

Abstract
Innate immune system is considered the first line of defense during viral invasion, with the wealth of the literature demonstrating innate immune control of diverse viruses during acute infection. What is far less clear is the role of innate immune system during chronic virus infections. This short review focuses on alpha- and gammaherpesviruses, two highly prevalent herpesvirus subfamilies that, following a brief, once in a lifetime period of acute lytic infection, establish life-long latent infection that is characterized by sporadic reactivation in an immunocompetent host. In spite of many similarities, these two viral families are characterized by distinct cellular tropism and pathogenesis. Here we focus on the published in vivo studies to review known interactions of these two viral subfamilies with the innate immunity of the intact host, both during acute and, particularly, chronic virus infection.
Keywords: innate immune response, gammaherpesvirus, alphaherpesvirus, EBV, KSHV, HSV, MHV68
Alphaherpesvirus and gammaherpesvirus overview.
Herpesviruses are ancient viruses that have coevolved with their hosts and, following exposure, establish a lifelong infection which is never cleared. The Herpesviridae family contains three subfamilies, alpha-, beta-, and gammaherpesviruses, which are all large (100–200 nm), linear double-stranded DNA (120–250 kb) viruses [1]. This review focuses on the alpha-(herpes simplex viruses (HSV-1 and HSV-2)) and gammaherpesvirus (human Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV), and murine gammaherpesvirus-68 (MHV68) also known as murid herpesvirus 4) subfamilies. The choice of these two subfamilies was driven, in part, by the anatomical differences in establishing chronic infection: neurotropic nature of latent alphaherpesvirus infection versus systemic distribution of the latent gammaherpesvirus reservoir. The current review is further focused on the published studies of an intact host infection that reveal the complexity of virus-host interactions that is not fully captured by in vitro studies.
Alphaherpesviruses.
HSV-1 and HSV-2 share 83% homology in the coding regions. HSV-1 infects approximately 55% of the U.S. population and clinically manifests as vesicular lesions, mostly of the oral mucosa. Infection of the conjunctiva leads to a recurrent keratoconjunctivitis, a leading infectious cause of blindness worldwide [2]. HSV-2 infects around 20% of the U.S. population and is the common cause of genital disease [3]. For both HSV-1 and HSV-2, latent infection is established in the ganglia innervating the site of the original inoculation, with HSV-1 typically residing in the trigeminal and HSV-2 the sacral ganglia. Reactivation from latency delivers infectious virions in proximity to the original site of inoculation, with subsequent lytic amplification and transmission to a naïve host [4]. Systemic dissemination of HSV is uncommon but can occur in immunocompromised individuals leading to high morbidity and mortality.
HSV-1 and HSV-2 have a sufficiently broad host range to allow infection of laboratory animals. While HSV infection of genetically tractable mouse models recapitulates many features of acute infection in humans, spontaneous HSV reactivation in mice happens at much lower levels than in humans, an important caveat of this animal model. Other animal models of HSV infection, such as rabbits and guinea pigs, do spontaneously reactivate HSV [5] but, unfortunately, lack the robust host genetic tools available in the murine system. Studies in mouse models utilize multiple routes of HSV infection to mimic diverse aspects of viral pathogenesis observed in humans (summarized in Table 1), with most publications focused on the first 5–7 days of HSV infection during which acute viral replication occurs at the inoculation site and the innervating ganglia [5].
Table.1.
| Virus | Rout of infection | Dosages (pfu) | Publication |
|---|---|---|---|
| HSV-1 | Corneal | (10^3, 10M, 10^6, 2×10^6)/eye | 37, 38, 44, 56, 57, 58, 67, 68 |
| HSV-1 | Footpad injection | 2×10^6 | 29 |
| HSV-1 | Intracranial | 10^2, 10^4, 10^5 | 45, 55, 57 |
| HSV-1 | Intranasal | 10^6 | 28 |
| HSV-1 | Intraperitoneal | 5×10^7, 10^9 | 23,40 |
| HSV-1 | Intravenous | 10^7 | 39, 53, 54, 55 |
| HSV-2 | Intravaginal | 2×10^3, 6.7×10^4 | 25, 37, |
| MHV68 | Intranasal | 10^2, 5×10^2, 10^3, 4×10^3, 5×10^4, 10^5, 2×10^5, 4×10^5, 10^2, 2×10^6, 4×10^6 | 30–33, 46, 47, 60–62, 72, 73 |
| MHV68 | Intraperitoneal | 5×10^5, 10^6 | 33,41 |
| MHV68 | Intravenous | 2×10^6 | 32 |
Gammaherpesviruses.
EBV and KSHV are highly prevalent human gammaherpesviruses, with over 95% of adults worldwide harboring life-long infection with the former. Unlike alphaherpesviruses, gammaherpesviruses establish systemic infection of lymphoid cells and are associated with the development of cancers, including B cell lymphomas [6,7]. EBV infects naïve B cells and usurps B cell differentiation by inducing a robust germinal center response that includes both virus-infected and uninfected B cells, leading to passive expansion of the viral latent reservoir via cellular proliferation [8,9]. Subsequent B cell differentiation produces EBV-infected memory B cells, a reservoir of long-term infection, or plasma cells that reactivate the latent virus [10,11]. While KSHV infection of human B cells remains less understood, existing studies indicate that KSHV may employ similar mechanisms of B cell manipulation. Many EBV-driven B cell lymphomas are of germinal center or post germinal center origin, highlighting an unintended consequence of the infection of germinal center B cells [12,13]. Importantly, increased viral reactivation often precedes tumorigenesis [14,15].
The highly restricted species specificity of human EBV and KSHV has limited in vivo studies of the natural host to rare polymorphisms and mutations that unveil the pathogenicity of these viruses [16]. In contrast, the natural rodent gammaherpesvirus, MHV68, represents a tractable animal model of chronic gammaherpesvirus infection that has provided a wealth of information about the virus-host interactions in the intact host. MHV68 is genetically and biologically similar to EBV and KSHV, establishes latency in B cells, and drives lymphomas in immunocompromised hosts [17–19]. Intranasal and intraperitoneal are the most common routes of infection, with the spleen and peritoneal cavity representing the two sites of MHV68 latency examined in most studies. Acute MHV68 replication is cleared within 9–12 days with the peak of viral latency in the spleen observed at 16 days post infection [20].
Innate immune system of the host has traditionally been perceived as the “first line of defense” and, as such, has been almost exclusively examined in the context of acute virus replication in vivo. In contrast, studies of chronic virus infections place a major emphasis on defining parameters of adaptive immune responses, with innate immune factors mostly disregarded. Importantly, the few paradigm shifting studies that had examined innate immune pathways during lymphocytic choriomeningitis virus infection [21,22], have highlighted the continuing role of the host innate immunity in modifying viral control and pathogenesis in a chronically infected host. This review focuses on comparing innate immune mechanisms relevant for the control of alpha- and gammaherpesvirus infections during both acute, and, more importantly, chronic phase of infection of an intact host.
Sensing herpesvirus infection.
TLRs:
There is a myriad of virus pattern sensors whose expression and function are further modified in the cell type-dependent manner. Due to the high complexity, this review focuses on the sensors that were examined during alpha- or gammaherpesvirus infection of the intact host (Fig. 1). One such group of sensors, Toll like receptors (TLRs) [23], are present on the cell surface (TLR2) or in endosomes (TLRs 3, 7, and 9). TLR2 is a promiscuous sensor of pathogens; the exact viral moieties recognized by this sensor remain poorly understood [24]. In contrast, the ligands for the other TLRs are better defined, such as dsRNA for TLR3, ssRNA for TLR7, and CpG for TLR9 [24].
Figure 1. Innate immune mechanisms modifying alpha- and gammaherpesvirus infections of the intact host.
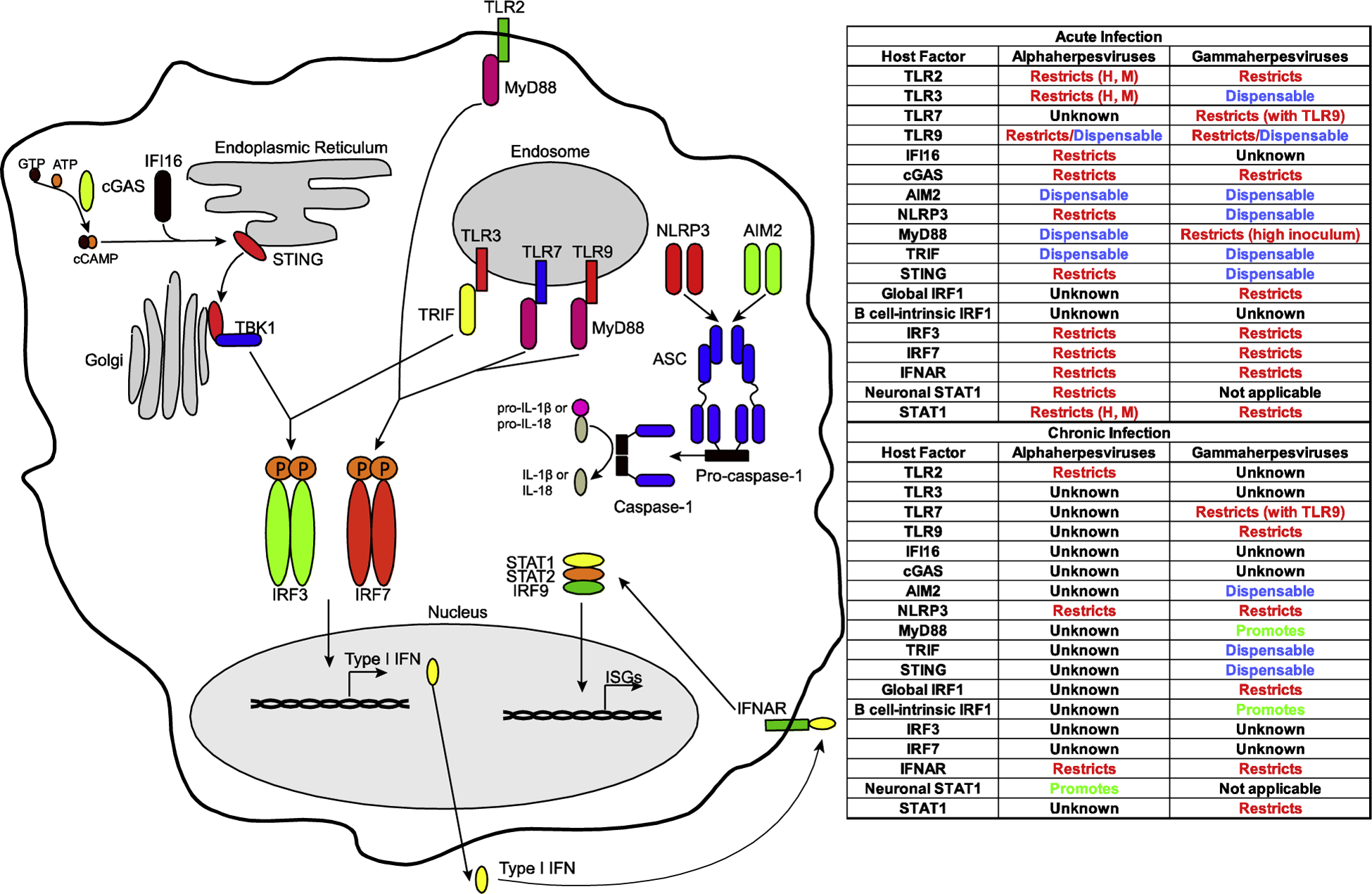
Table represents changes in viral parameters of infection observed in animal models of global or cell type-specific host factor deficiency. H, M indicates that the same viral phenotypes were observed in human genetic deficiencies and animal models.
TLRs in HSV infection.
The role of TLR2 during HSV-1 infection is nuanced. In animal models, significantly fewer TLR2−/− mice succumbed to intraperitoneal infection with HSV-1. TLR2−/− mice displayed decreased systemic levels of pro-inflammatory cytokines and fewer inflammatory lesions in the brain, despite slightly increased brain HSV-1 titers [25], suggesting that TLR2 primarily mediates immunopathology during acute HSV-1 infection. Intriguingly, specific polymorphisms in intron 1 of human TLR2 are associated with increased HSV-2 shedding and lesion frequency over a monitored 60 day period [26], indicating a role during chronic infection. Unfortunately, in what will become a trend in this review, far less is understood about the role of TLRs during chronic HSV infection of animal models.
TLR3−/− mice display increased disease score, including hind limb paralysis, and a significant increase in the viral titers during acute HSV-2 infection. Loss of TLR3 in astrocytes renders these more permissive to HSV-2 replication [27]. Highlighting significance of TLR3, TLR3 deficiency in humans results in childhood HSV encephalitis [28,29]. The role of the other endosomal TLRs, TLR7 & TLR9, is less defined. Nothing is known about TLR7 during HSV infection. TLR9 restricts acute HSV-1 infection following intranasal infection [30], but has no impact on HSV-1 replication following a footpad route of inoculation [31], indicating a route of inoculation-dependent restriction of acute HSV-1 infection by TLR-9. The role of TLR7 or TLR9 in human HSV infection is not known.
TLRs in gammaherpesvirus infection.
There is a paucity of reports linking TLR polymorphisms in humans to EBV or KSHV-driven disease, thus, TLR-gammaherpesvirus interaction has been defined using the MHV68 model. While expression of TLR2 attenuates acute MHV68 replication in part by increasing type I interferon (IFN) expression [32], TLR3, in contrast to that observed for HSV, is dispensable for the control of acute MHV68 infection [33]. TLR7 expression has no impact on acute MHV68 infection [34], while TLR9 attenuates acute MHV68 infection in the route of infection-dependent manner, similar to that observed for HSV. TLR9 deficiency has no effect on acute MHV68 titers following intranasal inoculation [34,35], however, lack of TLR9 leads to increased MHV68 acute titers following intraperitoneal or intravenous inoculation. Interestingly, a combined deficiency of TLR7 and TLR9 along with the loss of host protein UNC93B, which is required for endosomal TLR signaling, results in significantly higher acute MHV68 titers in several organs.
In the context of chronic infection, loss of TLR9 results in increased MHV68 reactivation and MHV68 DNA copy number [34,35]. Similar to acute infection, while chronic MHV68 infection is well-controlled in TLR7−/− mice, loss of both TLR7 and TLR9 leads to an even greater increase in reactivation and viral copy numbers as compared to TLR9 deficiency alone [34], highlighting non-overlapping functions of TLR7 and TLR9. Interestingly, administration of LPS or CpG to chronically infected mice induces MHV68 reactivation [36], however, the TLR dependence of this phenotype is unknown.
IFI16 and cGAS:
Other relevant viral sensors include interferon-inducible protein 16 (IFI16), which can recognize cytosolic and nuclear DNA [37] and cyclic GMP-AMP synthase (cGAS), which senses cytosolic DNA. Activation of cGAS by cytosolic DNA produces cyclic GMP-AMP (cGAMP) that engages Stimulator of IFN Genes (STING) [38]. Both cGAS and IFI16 engage STING to induce type I IFN expression (Fig. 1).
IFI16 and cGAS in HSV infection.
While well-described in tissue culture, the in vivo role of IFI16 during HSV infection remains enigmatic, due to the divergence of human and mouse IFI16 orthologues. Knockdown of mouse orthologue p204 in corneal epithelium led to mild increase in the titers of HSV-1 in the cornea [39] offering some insight into the role of this sensor in the human infection. Polymorphisms of human IFI16 or cGAS have not yet been associated with HSV- or EBV/KSHV-driven disease in humans.
In contrast to IFI16, the role of cGAS is well defined in mouse models of acute HSV infection. All cGAS−/− mice died within 6 days of HSV-1 infection, accompanied by increased HSV-1 titers in the brain, reduced type I IFN, and severe disease, [40,41]. Intriguingly, the true extent of cGAS anti-HSV-1 activity may be masked by HSV-1 tegument protein, UL37, which deaminates cGAS, rendering it unable to sense dsDNA, [42]. Severely attenuated phenotype of HSV-1 mutant encoding catalytically dead UL37 was completely rescued in cGAS−/− mice, offering an elegant evidence for the physiologically relevant viral-host antagonism [42].
IFI16 and cGAS in gammaherpesvirus infection.
As mentioned in the previous section, nothing is known about the role of IFI16 during gammaherpesvirus infection of an intact host. In contrast and similar to HSV, cGAS attenuates acute MHV68 infection [43]. Interestingly, the role of MHV68 protein orf52, a homologue of KSHV cGAS antagonist [44], has not been resolved in vivo. As with alphaherpesvirus infection, the impact of cGAS on chronic gammaherpesvirus infection is unknown.
Inflammasome:
The inflammasome complex is assembled in the cytosol upon sensing of microbial patterns, with subsequent ASC-dependent recruitment and activation of caspase-1 or caspase-11 that mediate cleavage of pro IL-1β and pro IL-18 into their active forms [45] (Fig. 1). Relevant inflammasome associated sensors include AIM2 (cytosolic DNA) and NLRP3 (cytosolic ATP and other motifs).
Inflammasome in HSV infection.
HSV-1 infection of NLRP3−/− mice results in early onset severe disease, with slightly increased viral titers [46]. Loss of AIM2 has no impact on acute HSV-1 infection, an intriguing absence of phenotype which could reflect the efficient antagonism of AIM2 by HSV-1 tegument protein VP22 [47]. The role of inflammasome in chronic HSV infection is less clear, as loss of NLRP3 resulted in greater immunopathology at 15 days post HSV-1 infection [46].
Inflammasome in gammaherpesvirus infection.
Acute MHV68 infection is not affected by combined caspase1/11 deficiency [48] or loss of AIM2[49]. Further, loss of caspase1/11 and, presumably, generation of biologically active IL-1β has no impact on chronic MHV68 infection [48]. Unexpectedly, deficiency of IL-1R1, a receptor engaged by IL-1α and IL-1β, attenuates MHV68 reactivation [50], highlighting the proviral functions of either inflammasome-independent IL-1α or non-canonical processing of IL-1β. While loss of AIM2 has no effect on chronic MHV68 infection, deficiency of NLRP3 or ASC resulted in a slight (less than 2-fold), statistically significant increase in MHV68 genome copy numbers in long-term infected spleens (60 days; [49]), a time point beyond that examined in caspase1/11 deficient mice [48]. Importantly, human deficiency of XIAP, a multifunctional protein that also suppresses NLRP3 inflammasome [51], results in severe EBV-driven hemophagocytic lymphohistiocytosis [52]. Further, EBV and KSHV attenuate NLRP3 inflammasome activation in vitro [53,54], highlighting the importance of additional studies of inflammasome during chronic infection.
Connecting sensors to IFN transcription.
MyD88 and TRIF in HSV infection.
MyD88 and TRIF are important adapter proteins downstream of TLRs and contribute to the induction of type I interferons (IFN) [45] (Fig. 1). Both adapters play a relatively minor role during acute HSV infection in spite of the significant antiviral role of TLR3 during acute HSV infection. Specifically, parameters of acute HSV-1 infection in MyD88−/−, TRIF−/−, and MyD88−/−TRIF−/− mice were either similar to that of control mice [39] or produced a minor (10%) increase in mortality [55]. The role of MyD88 and TRIF in chronic HSV-1 infection is not defined.
MyD88 and TRIF in gammaherpesvirus infection.
Similar to acute HSV infection, TLR adapters MyD88 and TRIF have minimal impact during acute MHV68 infection. While TRIF or MyD88 deficiency did not affect acute MHV68 replication following low dose inoculation [33], a 100-fold higher inoculation dose of MHV68 led to loss of viral control and reduction in type 1 IFN in the lungs of MyD88−/− mice [32].
Intriguingly during chronic infection, MyD88 supports the establishment of MHV68 latency, via promoting MHV68-driven germinal center response and B cell differentiation [33]. Because MyD88 participates in several signaling networks, it is not clear whether the proviral role of this adaptor during chronic MHV68 infection is exclusively due to the TLR signaling pathways.
STING in HSV and gammaherpesvirus infections.
The adapter STING, which is activated by cGAS and IFI16 to induce type I IFN, is profoundly antiviral during acute HSV-1 infection: STING expression ensures host survival, particularly following infection with neuroinvasive McKrae strain, and attenuates viral replication and disease scores [40,41,56,57]. In the context of McKrae HSV-1 infection, STING is required for the upregulation of tetherin, which impedes HSV-1 neuroinvasion [58]. Chronic HSV infection has not been explored in STING−/− mice. In contrast to the critical role of STING during HSV-1 infection, STING deficiency has minimal if any effect on acute MHV68 titers and no measurable effect during chronic infection [49].
Activation of IFN.
Sensing of herpesviruses converges upon activation of the transcription factors NF-κB, IFN regulatory factor (IRF) 3, and IRF7, with subsequent expression of type I IFN and inflammatory cytokines [23]. For example, engagement of STING leads to its relocalization together with TANK- binding kinase 1 (TBK-1) from endoplasmic reticulum to the Golgi complex where TBK-1 phosphorylates IRF3 and IRF7, leading to the production of type I IFN [45] (Fig. 1).
IRFs in HSV infection.
Of the nine IRFs, IRF3 and IRF7 are the primary mediators of type I IFN expression downstream of multiple viral sensors. Interestingly, while IRF3 expression was dispensable for the control of HSV-1 replication in the cornea, IRF3 played a critical role in limiting HSV-1 spread through the central nervous system (CNS), with increased mortality, HSV-1 titers, and a significant drop in type I IFN expression within the CNS of IRF3−/− mice [55,59,60]. In contrast, IRF7 expression was important for the control of both peripheral and CNS HSV-1 infection. All IRF7−/− mice succumbed to infection accompanied by severe disease and significantly higher viral titers in the serum, cornea, trigeminal ganglia, and CNS when compared to IRF3−/− and control mice [60]. Combined deficiency of IRF-3 and IRF-7 revealed overlapping functions of these transcription factors in the control of HSV-1 infection [60].
Unfortunately, as with many other aspects of the innate immune system, the role of IRF3 and IRF7 is entirely unexplored in chronic HSV infection.
IRFs in gammaherpesvirus infection.
Similar to HSV-1, increased MHV68 acute titers were found in the lungs of IRF3−/− and IRF7−/− mice [61]. Interestingly, another member of the IRF family, IRF1, which is dispensable for expression of type I IFN during MHV68 infection [61,62], also attenuated acute MHV68 replication.
While the roles of IRF3 and IRF7 in chronic MHV68 infection are unknown, global loss of IRF1 showed that IRF1 is critical for the attenuation of chronic MHV68 infection and MHV68- but not LCMV-driven germinal center response [63]. Further, IRF1 protein levels are selectively decreased in EBV positive post transplant lymphoproliferative disease, suggesting that IRF1 may act as a tumor suppressor during EBV lymphomagenesis [63]. Surprisingly, while investigating the cell-type-dependent roles of IRF1, we have recently shown that B cell-intrinsic IRF-1 expression supports the establishment of MHV68 latency and MHV68-driven germinal center response, in part by increasing levels of activated tyrosine phosphatase SHP1, which plays a B cell-intrinsic proviral function during MHV68 infection [64,65]. These exciting results highlight pleiotropic, cell type-dependent roles of IRF1 during chronic gammaherpesvirus infection. Finally, another IRF family member, IRF2, was shown to directly suppress MHV68 latent gene expression [66].
Type I IFN signaling.
The culmination of viral sensing by the innate immune system is the release of type I IFN and its engagement of the type I IFN receptor (IFNAR). Activation of IFNAR leads to the assembly of the STAT1-STAT2-IRF-9 complex and transcription of hundreds of IFN stimulated genes (ISGs), with only a handful of ISGs attenuating replication of any particular virus, based on high throughput screens of antiviral activity [67] (Fig. 1).
IFNAR and STAT1 in HSV infection.
The STAT1-containing complex downstream of IFNAR is critical for the expression of ISGs. STAT1 deficiency in humans leads to disseminated HSV-1 infection and recurrent encephalitis [68]. Not surprisingly, IFNAR−/− mice manifested increased HSV-1 titers in the cornea and trigeminal ganglia [69,70]. Interestingly, while loss of type II IFN signaling (that also engages STAT1) had no impact on acute HSV-1 titers, combined type I and II IFN signaling deficiency resulted in high mortality and systemic HSV-1 dissemination, indicating that type II IFN signaling is needed for the control of systemic HSV-1 spread in the absence of IFNAR [70]. Similarly, loss of STAT1 led to significantly higher HSV titers, disease scores, and decreased survival [71]. Mice with neuronal-specific STAT1 deficiency rapidly succumbed to HSV-1 corneal infection, indicating that IFN-mediated control of HSV replication in peripheral tissues is not enough to ensure host survival [72].
Due to the critical role of type I IFN signaling during acute infection, the studies of chronic infection are limited to either low inoculum dose that leads to some survivors (for global genetic deficiency) or genetic models of cell type-specific deficiencies. Interestingly, and in contrast to the phenotypes observed during acute HSV-1 infection, mice with neuron-specific STAT1 deficiency had similar levels of latent HSV-1 genomes in the trigeminal ganglia with decreased viral reactivation upon ex vivo culture. This unexpected proviral phenotype was attributed to increased neuronal death in the absence of STAT1 [73].
IFNAR and STAT1 in gammaherpesvirus infection.
Similar to acute HSV infection, increasing doses of MHV68 inoculum led to decreased survival of IFNAR−/− mice with corresponding increase in acute viral titers in multiple organs [74,75]. Further, STAT1−/− mice were profoundly susceptible to acute MHV68 infection, as inoculation with 100 PFU of MHV68 resulted in all STAT1−/− mice succumbing to infection, as compared to 50% survival of IFNAR−/− animals under the same conditions [75].
Chronically infected IFNAR−/− mice display a dramatic increase in the levels of persistent MHV68 replication which is not fully controlled in survivors until day 21–28 post infection [66,75]. Importantly, MHV68 reactivation was increased in IFNAR−/− mice at 28 days post infection, and depletion of type I IFN in wild type mice at 21–28 days post infection stimulated MHV68 reactivation [75], indicating an important and sustained role of type I IFN in the suppression of chronic gammaherpesvirus infection. Of interest, MHV68 has evolved STAT1 binding elements in the promoter of RTA, a lytic switch protein; direct, STAT1-mediated repression of RTA and subsequent lytic replication is likely to play a role during both acute and chronic infection [76].
Conclusions and future perspectives.
We hope that, in addition to the literature discussion, this review highlights important issues to be explored in the future studies. Foremost of those is the traditional perception of innate immune responses being limited to the acute phase of virus infection. In contrast, emerging studies of herpesvirus and other chronic virus infections indicate that innate immune factors continue to exert their influence beyond the initial encounter with the virus. The role of classical innate immune system during chronic herpesvirus infection is an exciting and grossly understudied aspect of the antiviral immune response that calls for much more attention than it has been given. The interaction between classical innate immunity and viruses may be much more nuanced during chronic infection, as evidenced by the observation that neuron-specific deficiency of STAT1 decreases the efficiency of HSV-1 reactivation [73]. This observation and reports of proviral IFN function during chronic LCMV infection [21,22,77] raise an intriguing possibility that herpesviruses may usurp innate immune factors to facilitate aspects of chronic infection and regulate disease development. Finally, innate immune responses have to be fine-tuned in the context of individual virus infection. It is clear that, even for related alpha- and gammaherpesviruses, a unique combination of innate immune mechanisms controls the individual infection, a combination that is further modified by the infectious dose, route of infection, tissue- and cell type-specific immunological milieu, and myriad of other host and viral factors that have not been considered. Thus, one size does not fit all when it comes to predicting the effects of a given innate immune mechanism on acute and chronic virus infection.
Acknowledgments.
The space restriction of this review has, unfortunately, precluded discussion of many outstanding studies dissecting alpha- and gammaherpesvirus interactions with the innate immunity of the host. This work was supported by the American Cancer Society Postdoctoral Award (134165-PF-19-176-01-MPC, C.N.J.); CA183593 and CA203923 (V.L.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
This manuscript has not been submitted elsewhere and the authors declare no conflict of interest.
References.
- 1.McGeoch DJ, Rixon FJ, Davison AJ: Topics in herpesvirus genomics and evolution. Virus Res 2006, 117:90–104. [DOI] [PubMed] [Google Scholar]
- 2.Liesegang TJ: Herpes simplex virus epidemiology and ocular importance. Cornea 2001, 20:1–13. [DOI] [PubMed] [Google Scholar]
- 3.Whitley RJ, Roizman B: Herpes simplex virus infections. The Lancet 2001, 357:1513–1518. [DOI] [PubMed] [Google Scholar]
- 4.Simmons A: Clinical manifestations and treatment considerations of herpes simplex virus infection. J Infect Dis 2002, 186 Suppl 1:S71–77. [DOI] [PubMed] [Google Scholar]
- 5.Kollias CM, Huneke RB, Wigdahl B, Jennings SR: Animal models of herpes simplex virus immunity and pathogenesis. J Neurovirol 2015, 21:8–23. [DOI] [PubMed] [Google Scholar]
- 6.Cesarman E: Gammaherpesviruses and lymphoproliferative disorders. Annu Rev Pathol 2014, 9:349–372. [DOI] [PubMed] [Google Scholar]
- 7.Jha HC, Banerjee S, Robertson ES: The Role of Gammaherpesviruses in Cancer Pathogenesis. Pathogens 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roughan JE, Thorley-Lawson DA: The intersection of Epstein-Barr virus with the germinal center. J. Virol 2009, 83:3968–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sangster MY, Topham DJ, D’Costa S, Cardin RD, Marion TN, Myers LK, Doherty PC: Analysis of the virus-specific and nonspecific B cell response to a persistent B-lymphotropic gammaherpesvirus. J. Immunol 2000, 164:1820–1828. [DOI] [PubMed] [Google Scholar]
- 10.Babcock GJ, Decker LL, Freeman RB, Thorley-Lawson DA: Epstein-barr virus-infected resting memory B cells, not proliferating lymphoblasts, accumulate in the peripheral blood of immunosuppressed patients. J. Exp. Med 1999, 190:567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laichalk LL, Thorley-Lawson DA: Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. Journal of Virology 2005, 79:1296–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuppers R: B cells under influence: transformation of B cells by Epstein-Barr virus. Nat. Rev. Immunol 2003, 3:801–812. [DOI] [PubMed] [Google Scholar]
- 13.Hezaveh K, Kloetgen A, Bernhart SH, Mahapatra KD, Lenze D, Richter J, Haake A, Bergmann AK, Brors B, Burkhardt B, et al. : Alterations of microRNA and microRNA-regulated messenger RNA expression in germinal center B-cell lymphomas determined by integrative sequencing analysis. Haematologica 2016, 101:1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meerbach A, Wutzler P, Hafer R, Zintl F, Gruhn B: Monitoring of Epstein-Barr virus load after hematopoietic stem cell transplantation for early intervention in post-transplant lymphoproliferative disease. J. Med. Virol 2008, 80:441–454. [DOI] [PubMed] [Google Scholar]
- 15.Gluck B, Schmidtke M, Walther M, Meerbach A, Wutzler P: Simvastatin treatment showed no prophylactic effect in influenza virus-infected mice. J. Med. Virol 2013, 85:1978–1982. [DOI] [PubMed] [Google Scholar]
- 16.Houldcroft CJ, Kellam P: Host genetics of Epstein-Barr virus infection, latency and disease. Rev Med Virol 2015, 25:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Efstathiou S, Ho YM, Hall S, Styles CJ, Scott SD, Gompels UA: Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J. Gen. Virol 1990, 71:1365–1372. [DOI] [PubMed] [Google Scholar]
- 18.Tarakanova VL, Suarez FS, Tibbetts SA, Jacoby M, Weck KE, Hess JH, Speck SH, Virgin HW: Murine gammaherpesvirus 68 infection induces lymphoproliferative disease and lymphoma in BALB b2 microglobulin deficient mice. J Virol 2005, 79:14668–14679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Virgin HW, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH: Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol 1997, 71:5894–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barton E, Mandal P, Speck SH: Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annual review of immunology 2011, 29:351–397. [DOI] [PubMed] [Google Scholar]
- 21.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB: Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG: Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340:202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawai T, Akira S: The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature Immunology 2010, 11:373. [DOI] [PubMed] [Google Scholar]
- 24.Akira S, Uematsu S, Takeuchi O: Pathogen recognition and innate immunity. Cell 2006, 124:783–801. [DOI] [PubMed] [Google Scholar]
- 25.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW: Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 2004, 101:1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bochud PY, Magaret AS, Koelle DM, Aderem A, Wald A: Polymorphisms in TLR2 are associated with increased viral shedding and lesional rate in patients with genital herpes simplex virus Type 2 infection. J Infect Dis 2007, 196:505–509. [DOI] [PubMed] [Google Scholar]
- 27.Reinert LS, Harder L, Holm CK, Iversen MB, Horan KA, Dagnaes-Hansen F, Ulhoi BP, Holm TH, Mogensen TH, Owens T, et al. : TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J Clin Invest 2012, 122:1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, et al. : TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317:1522–1527. [DOI] [PubMed] [Google Scholar]
- 29.Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, Anguiano E, Sancho-Shimizu V, Lorenzo L, Pauwels E, et al. : Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med 2011, 208:2083–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lima GK, Zolini GP, Mansur DS, Freire Lima BH, Wischhoff U, Astigarraga RG, Dias MF, das Gracas Almeida Silva M, Bela SR, do Valle Antonelli LR, et al. : Toll-like receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection. Am J Pathol 2010, 177:2433–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M: Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2004, 103:1433–1437. [DOI] [PubMed] [Google Scholar]
- 32.Michaud F, Coulombe F, Gaudreault E, Kriz J, Gosselin J: Involvement of TLR2 in recognition of acute gammaherpesvirus-68 infection. PLoS. ONE 2010, 5:e13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gargano LM, Moser JM, Speck SH: Role for MyD88 signaling in murine gammaherpesvirus 68 latency. J. Virol 2008, 82:3853–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bussey KA, Murthy S, Reimer E, Chan B, Hatesuer B, Schughart K, Glaunsinger B, Adler H, Brinkmann MM: Endosomal Toll-Like Receptors 7 and 9 Cooperate in Detection of Murine Gammaherpesvirus 68 Infection. Journal of Virology 2019, 93:e01173–01118. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study found that during acute MHV68 infection loss of both TLR7 and TLR9 as well as host protein UNC93B resulted in higher acute viral titers. Further, during chronic MHV68 infection loss of the two TLRs led to an even greater increase in viral reactivation compared to TLR9 deficiency alone.
- 35.Guggemoos S, Hangel D, Hamm S, Heit A, Bauer S, Adler H: TLR9 contributes to antiviral immunity during gammaherpesvirus infection. J. Immunol 2008, 180:438–443. [DOI] [PubMed] [Google Scholar]
- 36.Gargano LM, Forrest JC, Speck SH: Signaling through Toll-like receptors induces murine gammaherpesvirus 68 reactivation in vivo. J. Virol 2009, 83:1474–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, et al. : IFI16 is an innate immune sensor for intracellular DNA. Nature immunology 2010, 11:997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu J, Chen ZJ: Innate Immune Sensing and Signaling of Cytosolic Nucleic Acids. Annual Review of Immunology 2014, 32:461–488. [DOI] [PubMed] [Google Scholar]
- 39.Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJJ: Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal immunology 2012, 5:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reinert LS, Lopušná K, Winther H, Sun C, Thomsen MK, Nandakumar R, Mogensen TH, Meyer M, Vægter C, Nyengaard JR, et al. : Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nature communications 2016, 7:13348–13348. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study showed that the cGAS-STING pathway is required for type 1 IFN production and further antiviral programs in microglia during HSV-1 infection of the central nervous system.
- 41.Li X-D, Wu J, Gao D, Wang H, Sun L, Chen ZJ: Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science (New York, N.Y.) 2013, 341:1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Zhao J, Xu S, Li J, He S, Zeng Y, Xie L, Xie N, Liu T, Lee K, et al. : Species-Specific Deamidation of cGAS by Herpes Simplex Virus UL37 Protein Facilitates Viral Replication. Cell host & microbe 2018, 24:234–248.e235. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study found that the HSV tegument protein UL37 deaminates cGAS in both mice and humans, which results reduced innate immune activation and antagonize cGAS-mediated immune response. Deamidation of cGAS results in loss of dsDNA sensing and restriction of HSV-1 infection. Further, deamidation prevents cGAMP synthesis.
- 43.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, et al. : Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505:691–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li W, Avey D, Fu B, Wu JJ, Ma S, Liu X, Zhu F: Kaposi’s Sarcoma-Associated Herpesvirus Inhibitor of cGAS (KicGAS), Encoded by ORF52, Is an Abundant Tegument Protein and Is Required for Production of Infectious Progeny Viruses. J Virol 2016, 90:5329–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rathinam VA, Fitzgerald KA: Innate immune sensing of DNA viruses. Virology 2011, 411:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gimenez F, Bhela S, Dogra P, Harvey L, Varanasi SK, Jaggi U, Rouse BT: The inflammasome NLRP3 plays a protective role against a viral immunopathological lesion. J Leukoc Biol 2016, 99:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maruzuru Y, Ichinohe T, Sato R, Miyake K, Okano T, Suzuki T, Koshiba T, Koyanagi N, Tsuda S, Watanabe M, et al. : Herpes Simplex Virus 1 VP22 Inhibits AIM2-Dependent Inflammasome Activation to Enable Efficient Viral Replication. Cell Host Microbe 2018, 23:254–265.e257. [DOI] [PubMed] [Google Scholar]; *This study showed that the HSV-1 tegument protein VP22 blocks the oligomerization of AIM-2 to prevent inflammasome activation.
- 48.Cieniewicz B, Dong Q, Li G, Forrest JC, Mounce BC, Tarakanova VL, van der Velden A, Krug LT: Murine Gammaherpesvirus 68 Pathogenesis Is Independent of Caspase-1 and Caspase-11 in Mice and Impairs Interleukin-1beta Production upon Extrinsic Stimulation in Culture. J Virol 2015, 89:6562–6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun C, Schattgen SA, Pisitkun P, Jorgensen JP, Hilterbrand AT, Wang LJ, West JA, Hansen K, Horan KA, Jakobsen MR, et al. : Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. Journal of immunology (Baltimore, Md. : 1950) 2015, 194:1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Darrah EJ, Jondle CN, Johnson KE, Xin G, Lange PT, Cui W, Olteanu H, Tarakanova VL: Conserved gammaherpesvirus protein kinase selectively promotes irrelevant B cell responses. J Virol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lawlor KE, Feltham R, Yabal M, Conos SA, Chen KW, Ziehe S, Grass C, Zhan Y, Nguyen TA, Hall C, et al. : XIAP Loss Triggers RIPK3- and Caspase-8-Driven IL-1beta Activation and Cell Death as a Consequence of TLR-MyD88-Induced cIAP1-TRAF2 Degradation. Cell Rep 2017, 20:668–682. [DOI] [PubMed] [Google Scholar]
- 52.Latour S, Winter S: inherited immunodeficiencies with High Predisposition to epstein–Barr virus-Driven Lymphoproliferative Diseases. Frontiers in Immunology 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study found that deficency of human XIAP predisposes an individual to EBV-driven lymphomas.
- 53.Haneklaus M, Gerlic M, Kurowska-Stolarska M, Rainey AA, Pich D, McInnes IB, Hammerschmidt W, O’Neill LA, Masters SL: Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J Immunol 2012, 189:3795–3799. [DOI] [PubMed] [Google Scholar]
- 54.Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, Ting JP, Damania B: Discovery of a viral NLR homolog that inhibits the inflammasome. Science 2011, 331:330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, et al. : IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434:772–777. [DOI] [PubMed] [Google Scholar]
- 56.Ishikawa H, Ma Z, Barber GN: STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461:788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parker ZM, Murphy AA, Leib DA: Role of the DNA Sensor STING in Protection from Lethal Infection following Corneal and Intracerebral Challenge with Herpes Simplex Virus 1. Journal of virology 2015, 89:11080–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Royer DJ, Carr DJJ: A STING-dependent innate-sensing pathway mediates resistance to corneal HSV-1 infection via upregulation of the antiviral effector tetherin. Mucosal immunology 2016, 9:1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study showed that during HSV-1 infection, STING is needed for tetherin expression in the corneal epithelium and tetherin is needed to restrict local viral spread and neuroinvasion.
- 59.Menachery VD, Leib DA: Control of herpes simplex virus replication is mediated through an interferon regulatory factor 3-dependent pathway. J. Virol 2009, 83:12399–12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murphy AA, Rosato PC, Parker ZM, Khalenkov A, Leib DA: Synergistic control of herpes simplex virus pathogenesis by IRF-3, and IRF-7 revealed through non-invasive bioluminescence imaging. Virology 2013, 444:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mboko WP, Rekow MM, Ledwith MP, Lange PT, Schmitz KE, Anderson S, Tarakanova VL: IRF-1 and type I interferon cooperate to control acute gammaherpesvirus infection. J. Virol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study found that IRF-1, 3, & 7 all play an antiviral role during acute MHV68 infection in the lungs.
- 62.Mboko WP, Mounce BC, Emmer J, Darrah E, Patel SB, Tarakanova VL: Interferon regulatory factor-1 restricts gammaherpesvirus replication in primary immune cells. J. Virol 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mboko WP, Olteanu H, Ray A, Xin G, Darrah EJ, Kumar SN, Kulinski JM, Cui W, Dittel BN, Gauld SB: Tumor suppressor interferon-regulatory factor 1 counteracts the germinal center reaction driven by a cancer-associated gammaherpesvirus. Journal of virology 2016, 90:2818–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study showed that IRF-1 is needed to regulate the germinal center response during chronic MHV68 infection.
- 64.Jondle CN, Johnson KE, Uitenbroek AA, Sylvester PA, Nguyen C, Cui W, Tarakanova VL: B cell-intrinsic expression of Interferon Regulatory Factor 1 supports chronic murine gammaherpesvirus 68 infection. Journal of Virology 2020:JVI.00399–00320. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study found that contrary to what was observed in global IRF-1 deficency, where IRF-1 plays an antiviral role by controlling the germinal center response during chronic MHV68 infection, B cell-intrinsic IRF-1 expression supports chronic MHV68 infection.
- 65.Johnson KE, Lange PT, Jondle CN, Volberding PJ, Lorenz UM, Cui W, Dittel BN, Tarakanova VL: B Cell-Intrinsic SHP1 Expression Promotes the Gammaherpesvirus-Driven Germinal Center Response and the Establishment of Chronic Infection. J Virol 2019, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mandal P, Krueger BE, Oldenburg D, Andry KA, Beard RS, White DW, Barton ES: A gammaherpesvirus cooperates with interferon-alpha/beta-induced IRF2 to halt viral replication, control reactivation, and minimize host lethality. PLoS. Pathog 2011, 7:e1002371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM: A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472:481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, et al. : Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet 2003, 33:388–391. [DOI] [PubMed] [Google Scholar]
- 69.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW: Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med 1999, 189:663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luker GD, Prior JL, Song J, Pica CM, Leib DA: Bioluminescence imaging reveals systemic dissemination of herpes simplex virus type 1 in the absence of interferon receptors. J Virol 2003, 77:11082–11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pasieka TJ, Cilloniz C, Lu B, Teal TH, Proll SC, Katze MG, Leib DA: Host responses to wild-type and attenuated herpes simplex virus infection in the absence of Stat1. Journal of virology 2009, 83:2075–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosato PC, Leib DA: Neuronal Interferon Signaling Is Required for Protection against Herpes Simplex Virus Replication and Pathogenesis. PLoS pathogens 2015, 11:e1005028–e1005028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rosato PC, Katzenell S, Pesola JM, North B, Coen DM, Leib DA: Neuronal IFN signaling is dispensable for the establishment of HSV-1 latency. Virology 2016, 497:323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dutia BM, Allen DJ, Dyson H, Nash AA: Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology 1999, 261:173–179. [DOI] [PubMed] [Google Scholar]
- 75.Barton ES, Lutzke ML, Rochford R, Virgin HW: Alpha/beta interferons regulate murine gammaherpesvirus latent gene expression and reactivation from latency. Journal of Virology 2005, 79:14149–14160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goodwin MM, Canny S, Steed A, Virgin HW: Murine gammaherpesvirus 68 has evolved gamma interferon and stat1-repressible promoters for the lytic switch gene 50. J. Virol 2010, 84:3711–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fallet B, Narr K, Ertuna YI, Remy M, Sommerstein R, Cornille K, Kreutzfeldt M, Page N, Zimmer G, Geier F, et al. : Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Sci Immunol 2016, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]