

Abstract
Background
Alport syndrome (ATS) is a hereditary nephritis with hereditary and clinical heterogeneity; the early clinical symptoms are atypical, which can easily lead to misdiagnosis. The proband, a 6‐year‐old girl, was found to have microscopic hematuria, proteinuria, and visual impairment at about 5 years old; the results of renal pathological examination revealed mesangial hyperplasia and IgA deposition. The proband's father exhibited gross hematuria, eye swelling, and bilateral hearing loss after the age of 5, renal function progressively decreased, and he underwent right renal allograft at the age of 23 due to renal failure. The proband and her father were clinically diagnosed as IgA nephropathy and chronic glomerulonephritis, respectively.
Methods
For proband, targeted exome capture sequencing was performed using the Targeted Exome Capture Kit; this kit targets 162 genes known to cause renal diseases. The identified mutation was confirmed and analyzed for cosegregation by Sanger sequencing in other family members whose gDNA was available.
Results
Targeted exome capture sequencing revealed a novel heterozygous variant (NM_000495, c.697delG, p.G233fs) in the COL4A5 gene of the proband; the variant was inherited from her father. The variant was likely pathogenic according to the criteria of the American College of Medical Genetics and Genomics.
Conclusion
In this study, we first report a c.697delG mutation of COL4A5 in two patients presumed IgA nephropathy and chronic glomerulonephritis. This study emphasizes on the diagnostic value of next‐generation sequencing for hereditary kidney diseases to help in their timely and cost‐effective diagnosis, determine appropriate treatments, and promote genetic counseling.
Keywords: alport syndrome, COL4A5, next‐generation sequencing, novel mutation, targeted exome capture sequencing
1. INTRODUCTION
Alport syndrome (ATS) is a hereditary nephritis characterized by persistent microscopic hematuria, recurrent gross hematuria, proteinuria, and progressive renal dysfunction. Additionally, it is often accompanied by ocular anomalies and sensorineural deafness, affecting 1 in 5000‐50000 individuals. 1 , 2 ATS is a heterogeneous disease caused by mutations in genes encoding the alpha3, alpha4, or alpha5 chains of type IV collagen, the main structural component of basement membrane. Different types of mutations result indifferent inheritance patterns of ATS. Approximately 85% of ATS cases are X‐linked and about 15% are autosomal recessive, while autosomal‐dominant inheritance is rare. 3
X‐linked Alport syndrome‐1 (ATS1; OMIM 301050) is caused by mutation in the gene encoding the alpha‐5 chain of basement membrane collagen type‐IV (COL4A5; OMIM 303630) on Xq22. Male patients with ATS1 usually exhibit more severe clinical symptoms, faster disease progression, and earlier onset of renal failure than female patients. 4 , 5 , 6 Hearing loss was present in 67%‐79% of patients, but typical ocular abnormalities were not common in ATS1, accounting for about 30%‐40%. 7 , 8 Early clinical manifestations in ATS patients include only hematuria, with or without proteinuria. The early clinical symptoms and pathological findings of renal biopsy are atypical and non‐specific, making misdiagnosis in ATS patients common. 9 In the current study, we present 2 patients in a Chinese family with presumed diagnosis of IgA nephropathy and chronic glomerulonephritis that carried a frameshift mutation in COL4A5, which was discovered by next‐generation sequencing (NGS).
2. MATERIALS AND RESULTS
2.1. Patient
The proband (case IV‐1) was a 6‐year‐old girl, delivered by cesarean section at 38 weeks of gestation. At birth, the girl weighed 2755 g, length was 49 cm, and the Apgar score was normal. 10 Food intake and growth were also normal after birth. Eleven months ago, the subject underwent a clinical examination due to frequent urination. Microscopic hematuria (red blood cell >4 per high‐power visual field), proteinuria (urinary protein: 0.2‐1.0 g/L), increased urinary transferrin (0.38 mg/dL, reference value should be less than 0.2 mg/dL), increased urinary microalbumin (11.3 mg/dL, reference value should be less than 1.9 mg/dL), and increased ratio of urinary protein to creatinine (0.66, reference value should be less than 0.2) were observed using urine analysis. The values of urea and creatinine in serum were normal, and no significant abnormalities in both kidneys and ureters were observed using ultrasonography. Mild proliferation of mesangial cells and a thin glomerular basement membrane (less than 200 nm) were observed in the pathological examination of kidneys. Furthermore, IgA, IgG, and C1q tests using immunofluorescence staining of glomerular vascular wall were positive. The results of pathological examination are shown in Figure 1. Therefore, the subject was clinically diagnosed with IgA nephropathy. In addition, the patient's bilateral vision slightly decreased 6 months post‐examination.
Figure 1.
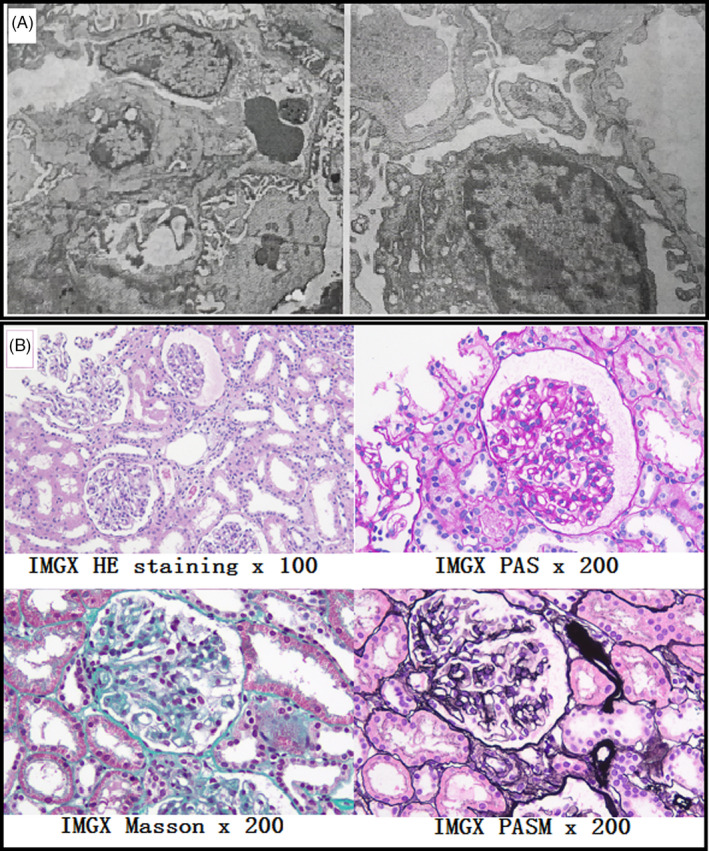
Renal pathological examination results of case IV‐1. Mild proliferation of mesangial cells and a thin glomerular basement membrane (less than 200 nm) were observed in the pathological examination of kidneys (A). Furthermore, IgA, IgG, and C1q tests using immunofluorescence staining of glomerular vascular wall were positive (B)
The proband's father (case III‐2) exhibited normal growth and development before the age of 5. However, after the age of 5, he exhibited gross hematuria, eye swelling, and bilateral hearing loss; later, he was clinically diagnosed with chronic glomerulonephritis. Renal function progressively decreased and the subject underwent right renal allograft at the age of 23 due to renal failure. Audiological examination revealed sensorineural deafness, more severe on the right side. At the time of our study, the subject wore hearing aids and communicated normally.
Other members of the family exhibited no clinical symptoms such as hematuria, proteinuria, hearing loss, and eye diseases. The proband's paternal grandfather (case II‐1) died of cerebral hemorrhage at the age of 60. The proband's paternal grandmother (case II‐2) was aged 57 and in good health that the time of our study. Maternal grandfather (case I‐1) and grandmother (case I‐2) of the proband's father died at the age of 70 and 80, respectively; however, the cause of their death was unclear. The pedigree of the family is shown in Figure 2. The study protocol was approved by the Local Ethics Committee of West China Second University Hospital of Sichuan University, and patient has provided informed consent for publication of the case.
Figure 2.
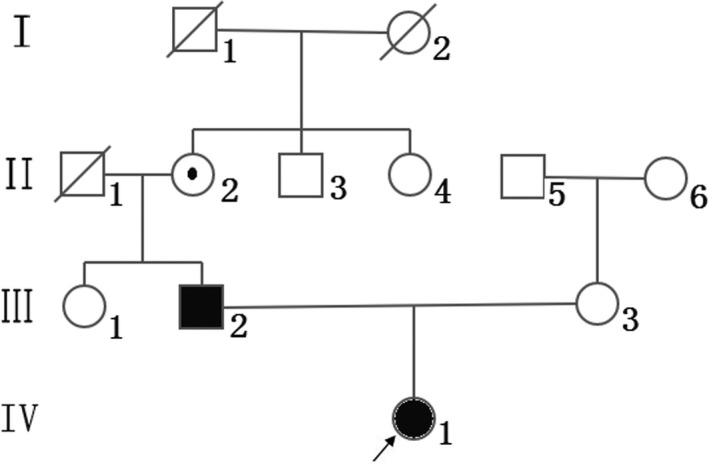
Pedigree of the present family. Squares represent males and circles represent females. Black symbols denote patients, and open symbols denote healthy individuals. The proband is indicated by an arrow. Circle‐enclosed dot indicates asymptomatic female carriers. Diagonal lines denote deceased individuals
2.2. Targeted exome capture sequencing and Sanger sequencing
Genomic DNA (gDNA) was isolated from fresh EDTA peripheral blood using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer's protocol. The gDNA concentration and purity were detected using a NanoDrop 2000 UV‐vis spectrophotometer (Thermo Fisher Scientific). For case IV‐1, targeted exome capture sequencing was performed using the MGP009 Targeted Exome Capture Kit (MyGenostics) according to the manufacturer's protocol; this kit targets 162 genes known to cause renal diseases (Appendix S1). The enrichment libraries were sequenced using a NextSeq 500 sequencer (Illumina). The clinical significance of the variants was interpreted according to the standards and guidelines for the interpretation of sequence variants recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. 11 Combined with the Sanger sequencing results and phenotypes of the other family members, we classified the pathogenicity of variants into five categories: benign variants, likely benign variants, uncertain significance, likely pathogenic variants, and pathogenic variants.
Targeted exome capture sequencing revealed 1280 genetic variants of case IV‐1, the data results of NGS meet the quality control standard, and the details are shown in Table 1. A novel heterozygous variant (NM_000495, c.697delG) in the COL4A5 gene was identified. The mutation was further confirmed by Sanger sequencing. Case III‐2 showed hemizygous mutation (delG) at the same site, and case II‐2 showed heterozygous mutation (G/delG) at the same site. The other healthy members (cases III‐1 and III‐3) showed no variation at this site. The results of Sanger sequencing are shown in Figure 3.
Table 1.
The quality control results of targeted next‐generation sequencing
| Data category | Results |
|---|---|
| Raw_data_bases (Mb) | 1239.63 |
| Clean_data_bases (Mb) | 1187.49 |
| Aligned_bases (Mb) | 1171.71 |
| Aligned (%) | 98.67 |
| Initial bases on target (n) | 544,632 |
| Base covered on target (n) | 544,093 |
| Coverage of target region (%) | 99.90 |
| Effective bases on target (n) | 436,541,708 |
| Fraction of effective bases on target (%) | 37.26 |
| Average sequencing depth on target (n) | 801 |
| Fraction of target covered with at least 20× (%) | 98.79 |
Figure 3.
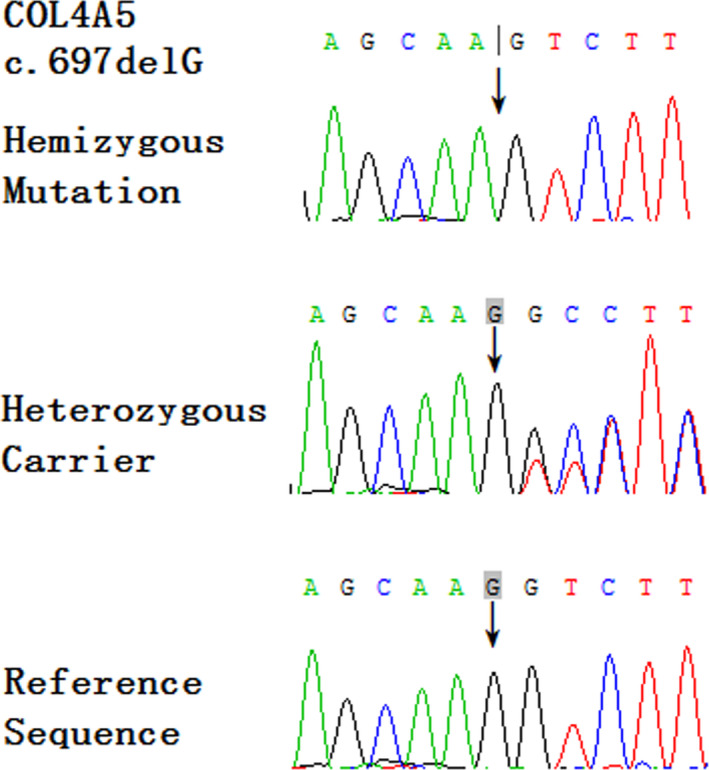
Sequence analysis of the COL4A5 gene in affected male (hemizygous mutation), affected female, and asymptomatic female carrier (heterozygous mutation) and other asymptomatic members (reference sequence). Arrows indicate the positions of the novel mutations (c.697). COL4A5 = collagen type‐IV alpha 5 chain
3. DISCUSSION
The COL4A5 gene contains 250 kb of genomic DNA and 51 exons, encoding a 6.5 kb transcript. 12 In the present study, we identified an inherited novel frameshift mutation in the exon 13 (c.697delG, p.G233fs) of a Chinese patient. The c.697delG mutation causes a substitution of the 233rd amino acid, changing it from glycine to valine and converting the 253rd codon to a stop codon. The COL4A5 gene encodes a protein (collagen alpha‐5(IV) chain) of 1685 amino acids (NM_000495). Frameshift mutation can often be assumed to disrupt gene function, leading to absence of the gene product due to lack of transcription or nonsense‐mediated mRNA decay of an altered transcript. 11 Therefore, the c.697delG mutation may cause collagen alpha‐5(IV) chain dysfunction, and the variant was likely pathogenic according to the criteria of the American College of Medical Genetics and Genomics. 11
Alport syndrome‐1 is a type of basement membrane nephropathy with clinical heterogeneity. Male patients usually develop end‐stage renal disease between the age of 20 and 30. They generally exhibit poor disease prognosis and often require dialysis and kidney transplantation. Various manifestations were observed with renal histopathology, and only mild and non‐specific manifestations were observed with light microscopy. Moreover, electron microscopic analysis exhibits atypical results in the early stage of the disease, while the immunohistochemical staining of type‐IV collagen alpha chains may reveal false‐positive results. Characteristic manifestations such as uneven thickness, stratification, and tearing of glomerular basement membrane are observed under electron microscopy only after significant disease progression. 13 Therefore, in the early stage of the disease, ATS is easily confused with other kidney diseases such as thin basement membrane nephropathy, focal segmental glomerulosclerosis, and IgA nephropathy, resulting in misdiagnosis or missed diagnosis.
In this family, the proband (case IV‐1) was misdiagnosed as IgA nephropathy due to its slight clinical manifestations such as microscopic hematuria and proteinuria, and pathological examinations revealing mesangial hyperplasia and IgA deposition. Case III‐2 was clinically diagnosed with glomerulonephritis at the age of 8. His condition progressively worsened and eventually developed into renal failure. Lack of timely genetical testing resulted in misdiagnosis for several years. The mutation (c.697delG) was inherited from case II‐2. She is 57 years old and exhibits no clinical symptoms such as gross hematuria, hearing loss, and eye lesions. No comparable clinical symptoms were observed in other members of the family. The difference in clinical phenotypes of X‐linked hereditary diseases is related to the inactivation of X chromosome. There is a positive correlation between the renal phenotypes and the mutant mRNA expression of COL4A5 in female patients with ATS1. Therefore, the severity of clinical symptoms varies among female carriers. 14 , 15 For asymptomatic carriers, we suggest clinicians to pay close attention to the clinical symptoms of the carriers. In addition, we recommend family members to undergo genetic testing and clinical examination for early identification and treatment of the affected family members, including prenatal diagnosis for identifying high‐risk fetuses.
For male ATS1 patients, in case of two different genes of MYH9, COL4A3, COL4A4, or COL4A5 having pathogenic mutations, the clinical manifestations are usually more serious compared with monogenic variants of 1 of these genes. However, there was no genotype–phenotype correlation in female ATS1, and no obvious modifier genes were detected in most of the clinically severe female ATS1 patients. 16 , 17 , 18 Moreover, the severity of mutations is directly associated with the severity of the disease in males with ATS1. Missense mutations that cause glycine substitution are usually associated with the less severe disease form, while frameshift mutations, nonsense mutations, and other mutations that lead to chain termination usually result in more severe phenotypes. 18 , 19 , 20 Therefore, for kidney diseases, especially for those with atypical clinical symptoms and pathological findings, we recommend early gene testing to facilitate clear diagnosis, identification of gene mutation types, analysis of prognosis, and formulation of appropriate treatment plans. The pathogenic genes COL4A3, COL4A4, and COL4A5 of ATS contain 52, 48, and 51 exons, respectively, and no mutation hotspots have been discovered. 19 , 20 The traditional Sanger sequencing method is expensive and time‐consuming for the detection of pathogenic gene mutations. Conversely, targeted exome capture sequencing has advantages in detecting pathogenic genes associated with hereditary kidney diseases owing to its low cost and high throughput. 21
4. CONCLUSIONS
Our study is the first to report an ATS‐causing c.697delG mutation on exon 13 of the COL4A5 gene. This study emphasizes on the diagnostic value of NGS for hereditary kidney diseases to help in their timely and cost‐effective diagnosis, determine appropriate treatments, and promote genetic counseling.
Supporting information
Appendix S1
Zhu Q, Zhou C, Wang J. A novel frame‐shift mutation of COL4A5 in a Chinese family with presumed IgA nephropathy and chronic glomerulonephritis. J Clin Lab Anal. 2020;34:e23558 10.1002/jcla.23558
Funding information
This study was funded by grants from the Program of Science and Technology Department of Sichuan Province (No. 2020YFS0095).
DATA AVAILABILITY STATEMENT
All the information about the case report is available from the corresponding author on reasonable request.
REFERENCES
- 1. Savige J. Alport syndrome: its effects on the glomerular filtration barrier and implications for future treatment. J Physiol. 2014;592(18):4013‐4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Levy M, Feingold J. Estimating prevalence in single‐gene kidney diseases progressing to renal failure. Kidney Int. 2000;58(3):925‐943. [DOI] [PubMed] [Google Scholar]
- 3. Kashtan CE. Alport syndrome: an inherited disorder of renal, ocular, and cochlear basement membranes. Medicine (Baltimore). 1999;78(5):338‐360. [DOI] [PubMed] [Google Scholar]
- 4. Temme J, Peters F, Lange K, et al. Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of X‐chromosomal and autosomal recessive Alport mutations. Kidney Int. 2012;81(8):779‐783. [DOI] [PubMed] [Google Scholar]
- 5. Muckova P, Wendler S, Rubel D, et al. Preclinical alterations in the serum of COL(IV)A3(‐)/(‐) mice as early biomarkers of Alport syndrome. J Proteome Res. 2015;14(12):5202‐5214. [DOI] [PubMed] [Google Scholar]
- 6. Vivante A, Calderon‐Margalit R, Skorecki K. Hematuria and risk for end‐stage kidney disease. Curr Opin Nephrol Hypertens. 2013;22(3):325‐330. [DOI] [PubMed] [Google Scholar]
- 7. Jais JP, Knebelmann B, Giatras I, et al. X‐linked Alport syndrome: natural history in 195 families and genotype‐ phenotype correlations in males. J Am Soc Nephrol. 2000;11(4):649‐657. [DOI] [PubMed] [Google Scholar]
- 8. Bekheirnia MR, Reed B, Gregory MC, et al. Genotype‐phenotype correlation in X‐linked Alport syndrome. J Am Soc Nephrol. 2010;21(5):876‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. International Alport Mutation Consortium , Savige J, Ars E, et al. DNA variant databases improve test accuracy and phenotype prediction in Alport syndrome. Pediatr Nephrol. 2014;29(6):971‐977. [DOI] [PubMed] [Google Scholar]
- 10. Casey BM, McIntire DD, Leveno KJ. The continuing value of the Apgar score for the assessment of newborn infants. N Engl J Med. 2001;3:467‐471. [DOI] [PubMed] [Google Scholar]
- 11. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou J, Leinonen A, Tryggvason K. Structure of the human type IV collagen COL4A5 gene. J Biol Chem. 1994;269(9):6608‐6614. [PubMed] [Google Scholar]
- 13. Heidet L, Gubler MC. The renal lesions of Alport syndrome. J Am Soc Nephrol. 2009;20(6):1210‐1215. [DOI] [PubMed] [Google Scholar]
- 14. Guo C, Van Damme B, Vanrenterghem Y, Devriendt K, Cassiman JJ, Marynen P. Severe alport phenotype in a woman with two missense mutations in the same COL4A5 gene and preponderant inactivation of the X chromosome carrying the normal allele. J Clin Invest. 1995;95(4):1832‐1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brown RM, Brown GK. X chromosome inactivation and the diagnosis of X linked disease in females. J Med Genet. 1993;30(3):177‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Strasser K, Hoefele J, Bergmann C, et al. COL4A5‐associated X‐linked Alport syndrome in a female patient with early inner ear deafness due to a mutation in MYH9 . Nephrol Dial Transplant. 2012;27(11):4236‐4240. [DOI] [PubMed] [Google Scholar]
- 17. Mencarelli MA, Heidet L, Storey H, et al. Evidence of digenic inheritance in Alport syndrome. J Med Genet. 2015;52(3):163‐174. [DOI] [PubMed] [Google Scholar]
- 18. Yamamura T, Nozu K, Fu XJ, et al. Natural history and genotype‐phenotype correlation in female X‐linked Alport syndrome. Kidney Int Rep. 2017;2(5):850‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knebelmann B, Breillat C, Forestier L, et al. Spectrum of mutations in the COL4A5 collagen gene in X‐linked Alport syndrome. Am J Hum Genet. 1996;59(6):1221‐1232. [PMC free article] [PubMed] [Google Scholar]
- 20. Crockett DK, Pont‐Kingdon G, Gedge F, Sumner K, Seamons R, Lyon E. The Alport syndrome COL4A5 variant database. Hum Mutat. 2010;31(8):E1652‐E1657. [DOI] [PubMed] [Google Scholar]
- 21. Zhu F, Li W, Li Z, Zhu H, Xiong J. Identification of a novel COL4A4 variant in compound‐heterozygous state in a patient with Alport syndrome and histological Findings Similar to Focal Segmental Glomerulosclerosis (FSGS). Front Genet. 2019;9:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
All the information about the case report is available from the corresponding author on reasonable request.