

Abstract
Background
The extremely high genetic heterogeneity of hearing loss due to diverse group of genes encoding proteins required for development, function, and maintenance of the complex auditory system makes the genetic diagnosis of this disease challenging. Up to now, 121 different genes have been identified for nonsyndromic hearing loss (NSHL), of which 76 genes are responsible for the most common forms of NSHL, autosomal recessive nonsyndromic hearing loss (ARNSHL).
Methods
After excluding mutations in the most common ARNSHL gene, GJB2, by Sanger sequencing, genetic screening for a panel of genes responsible for hereditary hearing impairment performed in 9 individuals with ARNSHL from unrelated Iranian consanguineous pedigrees.
Results
One compound heterozygote and eight homozygote variants, of which five are novel, were identified: CDH23:p.(Glu1970Lys), and p.(Ala1072Asp), GIPC3:p.(Asn82Ser), and (p.Thr41Lys), MYO7A:p.[Phe456Phe]; p.[Met708Val], and p.(Gly163Arg), TECTA:p.(Leu17Leufs*19), OTOF:c.1392+1G>A, and TRIOBP:p.(Arg1068*). Sanger sequencing confirmed the segregation of the variants with the disease in each family.
Conclusion
Finding more variants and expanding the spectrum of hearing impairment mutations can increase the diagnostic value of molecular testing in the screening of patients and can improve counseling to minimize the risk of having affected children for at risk couples.
Keywords: autosomal recessive nonsyndromic hearing loss (ARNSHL), Iranian consanguineous pedigrees, next‐generation sequencing (NGS), novel variants
In the present study, nine unrelated Iranian consanguineous families with at least one affected individual were tested by next‐generation sequencing (NGS) of 127 known deafness genes. In this report, one compound heterozygote and eight homozygote variants, of which five are novel, were identified: CDH23:p.(Glu1970Lys), and p.(Ala1072Asp), GIPC3:p.(Asn82Ser), and (p.Thr41Lys), MYO7A:p.[Phe456Phe]; p.[Met708Val], and p.(Gly163Arg),TECTA:p.(Leu17Leufs*19), OTOF:c.1392+1G>A, and TRIOBP:p.(Arg1068*). Sanger sequencing confirmed the segregation of the variants with the disease in each family.

1. INTRODUCTION
Hearing loss is the most common sensory disorder in humans. It is estimated that 360 million people worldwide are suffering from hearing loss. 1 The frequency of congenital deafness ranges from 1 to 2 per 1000 in Western countries, while in Iran it reaches to 1 in 166; in other words, the prevalence of deafness in Iran is estimated to be 2‐3 times higher than the other parts of the world. 2 , 3 Iran as one of the consanguinity belt countries, with 38.6% rate of consanguineous marriage which is culturally and socially favored, among the world's most heterogeneous populations, has received a great deal of attention as a potential risk factor for many autosomal recessive disorders including autosomal recessive nonsyndromic hearing loss (ARNSHL). 4 , 5
Genetic forms of deafness responsible for more than half of hearing loss cases have been shown to have diverse etiologies, and it is estimated that approximately 1% of all human genes are involved in the biology of hearing. 6 Congenital hearing loss is the second most common disorder following intellectual impairment in Iran. 3 Malfunctions of the cochlea and inner ear due to dysfunction of proteins involved in mechanisms related to the adhesion of hair cells, intracellular transport, neurotransmitter release, ionic homeostasis, and cytoskeleton of hair cells can cause hearing impairment. A defect in any part of these mechanisms can cause the disease. 7 The extremely genetic heterogeneity of deafness can be due to the complexity of the auditory system, which requires coordination of multiple processes controlled by the interaction of various proteins coded by several hundred genes. 6 , 8 Up to now, 121 genes have been implicated in the pathogenesis of nonsyndromic deafness in which about 76 genes have been reported to cause ARNSHL (https://hereditaryhearingloss.org/). Causative genes can be classified by their molecular function, homeostasis, hair cell structure, transcription factors, cytokinesis, extracellular matrix, mitochondrial, and other/unknown. 6 , 9
Previous studies have shown mutations in GJB2, SLC26A4, and TECTA genes as the most common cause of NSHL in the Iranian population followed by MYO15A, ILDR1, TMC1, PJVK, LRTOMT, MYO7A, OTOF, and MARVELD2. 10
In spite of tremendous heterogeneity, recently in a cohort of 302 GJB2‐negative Iranian probands with ARNSHL, over half of all genetic diagnoses (52%) have been shown to be due to the causative variants in only five genes (SLC26A4, MYO15A, MYO7A, CDH23, and PCDH15). 1 In the remaining pedigrees, mutations in 35 other genes including GIPC3, TECTA, OTOF, and TRIOBP were identified. 1
In the present study, 9 unrelated Iranian families with at least one affected individual who were negative for mutations in GJB2 were screened by next‐generation sequencing (NGS) for 127 known deafness genes. In this report, variants in 6 different genes including three variants in MYO7A, two variants in CDH23 and GIPC3, and one variant in TECTA, OTOF, and TRIOBP were identified.
2. MATERIALS AND METHODS
2.1. Patients and ethics statement
In this study, nine Iranian families with at least one hearing impaired member who was referred to the Department of Medical Genetics, DeNA Laboratory, Tehran, Iran, were investigated. All clinical data of hearing impaired patients in these families were obtained at DeNA Laboratory using a uniform questionnaire according to ACMG guidelines for the etiologic diagnosis of congenital hearing loss, included consanguinity and hearing status of the parents and siblings, age of onset, one or both ears deafness, syndromic or nonsyndromic deafness, presence of accompanying symptoms such as visual anomalies, endocrine abnormalities, thyroid disorders, skin problems, exposure to environmental factors like taking drugs or drinking alcohol during pregnancy, and intrauterine infections. 11 The hearing impaired individuals in these pedigrees had no obvious vestibular dysfunction, retinal degeneration, or report of other anomalies, suggesting that the families are suffering from nonsyndromic deafness. Evaluation of the deaf patients showed prelingual bilateral nonsyndromic sensorineural hearing loss in all cases. Medical investigations included otoscopy and physical examination by an otolaryngologist and a geneticist. According to audiological evaluations, the severity of deafness varied among patients, ranging from mild to profound (Table 1).
Table 1.
Clinical details of hearing impaired patients
| Family ID | Individual ID | Gender | Hearing impairment | Other clinical features | Family history | Consanguinity | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Onset | Type | Severity a | cochlear implants | |||||||
| Family 1 | IV1 | Male | Prelingual | Sensorineural | Bilateral | Severe | Yes | No | No | Yes |
| Family 2 | II2 | Female | Prelingual | Sensorineural | Bilateral | Severe | No | No | No | Yes |
| Family 3 | II4 | Female | Prelingual | Sensorineural | Bilateral | Severe | No | No | No | Yes |
| Family 4 | IV1 | Female | Prelingual | Sensorineural | Bilateral | Profound | Yes | No | No | Yes |
| Family 5 | IV6 | Male | Prelingual | Sensorineural | Bilateral | Moderate | No | No | Yes | Yes |
| Family 6 | IV5 | Female | Prelingual | Sensorineural | Bilateral | Profound | Yes | No | Yes | Yes |
| Family 7 | IV1 | Male | Prelingual | Sensorineural | Bilateral | Moderate | Yes | No | No | Yes |
| Family 8 | III1 | Female | Prelingual | Sensorineural | Bilateral | Severe to profound | No | No | No | Yes |
| Family 9 | III2 | Female | Prelingual | Sensorineural | Bilateral | Severe to profound | No | No | Yes | Yes |
The intensity of hearing loss is classified according to Shearer et al (1999). 45
In all cases, deaf patients had consanguineous normal parents, suggesting autosomal recessive deafness. Written informed consent for genetic testing was obtained from the adult patients or from their parents in case the patients were under 18 years of age. Some cases were sporadic, while other families had history of multiple affected members with hearing loss. Pedigrees are shown in Figure 1.
Figure 1.
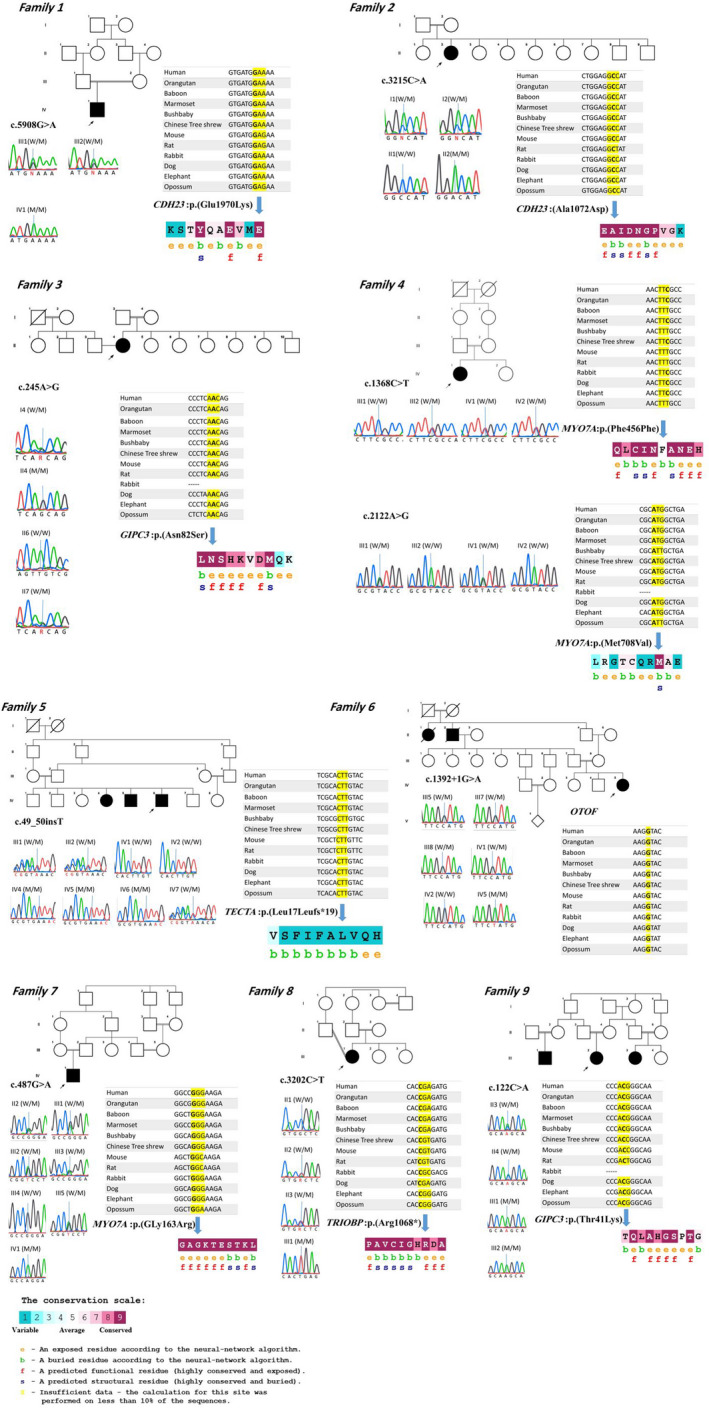
Representative pedigrees, sequence chromatograms confirming the mutations, cross‐species alignments, and ConSurf results of amino acids
2.2. DNA extraction
Blood samples were collected from families including 12 patients and 32 normal individuals (Table 2). Genomic DNAs were extracted from the peripheral blood of the patients and all available family members by the High Pure PCR template preparation kit (Roche: Product No. 11814770001).
Table 2.
List of identified mutations
| Family ID | Family member | Age | Affection status | Genotype | Gene | Chromosome location [GRCh37.p13] | Amino acid alternation | Reference |
|---|---|---|---|---|---|---|---|---|
| Family 1 | IV1 | 9 | Affected | AA |
CDH23 |
NC_000010.10:g.73548784C>T Exon43 |
p.(Glu1970Lys) | This study |
| III1 | 44 | Normal hearing | GA | |||||
| III2 | 40 | Normal hearing | GA | |||||
| Family 2 | II2 | 21 | Affected | AA |
CDH23 |
NC_000010.10:g.73468963G>T Exon26 |
p.(Ala1072Asp) | 26 |
| I1 | 60 | Normal hearing | CA | |||||
| I2 | 58 | Normal hearing | CA | |||||
| II1 | 30 | Normal hearing | CC | |||||
| Family 3 | II4 | 34 | Affected | GG |
GIPC3 |
NC_000019.9:g.3586512T>C Exon2 |
p.(Asn82Ser) | This study |
| I4 | 60 | Normal hearing | AG | |||||
| II6 | 33 | Normal hearing | AA | |||||
| II7 | 32 | Normal hearing | AG | |||||
| Family 4 | IV1 | 19 | Affected | CT |
MYO7A |
NC_000011.9:g.76873190G>A Exon13 |
p.(Phe456Phe) | This study |
| III1 | 48 | Normal hearing | CC | |||||
| III2 | 38 | Normal hearing | CT | |||||
| IV2 | 14 | Normal hearing | CT | |||||
| IV1 | 19 | Affected | AG |
MYO7A |
NC_000011.9:g.76886445T>C Exon18 |
p.(Met708Val) | This study | |
| III1 | 48 | Normal hearing | AG | |||||
| III2 | 38 | Normal hearing | AA | |||||
| IV2 | 14 | Normal hearing | AA | |||||
| Family 5 | IV6 | 32 | Affected | InsT/ InsT |
TECTA |
NC_000011.9:g120973423_120973424insA (NM_005422.2: c.49_50insT) Exon1 |
p. (Leu17Leufs*19) | This study |
| III1 | 57 | Normal hearing | N/ InsT | |||||
| III2 | 60 | Normal hearing | N/ InsT | |||||
| IV1 | 44 | Normal hearing | N/N | |||||
| IV2 | 42 | Normal hearing | N/N | |||||
| IV4 | 40 | Affected | InsT/ InsT | |||||
| IV5 | 38 | Affected | InsT/ InsT | |||||
| IV7 | 37 | Normal hearing | N/ InsT | |||||
| Family 6 | IV5 | 15 | Affected | AA |
OTOF |
NG_009937.1(NM_194248.3): c.1392+1G>A Intron13 |
– | 39 |
| III5 | 60 | Normal hearing | GA | |||||
| III7 | 50 | Normal hearing | GA | |||||
| III8 | 48 | Normal hearing | GA | |||||
| IV1 | 34 | Normal hearing | GA | |||||
| IV2 | 31 | Normal hearing | GG | |||||
| Family 7 | IV1 | 7 | Affected | AA |
MYO7A |
NC_000011.9:g.76867722C>T Exon6 |
p.(Gly163Arg) | 31 |
| II2 | 50 | Normal hearing | GA | |||||
| III1 | 25 | Normal hearing | GA | |||||
| III2 | 28 | Normal hearing | GA | |||||
| III3 | 31 | Normal hearing | GA | |||||
| III4 | 37 | Normal hearing | GG | |||||
| III5 | 34 | Normal hearing | GA | |||||
| Family 8 | III1 | 32 | Affected | TT |
TRIOBP |
NC_000022.10:g.38121765G>A Exon7 |
p.(Arg1068*) | 34 |
| II1 | 38 | Normal hearing | CC | |||||
| II2 | 56 | Normal hearing | CT | |||||
| II3 | 49 | Normal hearing | CT | |||||
| Family 9 | III2 | 16 | Affected | AA |
GIPC3 |
NC_000019.9:g. 3585717G>T Exon1 |
p.(Thr41Lys) | 29 |
| II3 | 41 | Normal hearing | CA | |||||
| II4 | 44 | Normal hearing | CA | |||||
| III1 | 7 | Affected | AA |
2.3. Targeted next‐generation sequencing and in silico analysis
All families were negative for mutations in GJB2. A custom‐designed NimbleGen chip capturing 127 genes involved in HL based on the deafness variation database (DVD) (http://deafness‐variationdatabase.org/letter) followed by next‐generation sequencing was employed to do genetic screening in proband in each family. List of the genes included in this panel is provided as Table S1. In general, the test examined >95% of the target genes with sensitivity >99%. Point mutations, microinsertion, deletion, and duplication (<20 bp) can be simultaneously detected by this targeted NGS panel. Reads were mapped to the reference human genome (GRCh37, UCSC hg19) using the Burrows‐Wheeler Aligner (http://bio‐bwa.sourceforge.net/). Single‐nucleotide variants (SNVs) and microinsertions‐deletions (indels) were called using SAMtools (http://samtools.sourceforge.net/), based on filtered variants with a mapping quality score of >20, and were annotated using ANNOVAR (http://www.openbioinformatics.org/annovar/). For analysis of the sequencing results, the international publicly available mutation and polymorphism databases such as 1000 Genomes Project (http://www.1000genomes.org/), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/), Exome Sequencing Project (ESP)(http://evs.gs.washington.edu/EVS/), and Deafness Variation Database (DVD) (http://deafness‐variationdatabase.org/letter) as well as BGI self‐developed local database were employed. 12 Only variants with a frequency below 1 percent were selected. Previously reported mutations that have been described in Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) as pathogenic or likely pathogenic were given the highest priority. 13 Prediction of the consequence of point mutations was obtained from at least three online databases, namely SIFT (https://sift.bii.a‐star.edu.sg/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (http://www.mutationtaster.org/). In case of intronic variants, Human Splicing Finder (HSF) (http://www.umd.be/HSF3/) which predicts the formation or disruption of splice donor sites, splice acceptor sites, exonic splicing silencer (ESS) sites, and exonic splicing enhancer (ESE) sites was utilized. 14 For further consideration, the frequency of the variants was checked out on the local database, Iranome (http://www.iranome.ir/). Also, ConSurf (http://www.consurf.tau.ac.il) was applied to check the evolutionary conservation in the region of the mutations (Figure 1).
2.4. Segregation analysis
The identified variants were confirmed by direct Sanger sequencing in patients and their all available family members to determine the variants segregation with the disease in these families. Primers surrounding region of the identified variant were designed using Primer3Plus (https://primer3plus.com/cgi‐bin/dev/primer3plus.cgi) web‐based server [PCR conditions and primer sequences are available upon request]. Consequently, DNA sequencing of the PCR products was performed on ABI 3130 with the ABI PRISM BigDye Terminator v. 3.1 sequencing kit (Applied Biosystems, USA). Sequencing chromatograms (Figure 1) were analyzed using CodonCode Aligner software version 8.0.2 (CodonCode Corp).
3. RESULTS
This study assessed a total of 9 ARNSHL Iranian families, 9 index cases and their 35 relatives, to confirm the diagnosis of the ARNSHL disease (Table 2). All the patients in this study had consanguineous parents and diagnosed with bilateral congenital sensorineural hearing loss. None of the patients displayed any additional symptoms apart from hearing loss. Targeted NGS of 127 hearing loss‐related genes was carried out in the nine probands. Possible causative variants in each family are summarized in Table 2.
A total of 10 variants in 6 distinct genes (CDH23, GIPC3, MYO7A, TRIOBP, TECTA, and OTOF) in 9 recessive pedigrees (Table 2) were identified. Among them, five variants were previously reported and the other 5 variants were novel. The 10 identified variants included 6 missense, 1 nonsense, 1 intronic, 1 frameshift, and 1 synonymous variant which predicted to affect on splicing by Human Splicing Finder (Table 3).
Table 3.
Various online databases that used to predict the pathogenicity of the exonic variants
| Family 1 | Family 2 | Family 3 | Family 4 | Family 5 | Family 6 | Family 7 | Family 8 | Family 9 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | CDH23 | CDH23 bvn | GIPC3 | MYO7A | MYO7A | TECTA | OTOF | MYO7A | TRIOBP | GIPC3 |
| Nucleic acid alternation | c.5908G>A | c.3215C>A | c.245A>G | c.1368C>T | c.2122A>G | c.49_50insT | c.1392+1G>A | c.487G>A | c.3202C>T | c.122C>A |
| Mutation type | Nonsynonymous | Nonsynonymous | Nonsynonymous | Synonymous | Nonsynonymous | Frameshift | Intronic | Nonsynonymous | Stop Gain | Nonsynonymous |
| Mutation function (ACMG) | Variant of uncertain significance | Variant of uncertain significance | Variant of uncertain significance | Variant of uncertain significance | Variant of uncertain significance | Likely pathogenic | Likely pathogenic | Pathogenic | Pathogenic | Likely pathogenic |
| SIFT | Damaging | Damaging | Tolerated | – | Damaging | – | – | Damaging | – | Tolerated |
| Polyphen | Probable damaging | Probable damaging | Probable damaging | – | Benign | – | – | Probable damaging | – | Polymorphism |
| MutationTaster | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing | Disease causing |
| 1000 Genome | N.R | N.R | N.R |
Homozygous: 0 Heterozygous: 1 |
Homozygous: 0 Heterozygous: 2 |
N.R | N.R | N.R | N.R | N.R |
| EXAC all MAF | N.R | N.R | 0.000041 | 0.000223 | 0.000258 | N.R | N.R | N.R | 0.000008 | N.R |
| Varsome | Uncertain significance | Uncertain significance | Uncertain significance | Likely benign | Uncertain significance | Pathogenic | – | Pathogenic | Pathogenic | Uncertain significance |
| Spliceview | – | – | – | – | – | – | Mutant N.R | – | – | – |
| NetGene2 2.3 | – | – | – | – | – | – | Mutant N.R | – | – | – |
| BDGP | – | – | – | – | – | – | Mutant N.R | – | – | – |
| HSF | – | – | – |
New ESS site ESE site broken |
||||||
| Segregates in the family | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; ExAC, Exome Aggregation Consortium; MAF, Minor Allele Frequency; NR, not reported.
Sanger sequencing on available family members revealed that these variants segregate with the disease in each family (Table 2 and Figure 1). The in silico pathogenicity predictions for each variant using SIFT, Polyphen2, and MutationTaster software are shown in Table 3.
Family 1: DNA from a 9‐year‐old boy (IV‐1) with cochlear implants due to severe hearing loss was screened for mutations in HL genes by NGS. The sequencing of the total length of 619 167 bp was obtained with a coverage of 98.81%, an average depth of 293.97X, and a minimum depth of 30X. A novel missense variant, c.5908G>A; p.(Glu1970Lys), in exon 43 of CDH23 was identified. We showed that this variant is segregating with the disease in this family by investigating normal carrier parents (III‐1 and III‐2). This variant has not been reported in 1000 Genome and ExAC databases. Prediction of the consequence of this variant was disease‐causing by mutation tasting, damaging by SIFT, and probable damaging by Polyphen. Based on the ACMG guidelines, the c.5908G>A variant classified as variant of uncertain significance (VUS).
Family 2: DNA from a 21‐year‐old woman (II‐2) with severe hearing loss and no family history was screened for variants in HL genes by NGS. The sequencing of the total length of 620 604 bp was obtained with a coverage of 98.49%, an average depth of 351.7X, and a minimum depth of 30X. A previously reported missense variant, c.3215C>A; p.(Ala1072Asp), in exon 26 of CDH23 was identified. We showed that this variant is segregating with the disease in this family by investigating her normal parents (I‐1 and I‐2) and her sister (II‐1). This variant has not been reported in 1000 Genome and ExAC databases. Prediction of the consequence of this variant was disease‐causing by mutation tasting, damaging by SIFT, and probable damaging by Polyphen. This variant classified as VUS according to the ACMG recommendations.
Family 3: DNA from a 34‐year‐old woman (II‐4) with severe hearing loss was investigated for variants in HL genes by NGS. The sequencing of the total length of 619 167 bp was obtained with a coverage of 99.01%, an average depth of 354.28X, and a minimum depth of 30X. A novel missense, c.245A>G; p.(Asn82Ser), in exon 2 of the GIPC3 gene was identified, which was confirmed by Sanger sequencing. We showed that this variant is segregating with the disease in this family by investigating her normal mother (I‐4) and two sisters (II‐6 and II‐7). This variant has not been reported in 1000 Genome database. This variant was predicted to be disease‐causing by mutation tasting and probable damaging by Polyphen. It classified as VUS based on ACMG guidelines.
Family 4: DNA from a 19‐year‐old girl (IV‐1) characterized by profound hearing loss who received cochlear implants was screened for mutations in HL genes by NGS. The sequencing of the total length of 619 167 bp was obtained with a coverage of 99.13%, an average depth of 383.96X, and a minimum depth of 30X. Two novel variants, c.[1368C>T];[c.2122A>G], p.[Phe456Phe];p.[Met708Val] in the MYO7A gene, were identified. We investigated these variants in her normal hearing parents (III‐1 and III‐2) and sister (IV‐2) and could show segregation with the disease in this family. Both variants were predicted to be disease‐causing by mutation tasting and based on the ACMG guidelines classified as VUS. According to the HSF the synonymous variant, c.1368C>T predicted to affect on splicing by creation of a new ESS site and disruption of an ESE.
Family 5: DNA from a 32‐year‐old man (IV‐6) suffered from moderate hearing impairment, with two other affected siblings, was screened for mutations in HL genes by NGS. The sequencing of the total length of 619 167 bp was obtained with a coverage of 99.24%, an average depth of 420.44X, and a minimum depth of 30X. A novel insertion, c.49_50insT; p.(Leu17Leufs*19), in exon 1 of the TECTA gene was identified. We investigated this variant in her normal parents (III‐1 and III‐2), as well as his three normal (IV‐1, IV‐2 and IV‐7) and two affected siblings (IV‐4 and IV‐5), and therefore could show cosegregation of this variant with the disease in this family. This variant has not been reported in 1000 Genome and ExAC databases. Prediction of the consequence of variant was disease‐causing by mutation tasting. This variant classified based on ACMG guidelines as likely pathogenic.
Family 6: DNA from a 15‐year‐old girl (IV‐5) with profound deafness with cochlear implants was screened for mutations in HL genes by NGS. There was history of other affected individuals in the pedigree. The sequencing of the total length of 620 604 bp was obtained with a coverage of 99.27%, an average depth of 214.02X, and a minimum depth of 30X. A reported splice site variant, c.1392+1G>A, in the OTOF gene, was identified. This homozygote variant was absent in her normal parents (III‐7 and III‐8) and her 3 relatives (III‐5, IV‐1, and IV‐2). This variant has not been reported in 1000 Genome and ExAC databases. Prediction of the consequence of variant was disease‐causing by mutation tasting. This variant classified as likely pathogenic based on ACMG guidelines.
Family 7: DNA from a 7‐year‐old boy (IV‐1) with sporadic moderate hearing impairment who has cochlear implants was screened for mutations in HL by NGS. The sequencing of the total length of 620 604 bp was obtained with a coverage of 99.39%, an average depth of 272.86X, and a minimum depth of 30X. A previously reported missense variant, c.487G>A; p.(Gly163Arg), in the MYO7A gene, was identified. We showed segregation of this variant with the disease in this family by studying his normal parents (III‐1 and III‐2) and 4 relatives (II‐2, III‐3, III‐4, and III‐5). This variant has not been described in 1000 Genome and ExAC databases. Prediction of the consequence of variant was disease‐causing by mutation tasting, damaging by SIFT, and probable damaging by Polyphen. This variant was classified as pathogenic based on ACMG guidelines.
Family 8: DNA from a 32‐year‐old man (III‐1) with severe to profound bilateral sensorineural hearing impairment with congenital onset was screened for mutations in HL by NGS. There was no family history of deafness in this pedigree. The sequencing of the total length of 620 604 bp was obtained with a coverage of 98.92%, an average depth of 241.61X, and a minimum depth of 30X. A previously reported nonsense variant, c.3202C>T; p.(Arg1068*), in the TRIOBP gene, was identified. We showed segregation of this variant with the disease in this family by studying his normal parents (II‐2 and II‐3) and his consanguineous partner (II‐1). This variant has not been reported in 1000 Genome database. Prediction of the consequence of variant was disease‐causing by mutation tasting. This variant classified as pathogenic based on ACMG guidelines.
Family 9: DNA from a 16‐year‐old girl (III‐2) with severe to profound hearing impairment was screened for mutations in HL by NGS. The sequencing of the total length of 620 604 bp was obtained with a coverage of 98.92%, an average depth of 241.61X, and a minimum depth of 30X. A previously reported missense variant, c.122C>A; p.(Thr41Lys), in exon 1 of the GIPC3 gene, was identified. We showed segregation of this variant with the disease in this family by studying her normal parents (II‐3 and II‐4) and another additional patient in the pedigree (III‐1). This variant has not been reported in 1000 Genome and ExAC databases. Prediction of the consequence of variant was disease‐causing by mutation tasting. This variant classified as likely pathogenic based on ACMG guidelines.
4. DISCUSSION
Auditory processing originates in the cochlea of the inner ear, where sounds are detected by sensory hair cells and then transmitted to the central nervous system. The sound waves, after traveling through the external canal and middle ear, lead to the stimulation of hair cells of the organ of Corti by fluids movement inside the cochlea. Each hair cell detects a narrow range of sound frequencies. Information about the sounds including timing, frequency, and intensity is then transmitted through highly efficient ribbon synapses to the spiral ganglion neurons. A defect in any part of this procedure can cause hearing impairment. 15 There are many genes and loci which are involved in this process. Mutations in genes encoding cytoskeletal proteins, structural proteins, regulatory elements, ion channel, and transport proteins can lead to malfunctions of the cochlea and inner ear. 7 The congenital hearing loss affects the language and speech development followed by child's education. The early identification of deafness may assist with hearing aid or treatment of the disorder such as cochlear implantation at the earliest possible time which can improve speech and language development. 16 , 17
Nonsyndromic hearing loss is the second most common disorder after intellectual disability in Iran, affecting one in 16 individuals. This relatively high incidence of hearing loss may be explained by high consanguinity rate in Iran. 18 Consanguineous marriage is frequent among Asian, African, and Latin American communities due to various factors such as their tradition, culture, and religion. Large pedigrees are also frequent in these communities. 19 Consanguineous marriage in Middle Eastern countries is ranging from 20% to 70%. Iran with consanguinity rates of 38% of all marriages, ranging from 15.9% in the northern provinces to 47.0% in the eastern provinces, accounts as one of the countries with high levels of consanguinity. 5 Single gene autosomal recessive inheritance is responsible for the majority of hereditary hearing loss cases. 19 Consanguineous matings have long been known as a key etiologic factor in the prevalence of genetic disorders through making disease‐causing recessive genes, inherited from a common ancestor, homozygous. In other words, the probability of inheritaning a similar deleterious recessive allele from both parents increases. 16
It is confirmed in various reports that the deafness is more common among children of consanguineous marriages. In two epidemiological Saudi Arabian surveys, the prevalence of SNHL has been shown to be 66% and 36.6%, respectively, out of which about 45% and 47% of the children had consanguineous parents. 19 In an Indian case‐control study, the rates of affected children with consanguineous and nonrelated parents have been shown to be 48% and 28%, respectively. 17 Parental consanguinity was shown to be more common in Qatari families with hearing impaired patients compared to ones with normal hearing children, 60.5% versus 25.3%. 20 In a large‐scale study in Oman, it was found that 70% of the hearing impaired children had blood relative parents. 21 The parental consanguinity rate of hearing impaired patients was measured to be over 60% in several reports from Iran. 22 , 23 , 24 , 25
Nowadays with the advent of NGS, identification of molecular defects involved in HL has been accelerated.
We have studied nine Iranian families, comprising at least one affected individual with nonsyndromic bilateral autosomal recessive prelingual hearing loss. We assessed a total of 9 index cases and their 35 relatives, to confirm the diagnosis of the ARNSHL disease. All deaf probands were born to consanguineous parents. Targeted NGS of 127 hearing loss‐related genes was carried out in the 9 probands, which allowed us to detect 5 reported and 5 novel variants in 6 distinct deafness genes including CDH23, GIPC3, MYO7A, TRIOBP, TECTA, and OTOF.
A novel missense variant, c.5908G>A; p.(Glu1970Lys), in exon 43 and a previously reported missense mutation, 26 c.3215C>A; p.(Ala1072Asp), in exon 26 of CDH23 were identified in families 1 and 2, respectively. CDH23 encodes a putative calcium‐dependent adhesion molecule protein with 27 extracellular cadherin (EC) domains, a single transmembrane domain, and a short cytoplasmic domain. Cadherin 23 is required for proper morphogenesis of hair bundles of inner ear neurosensory cells. 27 Previous studies have revealed the importance of ethnic diversity of genetic variants in CDH23. Mutations in CDH23 are one of the most important pathogenic causes of autosomal recessive nonsyndromic hearing loss (DFNB12) in Iranian populations. 1 Mutations in the CDH23 gene are known to cause both Usher syndrome type 1D (USH1D) and nonsyndromic hearing loss (DFNB12). To date, at least 80 pathogenic variants of the CDH23 have been reported in familial or sporadic patients of USH1D and DFNB12 worldwide. Usually, pathogenic missense mutations in any domain of the protein can lead to DFNB12, whereas nonsense, splice site, and frameshift mutations can cause USH1D. 28 In this study, two missense homozygous variants, p.(Glu1970Lys) and p.(Ala1072Asp), in CDH23 affecting two highly conserved residues in the extracellular domains of EC19 and EC10, respectively, were detected (https://www.uniprot.org/).
A novel missense variant, c.245A>G; p.(Asn82Ser), in exon 2 and a previously reported missense mutation, 29 c.122C>A; p.(Thr41Lys), in exon 1 of the GIPC3 gene were identified in families 3 and 9, respectively. GIPC3 encodes a 312 amino acid protein that contains 3 domains: an N‐terminal GIPC homology domain (GH1), a central PDZ domain, and a C‐terminal GH2 domain. GIPC3 localizes to inner ear sensory hair cells and is important in peripheral auditory signal transmission. 29 The GH1, PDZ, and GH2 domains are well conserved among GIPC1, GIPC2, and GIPC3 orthologs. GIPC proteins are involved in the trafficking, signaling, and recycling of various transmembrane proteins. They regulate a variety of cellular processes including proliferation, planar cell polarity, cytokinesis, and migration. Dysregulation of GIPCs results in human pathologies, such as hearing loss and cancer. 30 The two homozygote variants identified in this study, p.(Asn82Ser) and p.(Thr41Lys), are affecting the GH1 domain. 29
Two novel variants, c.1368C>T; p.(Phe456Phe) and c.2122A>G; p.(Met708Val) in compound heterozygote state in family 4 and a previously reported missense mutation, 31 c.487G>A; p.(Gly163Arg), in family 7 were identified in the MYO7A gene. MYO7A encodes the actin‐binding motor protein, which is involved in differentiation, morphogenesis, and organization of cochlear hair cell bundles. 32 The myosin‐VIIa protein contains different domains including a myosin head‐like domain, which contains the crucial ATP‐binding site and actin‐bind site, five IQ motifs, a coiled‐coil region, two MyTH4 domains, two FERM domains, and a SH3 domain. Mutations in the MYO7A gene have been identified to be associated with nonsyndromic hearing loss (DFNB2, DFNA11) and Usher syndrome type 1B (USH1B). 33 Here, we identified compound heterozygous missense variants MYO7A:p.[Phe456Phe]; p.[Met708Val] and a homozygous variant, p.(Gly163Arg), in two Iranian families with nonsyndromic hearing loss which are affecting the myosin head‐like domain containing residues 1 to 729 of the protein. The synonymous variant; c.1368C>T; p.(Phe456Phe) predicted by HSF to affect splicing by creation of a new ESS site and disruption of an ESE. The p.(Gly163Arg) variant also affects the ATP‐binding site (158‐165). 33
A previously reported nonsense variant, 34 c.3202C>T; p.(Arg1068*), in exon 7 of the TRIOBP gene was identified in family 8. TRIOBP encodes a filamentous actin‐binding protein that has been identified as the gene for DFNB28 deafness. TRIOBP variants are not a common cause of HL. To date, over 30 point mutations have been reported in the TRIOBP gene. Previous studies have suggested exon 7 of TRIOBP as a hotspot for mutations, probably due to presence of repetitive sequences. 32 , 34 The protein contains two types of domains, N‐terminal pleckstrin homology (PH) and C‐terminal coiled‐coil. Studies have revealed that TRIOBP directly binds and stabilizes the F‐actin structures, presumably via their nonconventional actin‐binding sites. 34 TRIOBP protein has multiple roles in the organization of actin‐cytoskeleton, proper centrosomal localization and segregation of chromosomes during cell division, and cell cycle regulation. 34
The proband and his affected siblings in family 5 carried a novel likely pathogenic insertion, c.49_50insT; p.(Leu17Leufs*19), in exon 1 of the TECTA gene which was absent in the unaffected members of the family. TECTA gene, located on 11q22‐q24, has been implicated both in autosomal dominant (DFNA) and autosomal recessive (DFNB) forms of nonsyndromic hearing loss. The gene comprises 23 exons which encodes one of the major noncollagenous glycoproteins of the tectorial membrane, alpha‐tectorin. The protein is a part of the noncellular matrix which lies over the stereocilia of the cochlear hair cells and is critical both for the mechanical amplification of sound and its transmission to the inner hair cells. Mutations in various parts of alpha‐tectorin lead to deafness at different frequencies. Studies on different populations have shown that alpha‐tectorin is among the top 10 genes responsible for ARNSHL. Previous studies have shown that TECTA mutations account for about 4.13% of ARNSHL among GJB2 negative Iranian families. 35 , 36 TECTA is associated with a moderate‐to‐severe audio profile. 35 Mainly missense mutations of TECTA cause ADNSHL (DFNA8/12), while the majority of autosomal recessive NSHL (DFNB21) variants are truncating and most likely loss‐of‐function mutations. 36 , 37 The variant reported in this study is an insertion, c.49_50insT; p.(Leu17Leufs*19), within the signal peptide of alpha‐tectorin (https://www.uniprot.org/uniprot/O75443). Mutations in signal peptides can affect directing of proteins to their proper cellular and extracellular locations and the translocation of proteins across the cytoplasmic membrane. 38
The c.1392+1G>A variant in the OTOF gene, which we found in the proband of family 6, has been recently reported as a pathogenic splice site variant in another Iranian family. 39 This variant is affecting the donor splice site of intron 13, in which G nucleotide is replaced by A. Splice site software tools and MutationTaster, Table 3, predicted that this variant causes lose of donor splice site and leading to intron retention. 38 Previous studies have shown that a single amino acid change, even in nonconserved residues, in 1 C2 domain severely affects protein stability and localization. This could explain the profound deafness phenotype due to the severe effect of the splice site variant on C2C and downstream domains of the protein. 39 The OTOF gene (DFNB9) is mainly expressed in cochlear inner hair cells and is necessary for synaptic exocytosis at the auditory ribbon synapse. Because of the expected good outcomes of cochlear implantation for patients with OTOF mutations, it is important to perform mutation screening for OTOF to select the appropriate intervention. 40 To date, more than 100 pathogenic variants including missense, nonsense, frameshift, splice site, deletion, and duplication have been found in various populations, nearly a third of which are from the Middle East, especially Pakistan and Turkey. Studies in Iran suggested that ARNSHL due to OTOF gene mutations ranges from 0.7% to 2.6%. There are few reports of splice site mutations. 39 Hearing loss due to OTOF mutations is characterized by abnormal inner hair cell function and dyssynchrony of neural transmission of the auditory signal from the inner ear to the auditory nerve and brainstem. 41 The OTOF gene, located on chromosome 2p23.1, encodes a membrane‐anchored cytosolic protein, otoferlin, with several isoforms. 42 , 43 It is believed that variants affecting the long isoform of this gene cause ARNSHL. 41 The long isoform, which is thought to be required for normal hearing, involves 48 coding exons which contains six C2 domains (C2A‐C2F) and a transmembrane domain (TM). 43 , 44 Otoferlin plays a role in the calcium‐dependent fusion of vesicles to the plasma membrane. 43
5. CONCLUSION
In this study, a total of 10 variants in the patients were identified. Among them, five mutations were previously reported and the other five variants were novel. Accurate identification of causative mutations plays a key role in affected families to offer them preimplantation genetic diagnosis (PGD), prenatal diagnosis (PND), or further therapy strategies. Besides finding more mutations and new genes provides the possibility to do further studies on the pathophysiology of this disease and identify the involved pathways and mechanisms.
AUTHORS' CONTRIBUTIONS
MG and FB conceived and designed the experiments. MG and F.B contributed to data collection. FB and MG wrote the paper. MG, SY. S, and M. M supervised the work. All authors read and approved the final manuscript.
Supporting information
Tab S1
ACKNOWLEDGMENTS
We thank the families for their contribution to this study. This research received no specific grant from any funding agency, commercial, or not for profit sectors.
Bitarafan F, Seyedena SY, Mahmoudi M, Garshasbi M. Identification of novel variants in Iranian consanguineous pedigrees with nonsyndromic hearing loss by next‐generation sequencing. J Clin Lab Anal. 2020;34:e23544 10.1002/jcla.23544
REFERENCES
- 1. Sloan‐Heggen CM. Precision health and deafness–optimizing genetic diagnosis. Iowa, University of Iowa. Epub ahead of print May 2018. [Google Scholar]
- 2. Mahdieh N, Rabbani B, Wiley S, Akbari MT, Zeinali S. Genetic causes of nonsyndromic hearing loss in Iran in comparison with other populations. J Hum Genet. 2010;55:639‐648. [DOI] [PubMed] [Google Scholar]
- 3. Sloan‐Heggen CM, Babanejad M, Beheshtian M, et al. Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. J Med Genet. 2015;52:823‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beheshtian M, Babanejad M, Azaiez H, et al. Heterogeneity of hereditary hearing loss in Iran: a comprehensive review. Arch Iran Med. 2016;19:720‐728. [PMC free article] [PubMed] [Google Scholar]
- 5. Ajalloueyan M, Amirsalari S, Hassanalifard M, et al. Hearing loss prevalence and risk factors among Iranian deaf children: the first report from Iran. J Clin Neonatol. 2014;3:191. [Google Scholar]
- 6. Schrijver I. Hereditary non‐syndromic sensorineural hearing loss transforming silence to sound. J Mol Diagnostics. 2004;6:275‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Egilmez OK, Kalcioglu MT. Genetics of nonsyndromic congenital hearing loss. Scientifica (Cairo). 2016;2016:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao X, Dai P. Impact of next‐generation sequencing on molecular diagnosis of inherited non‐syndromic hearing loss. J Otol. 2014;9:122‐125. [Google Scholar]
- 9. Zhong LX, Kun S, Jing Q, Jing C, Denise Y. Non‐syndromic hearing loss and high‐throughput strategies to decipher its genetic heterogeneity. J Otol. 2013;8:6‐24. [Google Scholar]
- 10. Najmabadi H, Kahrizi K. Genetics of non‐syndromic hearing loss in the Middle East. Int J Pediatr Otorhinolaryngol. 2014;78:2026‐2036. [DOI] [PubMed] [Google Scholar]
- 11. Nance WE, Arnos KS, Carey JC, et al. Genetics evaluation guidelines for the etiologic diagnosis of congenital hearing loss. Genet Med. 2002;4:162‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang R, Chen S, Han P, et al. Whole exome sequencing identified a homozygous novel variant in CEP290 gene causes Meckel syndrome. J Cell Mol Med. 2020;24:1906‐1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han P, Wei G, Cai K, et al. Identification and functional characterization of mutations in LPL gene causing severe hypertriglyceridaemia and acute pancreatitis. J Cell Mol Med. 2020;24:1286‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Desmet FO, Hamroun D, Lalande M, et al. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Appler JM, Goodrich LV. Connecting the ear to the brain: molecular mechanisms of auditory circuit assembly. Prog Neurogibol. 2011;93:488‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zakzouk S, El‐Sayed Y, Bafaqeeh SA. Consanguinity and hereditary hearing impairment among Saudi population. Ann Saudi Med. 1993;13:447‐450. [DOI] [PubMed] [Google Scholar]
- 17. Shrikrishna BH, Deepa G. Study of association of family history and consanguinity with congenital hearing loss. Int J Otorhinolaryngol Head Neck Surg. 2016;2:61. [Google Scholar]
- 18. Ghasemnejad T, Shekari Khaniani M, Zarei F, et al. An update of common autosomal recessive non‐syndromic hearing loss genes in Iranian population. Int J Pediatr Otorhinolaryngol. 2017;97:113‐126. [DOI] [PubMed] [Google Scholar]
- 19. Zakzouk S. Consanguinity and hearing impairment in developing countries: a custom to be discouraged. J Laryngol Otol. 2002;116:811‐816. [DOI] [PubMed] [Google Scholar]
- 20. Bener A, Eihakeem AAM, Abdulhadi K. Is there any association between consanguinity and hearing loss. Int J Pediatr Otorhinolaryngol. 2005;69:327‐333. [DOI] [PubMed] [Google Scholar]
- 21. Al KM, Patton MA. Consanguinity and deafness in Omani children. Int J Audiol. 2008;47:30‐33. [DOI] [PubMed] [Google Scholar]
- 22. Ajallouyan M, Radfar S, Nouhi S, et al. Consanguinity among parents of Iranian deaf children. Iran Red Crescent Med J. 2016;18(11):e22038 10.5812/ircmj.22038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amirsalari S, Saburi A, Radfar S, et al. Hearing loss prevalence and risk factors among Iranian deaf children: the first report from Iran. J Clin Neonatol. 2014;3:191. [Google Scholar]
- 24. Amini SR, Kamali M. Consanguineous marriage among the parents of hearing impaired students in Mashhad. Iran Rehabil J. 2010;8:36‐39. [Google Scholar]
- 25. Hajilari M, Oladnabi M, Kianmehr A, Taziki MH, Abdollahi FZ. Hereditary hearing loss and consanguinity in Turkmen population of Iran: a retrospective study. Int J Pediatr. 2019;7:10323‐10334. [Google Scholar]
- 26. Duman D, Sirmaci A, Cengiz FB, Ozdag H, Tekin M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet Test Mol Biomarkers. 2011;15:29‐33. [DOI] [PubMed] [Google Scholar]
- 27. Woo HM, Park HJ, Park MH, et al. Identification of CDH23 mutations in Korean families with hearing loss by whole‐exome sequencing. BMC Med Genet. 2014;15:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu S, Sun F, Zhang J, et al. Genetic etiology study of ten chinese families with nonsyndromic hearing loss. Neural Plast. 2018;2018:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramzan K, Al‐Owain M, Allam R, et al. Homozygosity mapping identifies a novel GIPC3 mutation causing congenital nonsyndromic hearing loss in a Saudi family. Gene. 2013;521:195‐199. [DOI] [PubMed] [Google Scholar]
- 30. Katoh M. Functional proteomics, human genetics and cancer biology of GIPC family members. Exp Mol Med. 2013;45:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Atik T, Onay H, Aykut A, et al. Comprehensive analysis of deafness genes in families with autosomal recessive nonsyndromic hearing loss. PLoS ONE. 2015;10:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shang H, Yan D, Tayebi N, et al. Targeted next‐generation sequencing of a deafness gene panel (MiamiOtoGenes) analysis in families unsuitable for linkage analysis. Biomed Res Int. 2018;2018:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rong W, Chen X, Zhao K, et al. Novel and recurrent MYO7A mutations in Usher syndrome type 1 and type 2. PLoS ONE. 2014;9(5):e97808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pollak A, Lechowicz U, Murcia Pieńkowski VA, et al. Whole exome sequencing identifies TRIOBP pathogenic variants as a cause of post‐lingual bilateral moderate‐to‐severe sensorineural hearing loss. BMC Med Genet. 2017;18:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Asgharzade S, Tabatabaiefar MA, Modarressi MH, et al. A novel TECTA mutation causes ARNSHL. Int J Pediatr Otorhinolaryngol. 2017;92:88‐93. [DOI] [PubMed] [Google Scholar]
- 36. Behlouli A, Bonnet C, Abdi S, et al. A novel biallelic splice site mutation of TECTA causes moderate to severe hearing impairment in an Algerian family. Int J Pediatr Otorhinolaryngol. 2016;87:28‐33. [DOI] [PubMed] [Google Scholar]
- 37. Alasti F, Sanati MH, Behrouzifard AH, et al. A novel TECTA mutation confirms the recognizable phenotype among autosomal recessive hearing impairment families. Int J Pediatr Otorhinolaryngol. 2008;72:249‐255. [DOI] [PubMed] [Google Scholar]
- 38. Hiller K, Grote A, Scheer M, Munch R, Jahn D. PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res. 2004;32:375‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tabatabaiefar MA, Pourreza MR, Tahmasebi P, et al. A Novel pathologic variant in OTOF in an Iranian family segregating hereditary hearing loss. Otolaryngol Neck Surg. 2018;158:1084‐1092. [DOI] [PubMed] [Google Scholar]
- 40. Iwasa Y, Nishio S, Yoshimura H, et al. OTOF mutation screening in Japanese severe to profound recessive hearing loss patients. BMC Med Genet. 2013;14:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Lu Y, Cheng J, Zhang L, Han D, Yuan H. Novel OTOF gene mutations identified using a massively parallel DNA sequencing technique in DFNB9 deafness. Acta Otolaryngol. 2018;138:865‐870. [DOI] [PubMed] [Google Scholar]
- 42. Mahdieh N, Shirkavand A, Rabbani B, et al. Screening of OTOF mutations in Iran: a novel mutation and review. Int J Pediatr Otorhinolaryngol. 2012;76:1610‐1615. [DOI] [PubMed] [Google Scholar]
- 43. Xia H, Huang X, Xu H, et al. An OTOF frameshift variant associated with auditory neuropathy spectrum disorder. Curr Genomics. 2017;19:370‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choi B, Ahmed Z, Riazuddin S, et al. Identities and frequencies of mutations of the otoferlin gene ( OTOF ) causing DFNB9 deafness in Pakistan. Clin Genet. 2009;75:237‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shearer AE, Hildebrand MS, Smith RJH. Hereditary hearing loss and deafness overview 1999 Feb 14 [Updated 2017 Jul 27]. In: Adam MP, Ardinger HH, Pagon RA, et al. eds. GeneReviews® [Internet]. Seattle, WA: University of Washington; 1993. ‐2020. https://www.ncbi.nlm.nih.gov/books/NBK1434/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tab S1