (Scheme 3). Thus, the aryl hydroxide species generates aryl hydrazine and is catalytically competent.
Scheme 3.

Reaction of arylpalladium(II) hydroxide complex 5 with hydrazine.
Further studies revealed whether the aryl hydrazido complex formed from chloride complex 4 by path A or B (Figure 2), or a combination of them. We studied the viability of these pathways by conducting stoichiometric reactions of arylpalladium chloride complex 4 with hydrazine monohydrate in the presence of KOH. The conversion of chloride 4 to hydroxide 5 does not reach equilibrium from reaction of the soluble palladium halide and the insoluble hydroxide, but conversion of chloride 4 to hydroxide 5 with the soluble Bu4NOH quantitatively generated aryl hydroxide 5 at room temperature within 5 minutes in THF (Scheme 4a). In contrast, aryl hydroxide 5 does not react with 5 equiv of [TBA]Cl in a 5:1 mixture of THF and acetonitrile at room temperature (Scheme 4c).[21] Thus, the equilibrium between chloride complex 4 and aryl hydroxide 5 strongly favors complex 5. This salt metathesis reactions with the alkali metal hydroxides appear to occur in organic solvent because hydrazine and water aid the dissolution of KOH.
Scheme 4.

Reactions of arylpalladium(II) chloride complex 4 with hydroxide sources and reactions of arylpalladium(II) hydroxide complex with [TBA]Cl (TBA=tetrabutylammonium).
The reaction between chloride complex 4, N2H4•H2O, PPh3, and KOH was monitored by 31P NMR spectroscopy. Chloride 4 converted to approximately 31% hydroxide 5 upon mixing. After 50 min at 35 °C, the aryl hydrazine (89%) and the Pd(0) complex (CyPF-tBu)Pd(PPh3) formed. The reaction did not follow an exponential decay (data points labeled X, Figure 3). Consistent with pathway A, the concentration of hydroxide 5 initially increased and then decreased. The conversion of chloride 4 to hydroxide 5 and subsequent reaction with hydrazine with a rate similar to that of the reaction of the chloride complex with potassium hydroxide would increase the concentration of the aryl hydroxide complex at the outset of the reaction, followed by a decay of the aryl hydroxide compound. Simulation of this scenario to the experimental data using the modelling program COPASI[22] gave an excellent fit (Figure 3), which is consistent with our observed arylpalladium(II) chloride and an arylpalladium(II) hydroxide complex as the catalyst resting state (Scheme 1). Concurrent reaction through Paths A and B (Figure 2) cannot be excluded by fitting the predicted and experimental data. Thus, further experiments revealed whether hydrazine reacts with the hydroxo or the chloro complex on the major pathway to aryl hydrazine.
Figure 3.


Time course of reaction between (CyPF-tBu)Pd(p-tolyl)Cl (4), hydrazine monohydrate, and KOH monitored by 31P NMR spectroscopy with PMes3 as an internal standard and fitted curves obtained from COPASI. The circle trace corresponds to the concentration of 4, the multiplication sign trace corresponds to the concentration of (CyPF-tBu)Pd(p-tolyl)(OH) (5), and the diamond trace corresponds to the concentration of (CyPF-tBu)Pd(PPh3).
Competition Experiments Involving the Resting States of the Catalyst
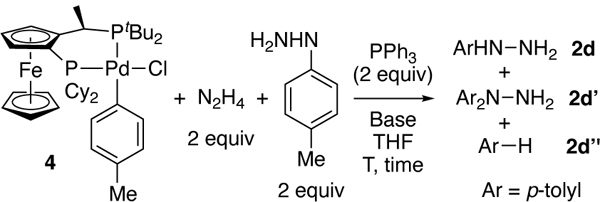
To assess the potential intermediacy of the observed Pd(II) species further, we conducted competition experiments. We compared the selectivity from catalytic and stoichiometric reactions of chloride complex 4 or aryl hydroxide complex 5 with the combination of hydrazine and aryl hydrazine. The catalytic reaction of 1-tBu-4-chlorobenzene with one equiv. of N2H4•H2O and 4-methylphenyl hydrazine formed a 5:1 ratio of 4-tBu-phenyl hydrazine to the 1,1-diaryl hydrazine at 69% conversion of the chloroarene (Scheme 5).
Scheme 5.

Competition experiment between hydrazine and p-tolyl hydrazine under catalytic conditions.
In contrast, the stoichiometric reaction of aryl hydroxide 5 with 2 equiv. of hydrazine and p-tolyl hydrazine formed nearly equal amounts of the two coupled products (Scheme 6). This result does not match the high selectivity for monoarylation in the catalytic reaction (Table 3 and Scheme 5). This low selectivity implies that the primary pathway forming aryl hydrazine involves a Pd complex other than hydroxide 5, even though 5 is kinetically competent to be an intermediate (Scheme 3).
Scheme 6.

Competition experiment between hydrazine and p-tolyl hydrazine with aryl hydroxide complex 5.
Table 3.
Competition experiments between hydrazine and p-tolyl hydrazine with chloride complex 4.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Base (equiv) | T (°C) | time (min) | Yield (%)[a] | ||
| 2d | 2d’ | 2d” | ||||
| 1 | NaOtBu (1.3) | 25 | 10 | 93 | 4 | 3 |
| 2 | LiHMDS (1.3) | 25 | 10 | 87 | 6 | 8 |
| 3 | KOH (1.3) | 65 | 180 | 35 | 34 | 30 |
Yield determined by 1H NMR spectroscopy.
In contrast to hydroxide 5, chloride 4 reacted with the same mixture of hydrazine and aryl hydrazine in the presence of NaOtBu or LiHMDS with high selectivity to form monoaryl hydrazine (Table 3, entries 1–2). Although chloride 4 could react with NaOtBu to form an arylpalladium alkoxide,[23] which could react with hydrazine, a salt metathesis between 4 and LiHMDS is unlikely, due to the steric bulk of the silylamide and the ancillary ligands of 4. Because the selectivity of the reaction in the presence of LiHMDS is nearly identical to that of NaOtBu, we propose that the aryl hydrazine forms from chloride complex 4, hydrazine, and NaOtBu or LiHMDS as base occurs by path B in Figure 2 and that reactions by this pathway are highly selective for the monoarylation of hydrazine.
The reactions with KOH in place of these bases, again, generated diaryl hydrazine and monoaryl hydrazine in approximately equal amounts and in nearly equal yield with toluene (Table 3, entry 3). The difference between this result and that with the soluble bases is discussed further in this manuscript, but we note here that the concentration of hydrazine in these experiments is necessarily lower than that in the catalytic process and that the results of reactions with a heterogeneous base that changes physical properties during the catalytic process are less conclusive than those of reactions with fully soluble components.
Because the selectivity of hydroxide 5 for reaction with hydrazine over aryl hydrazine was lower than that of the reactions of chloride 4 with the bases that ensure reaction through path B, we propose that monoaryl hydrazine is predominantly produced in the catalytic process by Path B. If so, then the deprotonation of hydrazine coordinated to chloride complex 4 by hydroxide base (Figure 2, Path B) must be faster than the reaction of aryl hydroxide 5 with hydrazine (Figure 2, Path A), and the formation of the diaryl hydrazine products in the catalytic reaction would originate from the reaction between aryl hydrazine and an aryl hydroxo complex.
To understand the relative rates of the reactions of chloride 4 and hydroxide 5, the effect of water on the reaction of 5 with hydrazine must be noted. The reaction of hydrazine hydrate with 5 was much slower than that of anhydrous hydrazine. The reaction with hydrazine monohydrate (Figure 4c) occurred with kobs=9.53(8)×10−5 s−1, while that with anhydrous hydrazine (Figure 4b) occurred with kobs=1.55(4)×10−3 s−1, which is approximately 15 times faster than that with hydrazine hydrate. For comparison, the reaction of chloride complex 4 with 10 equiv. of hydrazine monohydrate and 10 equiv. of KOH (Figure 4a) occurred with kobs=2.9(1)×10−4 s−1, which lies between the rate constants of the two reactions of the hydroxo complex.[24]
Figure 4.

Comparison of rates of product formation for the reaction of arylpalladium(II) complexes 4 and 5. (a) 4 (39.3 mM) with KOH (10 equiv), hydrazine monohydrate (10 equiv), and PPh3 (2 equiv); (b) 5 (39.3 mM) with anhydrous hydrazine (10 equiv) and PPh3 (2 equiv); (c) 5 (39.3 mM) with hydrazine monohydrate (10 equiv) and PPh3 (2 equiv) in 600 μL of THF-d8 at 35 °C.
A closer examination of the effect of water on the rate of the reaction between 5 and hydrazine indicated that this reaction was inverse second-order in water (Figure 5). We propose that aryl hydroxide 5 forms a hydrogen bonding network containing multiple waters interacting with the hydroxide ligand, as observed in its solid-state structure (see SI), and reversibly dissociates two water molecules to reveal a bare palladium hydroxide that reacts with hydrazine. Arylpalladium(II) hydroxides have been demonstrated to form strong hydrogen bonding networks with water, even in solution.[25] Together, these data imply that the inhibition of the reaction between N2H4 and hydroxide 5 by water causes the products to result from reaction by Path B in Figure 2.
Figure 5.

Dependence of the reciprocal of the observed rate constant 1/kobs (s−1) on [H2O]2 (0–0.155 M2) with [5] = 0.0393 M in THF-d8 at 35 °C.
Finally, the selectivity for monoarylation and diarylation products depended on the reaction temperature. The selectivity of the reaction of 4-chlorotoluene at 100 °C after 15.5 h with 800 ppm of catalyst was 91:9 at 96% conversion (Figure 6b). This value is higher than the 81:19 ratio at 89% conversion with 5 mol% catalyst at 60 °C for 50 min (Figure 6a). While there are many variables concerning this reaction with a heterogeneous base at varying concentrations and temperatures, the higher selectivity at higher temperature could result from the combination of the greater stability of the arylpalladium halide complex than hydroxo complex and their selectivity presented above. Aryl hydroxide 5 is unstable at high temperatures (>80 °C), causing its population to be lower at the temperature of the reaction with low catalyst loadings and reducing its contribution to the product distribution.[26]
Figure 6.

Time courses of the Pd-catalyzed C–N coupling reaction of p-chlorotoluene (1d) and hydrazine with (a) 5 mol% of Pd catalyst at 60 °C for 50 min; (b) 800 ppm of Pd catalyst at 100 °C for 15.5 h.
Determination of the Rate-Limiting Step of Catalysis
Having established that the catalytic process occurs primarily by reaction of hydrazine with arylpalladium chloride complexes (such as 4), we sought to determine the rate-limiting step. The initial rates imply that the rate-limiting step could be deprotonation of hydrazine to give an arylpalladium(II) hydrazido species or reductive elimination of aryl hydrazine after reversible generation of the arylpalladium hydrazido species. If deprotonation of hydrazine is rate limiting, then a primary KIE would be observed for reactions with hydrazine and hydrazine-d4. If reductive elimination of aryl hydrazine is rate-limiting, then a secondary or equilibrium KIE would be observed. Initial rates of the reaction of hydrazine-d4 monodeuterate and potassium deuteroxide as base with 1b as chloroarene versus those with protiated hydrazine monohydrate, and potassium hydroxide revealed a primary KIE of 3.7(7). This value is consistent with rate-limiting deprotonation of hydrazine (Scheme 7).
Scheme 7.

KIE of the reaction of 1b with hydrazine and KOH from reaction in separate vessels.
Synthesis and Reactivity of an Arylpalladium(II) Hydrazido Complex
Our mechanistic proposal hinges on an arylpalladium(II) hydrazido intermediate that undergoes reductive elimination to generate aryl hydrazine. Because such complexes have not been synthesized, the relative rates for reductive elimination from the parent hydrazido complex versus other steps on the catalytic cycle are not known. Aryl palladium(II) complex 8 containing the unsubstituted hydrazido ligand was synthesized from 4-methoxyphenylpalladium(II) bromide 7, hydrazine, and NaOtBu in thawing THF (Scheme 8). The 31P NMR spectrum at −30 °C consisted of two doublets at 59.1 ppm and 41.7 ppm with J = 42 Hz. A broad resonance at 2.48 ppm in the 1H NMR spectrum, integrating to three protons corresponded to the hydrazido ligand. This assignment was supported by a 1H-15N HSQC experiment.
Scheme 8.

Synthesis of arylpalladium(II) hydrazido complex 8 and subsequent reductive elimination to obtain aryl hydrazine.
The dynamics of the hydrazide ligand were probed by forming the 15N-labelled hydrazido complex. NMR spectroscopic data at −60 °C were similar to those at −30 °C. The broad singlet at 2.48 ppm in the 1H NMR spectrum for the unlabelled compound was observed as two broad singlets (J = 64 Hz) for the 15N labelled compound, due to 1J(1H,15N) coupling.[27] The 31P NMR spectrum of the labelled compound contained a 20 Hz coupling between the hydrazido ligand and the di-tert-butylphosphino moiety of CyPF-tBu located trans to it.[28,29] At the same temperature, the 15N INEPT NMR spectrum of 8 consisted of a single doublet at 77.2 ppm with J = 8.5 Hz, which is similar to reported 1J(15N, 15N) coupling constants.[30] The trans2J(15N,31P) coupling of 20 Hz was not detected in the 15N NMR spectrum, we propose, due to fast exchange of the two nitrogen sites in the hydrazido ligand (vide infra).[31]
Warming of the hydrazido complex 8 to room temperature generated palladium black and p-methoxyphenyl hydrazine within 2 min. This rapid reductive elimination at room temperature precluded isolation of the hydrazido complex. Generation of this complex in the presence of PPh3 and warming to room temperature gave (CyPF-tBu)Pd(PPh3) in 91% yield, along with p-methoxyphenyl hydrazine in 65% yield (Scheme 8).
Proposed Catalytic Cycle
Our data are consistent with a catalytic cycle involving a rate-limiting reaction between hydrazine bound to an arylpalladium(II) halide complex and base to generate an arylpalladium(II) hydrazido complex, which undergoes reductive elimination to give aryl hydrazine (Scheme 9). Independent synthesis and reactions of the aryl palladium(II) hydroxide, chloride, and hydrazido complexes demonstrate that the chloride and hydroxide complexes form aryl hydrazine by reaction with base and hydrazine or hydrazine alone, and the hydrazido complex generates aryl hydrazine by reductive elimination. The observation of the chloride and hydride complexes together as resting state, first-order dependence on the concentration of palladium catalyst and hydrazine and zero-order in concentration of aryl halide, together with the primary KIE for reaction with deuterated hydrazine, are fully consistent with this mechanism. The selectivities of the stoichiometric reactions of the hydroxo complex with hydrazine and inhibition of its reaction with hydrazine by water rule out its intermediacy in the catalytic reaction, despite its apparent kinetic competence.
Scheme 9.
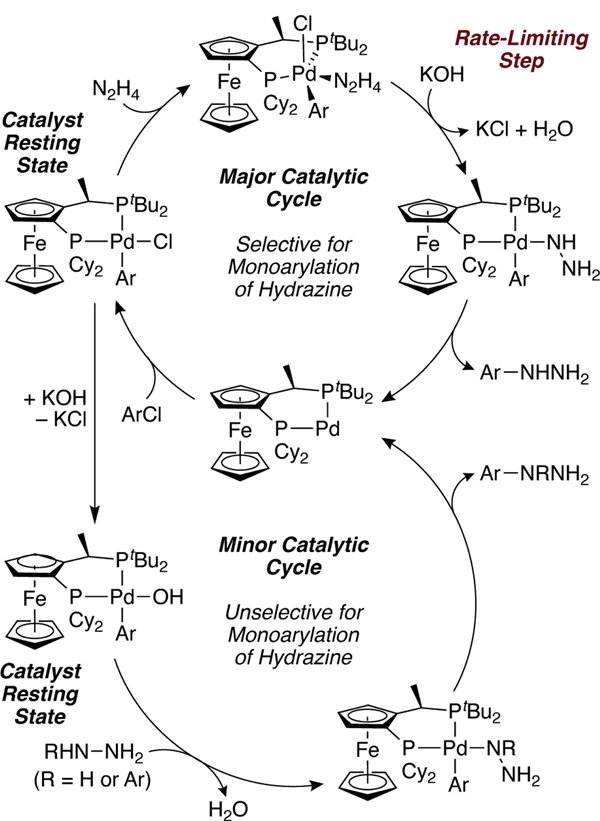
Proposed catalytic cycle for the synthesis of aryl hydrazine.
Conclusion
The coupling between hydrazine and (hetero)aryl chlorides and bromides catalyzed by palladium and the Josiphos ligand CyPF-tBu generates aryl hydrazines with broad scope at low loadings of catalyst (100 ppm to 1 mol%) and with an inexpensive hydroxide base. Under these conditions, several aryl hydrazines that are intermediates to agrochemicals were synthesized. The unusual reagent, high turnover numbers, high selectivity for monoarylation, and simple base warranted detailed mechanistic studies. These mechanistic studies showed a complex set of factors that control the rates and selectivity and are consistent with a pathway by which hydrazine, base, and an arylpalladium(II) chloride complex react in the rate-limiting step to generate an arylpalladium(II) hydrazido complex. An arylpalladium(II) hydrazido complex, the first example of a group 10 transition-metal complex bearing a parent hydrazido ligand, was prepared, and studies on its reactivity demonstrated that it undergoes reductive elimination to generate aryl hydrazine below room temperature. Future developments in this area will involve the design of improved systems that catalyze C–N coupling reactions with additional classes of reagents that contain N-heteroatom bonds.
Supplementary Material
Acknowledgements
The authors thank BASF and NIH (R35GM130387) for support of this work along with the Small Molecule X-ray Crystallography Facility (S10-RR027172), the Central California 900 MHz NMR Facility (NIH-GM68933), the College of Chemistry Nuclear Magnetic Resonance Facility (NIH S10-OD024998), and the Catalysis Facility of Lawrence Berkeley National Laboratory, which is supported by the Director, Office of Science, of the US Department of Energy under contract no. DE-AC02-05CH11231. The authors thank Dr. Nicholas Settineri for X-ray crystallographic analyses, and Dr. Hasan Celik for assistance with NMR spectroscopy.
Footnotes
Institute and/or researcher Twitter usernames: @hartwiggroup
Supporting information for this article is given via a link at the end of the document
References
- [1].a) Katrizky AR, Pozharskii AF, Handbook of Heterocyclic Chemistry, 2nd ed (Eds.: Katrizky AR, Pozharskii AF), Pergamon, Oxford, 2000; [Google Scholar]; b) Robinson B, Chem. Rev 1963, 63, 373–401; [Google Scholar]; c) Gaikwad DD, Chapolikar AD, Devkate CG, Warad KD, Tayade AP, Pawar RP, Domb AJ, Eur. J. Med. Chem 2015, 90, 707–731; [DOI] [PubMed] [Google Scholar]; d) Karrouchi K, Radi S, Ramli Y, Taoufik J, Mabkhot YN, Al-aizari FA, Ansar M, Molecules, 2018, 23, 134–218; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Potts KT, Chem. Rev 1961, 61, 87–127. [Google Scholar]
- [2].a) Habeeb AG, Rao PNP, Knaus EE, J. Med. Chem 2001, 44, 3039–3042; [DOI] [PubMed] [Google Scholar]; b) Kotagiri VK, Suthrapu S, Reddy JM, Rao CP, Bollugoddu V, Bhattacharya A, Bandichhor R, Org. Process Res. Dev 2007, 11, 910–912; [Google Scholar]; c) Baumann M, Baxendale IR, Ley SV, Nikbin N, Beilstein J. Org. Chem 2011, 7, 442–495; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Earley F, Sauter H, Rheinheimer J, Rieck H, Coqueron P-Y, Whittingham WG, Walter H in Modern Crop Protection Compounds, Vol. 1 (Eds.: Krämer W, Schirmer U, Jeschke P, Witschel M), Wiley-VCH Verlag & Co. KGaA, Weinheim, 2011, pp. 559–691; [Google Scholar]; e) Babczinski P, Watanabe Y, Nakatani M, Yoshimura T, Hanai R, Tanetani Y, Shimizu T in Modern Crop Protection Compounds, Vol. 1 (Eds.: Krämer W, Schirmer U, Jeschke P, Witschel M), Wiley-VCH Verlag & Co. KGaA, Weinheim, 2011, pp. 305–337; [Google Scholar]; f) Theodoridis G, Leibl R, Zagar C in Modern Crop Protection Compounds, Vol. 1 (Eds.: Krämer W, Schirmer U, Jeschke P, Witschel M), Wiley-VCH Verlag & Co. KGaA, Weinheim, 2011, pp. 163–195. [Google Scholar]
- [3].Schirmann J-P, Bourdauducq P. in Ulmann’s Encyclopedia of Industrial Chemistry, John and Wiley and Sons, New York, 2001. [Google Scholar]
- [4].a) For selected examples, see the following references; Charier S, Ruel O, Baudin J-B, Alcor D, Allemand J-F, Meglio A, Jullien L, Angew. Chem. Int. Ed 2004, 43, 4785–4788; Angew. Chem. 2004, 116, 4889–4892; [DOI] [PubMed] [Google Scholar]; b) Cho H-J; Kim M-Y; Um I-H, Bull. Korean Chem. Soc 2014, 35, 2448–2452; [Google Scholar]; c) Cho H-J, Um I-H, Bull. Korean Chem. Soc 2014, 35, 2371–2374; [Google Scholar]; d) Kumar GS, Zeller M, Gonnade RG, Prasad KJR, Tetrehedron Lett. 2014, 55, 4240–4244; [Google Scholar]; e) Wilsily A, Mennen SM, Cosbie A, Milne JE, Org. Process Res. Dev 2017, 21, 1286–1293; [Google Scholar]; f) Sánchez B, Calderón C, Tapia RA, Contreras R, Campodónico PR, Front. Chem 2018, 6, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Hartwig JF, Acc. Chem. Res 2008, 41, 1534–1544; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ruiz-Castillo P, Buchwald SL, Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Lundgren RJ, Stradiotto M, Angew. Chem. Int. Ed 2010, 49, 8686–8690; [DOI] [PubMed] [Google Scholar]; b) Crawford SM, Lavery CB, Stradiotto M, Chem. Eur. J 2013, 19, 16760–16771; [DOI] [PubMed] [Google Scholar]; c) DeAngelis A, Wang D-H, Buchwald SL, Angew. Chem. Int. Ed 2013, 52, 3434–3437; Angew. Chem. 2013, 125, 3518–3521. [DOI] [PubMed] [Google Scholar]
- [7].a) Wang Y, Shi Y-F, Chen Y-B, Wu L-M, J. Solid State Chem 2012, 191, 19–26; [Google Scholar]; b) Carrasco S, Martín-Matute B, Eur. J. Inorg. Chem 2019, 1951–1955. [Google Scholar]
- [8].Schlummer B, Scholz U, Adv. Synth. Catal 2004, 346, 1599–1626. [Google Scholar]
- [9].a) Kurandina DV, Eliseenkov EV, Ilyin PV, Boyarskiy VP, Tetrahedron 2014, 70, 4043–4048; [Google Scholar]; b) Kumar SV, Ma D, Chin. J. Chem 2018, 36, 1003–1006. [Google Scholar]
- [10].Littke AF, Fu GC, Angew. Chem. Int. Ed 2002, 41, 4176–4211. [DOI] [PubMed] [Google Scholar]
- [11] For mechanistic studies in palladium-catalyzed C–N cross-coupling reactions with nitrogen nucleophiles rather than hydrazine, see: a).Hartwig JF, Acc. Chem. Res 1998, 31, 852–860; [Google Scholar]; b) Hartwig JF, Nature 2008, 455, 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Frenández-Rodríguez MA, Shen Q, Hartwig JF, Chem. Eur. J 2006, 12, 7782–7796; [DOI] [PubMed] [Google Scholar]; b) Shen Q, Ogata T, Hartwig JF, J. Am. Chem. Soc 2008, 130, 6586–6596; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Frenández-Rodríguez MA, Hartwig JF, J. Org. Chem 2009, 74, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Shen Q, Shekhar S, Stambuli JP, Hartwig JF, Angew. Chem. Int. Ed 2005, 44, 1371–1375; [DOI] [PubMed] [Google Scholar]; b) Shen Q, Hartwig JF, J. Am. Chem. Soc 2006, 128, 10028–10029; [DOI] [PubMed] [Google Scholar]; c) Green A, Hartwig JF, Org. Lett 2014, 16, 4388–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alsabeh PG, Lundgren RJ, Longobardi LE, Stradiotto M, Chem. Commun 2011, 47, 6936–6938. [DOI] [PubMed] [Google Scholar]
- [15].Stradiotto et al. reported that 4-phenylbromobenzene instead of 4-phenylchlorobenzene showed incomplete conversion and lower yield of the desired hydrazine under [Pd(cinnamyl)Cl]2 and MorDalPhos catalytic conditions. For details, see ref. 6a.
- [16]. [To test if our system could catalyze the C–N coupling reaction between 3,5-dimethylpyrazole and an aryl halide, thereby providing a false positive result, a control reaction with 3,5-dimethylpyrazole, instead of hydrazine, and chlorobenzene was performed (see Supporting Information for details), and the N-aryl pyrazole was not detected by 1H NMR spectroscopy or GC-MS analysis. Thus, the pyrazoles formed by the reactions in Table 2 result from the condensation of the aryl hydrazine with acetylacetone and not by a C–N coupling reaction between an aryl halide and 3,5-dimethylpyrazole.]
- [17].The reactions with 2-chloropyridine and with 2-chloroquinoline occur by uncatalyzed SNAr reactions with hydrazine hydrate to afford the corresponding aryl hydrazines.
- [18].Hamprecht G, Witschel M, Hawkes TR, Edmunds AJF, Morris JA, van A Almsick in Modern Crop Protection Compounds, Vol. 1 (Eds.: Krämer W, Schirmer U, Jeschke P, Witschel M), Wiley-VCH Verlag & Co. KGaA, Weinheim, 2011, pp. 197–276. [Google Scholar]
- [19].Yang Q, Li X, Lorsbach BA, Muhuhi JM, Roth GA, Gray K, Podhorez DE, Org. Process Res. Dev 2019, 23, 2133–2141. [Google Scholar]
- [20].The disparity in these yields is likely due to the hydrolysis of the ester, nitrile, or amide functional groups in 1i, 1x, and 1y, repectively, or by the condensation of hydrazine onto the ketone in 1h under basic conditions during the course of the reaction of the hydrazine product and acetylacetone.
- [21].At room temperature, aryl hydrazine was not formed in these experiments.
- [22].Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U, Bioinformatics 2006, 22, 3067–3074. [DOI] [PubMed] [Google Scholar]
- [23].Alvaro E, Hartwig JF, J. Am. Chem. Soc 2009, 131, 7858–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. [The reaction represented by the circle trace in Figure 4 was conducted in an NMR tube. Because this heterogeneous reaction was not stirred, the rate of this reaction is expected to be even faster with stirring, as in the case of the catalytic reaction.]
- [25].Fulmer GR, Herndon AN, Kaminsky W, Kemp RA, Goldberg KI, J. Am. Chem. Soc 2011, 133, 17713–17726. [DOI] [PubMed] [Google Scholar]
- [26].More quantitatively, the decomposition of aryl hydroxide 5 at 100 °C in a 7:3 (v/v) mixture of 1,4-dioxane and THF by 31P NMR spectroscopy with p-chlorotoluene occurred with a rate constant of 1.8×10−4 s−1, which corresponds to a half-life of about 1 h versus the approximately 8 h time of the overall reaction. In contrast, chloride complex 4 underwent just 5% conversion at 100 °C after 12 h in the same mixture of 1,4-dioxane and THF. For experimental details, see the supporting information.
- [27].Hesse M, Meier H, Zeeh B, Spectroscopic Methods in Organic Chemistry, 2nd ed., Thieme Chemistry, New York, 2007, p.237. [Google Scholar]
- [28].Klinkenberg JL, Hartwig JF, J. Am. Chem. Soc 2010, 132, 11830–11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Motschi H, Pregosin PS, Venanzi LM, Helv. Chem. Acta 1979, 62, 667–677. [Google Scholar]
- [30].a) Berkhoudt T, Jakobsen HJ, J. Magn. Reson 1982, 50, 323–327; [Google Scholar]; b) Perera SA, Gregušová A, Bartlett RJ, J. Phys. Chem. A 2009, 113, 3197–3201. [DOI] [PubMed] [Google Scholar]
- [31].a) For a related exchange of the metal from one nitrogen to the other of a hydrazido complex, see Schrock RR, Liu AH, O’Regan MB, Finch WC, Payack JF, Inorg. Chem 1988, 27, 3574–3583. [Google Scholar]; b) for a more detailed discussion of the exchange involving the hydrazido ligand in complex 8, including the NMR data, see the supporting information. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.