

Abstract
Gram-negative bacteria have a cell envelope that comprises an outer membrane (OM), a peptidoglycan (PG) layer and an inner membrane (IM)1. The OM and PG are load-bearing, selectively permeable structures that are stabilized by cooperative interactions between IM and OM proteins2,3. In E. coli, Braun’s lipoprotein (Lpp) forms the only covalent tether between the OM and PG and is crucial for cell envelope stability4 but most other Gram-negative bacteria lack Lpp so it has been assumed that alternative mechanisms of OM stabilization are present5. We use a glycoproteomic analysis of PG to show that β-barrel OM proteins are covalently attached to PG in several Gram-negative species, including Coxiella burnetii, Agrobacterium tumefaciens and Legionella pneumophila. In C. burnetii, we found that four different types of covalent attachments occur between OM proteins and PG, with tethering of the β-barrel OM protein BbpA becoming most abundant in stationary phase and tethering of the lipoprotein LimB similar throughout the cell-cycle. Using a genetic approach, we demonstrate that the cell-cycle dependent tethering of BbpA is partly dependent on a developmentally regulated L,D transpeptidase (Ldt). We use our findings to propose a model of Gram-negative cell envelope stabilization that includes cell-cycle control and an expanded role for Ldts in covalently attaching surface proteins to PG.
The tripartite cell envelope of Gram-negative bacteria is composed of an OM, a PG layer, and an IM1. The OM is decorated with membrane-spanning β-barrel proteins that regulate flux of nutrients and hydrophilic antibiotics into the bacterial cell6,7. PG lies beneath the OM and is an elastic, mesh-like polymer that protects bacteria from osmotic lysis. It is composed of repeating disaccharide-pentapeptide subunits called muropeptides that consist of β−1,4-linked N-acetylglucosamine and N-acetylmuramic acid sugars substituted by peptide stems containing L-and D-amino acids (Fig. 1a). In Enterobacteriaceae, Vibrionaceae, and Pseudomonaceae, the cell envelope is stabilized by multiple IM and OM components including the highly abundant β-barrel OM protein OmpA, Peptidoglycan-associated lipoprotein (Pal), and Lpp. Both OmpA and Pal are OM proteins that non-covalently interact with PG via conserved PG binding motifs at their C-termini8–10. Lpp is an abundant 5.8 kDa helical lipoprotein that forms the only physical tether between the OM and PG. The triacylated N-terminus is embedded in the OM and the C-terminal lysine is covalently attached to meso-diaminopimelic acid (mDAP) of the PG stem peptide by Ldts4,5,11–14. Lpp functions as a molecular caliper to maintain the periplasmic distance between the OM and IM. Disruption of this spacing perturbs assembly of periplasmic-spanning structures as well as transduction of stress signals from the extracellular environment into the bacterial cell15,16. Lpp therefore plays a critical role in cell envelope structure and function in E. coli and closely related bacteria. Interestingly, most Gram-negative bacteria lack lpp orthologs, and the mechanism underpinning cell envelope stabilization in these organisms is an open question. Decades ago, PG-associated proteins were reported in the environmental pathogens C. burnetii and L. pneumophila, hinting at molecular determinants of OM stabilization in Lpp-deficient bacteria17–21. These organisms, along with Lpp- Agrobacterium tumefaciens, replicate in eukaryotic host cells to cause disease but also express unique physiologies that allow them to persist in environmental reservoirs22–24. These features make them attractive systems to investigate alternative mechanisms of cell envelope stabilization. For example, C. burnetii alternates between a replicating, exponential phase large cell variant (LCV) and a non-replicating, stationary phase small cell variant (SCV) during intracellular growth. The SCV has an unusually thick cell envelope, exhibits extreme resistance to physical and chemical disruption, and is hypothesized to be responsible for the long-term environmental stability of the pathogen24. Here, we use an automated glycoproteomic analysis of PG to demonstrate that OM β-barrel proteins are covalently attached to PG of C. burnetii and other ecologically diverse Lpp-deficient proteobacteria. In C. burneti, this attachment stabilizes the OM and is developmentally programmed.
Fig. 1. β-barrel OM proteins are covalently attached to PG and form a tight tether in C. burnetii.
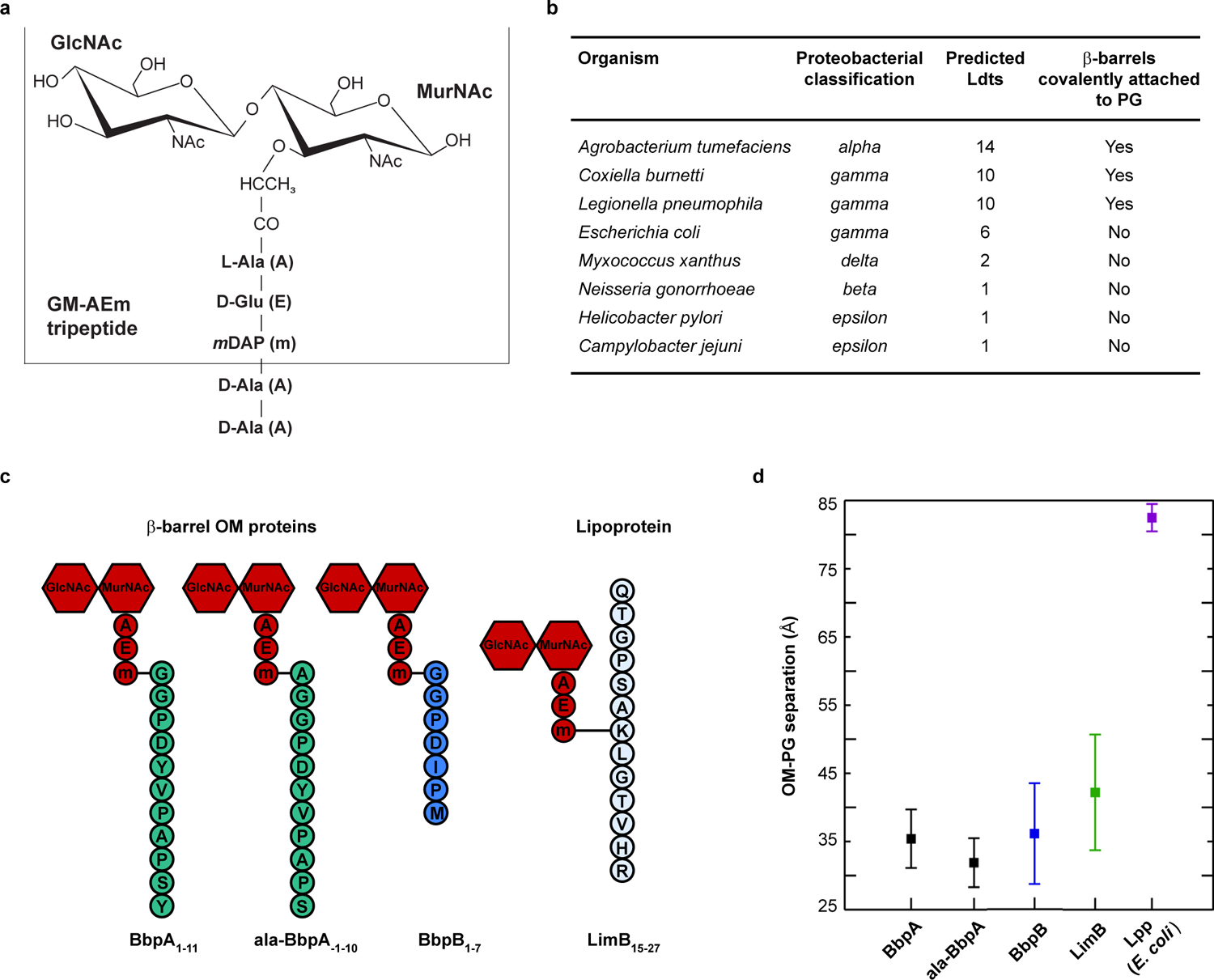
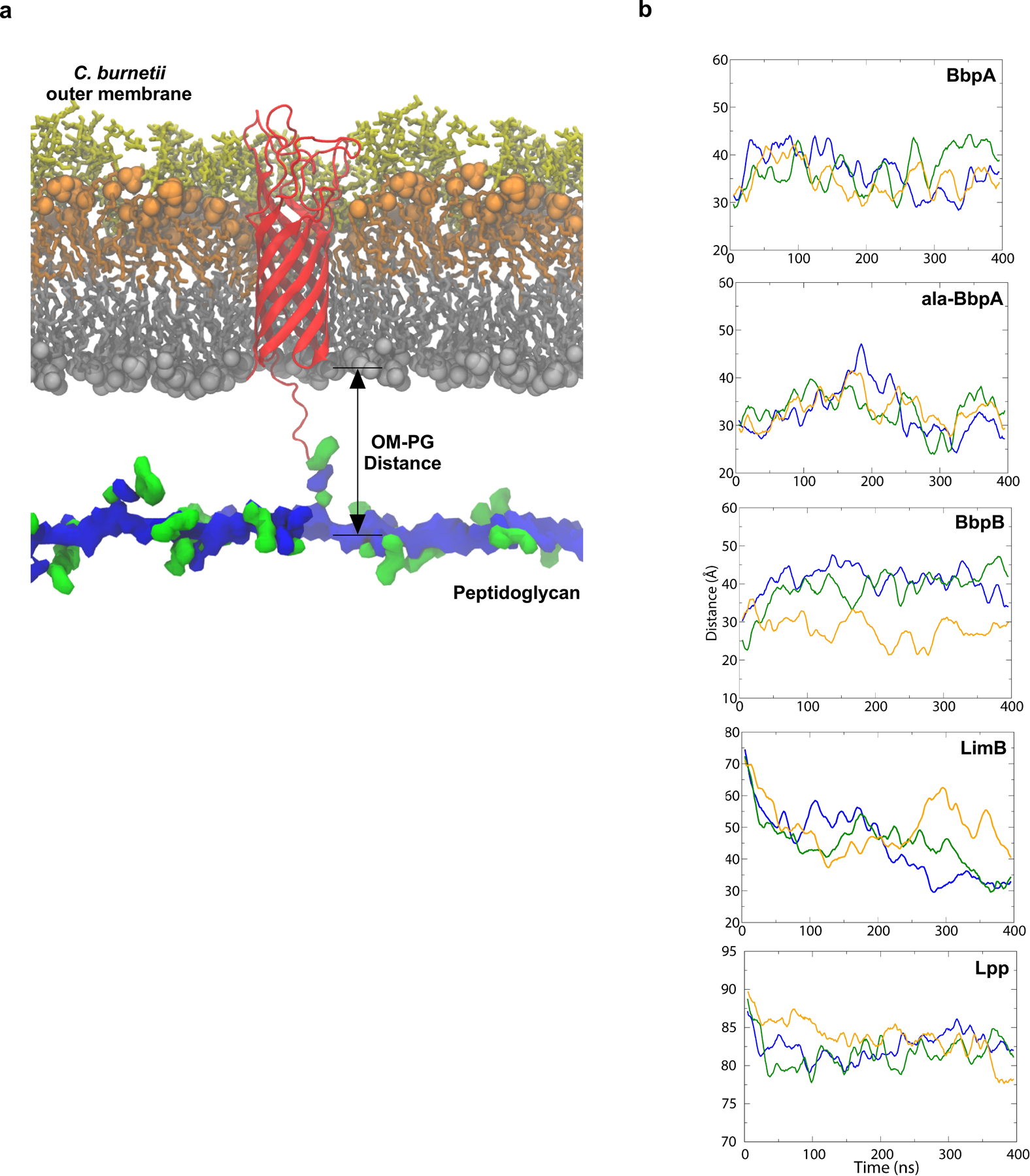
a. Schematic of the canonical Gram-negative muropeptide structure consisting of β−1–4 linked N-acetylglucosamine-N-acetylmuramic acid sugars [GlcNAc-MurNAc (GM)] attached to pentapeptide stems composed of L-alanine (A), γ-D-glutamic acid (E), meso-diaminopimelic acid (m), D-alanine (A), and D-alanine (A). The GM-AEm tripeptide moiety is outlined b. β-barrel OM proteins covalently attached to PG were identified in several proteobacteria. The number of predicted Ldts in each organism is shown. c. Schematic of the four covalently attached peptides identified in PG harvested from C. burnetii. Peptides are attached to the mDAP residue of the GM-AEm tripeptide moiety (red). d. Molecular dynamics simulation of C. burnetti OM protein-tethered models. The distance in angstroms between the centers-of-mass of the PG and the phosphorus atoms of the inner leaflet of the OM was measured for three independent 400 ns simulations of BbpA, ala-BbpA, BbpB, LimB, and E. coli Lpp. Distances are shown as averages with standard deviations.
First, to identify elements that mediate OM-PG tethering, we harvested PG from 8 proteobacterial species grown to stationary phase and analyzed PG using an unbiased tandem mass spectrometry approach25. A general search of the PG MS/MS data using species-specific proteomic databases identified the signature lysyl-arginyl dipeptide from Lpp covalently attached to PG in E. coli26. No peptides of appreciable length were found covalently attached to PG in Campylobacter jejuni, Neisseria gonorrhoeae, Helicobacter pylori, or Myxococcus xanthus, whereas peptides from 8-stranded or 16-stranded β-barrel OM proteins were covalently attached to PG in C. burnetii, L. pneumophila, and A. tumefaciens. Six β-barrel OM proteins were attached to PG in A. tumefaciens. Up to three possible β-barrel OM proteins were attached to PG in L. pneumophila. Two β-barrel OM proteins and a unique lipoprotein (LimB27) were attached to PG in C. burnetii [(hereafter, Bbp for β-barrel protein; BbpA (CBU0307), BbpB (CBU0311)] (Fig. 1b, c, Extended Data Fig. S1, 2, 3, 4). With a few exceptions, these β-barrel proteins exhibit little sequence similarity; however, invariant N-terminal residues are observed in a species-specific manner at the predicted signal peptide cleavage site. L. pneumophila and C. burnetii β-barrel proteins have conserved N-terminal glycyl residues whereas A. tumefaciens β-barrel proteins have conserved alanyl residues. These residues are also predicted to localize to the periplasm and were subsequently identified as the tethering residues to PG (Extended Data Fig. S1, 5). Using a complementary and modified search approach, we also identified alternative sites of PG attachment on −1 alanyl residues preceding the signal peptide cleavage site of C. burnetii BbpA (hereafter ala-BbpA) and A. tumefaciens ATU1020 and/or ATU1021, thereby representing two distinct linkages to the same protein(s) (Fig. 1c, Extended Data Fig. S1, 2, 3). Our proteomics search also identified that the C. burnetii-specific lipoprotein LimB is covalently attached to the mDAP residue of PG via an internal lysine residue (Lys21). This PG attachment is presumed to occur via the ε-amino group of lysine, similar to Lpp (Fig. 1c, Extended Data Fig. S1, 2)27,28. Non-covalent interactions between OM proteins and PG may stabilize the cell envelope in bacteria where proteins were not found covalently attached to PG8–10. Alternatively, covalent interactions between lipoproteins or other OM proteins that have tryptic cleavage sites close to the PG attachment site may preclude their identification. Collectively, these studies identified multiple proteins covalently bound to PG in many Gram-negative proteobacteria that do not encode an lpp ortholog.
Computational modeling and molecular dynamics simulation (MD) can be used to model molecular interactions and structural dynamics within the bacterial cell envelope29. In C. burnetii, the OM and PG layers are difficult to resolve by cryo-EM due to the pleomorphic nature of the bacterium and the unusually thick cell envelope of the SCV30. Therefore, we generated atomistic models of OM-PG tethering mediated by BbpA, BbpB, or LimB and used 400-ns MD simulations to estimate the distances between the OM and PG2,29. Interestingly, the OM-PG distance for all C. burnetii β-barrel and LimB tethers was consistently smaller than the OM-PG distance for E. coli Lpp (Fig. 1d, Extended Data Fig. S6, 7). These data suggest that β-barrel tethering confers unique transenvelope architecture and that critical periplasmic-spanning protein complexes, such as type IV secretion systems and OM signaling complexes of C. burnetii, have co-evolved with the architecture of the β-barrel tethered envelope15,16.
C. burnetii, A. tumefaciens, and L. pneumophila encode numerous Ldts (≥10) (Extended Data Fig. S8)31,32. In E. coli, six Ldts carry out distinct yet partially redundant roles during cell wall remodeling: two enzymes catalyze the formation of non-canonical L,D cross-linking of PG and four fulfill an auxiliary role in covalently attaching Lpp to PG13,14,33. Several genes belonging to the Ldt family are highly up-regulated in C. burnetii upon transition to stationary phase31. PG-bound BbpA is considerably more abundant during stationary phase while PG-bound LimB is present at similar levels during both exponential and stationary phase (Fig. 2a, b). We therefore sought to determine if the C. burnetii stationary phase-specific Ldts serve a similar functional role in covalently attaching β-barrel OM proteins to PG. To this end, three stationary phase ldts were deleted (Extended Data Fig. S8) and strains characterized. A higher molecular weight PG-bound isoform of BbpA was identified in all strains except the Δldt2 mutant (Fig. 3a). Consistent with these results, extracted ion chromatograms (XIC) representative of PG-bound BbpA and PG-bound BbpB, but not ala-BbpA, were also identified in PG from each strain except the Δldt2 mutant (Fig. 3b, Extended Data Fig. S9). Pronounced OM vesiculation was observed in the Δldt2 mutant, a phenotype analogous to the E. coli Δlpp mutant (Fig. 3c, d)34. Additionally, the mutant displayed a delayed lag phase, suggesting diminished OM tethering negatively impacts metabolic fitness and the onset of replication (Extended Data Fig. S10)35. Increased vesiculation was also observed in the Δldt3 mutant without a significant decrease in OM-tethering by BbpA and BbpB (Extended Data Fig. S9). However, this does not exclude the possibility that Ldt3 plays a role in OM stabilization independent of these proteins. Collectively, these data indicate that ldt2 is the gene involved in covalent attachment of BbpA and BbpB to PG, which in turn stabilizes the OM during stationary phase. A model depicting these results is shown in Fig. 4. The conserved N-terminal glycyl-glycyl-prolyl-aspartyl motif of BbpA and BbpB is likely important for acceptor substrate recognition by Ldt2. However, the presence of alanyl-linked BbpA (ala-BbpA) in the Δldt2 mutant suggests an enzyme with acceptor substrate specificity different from Ldt2 cross-links BbpA to PG via the −1 alanyl residue preceding the signal peptide cleavage site. C. burnetii transitions from a typical exponential phase Gram-negative cell into a hardy spore-like form during stationary phase30,31. Thus, increased β-barrel tethering by Ldts during stationary phase is likely critical for enhanced stability of C. burnetii and potentially other bacteria that employ this mechanism of OM stabilization.
Fig. 2. β-barrel tethering in C. burnetii is abundant in stationary phase.
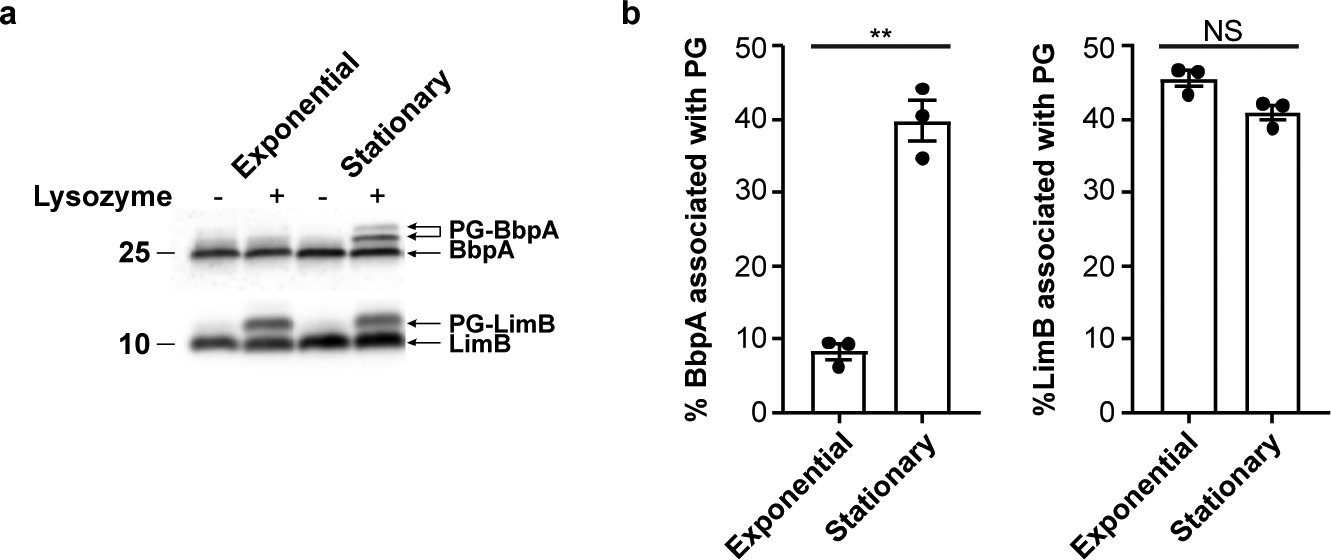
a. Immunoblots of whole cell lysate from exponential phase (large cell variant) and stationary phase (small cell variant) C. burnetii, lysozyme-digested or undigested, and probed with BbpA (top) or LimB (bottom) specific antibodies. Molecular weight markers are shown to left of the blot. b. Quantification of immunoblots from panel a. The plot depicts means ± standard error of the mean of lysozyme mobilized BbpA or LimB signal expressed as a percentage of total BbpA or LimB signal of n=3 independent experiments. Statistical significance was calculated using an unpaired two-sided Student’s t test, **P = 0.000016; NS, not significant.
Fig. 3. The L,D transpeptidase ldt2 is required for covalent attachment of BbpA and BbpB to PG.
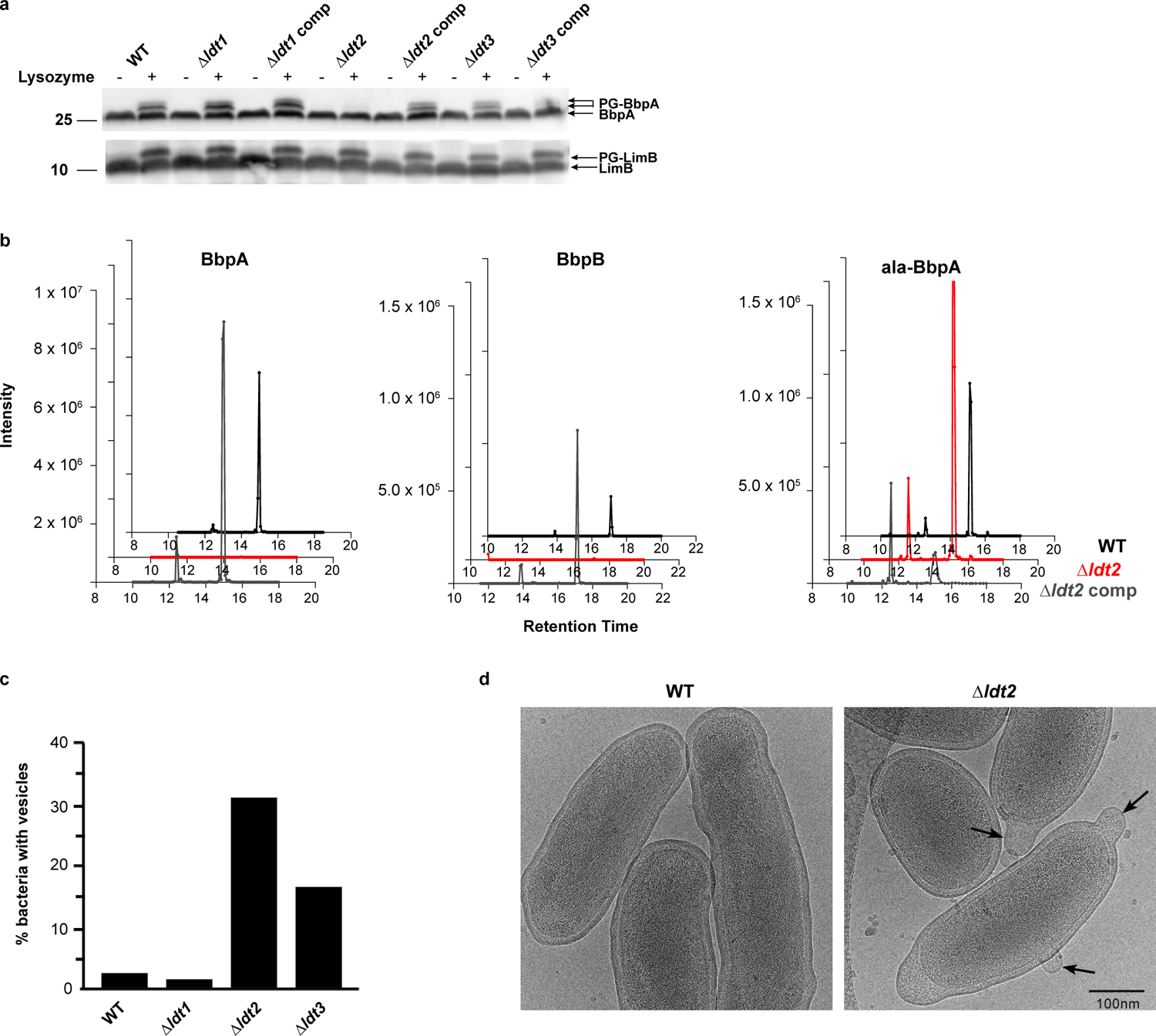
a. Immunoblots of stationary phase whole cell lysate from wild-type (WT), Δldt, and complement strains, lysozyme-digested or undigested, and probed with BbpA (top) or LimB (bottom) specific antibodies. Data are representative of n=3 independent experiments. Molecular weight markers are shown to left of the blot. b. Extracted Ion Chromatograms (XIC) of precursor masses (m/z) corresponding to PG-BbpA (AEmGGPDYVPAPS, m/z = 906.909, z = 2), PG-ala-BbpA (AEmAGGPDYVPAPS, m/z = 942.427, z = 2), PG-BbpB (AEmGGPDIPM, m/z = 769.843, z = 2) identified in PG from WT, Δldt2, and Δldt2 complement strains. c. WT and Δldt deletion mutants were grown in ACCM-D for 14 days and imaged by cryo-EM. OM vesiculation was quantified by imaging ≥50 bacterial cells per strain and expressed as the percentage of bacteria with vesicles attached to the OM. d. Ultrastructure of WT and the Δldt2 deletion mutant imaged by cryo-EM. Arrows denote OM vesiculation.
Fig. 4.
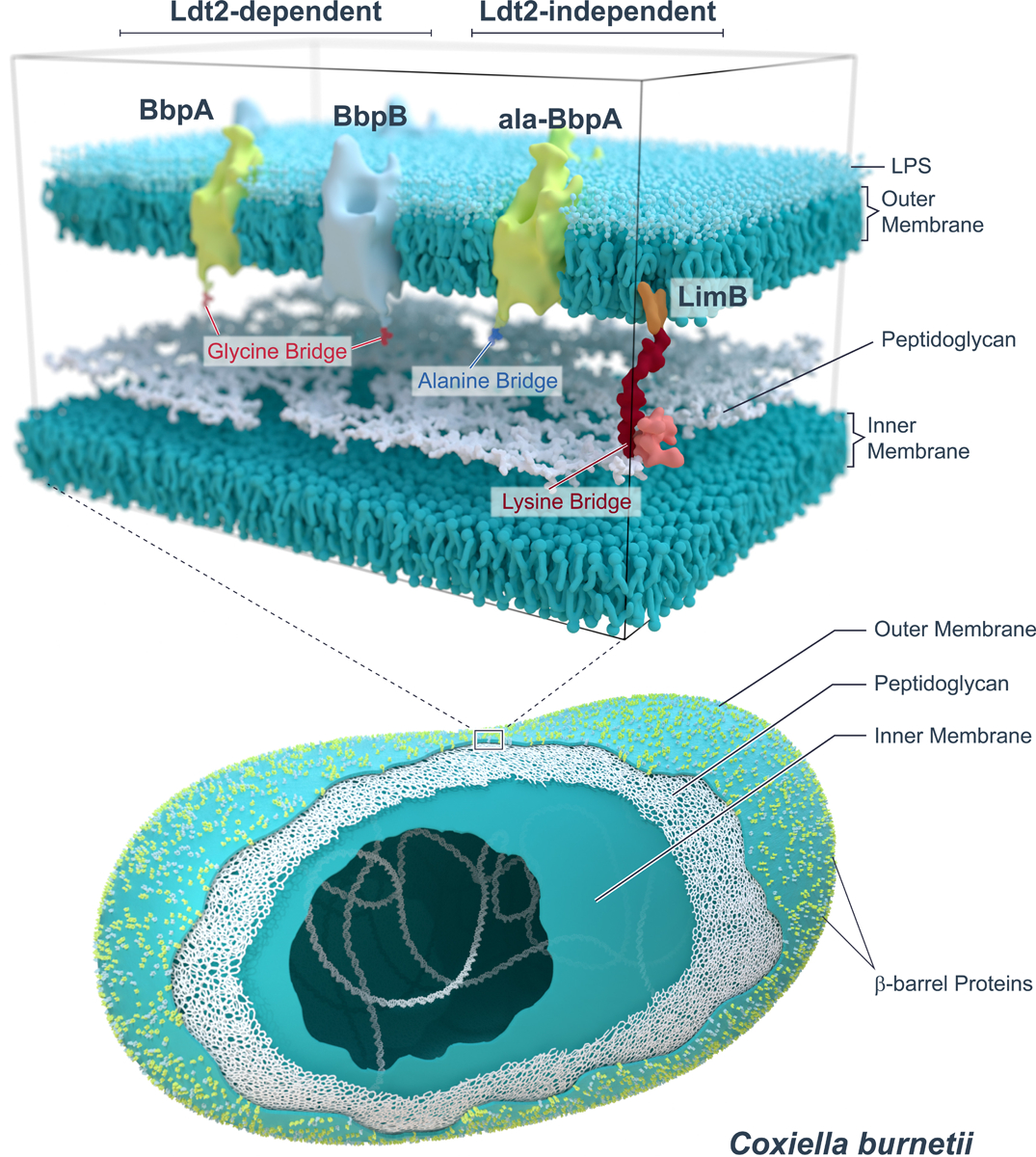
Schematic showing Ldt2-dependent and -independent covalent attachments of OM proteins to C. burnetii PG.
The cell-cycle independent PG-bound isoform of LimB was identified in all ldt knockout strains, indicating PG-bound LimB is formed independent of ld1, ldt2, ldt3 (Fig. 3a). Approximately 40% of total LimB is covalently attached to PG throughout the cell cycle (Fig. 2a, b). LimB is a Coxiella-specific lipoprotein with a cysteine and histidine rich C-terminus that binds metal cations27. It is hypothesized to serve as a surface receptor for metal acquisition in Coxiella. Our data demonstrate LimB also serves a structural role in the C. burnetii cell envelope by tethering the lipoprotein to the PG layer. How covalent attachment of LimB to PG impacts its metal binding activity is unknown. The site of PG attachment (Lys21) is distinct from the metal binding C-terminal domain (His26-His45), so both PG and metal binding could occur concurrently. Alternatively, metal-binding could be specific to the fraction that remains unbound to PG.
Our findings reveal that β-barrel OM proteins function to tether the OM in selected proteobacteria. β-barrel OM proteins are among the most abundant, variable, and immunogenic proteins found on the bacterial surface7. They modulate bacterial physiology, host-pathogen interactions, and transport small molecules across the OM, including hydrophilic antibiotics such as β-lactams and fluoroquinolones36. Moreover, bacteria that alternate between host and terrestrial environments and survive extreme fluctuations in pH, temperature, and osmolarity, are hypothesized to have multiple OM tethers5. Here, we report that multiple β-barrel OM proteins are covalently attached to PG during stationary phase in various environmental pathogens. The identification of Ldt-mediated attachment of β-barrel proteins to PG in C. burnetii serves to broaden our understanding of the diversity of substrates recognized by these important antibiotic targets. Carbapenems inhibit Ldts, so determining the catalytic and cell-cycle specificities of Ldts in Gram-negative pathogens will have important implications for the treatment of bacterial infections33,37–39. The increased abundance of β-barrel tethering in C. burnetii during stationary phase points to a model in which a structural determinant has a role in the formation of growth-arrested proteobacterial cells.
The physiological importance of β-barrel tethering in these environmental bacteria remains unknown. But it is tempting to speculate that this mechanism contributes to cell-envelope stabilization during growth-arrest and long-term survival of these organisms in the environment. In fact, most of earth’s microbial biomass exists in a growth-arrested state, yet very little is known about cell envelope structure, maintenance, and modification during this physiological condition40. We hope that these findings will usher in further research into Gram-negative cell envelope structure and physiology in a more ecologically diverse assemblage of bacteria.
Methods
Bacterial strains and growth conditions.
C. burnetii Nine Mile phase II (clone 4, RSA439) was used in this study. C. burnetii Δldt1, Δldt2, Δldt3, and complement strains, were created using previously reported methods41. Exponential phase large cell variants (LCVs) and stationary phase small cell variants (SCVs) of C. burnetii were generated by culture in ACCM-D for 6 and 14 days, respectively42. C. burnetii peptidoglycan was purified from 3.5 liters of log phase culture (LCV, ~5 ×107 genome equivalents/mL) or 500 mL of stationary phase culture (SCV, ~1 ×109 genome equivalents/mL) and digested with mutanolysin to solubilize muropeptides as previously described31. Muropeptides were resuspended in a final volume of 200 μL.
Culture conditions used to propagate the remaining bacterial strains used in this study are described below. L. pneumophila strain JR32 was inoculated into AYE broth from a 2-day heavy patch and incubated at 37°C with shaking until stationary phase was reached. A. tumefaciens C58 was grown to stationary phase in AB minimal media at pH 5.543. C. jejuni NCTC 11168 was grown in Muller-Hinton broth, supplemented with 20 mM L-serine and 10 μg/ml vancomycin, under standard microaerobic conditions at 42°C in 10% v/v O2, 5% v/v CO2, 85% v/v N2 for approximately 18 hours to reach early stationary phase. M. xanthus DK1622 was grown in CTT broth (10 g casitone, 10 mM Tris pH 8.0, 8 mM MgSO4, 1 mM K2HPO4 pH 8.0 per liter). Cultures were incubated at 32°C with shaking (200 rpm) and harvested at logarithmic phase. H. pylori 26695 was grown in BHI broth (Oxoid) with 10% fetal bovine serum (Eurobio) and addition of 1x antibiotic cocktail [500x antibiotic cocktail stock contains, polymyxin B - 155 mg/L, vancomycin - 6.25 g/L, trimethoprim - 3.125 g/L, amphotericin B - 1.25 g/L, DMSO - 2.5% (v/v)]. Cultures were incubated at 37°C in 6% O2, 10% CO2, 84% N2 until they reached late exponential phase (OD = 0.75) or coccoid phase (72 hours). N. gonorrhoeae MS11 was first streaked on chocolate agar (E & O Laboratories Limited, Scotland) and incubated overnight at 37°C in 5% CO2. Isolated colonies were used to inoculate 30 mL BHI broth supplemented with 0.5% (w/v) yeast extract (Oxoid, UK) and 10 mM NaHCO3 and the cultures incubated overnight at 37°C with shaking (180 rpm). The culture was used to inoculate 500 mL of BHI broth which was cultured overnight until stationary phase was reached. Peptidoglycan extraction and purification were done as previously described31.
LC-MS/MS data acquisition.
For the quantification of PG-bound proteins from C. burnetii ldt knockout and complement strains, samples were analysed online using an Ultimate 3000 RSLC nano LC LC system (Dionex) coupled to an LTQ Orbitrap Elite Hybrid mass spectrometer (Thermo) equipped with an EasySpray ion source. Desalted PG samples (200 ng) were separated using an EasySpray C18 capillary column (150 μm × 150 mm, 2 μm particle size - PN ES806). PG-bound peptides were eluted using water with 0.1% (v/v) formic acid (buffer A) and 80% acetonitrile with 0.1% (v/v) formic acid (buffer B) at a flow rate of 1.5 μL/min with the following gradient: 3% buffer B for 5 minutes, 50% buffer B for 15 minutes, 95% buffer B for 5 minutes, then 3% buffer B for 5 minutes. The mass spectrometer operated in a standard data-independent acquisition mode controlled by Xcalibur 3.0 and LTQ tuneplus 2.7. The instrument was operated with a cycle of one MS (in Orbitrap) acquired at a resolution of 60,000 from an m/z range of 375 to 1600. The top 10 most abundant multiply charged ions (2+ and higher) were subjected to higher energy collisional dissociation (HCD) fragmentation (isolation window 2.50 m/z, normalised collision energy = 30, activation time 0.1 ms). An FTMS ACG target value of 1×106 and HCD target value of 50,000 were used. Dynamic exclusion was enabled with a repeat duration of 30 seconds, an exclusion list of 500, and an exclusion duration of 45 seconds. A lock mass using dodecamethylcyclohexasiloxane (m/z = 445.12003) was enabled for all experiments.
For identification of proteins covalently attached to PG, data were collected using an Orbitrap-Fusion Tribrid mass spectrometer (Thermo-Electron) equipped with an EASY-Spray Ion Source and an Easy-nLC 1000 liquid chromatography system. Nano-LC was conducted using PepMap C18 columns (the analytical column: particle size 2 μm, length 25 cm, inner diameter 75 μm, the trap column: particle size 3 μm, length 2 cm, inner diameter 75 μm), operating at a 300 nL/min flow rate with mobile phases A: water, 0.1% (v/v) formic acid and B: 80%/20% (v/v) acetonitrile /water, 0.1% formic acid. The analytical column temperature was set at 50°C. After a 5 μL sample injection, MS/MS data were acquired during a 75 min step gradient: 0 to 40% B for 64 min, 40 to 100% B for 6 min, held at 100% B for 5 min. MS was operated in standard data dependent acquisition mode. First, the Orbitrap FTMS was used to acquire a full MS scan of all ions with an m/z range from 200–1400 at a resolution of 120,000 using dodecamethylcyclohexasiloxane (m/z = 445.12003) as an internal calibrant. The top five most intense precursor ions were subjected to both electron transfer dissociation (ETD) and HCD with precursors isolated by the quadrupole mass filter and scanned by the Orbitrap at a resolution of 60,000. For HCD, the precursor was excited at the collision energy at 25%. For ETD, the charge-dependent reaction time was utilized.
MS/MS data analysis.
Protein Metrics Byos version 3.5–4.0 was used to identify and compute extracted ion chromatograms (XIC) for PG-bound proteins. Identifications were made using Byonic module version 3.5.3 and XIC’s were computed using Byologic version 3.5.4. A FASTA database containing the peptide sequences for β-barrel proteins from C. burnetii BbpA (CBU0307), ala-BbpA (ala-CBU0307), BbpB (CBU0311) was assembled with the PG tripeptide monomer (GM-AEm) concatenated to the N-terminus of each mature sequence (signal peptide removed). A fixed modification of +72.0848 was added to J (an unused letter which Byonic sets to 100.0000 Da) to represent mDAP (mass = 172.0848). A variable modification of 480.1961 (GlcNAc-MurNAc) was enabled on the peptide N-terminus. Search tolerances of 10 ppm for precursor mass and 20 ppm for fragment mass tolerance were set. The manual score cut off was set to 0 and a protein false discovery rate of 1%. An m/z window of 20 ppm, Apex search window of 2 minutes, and XIC area window of 1 minute were set for XIC extraction using Byologic. Peptides without a PG modification were discarded. All remaining identified peptides where then manually inspected to ensure XIC integration was correctly computed.
Identification of alternative sites of PG attachment.
Unbiased searches were performed using Byonic module 3.6.0 against bacterial proteomes with variable modifications of 480.196 (GlcNAc-MurNAc) + 372.1645 (AE-mDAP) or 438.185 (GlcN-MurNAc) + 372.1645 (AE-mDAP) permitted once per peptide on any residue within the peptide. Searches were performed using non-specific cleavage parameters. Precursor mass tolerance was set at 8 ppm and fragment mass tolerance was set to 20 ppm for ETD and HCD fragmentation. A score cut-off of 200 was applied and peptides containing modifications scoring above this were examined and spectra were extracted.
Immunoblots.
Bacterial pellets were harvested from exponential and stationary phase bacteria and resuspended in phosphate buffered saline (PBS) containing 0.5% Triton-X. Lysozyme was added at 100 μg/ml and cell suspensions were incubated at 37°C for 2 hours. Lysozyme-treated samples were separated by SDS-PAGE using 12% Criterion XT Bis-Tris precast gels (Bio-Rad) and transferred to an Immobilon-P polyvinylidene difluoride membrane (MilliporeSigma). Membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 (TBST) and 5% milk, then incubated with α-BbpA or α-LimB antibody. The α-BbpB antibody exhibits cross-reactivity with BbpA; therefore, immunoblot results were not included. Immunoblots were washed with TBST and incubated with SuperSignal West Femto maximum sensitivity substrate (ThermoFisher). Immunoblots were analyzed on an Azure c600 imaging system (Azure Biosystems) and immunoreactive bands were quantitated using Image Studio Lite (Li-Cor Biotechnology). The α-BbpA monoclonal antibody (K64) and α-LimB polyclonal antibody were kind gifts from James Samuel (Texas A & M University) and Micheal Minnick (University of Montana), respectively.
Cryo-EM.
Stationary phase C. burnetii samples were frozen on a glow discharged Quantifoil™ R 2/2 grid using a Leica EM GP plunge freezer (Leica Microsystems, Wetzlar, Germany) set to a relative humidity of 100%. Grids were mounted in an autogrid assembly and cryo 2D images collected using an FEI Titan Krios 300kV FEG-TEM and an FEI Falcon II CMOS direct electron detection camera. Low dose images of 13e/A2 were captured at 6 μm defocus with a nominal magnification of 29 K which corresponds to a pixel size of 2.87 Å using a 70 μm objective aperture. OM vesiculation was quantified by imaging ≥50 bacterial cells per strain and is expressed as the percentage of bacteria with vesicles clearly attached to the bacterial OM.
Structural modeling.
Sequences used for structural modeling of BbpA (Q83EL2), BbpB (Q83EK8), and LimB (B5QSB9) are shown (Extended Data Fig. 6). For structural modeling of BbpA, HHpred from the Bioinformatics Toolkit was used to search for suitable structural templates44,45. The top hit (only 20% identity) was the 1.65 Å X-ray crystal structure of OmpA from E. coli (PDB entry 1QJP)46. Because of the low sequence identity, coevolutionary information was also used to facilitate structural modeling. Sequence homologs were searched and coevolution analysis was performed using the GREMLIN web server to identify alternative templates, and to generate residue-residue contact restraints for Rosetta structural modeling47,48. Briefly, GREMLIN used hhblits to perform an iterative hidden Markov model (HMM)-based search of the UniProt20 database for homologous sequences to BbpA49,50. The resulting multiple sequence alignment (MSA) was filtered to ensure that no two sequences were more than 90% identical and all sequences covered at least 75% of the query sequence. The MSA was further modified to remove columns that contained more than 25% gaps. The final MSA generated by GREMLIN included 3,376 homologous sequences to BbpA, which corresponds to an effective number (neff) of 2,440.6 sequences after filtering at an 80% sequence identity threshold. Accurate structural models can be generated by incorporating coevolutionary information provided that the quantity Nf is greater than 64 (Nf is defined as neff divided by the square root of the sequence length)51. For BbpA, this quantity was 175.7. The top X-ray structure identified using this approach was also OmpA from E. coli (PDB entry 1QJP). Thus, the coevolutionary and HHpred search approaches are in agreement. An initial threaded model was made on the basis of residue-residue contact predictions using the program map_align51. The threaded model was used in combination with coevolution-derived restraints (100,000 map_align models) and the final model was selected on the basis of the Rosetta score and overall cylindricity of the model.
For structural modeling of BbpB, a preliminary search with GREMLIN did not identify enough sequence homologs to enable a coevolution analysis. Thus, we used HHpred to search for suitable templates of BbpB to generate sequence alignments for threading. We then used RosettaCM to generate models with several of the top-scoring X-ray structures individually as templates52. Membrane span files were generated with Octopus, and membrane weights were applied during Rosetta modeling53. At least 5,000 models were generated for each template. The final BbpB model used for molecular dynamics simulation was selected from 50,000 models on the basis of its Rosetta energy and the positions of the extracellular loops54. This model was based on the 2.55 Å resolution X-ray crystal structure of the Neisserial surface protein A (NspA) from Neisseria meningitidis (PDB entry 1P4T, 19% identical to BbpB) as the template.
Molecular dynamics simulations.
To simulate the proteins in their native environment, a model of the C. burnetii Nine Mile phase II stationary phase, small cell variant outer membrane was constructed. The outer leaflet of the model is composed of 33 lipopolysaccharides, with lipid A modeled based on the chemical structure reported in55 and core oligosaccharide added to lipid A based on the structure reported in56. The following distribution of lipids for the inner leaflet was chosen based on the phospholipid analysis reported in57: 27% phosphatidylethanolamine, 25% cardiolipin (PVCL2), 11% Lyso-phosphatidylethanolamine, 4% Lyso-phosphotidylglycerol, and 2% phosphotidylglycerol. For Lpp, an E. coli outer membrane model described previously was used2. A previously constructed model of the E. coli cell wall was used for all simulations. We note that this model includes only 4–3 cross-linked peptides, which varies slightly from the C. burnetii small cell variant that employs a mixture of 4–3 and 3–3 cross-linked peptides2,31,58. All systems were constructed using VMD59. For BbpA and BbpB, the N-terminal glycine was bonded to a free diaminopimelate residue in the cell wall. For BbpA, a third system was built in which an alanine was added to the N-terminus prior to cross-linking it to the cell wall. For LimB, Lys21 was covalently bonded to a diaminopimelate residue on the cell wall. After initial equilibration, each of these systems were simulated in triplicate for 400 ns, giving a total of 6 μs.
MD simulation parameters.
All simulations were carried out using NAMD60. A time step of 2 fs was used, with short-range non-bonded interactions evaluated every time step and long-range electrostatics evaluated every other time step using the particle-mesh Ewald method61. Short-range interactions were cut off at 12 Å with a force-based switching function starting at 10 Å. Because the cell wall was built in a strained state, as expected for living cells, the area was held constant at the equilibrated area of the OM-protein system2. A temperature of 310 K was maintained using Langevin dynamics with a damping constant of 1.0 ps−1; a pressure of 1 atm was maintained using a Langevin piston. The CHARMM36m force field for proteins and CHARMM36 force field for lipids were used62,63.
Statistical analysis.
For all experiments, three technical replicates were used with n defined as the number of independent experiments performed. All data are presented with error bars defined as the mean ± standard error of the mean. Statistical analyses were performed using an unpaired two-sided Student’s t test to calculate P values with Prism software (GraphPad Software, Inc., La Jolla, CA). False-discovery rates were calculated based off of a q value of 1% using the Benjamini, Krieger, Yekutieli method.
Extended Data
Extended Data Fig. 1. PG modifications identified on proteobacterial OM proteins.
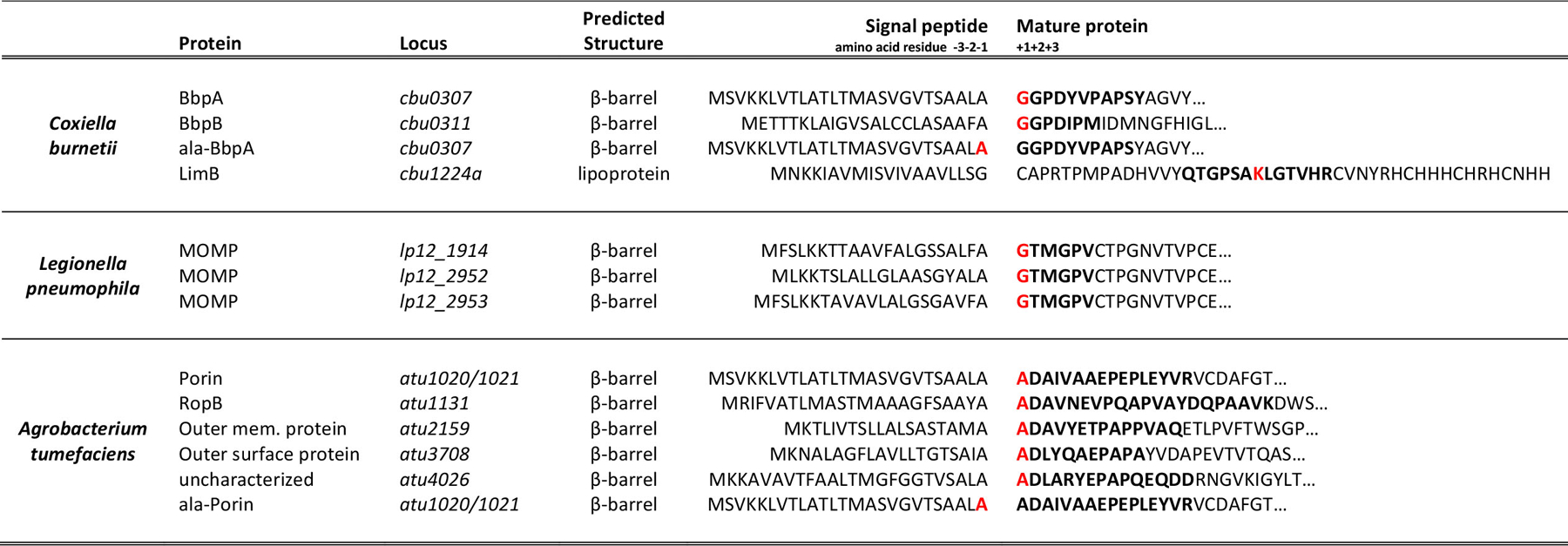
Proteins from A. tumefaciens, C. burnetii, and L. pneumophila are covalently attached to PG. Proteins identified in MS/MS data that contained a PG modification scoring higher than the first decoy and containing greater than 40% of the expected band y- ions were established as cut-off criteria. Predicted signal peptide cleavage sites of OM proteins covalently attached to PG from C. burnetii, L. pneumophila, and A. tumefaciens are shown. Amino acid numbering is based on the predicted signal peptide cleavage site. Peptide sequences that were found covalently attached to PG are bolded. Residuces with a PG tripeptide modification are also highlighted in red.
Extended Data Fig. 2. Automated spectrum assignment identifies PG modifications on C. burnetii OM proteins.
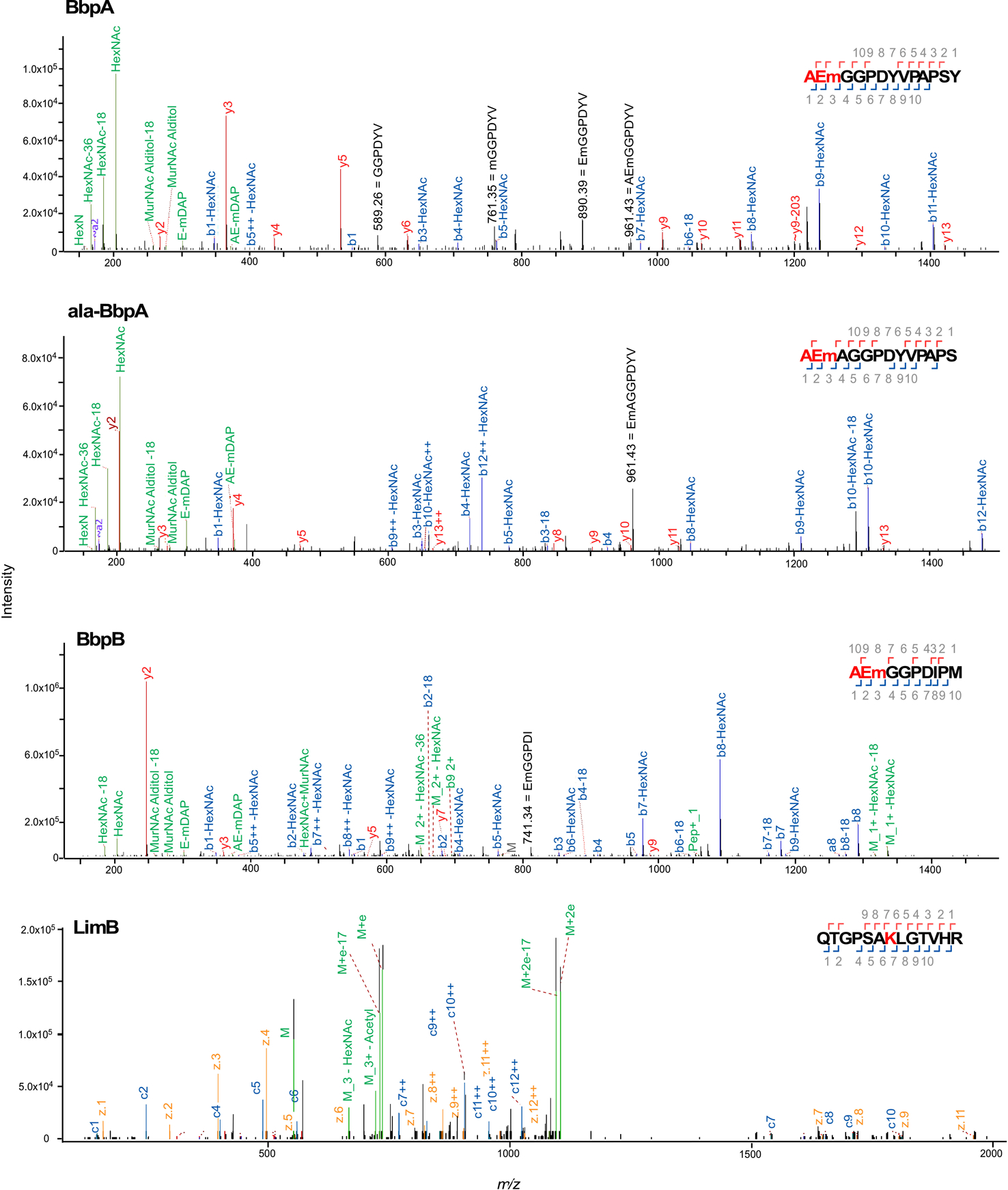
Representative MS/MS spectra for C. burnetii BbpA, ala-BbpA, BbpB β-barrel proteins and the lipoprotein LimB, covalently attached to mDAP (m) residues of PG. Spectra are shown as annotated by Byonic with manual annotations corresponding to internal fragments in black. J[+72.0848] has been replaced by m to represent mDAP. The PG tripeptide (AEm) is highlighted in red in the HCD spectra showing covalent attachment of PG to BbpA, ala-BbpA, and BbpB. The LimB Lys21 residue with PG tripeptide modification is highlighted in red in the ETD spectra showing covalent attachment of PG to LimB.
Extended Data Fig. 3. Automated spectrum assignment identifies PG modifications on β-barrel proteins from A. tumefaciens.
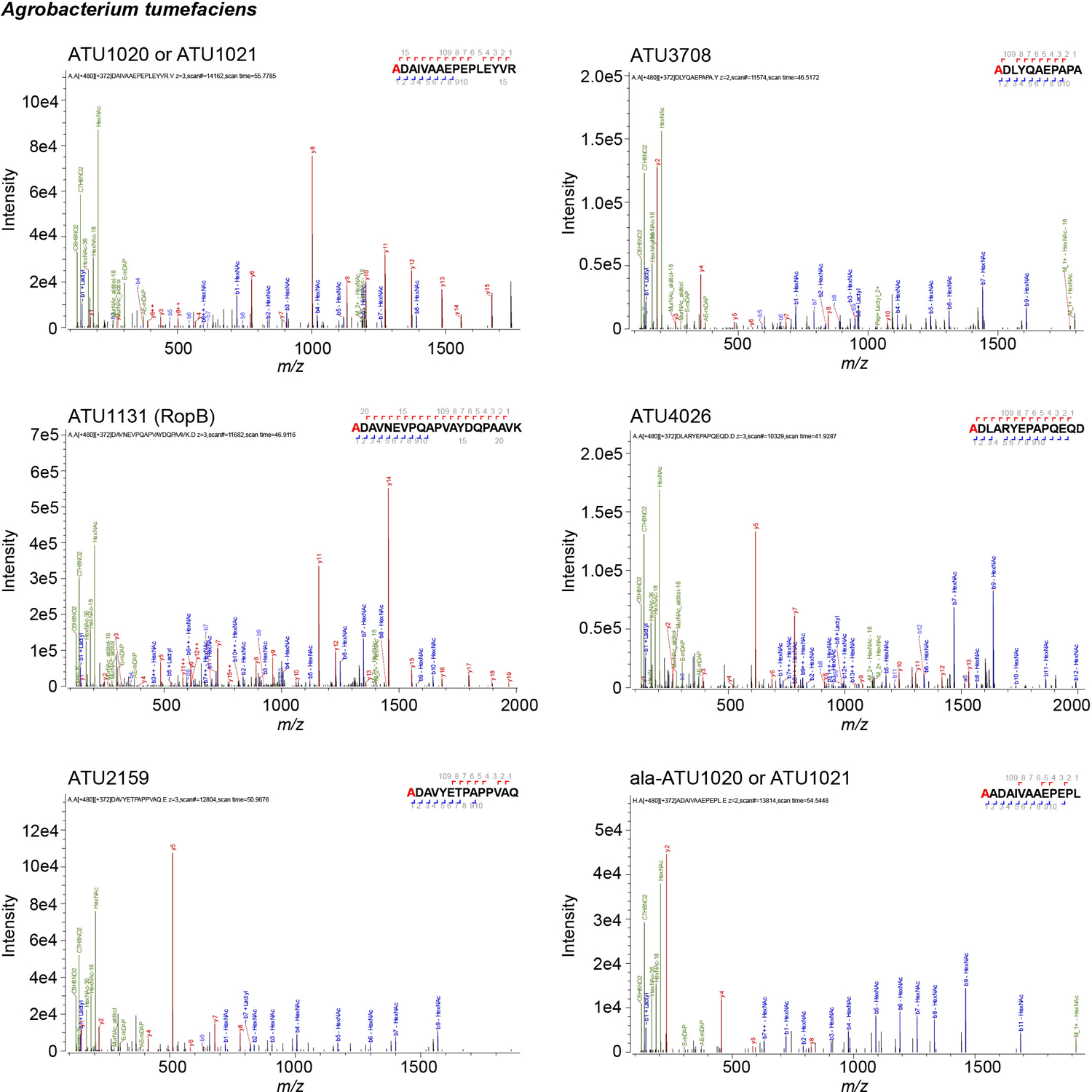
Representative MS/MS spectra for A. tumefaciens β-barrel proteins covalently attached to mDAP (m) residues of PG. Residues with PG tripeptide modification are highlighted in red. Spectra are shown as annotated by Byonic using an unbiased search approach.
Extended Data Fig. 4. Automated spectrum assignment identifies PG modifications on β-barrel proteins from L. pneumophila.
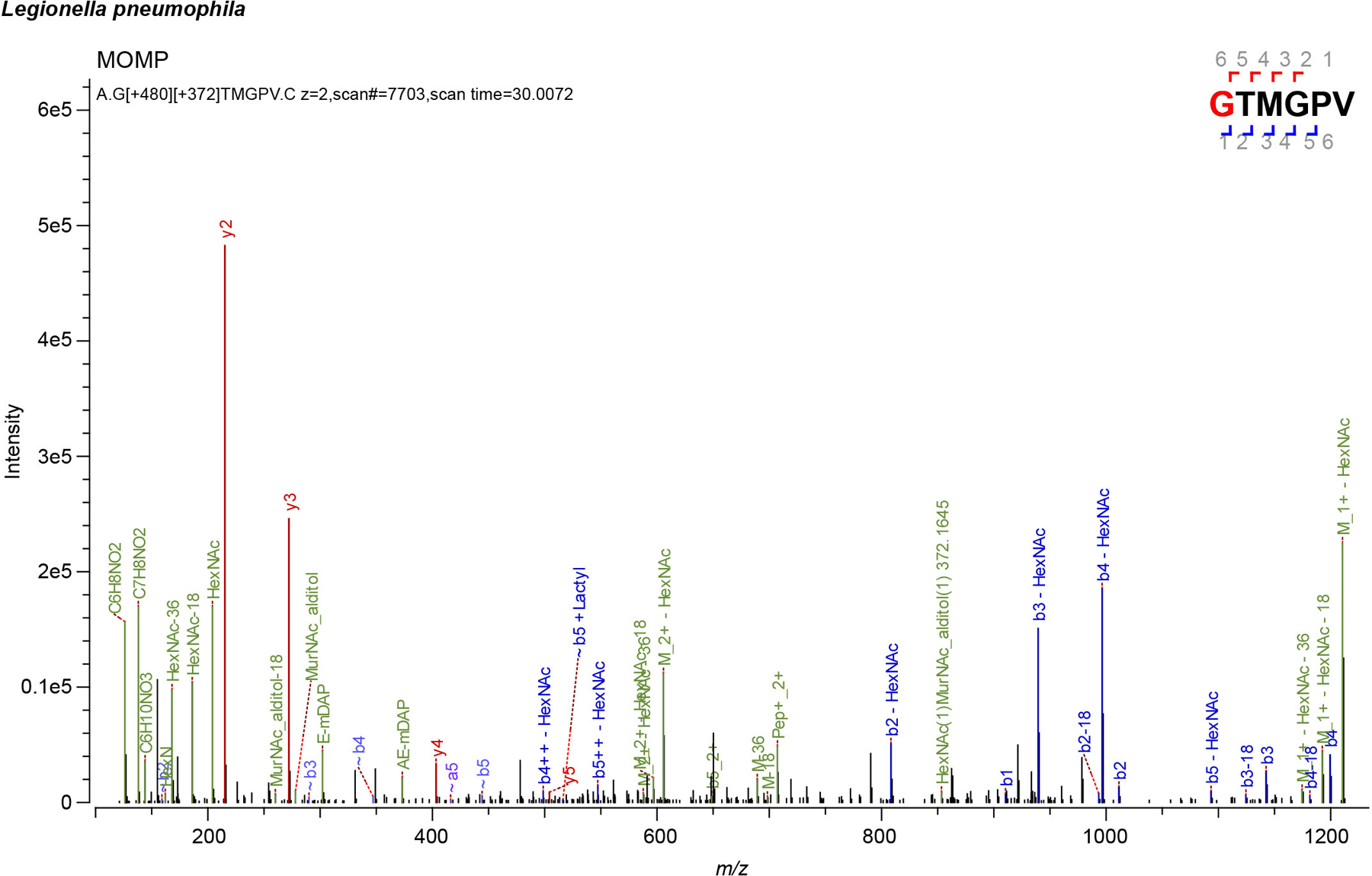
Representative MS/MS spectra for L. pneumophila Major Outer Membrane Protein (MOMP) covalently attached to mDAP (m) residues of PG. Residues with PG tripeptide modification are highlighted in red. Spectra are shown as annotated by Byonic using an unbiased search approach.
Extended Data Fig. 5. Structural topology of C. burnetii BbpA and BbpB.
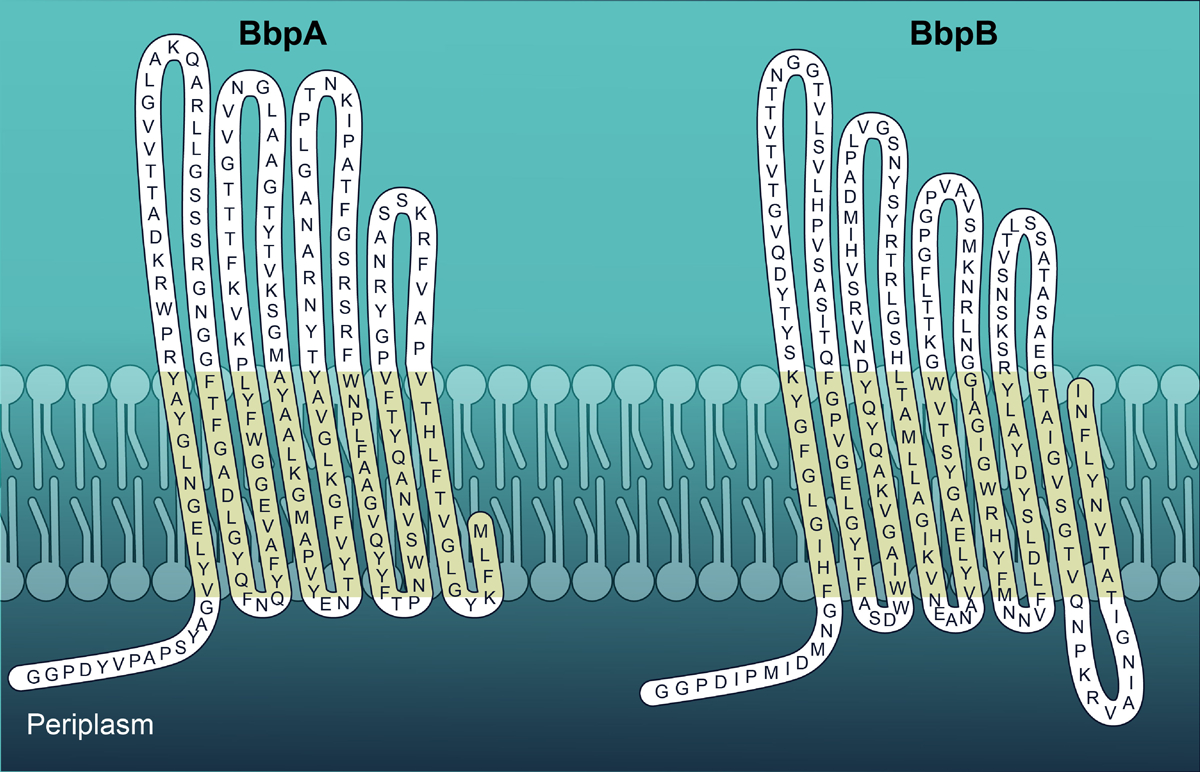
Predicted periplasmic, transmembrane, and extracellular domains of BbpA and BbpB using PRED-TMBB. BbpA and BbpB are depicted with periplasmic N-terminal regions. Similar topology is predicted in β-barrel proteins from A. tumefaciens and L. pneumophila that are covalently attached to PG.
Extended Data Fig. 6. Extended Data Figure S6. Structural modeling of C. burnetii OM proteins.

a. The protein sequences of BbpA, BbpB, and LimB are shown without N-terminal signal peptides. The disordered N-terminal domains of BbpA and BbpB were excluded from structural models but were included in subsequent molecular dynamics simulations (blue). Residues bound to PG in MD simulations are highlighted in red. b. The mature LimB was modeled as a random coil lacking any appreciable secondary structure. The three acyl tails at the N-terminus are shown in grey as is Lys21. c. Contact map predicted from coevolution analysis of BbpA. Contacts are shown in shades of blue (darker blue = higher probability) and contacts from E. coli OmpA (PDB entry 1QJP) are shown in gray. d. Predicted contacts mapped onto the final Rosetta model of BbpA. Contacts are color coded by Cα-Cα distance: green (<5 Å), yellow (5–10 Å), red (>10 Å).
Extended Data Fig. 7. β-barrels form a tight tether between the OM and PG.
Structural model of BbpA (red) in the C. burnetii OM. Structures of the cell envelope are colored as follows: inner leaflet of OM (grey), lipid A of LPS (orange), core oligosaccharides (yellow), glycan chains of PG (blue) peptide stems of PG (green). A similar model was generated for BbpB. b. Molecular dynamics simulation of C. burnetti OM-PG protein-tethered models. The distance in angstroms between the phosphorus atoms of the inner leaflet of the OM and PG layers was measured for three runs for BbpA, ala-BbpA, BbpB, LimB, and Lpp from E. coli. The solid lines are running averages. Distances measured in angstroms for run 1, run 2, and run 3 are shown.
Extended Data Fig. 8. Predicted or annotated Ldts in Proteobacteria used in this study.
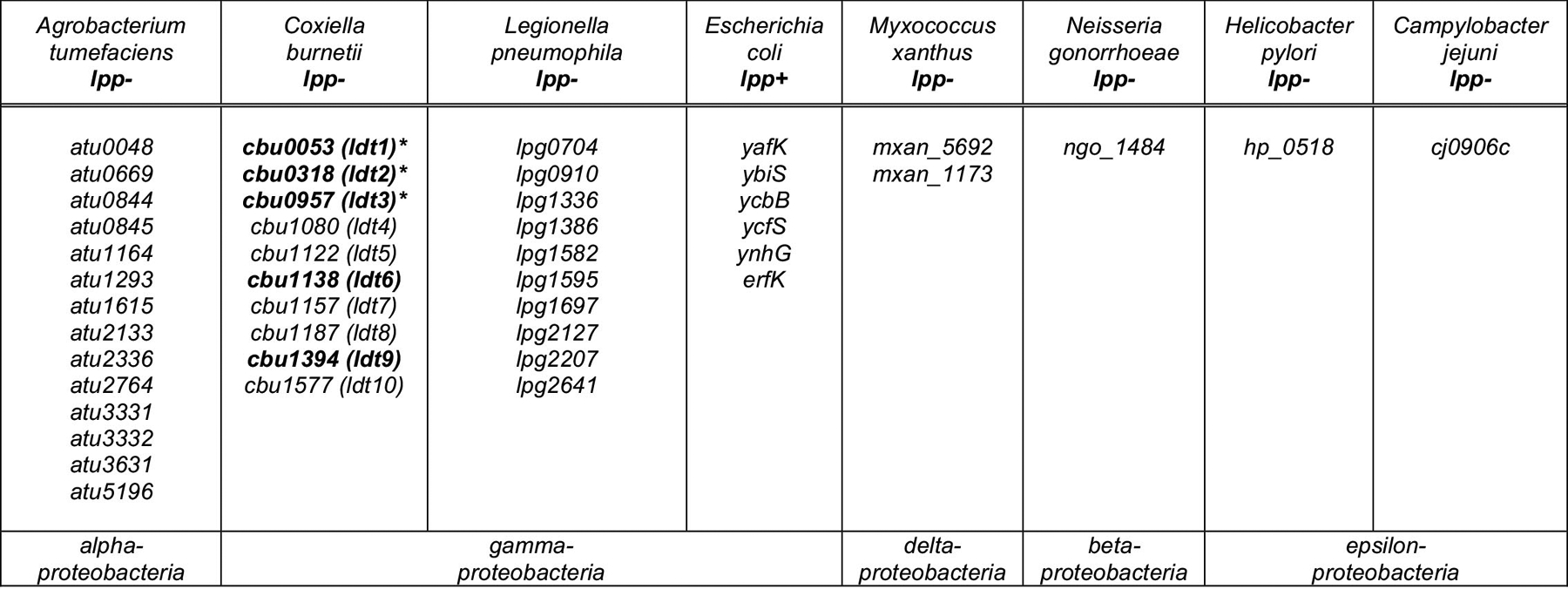
Predicted or annotated Ldts in E. coli (lpp+), C. burnetii, L. pneumophila, A. tumefaciens, M. xanthus, N. gonorrhoeae, H. pylori, and C. jejuni. The latter seven organisms lack lpp homologs. The C. burnetii ldt genes that were successfully inactivated in this study are denoted with an asterisk. C. burnetii ldts upregulated during stationary phase are in bold. The following ldts were previously annotated as enhanced entry genes (enh); cbu0053, cbu0318, cbu1122, cbu1138, and cbu1394.
Extended Data Fig. 9. The L,D transpeptidase ldt2 is required for covalent attachment of BbpA and BbpB to PG.
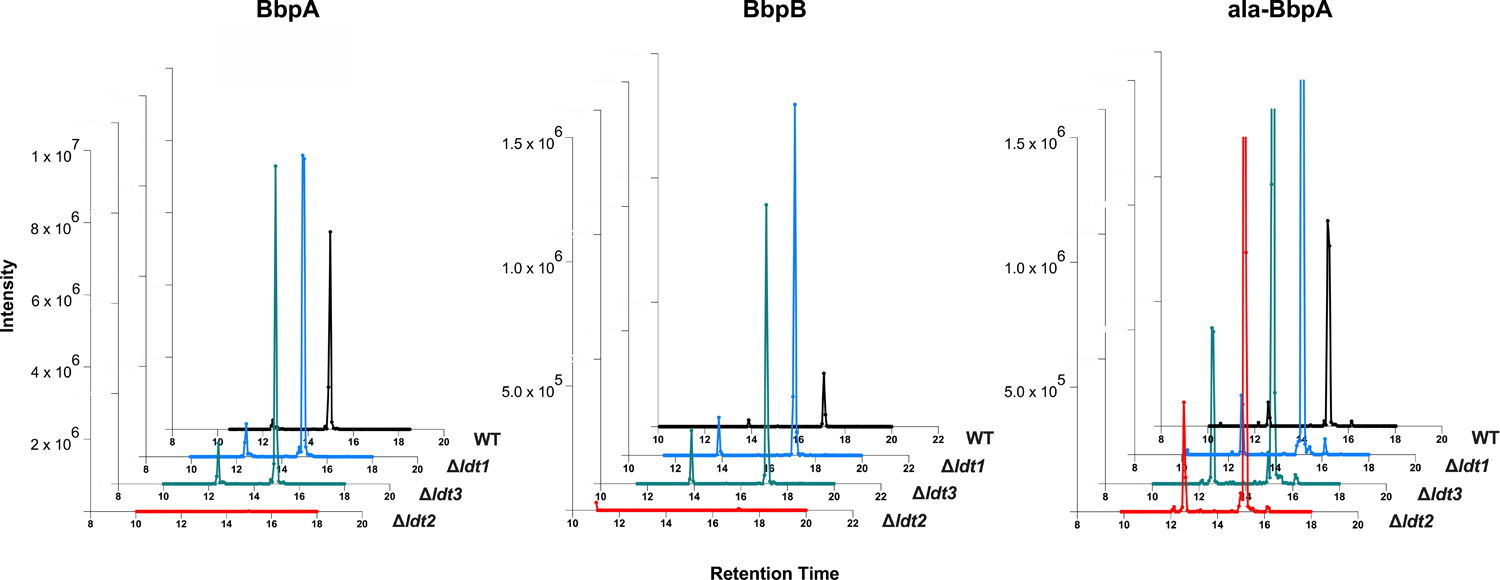
XIC analysis was performed on PG that was extracted from WT and Δldt mutant strains and analyzed by MS/MS. XIC’s of precursor masses (m/z) corresponding to PG-bound BbpA (AEmGGPDYVPAPS, m/z = 906.909, z = 2), ala-BbpA (AEmAGGPDYVPAPS, m/z = 942.427, z = 2), and BbpB (AEmGGPDIPM, m/z = 769.843, z = 2) are shown.
Extended Data Fig. 10. Onset of replication is delayed in the Δldt2 deletion mutant.
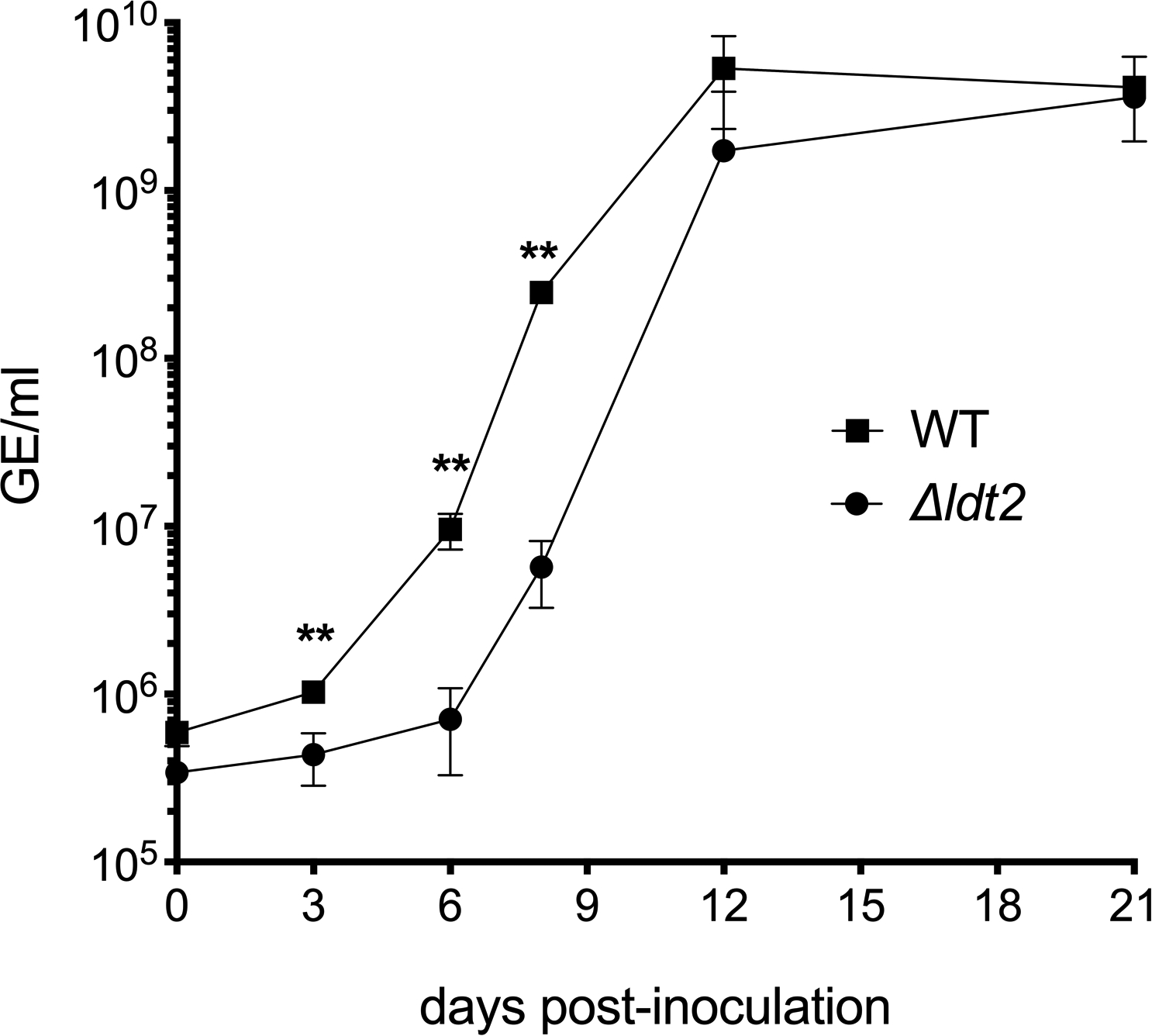
Growth of wild-type (WT) C. burnetii and a Δldt2 mutant strain in ACCM-D was assessed using qPCR to quantitate genome equivalents (GE) during a 21-day incubation. The results are expressed as the means from n=3 independent experiments. Error bars indicate the standard error of the mean, and asterisks indicate a statistically significant difference from WT C. burnetii. **P at 3, 6 and 9 days post-inoculation = 0.0046, 0.0027, and 0.0001, respectively. Statistical significance was calculated using a two-sided Student’s t test.
Supplementary Material
Acknowledgements:
The authors thank Motoshi Suzuki, Adelina E. Acosta Martin, and Mark Collins for their contribution to MS method development and Vinod Nair for cryo-EM. We also thank Ryan Kissinger for 3-dimensional modeling with assistance from Austin Athman, and Anita Mora for graphics support. We also thank Xavier De Bolle and Pierre Godessart for stimulating discussion, and John Zupan, Patricia Zambryski, David Kelly, Aidan Taylor, Jonathan Shaw, Ernesto Felix Diaz Parga, Richard Wheeler, Ivo Boneca, Muhamed-Kheir Taha, and Egbert Hoiczyk for bacterial cultures used for peptidoglycan extraction. PG Mass spectrometry analyses were performed by the biOMICS Facility of the Faculty of Science Mass Spectrometry Centre at the University of Sheffield and the Research Technologies Branch at the National Institutes of Health in Rockville, MD. Cryo-EM work was performed at the Research Technologies Branch at the National Institutes of Health in Hamilton, MT.
Funding: This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases (R.A.H. and S.A.P), the Medical Research Council grant MR/S009272/1 (S.M.), and the Biotechnology and Biological Sciences Research Council grant BBSRC BB/N000951/1 (S.M.). R.E.S. and A.V.P. are supported by a Biotechnology and Biological Sciences Research Council studentship (doctoral training program grant BB/M011151/1). J.C.G. acknowledges support from NIH under Grant No. R01-GM123169. C.J.C. is supported by a National Science Foundation (NSF) Graduate Research Fellowship under Grant No. 2017219379. J.M.P. was supported by the Laboratory Directed Research and Development program at Oak Ridge National Laboratory, which is managed by UT-Battelle, LLC, for the U.S. Department of Energy under Contract No. DE-AC05-00OR22725. This work used resources of the Compute and Data Environment for Science (CADES) at ORNL as well as the Extreme Science and Engineering Discovery Environment (XSEDE; allocation TG-MCB130173), which is supported by NSF Grant No. ACI-1548562.
Footnotes
Reporting summary: Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability: The MS data sets are available through the GlycoPOST repository (DOI: GPST000124)64. Cryo-EM micrographs used as data in Fig. 3c are available at Figshare under the URL: https://doi.org/10.6084/m9.figshare.1279250665. Uncropped immunoblot blot scans from which Figs. 2a and 3a were derived are available as source data. Molecular dynamics simulation data and additional raw data that support the findings of this study are available from the corresponding authors upon reasonable request.
Competing interests: Authors declare no competing interests.
References
- 1.Silhavy TJ, Kahne D & Walker S The bacterial cell envelope. Cold Spring Harb Perspect Biol 2, a000414, doi: 10.1101/cshperspect.a000414 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hwang H, Paracini N, Parks JM, Lakey JH & Gumbart JC Distribution of mechanical stress in the Escherichia coli cell envelope. Biochim Biophys Acta Biomembr 1860, 2566–2575, doi: 10.1016/j.bbamem.2018.09.020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rojas ER et al. The outer membrane is an essential load-bearing element in Gram-negative bacteria. Nature 559, 617–621, doi: 10.1038/s41586-018-0344-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asmar AT & Collet JF Lpp, the Braun lipoprotein, turns 50-major achievements and remaining issues. FEMS Microbiol Lett 365, doi: 10.1093/femsle/fny199 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Egan AJF Bacterial outer membrane constriction. Mol Microbiol 107, 676–687, doi: 10.1111/mmi.13908 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Pages JM, James CE & Winterhalter M The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat Rev Microbiol 6, 893–903, doi: 10.1038/nrmicro1994 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Galdiero S et al. Microbe-host interactions: structure and role of Gram-negative bacterial porins. Curr Protein Pept Sci 13, 843–854 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park JS et al. Mechanism of anchoring of OmpA protein to the cell wall peptidoglycan of the gram-negative bacterial outer membrane. FASEB J 26, 219–228, doi: 10.1096/fj.11-188425 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godlewska R, Wisniewska K, Pietras Z & Jagusztyn-Krynicka EK Peptidoglycan-associated lipoprotein (Pal) of Gram-negative bacteria: function, structure, role in pathogenesis and potential application in immunoprophylaxis. FEMS Microbiol Lett 298, 1–11, doi: 10.1111/j.1574-6968.2009.01659.x (2009). [DOI] [PubMed] [Google Scholar]
- 10.Samsudin F, Ortiz-Suarez ML, Piggot TJ, Bond PJ & Khalid S OmpA: A Flexible Clamp for Bacterial Cell Wall Attachment. Structure 24, 2227–2235, doi: 10.1016/j.str.2016.10.009 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Braun V & Rehn K Chemical characterization, spatial distribution and function of a lipoprotein (murein-lipoprotein) of the E. coli cell wall. The specific effect of trypsin on the membrane structure. Eur J Biochem 10, 426–438 (1969). [DOI] [PubMed] [Google Scholar]
- 12.Braun V Covalent lipoprotein from the outer membrane of Escherichia coli. Biochim Biophys Acta 415, 335–377 (1975). [DOI] [PubMed] [Google Scholar]
- 13.Magnet S et al. Identification of the L,D-transpeptidases responsible for attachment of the Braun lipoprotein to Escherichia coli peptidoglycan. J. Bacteriol 189, 3927–3931, doi: 10.1128/JB.00084-07 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magnet S, Dubost L, Marie A, Arthur M & Gutmann L Identification of the L,D-transpeptidases for peptidoglycan cross-linking in Escherichia coli. J Bacteriol 190, 4782–4785, doi: 10.1128/JB.00025-08 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen EJ, Ferreira JL, Ladinsky MS, Beeby M & Hughes KT Nanoscale-length control of the flagellar driveshaft requires hitting the tethered outer membrane. Science 356, 197–200, doi: 10.1126/science.aam6512 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asmar AT et al. Communication across the bacterial cell envelope depends on the size of the periplasm. PLoS Biol 15, e2004303, doi: 10.1371/journal.pbio.2004303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butler CA & Hoffman PS Characterization of a major 31-kilodalton peptidoglycan-bound protein of Legionella pneumophila. J Bacteriol 172, 2401–2407 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amano K & Williams JC Partial characterization of peptidoglycan-associated proteins of Legionella pneumophila. Journal of biochemistry 94, 601–606 (1983). [DOI] [PubMed] [Google Scholar]
- 19.Amano K & Williams JC Peptidoglycan of Legionella pneumophila: apparent resistance to lysozyme hydrolysis correlates with a high degree of peptide cross-linking. J Bacteriol 153, 520–526 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffman PS, Seyer JH & Butler CA Molecular characterization of the 28- and 31-kilodalton subunits of the Legionella pneumophila major outer membrane protein. J Bacteriol 174, 908–913 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amano K & Williams JC Sensitivity of Coxiella burnetii peptidoglycan to lysozyme hydrolysis and correlation of sacculus rigidity with peptidoglycan-associated proteins. J Bacteriol 160, 989–993 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barton IS, Fuqua C & Platt TG Ecological and evolutionary dynamics of a model facultative pathogen: Agrobacterium and crown gall disease of plants. Environ Microbiol 20, 16–29, doi: 10.1111/1462-2920.13976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliva G, Sahr T & Buchrieser C The Life Cycle of L. pneumophila: Cellular Differentiation Is Linked to Virulence and Metabolism. Front Cell Infect Microbiol 8, 3, doi: 10.3389/fcimb.2018.00003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minnick MF & Raghavan R Developmental biology of Coxiella burnetii. Advances in experimental medicine and biology 984, 231–248, doi: 10.1007/978-94-007-4315-1_12 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Bern M, Beniston R & Mesnage S Towards an automated analysis of bacterial peptidoglycan structure. Analytical and bioanalytical chemistry 409, 551–560, doi: 10.1007/s00216-016-9857-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braun V & Wolff H The murein-lipoprotein linkage in the cell wall of Escherichia coli. Eur J Biochem 14, 387–391, doi: 10.1111/j.1432-1033.1970.tb00301.x (1970). [DOI] [PubMed] [Google Scholar]
- 27.Battisti JM, Hicks LD & Minnick MF A unique Coxiella burnetii lipoprotein involved in metal binding (LimB). Microbiology 157, 966–976, doi: 10.1099/mic.0.046649-0 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braun V & Bosch V Sequence of the murein-lipoprotein and the attachment site of the lipid. Eur J Biochem 28, 51–69, doi: 10.1111/j.1432-1033.1972.tb01883.x (1972). [DOI] [PubMed] [Google Scholar]
- 29.Pavlova A, Hwang H, Lundquist K, Balusek C & Gumbart JC Living on the edge: Simulations of bacterial outer-membrane proteins. Biochim Biophys Acta 1858, 1753–1759, doi: 10.1016/j.bbamem.2016.01.020 (2016). [DOI] [PubMed] [Google Scholar]
- 30.McCaul TF & Williams JC Developmental cycle of Coxiella burnetii: structure and morphogenesis of vegetative and sporogenic differentiations. J. Bacteriol 147, 1063–1076 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandoz KM et al. Transcriptional profiling of Coxiella burnetii reveals extensive cell Wall remodeling in the small cell variant developmental form. PLoS One 11, e0149957, doi: 10.1371/journal.pone.0149957 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cameron TA, Anderson-Furgeson J, Zupan JR, Zik JJ & Zambryski PC Peptidoglycan synthesis machinery in Agrobacterium tumefaciens during unipolar growth and cell division. MBio 5, e01219–01214, doi: 10.1128/mBio.01219-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.More N et al. Peptidoglycan Remodeling Enables Escherichia coli To Survive Severe Outer Membrane Assembly Defect. MBio 10, doi: 10.1128/mBio.02729-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sonntag I, Schwarz H, Hirota Y & Henning U Cell envelope and shape of Escherichia coli: multiple mutants missing the outer membrane lipoprotein and other major outer membrane proteins. J Bacteriol 136, 280–285 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bertrand RL Lag Phase Is a Dynamic, Organized, Adaptive, and Evolvable Period That Prepares Bacteria for Cell Division. J Bacteriol 201, doi: 10.1128/JB.00697-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delcour AH Outer membrane permeability and antibiotic resistance. Biochim Biophys Acta 1794, 808–816, doi: 10.1016/j.bbapap.2008.11.005 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mainardi JL et al. A novel peptidoglycan cross-linking enzyme for a beta-lactam-resistant transpeptidation pathway. J Biol Chem 280, 38146–38152, doi: 10.1074/jbc.M507384200 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Sutterlin L, Edoo Z, Hugonnet JE, Mainardi JL & Arthur M Peptidoglycan cross-linking activity of L,D-transpeptidases from Clostridium difficile and inactivation of theses enzymes by beta-lactams. Antimicrob Agents Chemother, doi: 10.1128/AAC.01607-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao H, Patel V, Helmann JD & Dorr T Don’t let sleeping dogmas lie: new views of peptidoglycan synthesis and its regulation. Mol Microbiol 106, 847–860, doi: 10.1111/mmi.13853 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bergkessel M, Basta DW & Newman DK The physiology of growth arrest: uniting molecular and environmental microbiology. Nat Rev Microbiol 14, 549–562, doi: 10.1038/nrmicro.2016.107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beare PA, Larson CL, Gilk SD & Heinzen RA Two systems for targeted gene deletion in Coxiella burnetii. Appl Environ Microbiol 78, 4580–4589, doi: 10.1128/AEM.00881-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sandoz KM, Beare PA, Cockrell DC & Heinzen RA Complementation of Arginine Auxotrophy for Genetic Transformation of Coxiella burnetii by Use of a Defined Axenic Medium. Appl. Environ. Microbiol 82, 3042–3051, doi: 10.1128/AEM.00261-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cangelosi GA, Best EA, Martinetti G & Nester EW Genetic analysis of Agrobacterium. Methods Enzymol 204, 384–397, doi: 10.1016/0076-6879(91)04020-o (1991). [DOI] [PubMed] [Google Scholar]
- 44.Soding J Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960, doi: 10.1093/bioinformatics/bti125 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Zimmermann L et al. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J Mol Biol 430, 2237–2243, doi: 10.1016/j.jmb.2017.12.007 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Pautsch A & Schulz GE High-resolution structure of the OmpA membrane domain. J Mol Biol 298, 273–282, doi: 10.1006/jmbi.2000.3671 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Kamisetty H, Ovchinnikov S & Baker D Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc Natl Acad Sci U S A 110, 15674–15679, doi: 10.1073/pnas.1314045110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balakrishnan S, Kamisetty H, Carbonell JG, Lee SI & Langmead CJ Learning generative models for protein fold families. Proteins 79, 1061–1078, doi: 10.1002/prot.22934 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Remmert M, Biegert A, Hauser A & Soding J HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nature methods 9, 173–175, doi: 10.1038/nmeth.1818 (2011). [DOI] [PubMed] [Google Scholar]
- 50.UniProt C UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515, doi: 10.1093/nar/gky1049 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ovchinnikov S et al. Protein structure determination using metagenome sequence data. Science 355, 294–298, doi: 10.1126/science.aah4043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song Y et al. High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742, doi: 10.1016/j.str.2013.08.005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Viklund H & Elofsson A OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24, 1662–1668, doi: 10.1093/bioinformatics/btn221 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Vandeputte-Rutten L, Bos MP, Tommassen J & Gros P Crystal structure of Neisserial surface protein A (NspA), a conserved outer membrane protein with vaccine potential. J Biol Chem 278, 24825–24830, doi: 10.1074/jbc.M302803200 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Zamboni DS et al. Stimulation of toll-like receptor 2 by Coxiella burnetii is required for macrophage production of pro-inflammatory cytokines and resistance to infection. J Biol Chem 279, 54405–54415, doi: 10.1074/jbc.M410340200 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Narasaki CT & Toman R Lipopolysaccharide of Coxiella burnetii. Adv Exp Med Biol 984, 65–90, doi: 10.1007/978-94-007-4315-1_4 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Stead CM, Cockrell DC, Beare PA, Miller HE & Heinzen RA A Coxiella burnetii phospholipase A homolog pldA is required for optimal growth in macrophages and developmental form lipid remodeling. BMC Microbiol 18, 33, doi: 10.1186/s12866-018-1181-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gumbart JC, Beeby M, Jensen GJ & Roux B Escherichia coli peptidoglycan structure and mechanics as predicted by atomic-scale simulations. PLoS Comput Biol 10, e1003475, doi: 10.1371/journal.pcbi.1003475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38 (1996). [DOI] [PubMed] [Google Scholar]
- 60.Phillips JC et al. Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802, doi: 10.1002/jcc.20289 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Darden TAYDMP, Particle mesh Ewald LG: An N log N method for Ewald sums in large systems. J Comput Phys 98, 10089–10092 (1993). [Google Scholar]
- 62.Huang J et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nature methods 14, 71–73, doi: 10.1038/nmeth.4067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klauda JB et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114, 7830–7843, doi: 10.1021/jp101759q (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sandoz KM et al. β-barrel proteins tether the outer membrane in many gram-negative bacteria GlycoPOST, doi:GPST000124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sandoz KM et al. β-barrel proteins tether the outer membrane in many gram-negative bacteria Figshare, 10.6084/m6089.figshare.12792506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.