

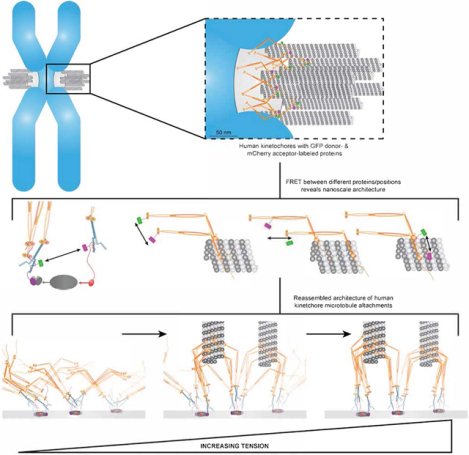
Summary
The nanoscale protein architecture of the kinetochore, a complex protein machine, plays an integral role in the molecular mechanisms underlying its functions in chromosome segregation. However, defining this architecture in human cells remains challenging because of the large size and compositional complexity of the kinetochore. Here, we use Förster Resonance Energy Transfer to reveal the architecture of individual kinetochore-microtubule attachments in human cells. We find that the microtubule-binding domains of the Ndc80 complex cluster at the microtubule plus-end. This clustering occurs only after microtubule attachment, and it increases proportionally with centromeric tension. Surprisingly, Ndc80 complex clustering is independent of the organization and number of its centromeric receptors. Moreover, this clustering is similar in yeast and human kinetochores despite significant differences in their centromeric organizations. These and other data suggest that the microtubule-binding interface of the human kinetochore behaves like a flexible “lawn” despite being nucleated by repeating biochemical subunits.
Graphical Abstract
eTOC Blurb:
Kukreja et al. use FRET microscopy to elucidate the nanoscale architecture of key human kinetochore proteins relative to the microtubule plus-end. Their data highlight a conserved organization of the microtubule-binding Ndc80 complex in the human and budding yeast kinetochores despite their significantly different centromeric foundations.
Introduction
To accurately segregate chromosomes, the kinetochore performs two functions. When unattached, it acts as a signaling hub to delay the onset of anaphase, but when attached to the plus-ends of spindle microtubules, it acts as a force generating machine. The nanoscale organization of kinetochore proteins relative to one another and relative to the microtubule plus-end, referred to here as the “architecture” of the kinetochore, plays key roles in the molecular mechanisms underlying both of these functions [1–4]. However, the architecture of the human kinetochore has not yet been defined. This is partly because the human kinetochore is compositionally complex and large, built from hundreds of protein molecules distributed upon a 200 nm diameter disk-like chromatin foundation known as the centromere. Furthermore, the human kinetochore changes in response to microtubule attachment and force [5–7], making its architecture intractable.
Because no currently available method can define kinetochore architecture, it must be synthesized from data defining four of its aspects: 1) the structures of kinetochore proteins, 2) their copy numbers, 3) their average localizations along the kinetochore-microtubule axis, and 4) their nanoscale distribution around and along the plus-end [8–10]. For the human kinetochore, data regarding the first three aspects are available [2, 5, 6, 11–13]. However, the nanoscale distributions of kinetochore proteins around microtubule plus-ends remains unknown. Here we apply Förster Resonance Energy Transfer (FRET) between fluorescently labeled kinetochore subunits to elucidate this aspect of the human kinetochore.
We designed FRET experiments to elucidate specific aspects of the human kinetochore’s architecture. One primary goal was to determine the organization of the microtubule-binding Ndc80 complex (Ndc80C) around the microtubule plus-end. Ndc80C forms end-on microtubule attachments, generates force, and regulates plus-end polymerization dynamics [14, 15]. The human kinetochore contains ~250 Ndc80C molecules and binds ~20 microtubule plus-ends, suggesting that ~12 Ndc80C molecules engage one microtubule [11, 16]. The nanoscale distribution of these molecules around the 25 nm diameter and along the longitudinal axis of the microtubule will influence the persistence of kinetochore-microtubule attachments [17]. The distribution of Ndc80C molecules is dictated by long, flexible centromere-bound protein linkages. Therefore, we extended our FRET analysis to members of the Constitutive Centromere-Associated Network (CCAN) of proteins involved in Ndc80C recruitment. Microtubule attachment- and tension-dependent changes in kinetochore architecture are at the heart of its ability to implement emergent mechanisms. Therefore, we also studied how the nanoscale distribution of Ndc80C changes in response to attachment, tension, and when its recruitment is perturbed. From our FRET data, we formulate a model of human kinetochore-microtubule attachments and contrast it with the yeast kinetochore.
Results
Implementation of a FRET imaging strategy to study kinetochore architecture
To determine protein proximities in HeLa kinetochores using FRET, we co-expressed EGFP (donor fluorophore, referred to as GFP) and mCherry (acceptor fluorophore) labeled kinetochore proteins (STAR Methods; [18]). To maximize the recruitment of labeled proteins to the kinetochore, we knocked down their endogenous, unlabeled counterparts using RNAi (Figure S1). Depending on the position of the donor and acceptor fluorophores (fused to either the C- or N-terminus of the selected proteins), we expected FRET to occur within a single protein complex (intra-complex), between neighboring complexes (inter-complex), or both (Figure 1A).
Figure 1. Design and implementation of a FRET imaging strategy to study kinetochore architecture.
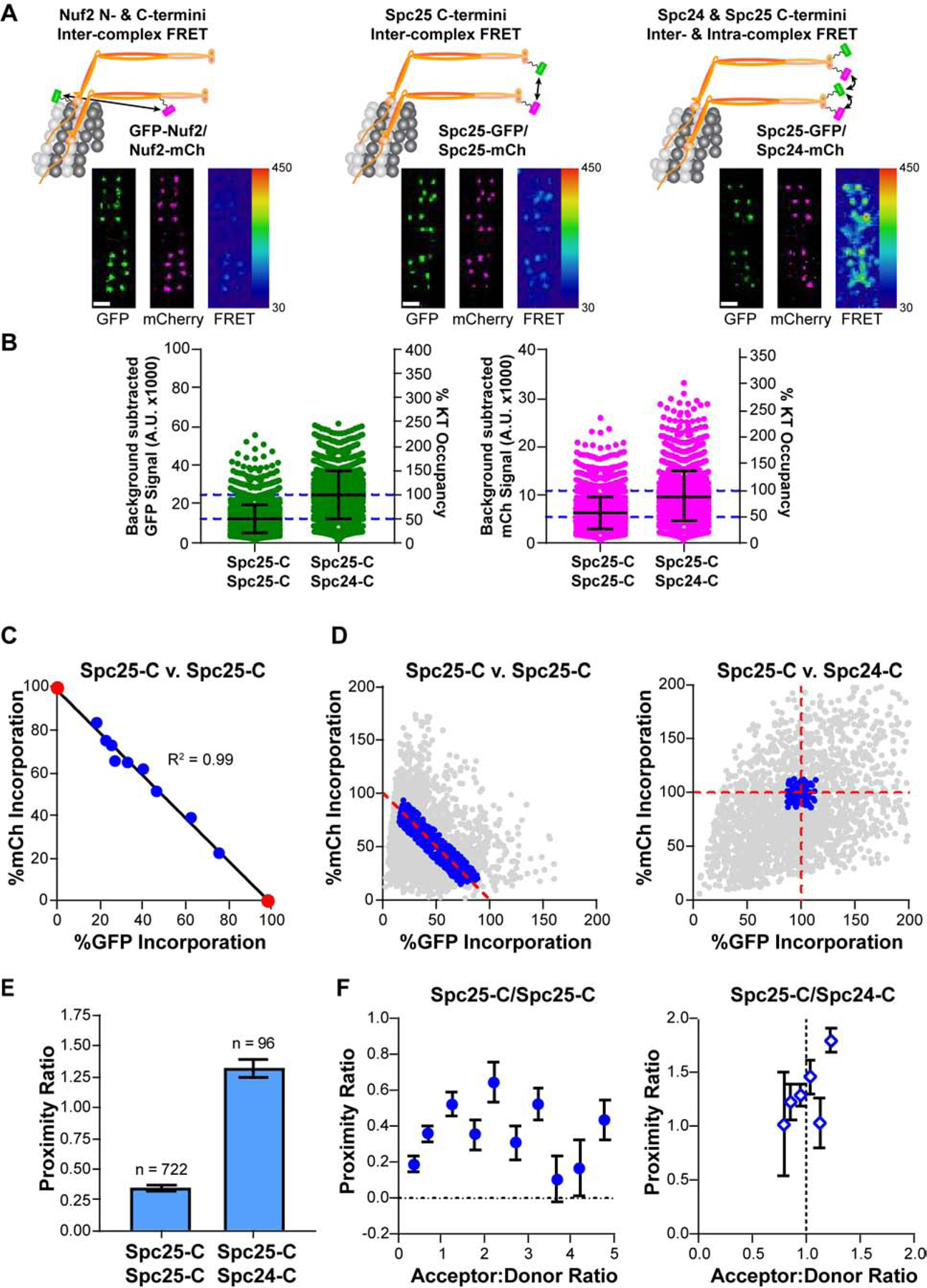
A) Top: Co-expression of GFP (FRET donor, green) and mCherry (mCh, FRET acceptor, magenta) fusions of the Ndc80 complex (Ndc80C, orange) reveal the proximity between adjacent Ndc80C molecules along the longitudinal axis (left), around the circumference of the microtubule (middle, right). For simplicity, only two Ndc80C molecules per microtubule are shown. Micrographs show representative metaphase plates from each cell line. FRET micrographs are scaled equivalently and pseudo-colored by the raw FRET values; GFP and mCherry micrographs are scaled for ease of viewing. Scale bar, 1 μm B) Background subtracted GFP and mCherry signals of individual kinetochores in cells expressing Spc25-GFP/Spc25-mCherry and Spc25-GFP/Spc24-mCherry after siRNA-mediated knockdown of endogenous Spc25 or both Spc25 and Spc24, respectively. The Y axis on the right shows the saturation level of the kinetochore by the GFP- and mCherry labeled subunit, estimated from [11] C) Correlation between Spc25-GFP and Spc25-mCherry signals measured from kinetochores from the data set in B. Data were binned by the ratio of mCherry to GFP fluorescence, and further normalized by the X- and Y-intercepts of their linear regression (black line; see main text). Measurements of cells expressing Spc25-GFP or Spc25-mCherry in isolation are marked by red circles. From left to right: n = 398, 379, 109, 108, 131, 145, 170, 212, 491, 320, 499. D) Normalized fluorescence signals for all kinetochores measured in Spc25-C/Spc25-C (left) and Spc25-C/Spc24-C (right) expressing cells. Only the data indicating complete saturation of the kinetochore by fluorophore-labeled proteins (blue circles) were used to measure FRET. Kinetochores from Spc25-C/Spc25-C expressing cells were further filtered by their acceptor to donor ratios (A:D) to include only the data within the range of 0.2 – 5 (see STAR Methods). All other values are excluded (gray circles) E) The proximity ratio for fully occupied, metaphase kinetochores in Spc25-C/Spc25-C and in Spc25-C/Spc24-C expressing cells. The number of kinetochores measured for each cell line is indicated above the bars. F) Dependence of the proximity ratio on the A:D. In the absence of competition, the proximity ratio clusters around an A:D that reflects the inherent stoichiometry of the two fluorophore-labeled subunits involved (Spc25-C/Spc24-C cells, right). Data are binned by A:D (mean ± SEM; for Spc25-C/Spc25-C, n = 150, 192, 93, 62, 37, 53, 50, 36, 24, 25; for Spc25-C/Sp24-C, n = 2, 13, 32, 30, 14, 5). In B), error bars are ± S.D. In C), E), and F) data represent the mean ± SEM In C), SEM error bars are too small to be seen. Data collected in B) - F) are from N ≥ 3 experiments. See also Figures S1 and S2, and Table S1.
For FRET to accurately reveal protein proximities, kinetochores must be saturated by donor- and acceptor-labeled proteins. However, in cells co-expressing GFP- and mCherry-labeled versions of Spc25, an Ndc80C subunit, we observed variability in kinetochore signals. This variability arises from several factors, including: chromosome-specific differences in kinetochore size [5, 7, 19–24], changes in fluorescence intensity that occur with depth from the coverslip, cell-to-cell variation in siRNA efficiency, and overlapping signals from neighboring kinetochores. To minimize the effects of this variability, we established a filtering scheme as follows.
We quantified GFP and mCherry fluorescence signals per kinetochore in cells co-expressing Spc25-GFP and Spc25-mCherry (Figure 1B). Because the kinetochore has a limited protein capacity, we expected the donor- and acceptor-labeled versions of Spc25 to compete for kinetochore binding. Indeed, the Spc25-GFP and Spc25-mCherry signals per kinetochore were anti-correlated (Figure 1C, blue circles). We performed linear regression of the data to determine the X- and Y-intercepts, which should correspond to intensities of kinetochores fully saturated with GFP or mCherry, respectively (Figure 1C, red circles; Figure S2A). We used these values with the copy number for Ndc80C per kinetochore to define the single molecule brightness of GFP and mCherry (STAR Methods; [11]). Using these single molecule brightness values, we converted the GFP and mCherry fluorescence intensities from each kinetochore into protein counts and retained only the measurements reflecting full kinetochore occupancy (Figure 1D, blue circles).
To quantify FRET, we determined the acceptor fluorescence due to FRET, which is known as ‘sensitized emission’. The sensitized emission for each kinetochore was calculated by subtracting the contributions of GFP bleed-through and mCherry cross-excitation from the measured FRET signal (Figure S2, STAR Methods). Because sensitized emission is directly proportional to the average FRET efficiency and the total number of FRET pairs, we normalized it with respect to the number of donor and acceptor molecules per kinetochore. This normalization renders a FRET metric referred to as the ‘proximity ratio’ and is proportional to the average FRET efficiency [25, 26].
Using this methodology, the average inter-complex distance between neighboring Ndc80C molecules at their centromeric ends (Spc25-C/Spc25-C FRET, where -C refers to fluorophores fused to the C-terminus) is <10 nm (Figure 1E). The higher FRET between Spc25 and Spc24 molecules indicates that these C-termini are more densely organized than Spc25-C/Spc25-C, consistent with their ~2 nm intra-complex separation [15]. We note that competition between donor- and acceptor-labeled Spc25 molecules in Spc25-C/Spc25-C expressing cells yields kinetochores with varying acceptor to donor ratios (A:D; Figure 1D). This effect introduces variation in the measured proximity ratio at a given kinetochore (Figure 1F). Accounting for A:D, however, does not significantly change the trends of our FRET data (Table S1).
Ndc80C molecules cluster around the microtubule and are staggered relative to one another
The nanoscale distribution of Ndc80C molecules around microtubule plus-ends governs kinetochore-microtubule attachments and the polymerization dynamics of attached microtubules [14, 17]. Current evidence suggests that Ndc80C molecules are collinear with the microtubule-kinetochore axis [13, 27], but their relative spacing and alignment are unknown. To reveal these aspects, we positioned fluorophores along Ndc80C’s length to measure inter-complex FRET. We chose three locations along the Ndc80C molecule: proximal to its microtubule-binding end (i.e., N-Nuf2, wherein N- denotes fluorophores fused to the N-terminus; we did not label the N-terminus of Hec1 because this affects Ndc80C function [28]), near the middle of its ~57 nm span (Nuf2-C, within its tetramerization domain), or near its centromeric end (Spc25-C). We detected FRET at all three positions, indicating that the average distance between adjacent Ndc80C molecules is <10 nm along its entire length (Figure 2A). Furthermore, the proximity ratio was higher at the microtubule-binding end (0.55 ± 0.02) and middle of Ndc80C (0.57 ± 0.03) than at its centromeric end (0.35 ± 0.02). Therefore, Ndc80C molecules are more tightly clustered on the microtubule lattice and at their tetramerization domains than at the ends which anchor them to the centromere.
Figure 2. Ndc80C molecules are clustered along their entire length and staggered along the microtubule lattice in metaphase kinetochores.
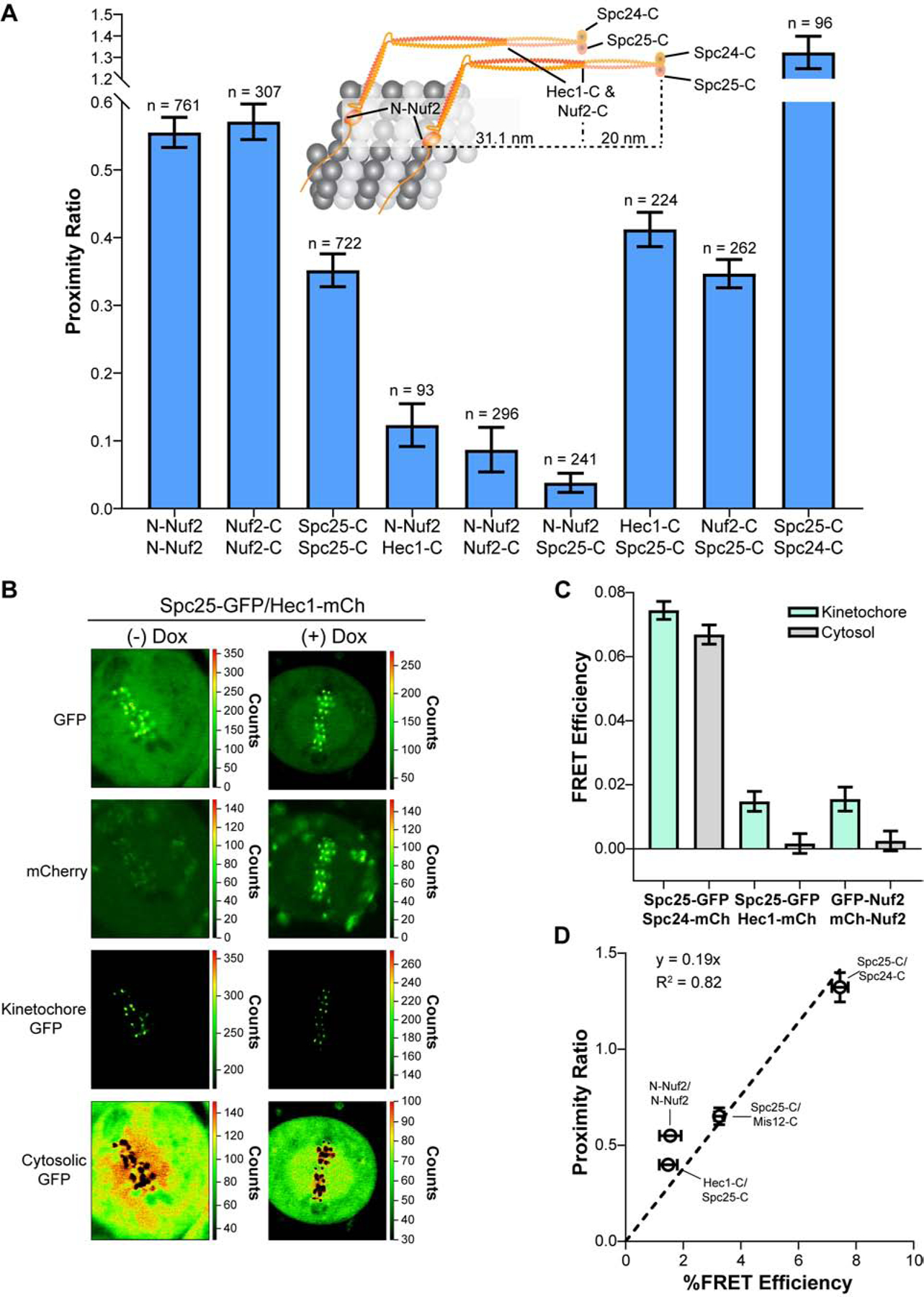
A) FRET measurements for Ndc80C subunits in microtubule-attached metaphase kinetochores. Cartoon depicts the position and approximate distance between different anchoring points for donor and acceptor fluorophores as determined from structural data of Ndc80C [15, 29–31]. For simplicity, only two Ndc80C molecules are shown. Bar graph displays the average proximity ratio ± SEM. The number of measurements is indicated above the bars. B) FLIM micrographs of Spc25-GFP/Hec1-mCherry HeLa cells. Doxycycline (Dox) induces the expression of Hec1-mCherry. All images are scaled by the number of photons/pixel (scale to the right of images). Intensity thresholding was used to separate kinetochore-localized from cytosolic GFP pixels (bottom two rows). Note that GFP signal bleeds into the mCherry channel. C) FRET efficiency of kinetochore-localized (light green bars) and cytosolic (gray bars) FRET pairs. Bars represent the average FRET efficiency ± SEM (n = 15, 14, and 13). D) Plot of the average proximity ratio versus the average FRET efficiency for the indicated FRET pairs (dashed line - linear regression). Error bars are ± SEM. The N-Nuf2/N-Nuf2 data point deviates from the trend, likely due to our inability to assess the A:D ratio on the FLIM setup. (N ≥ 3 experiments). See also Figure S3 and Table S1.
The clustering of Ndc80C molecules along their entire length suggests that adjacent molecules are aligned with one another as in the budding yeast kinetochore [3]. To reveal the extent of this alignment, we co-expressed two different fluorophore-tagged Ndc80C subunits specifically chosen to avoid intra-complex FRET. Within a single Ndc80C molecule, fluorophores at N-Nuf2/Spc25-C, N-Nuf2/Hec1-C, and Hec1-C/Spc25-C are separated by ~51, 31, and 20 nm, respectively [15, 29–31]. However, inter-complex FRET will arise if neighboring Ndc80C molecules are staggered along the kinetochore-microtubule axis such that the donor on one Ndc80C is within 10 nm of the acceptor on another. We detected very little FRET between the extremes of Ndc80C (N-Nuf2/Spc25-C; Figure 2A and Table S1). Interestingly, we detected low FRET at N-Nuf2/Hec1-C and higher FRET between Hec1-C/Spc25-C. These measurements were further confirmed by similar FRET values between N-Nuf2/Nuf2-C and Nuf2-C/Spc25-C (Figure 2A). Thus, a measurable fraction of Ndc80C molecules are staggered relative to one another along kinetochore-microtubule attachments. The extent of this staggering can be estimated by assuming that two Ndc80C molecules bind to the same protofilament. In such a scenario, two Ndc80C molecules would need to be staggered by at least 21 nm, but no greater than 30 nm, to allow for the FRET between N-Nuf2/Hec1-C and Hec1-C/Spc25-C (see the cartoon diagram in Figure 2A). If neighboring molecules are bound to adjacent protofilaments (~6.2 nm separation), then the staggering would need to be between ~23–28 nm. Of note, these same FRET pairs did not produce FRET in budding yeast kinetochores [3]. Thus, the staggered organization of Ndc80C molecules is a distinct feature of human kinetochore-microtubule attachments.
Fluorescence Lifetime Imaging confirms staggering of Ndc80C molecules
Concluding that Ndc80C molecules are staggered along the microtubule lattice assumes that the detected FRET occurs between adjacent complexes. To confirm this, we measured FRET from kinetochore-localized Ndc80C molecules and from molecules freely diffusing in the cytosol. If the observed FRET arises due to staggering, then it should be detected within kinetochores but not in the cytosol. Conversely, if FRET occurs intra-complex, then it should be detectable at both the kinetochore and within the cytosol. We used Fluorescence Lifetime IMaging (FLIM) to simultaneously measure FRET in both populations of kinetochore proteins. FLIM directly measures FRET efficiency from the decrease in the excited-state lifetime of the donor fluorophore due to the presence of an acceptor within 10 nm [32]. Since the donor fluorescence lifetime can be determined accurately even at low fluorophore concentration, we could separately quantify FRET between kinetochore-localized and cytosolic Ndc80C molecules (Figures 2B, S3, and STAR Methods).
We first tested the validity of this approach by measuring the FRET efficiency at N-Nuf2/N-Nuf2 and Spc25-C/Spc24-C. In the former case, FRET is inter-complex and should be detected only within kinetochores. In the latter, FRET is predominantly intra-complex and should occur within kinetochores and the cytosol [15]. Fluorescence lifetime measurements for these two FRET pairs confirmed our expectations (Figure 2C). For Hec1-C/Spc25-C, the pair from which we deduced Ndc80C staggering by our fluorescence intensity-based method, FLIM detected FRET only within kinetochores and not within the cytosol (Figure 2C). Thus, intra-complex FRET does not occur with the Hec1-C/Spc25-C pair, supporting the conclusion that adjacent Ndc80C molecules are staggered along kinetochore-microtubule attachments in human kinetochores.
As a final note, the FRET efficiencies measured via FLIM were directly proportional to the fluorescence intensity-based proximity ratios (Figure 2D). Thus, the proximity ratio reflects the average proximity between kinetochore subunits.
The Ndc80C recruitment linkages are sparsely distributed
The clustered and staggered organization of Ndc80C molecules in attached kinetochores may result from the spatial organization of its centromeric protein linkages. In human kinetochores, CenpC and CenpT recruit Ndc80C (Figure 3A, [33–39]). These proteins bind to the centromere using their C-terminal domains and extend flexible N-terminal domains to bind one Mis12 complex (Mis12C). Mis12C is a ~20 nm long linker/adaptor that binds one Ndc80C. Additionally, the CenpT N-terminal domain directly recruits up to two additional Ndc80C molecules [37]. Therefore, to better understand the spatial organization of Ndc80C we measured FRET between these linkages.
Figure 3. The protein linkages that tether Ndc80C to the centromere are sparsely distributed.
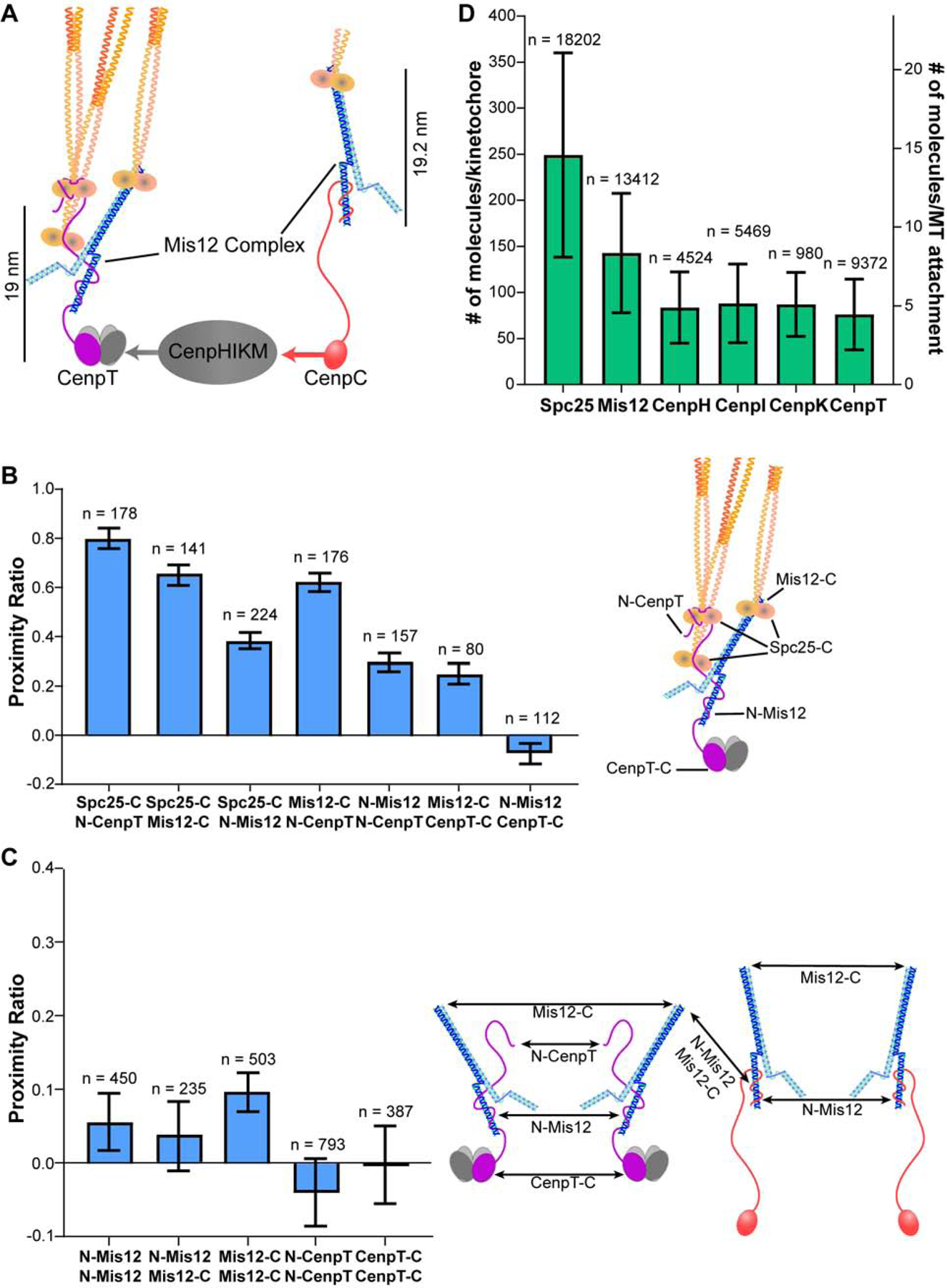
A) Diagram of the biochemical recruitment pathway for Ndc80C (cartoon clipped). B) FRET between proteins involved in Ndc80C recruitment. Diagram to the right shows the location of different fluorophore tags within the CenpT linkage. C) The lack of FRET between neighboring Mis12C molecules and between neighboring CenpT molecules suggests that Ndc80C linkages are ≥ 10 nm apart. The potential FRET pathways are indicated by arrows. D) Protein copy numbers for metaphase kinetochores, evaluated from unfiltered fluorescence signals of kinetochores in cells expressing GFP and/or mCherry labeled versions of the indicated subunits. Bar graphs in B) and C) display the average proximity ratio ± SEM. of fully occupied, metaphase kinetochores. Bar graph in D) displays the average number of molecules per kinetochore (left axis) or per microtubule (right axis, assuming 17.1 microtubules per human kinetochore) ± SD. The number of kinetochores measured for each cell line is indicated above the bars in B), C), and D). All data are from N ≥ 3 experiments. See also Figure S4 and Table S1.
FRET measurements characterizing the CenpT-Mis12C-Ndc80C linkage were consistent with its known organization [33, 37, 40] (Figure 3B). Next, since Mis12C serves as a convenient proxy for CenpC and CenpT (each binds only one Mis12C), we measured FRET between neighboring Mis12C molecules. At most, we detected weak inter-complex FRET between adjacent Mis12C molecules, irrespective of fluorophore placement (Figure 3C). Thus, adjacent Mis12C molecules are, on average, ≥10 nm apart. Interestingly, we did not detect FRET between fluorophores fused to either the C- or the N-terminus of CenpT (Figure 3C). We note that, although the copy number of CenpT is low (~80 molecules/kinetochore), the lower signal-to-noise ratio did not affect our ability to detect FRET (see STAR Methods and also Figures 5D and S6). Thus, neighboring CenpT molecules are spaced ≥10 nm apart. We did not include CenpC in these analyses due to technical difficulties. The absence of FRET between Mis12C molecules, however, indicates that the CenpC recruitment domains are also ≥10 nm apart.
Figure 5. Centromeric tension and microtubule dynamics promote Ndc80C clustering.
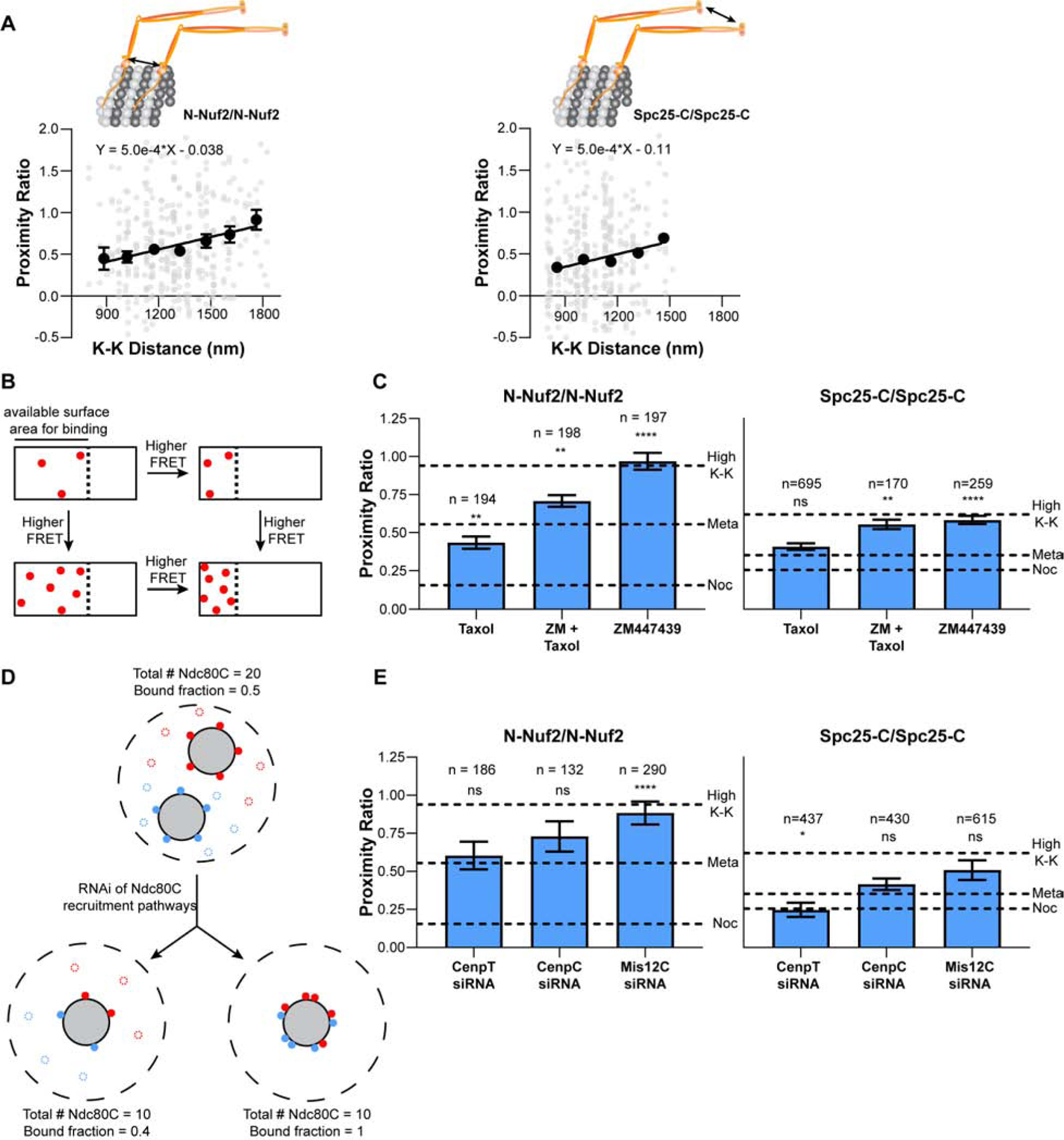
A) Correlation between the proximity ratio and sister kinetochore separation in metaphase for N-Nuf2/N-Nuf2 (left) and Spc25-C/Spc25-C (right) Proximity ratio measurements from fully occupied, metaphase kinetochores in either N-Nuf2/N-Nuf2 (left) or Spc25-C/Spc25-C (right). The unbinned data are in gray and the average value of each bin is shown in black. Bins were defined in ranges of 150 nm K-K distance and are the mean ± SEM. For N-Nuf2/N-Nuf2, n = 23, 52, 92, 83, 63, 30, 20 kinetochores; Pearson correlation coefficient = 0.17 for the unbinned data. For Spc25-C/Spc25-C, n = 82, 103, 108, 41, 14 kinetochores; Pearson correlation coefficient = 0.09 for the unbinned data. Solid line - linear regression. B) The diagram depicts the relationship between the binding density of Ndc80C and the FRET produced by a kinetochore. White rectangles represent the microtubule lattice, where the dashed line demarcates the region of the microtubule plus-end available for Ndc80C binding. Red dots represent bound Ndc80C molecules. C) N-Nuf2/N-Nuf2 and Spc25-C/Spc25-C FRET in response to the microtubule-stabilizing drug Taxol, the Aurora B kinase inhibitor ZM447439, or both. D) Cartoon depicting two potential effects of siRNA-mediated knockdown of Ndc80C recruitment pathways on the organization of Ndc80C molecules. The dashed outer circle denotes the entire kinetochore in an en face view. Blue and red circles indicate Ndc80C molecules linked to individual CenpA nucleosomes. Filled circles are molecules bound to microtubules (grey circles). Unfilled circles indicate unbound molecules. E) N-Nuf2/N-Nuf2 and Spc25-C/Spc25-C FRET after siRNA mediated knockdown of members of the Ndc80C recruitment pathways. In C) and E) data are the average proximity ratio ± SEM. The dashed lines indicate the average proximity ratio for untreated metaphase (Meta), nocodazole-treated (Noc), and for high tension kinetochores (High K-K). The number of kinetochores measured is indicated above the bars. All data are from N ≥ 3 experiments. Statistical significance between untreated, metaphase cells and each of the measurements in C) and E) was evaluated by the Mann Whitney test, ns = not significant; * = p<0.05; ** = p<0.01; *** = p<0.001; **** = p<0.0001. See also Figure S6 and Table S1.
An additional component that may influence the organization of Ndc80C is the CenpH, CenpI, CenpK, and CenpM (CenpHIKM) complex, a CCAN component [35, 41, 42]. CenpHIKM organizes centromeric chromatin and bridges CenpT with CenpC (Figure 3A). Consistent with this role, we found a ~1:1 stoichiometry between three of the four CenpHIKM subunits with CenpT (average for CenpH, CenpI, and CenpK = 86.3 ± 0.8 (S.E.M.) molecules; Figure 3D). FRET measurements between neighboring CenpI subunits revealed that, like CenpT and Mis12C, these subunits are also ≥10 nm apart (Figure S4). Additionally, most of the CenpHIKM subunits were proximal to the centromere and not within proximity of Ndc80C.
In sum, we find that the centromeric recruitment linkages for Ndc80C are ≥10 nm apart from one another. Nevertheless, Ndc80C molecules cluster and stagger along the microtubule lattice. Two factors may contribute to this Ndc80C organization. First, the multivalent recruitment of Ndc80C molecules by CenpT could place multiple Ndc80C molecules within 10 nm, allowing for both clustering and staggering. Second, the microtubule-binding domains of Ndc80C are ~50–70 nm from the CenpT and CenpC N-termini. Thus, even though these recruitment domains are not within FRET proximity, this span may allow distantly spaced Ndc80C molecules to bind near each other on the same plus-end.
Microtubule attachment clusters Ndc80C in both human and budding yeast kinetochores
As Ndc80C molecules cluster during microtubule attachment, we examined the role of microtubule-binding in Ndc80C organization by quantifying FRET in unattached kinetochores. For this, we destroyed the mitotic spindle by treating cells with nocodazole, a microtubule depolymerizing drug (Figures 4A, 4C & S5). In unattached kinetochores, inter-complex FRET between Ndc80C molecules reduced significantly (Figure 4B). The strongest decrease occurred at the microtubule-binding end (N-Nuf2), with smaller decreases near the tetramerization domain (Nuf2-C) and its centromeric end (Spc25-C). The reduced FRET unlikely arises from structural rearrangement within Ndc80C because Spc25-C/Spc24-C FRET showed only a modest decrease (Figure 4B). Thus, binding to the microtubule plus-end is the main reason for the clustering of the microtubule-binding domains of Ndc80C in metaphase kinetochores.
Figure 4. Microtubule attachment clusters Ndc80C in both human and budding yeast kinetochores.
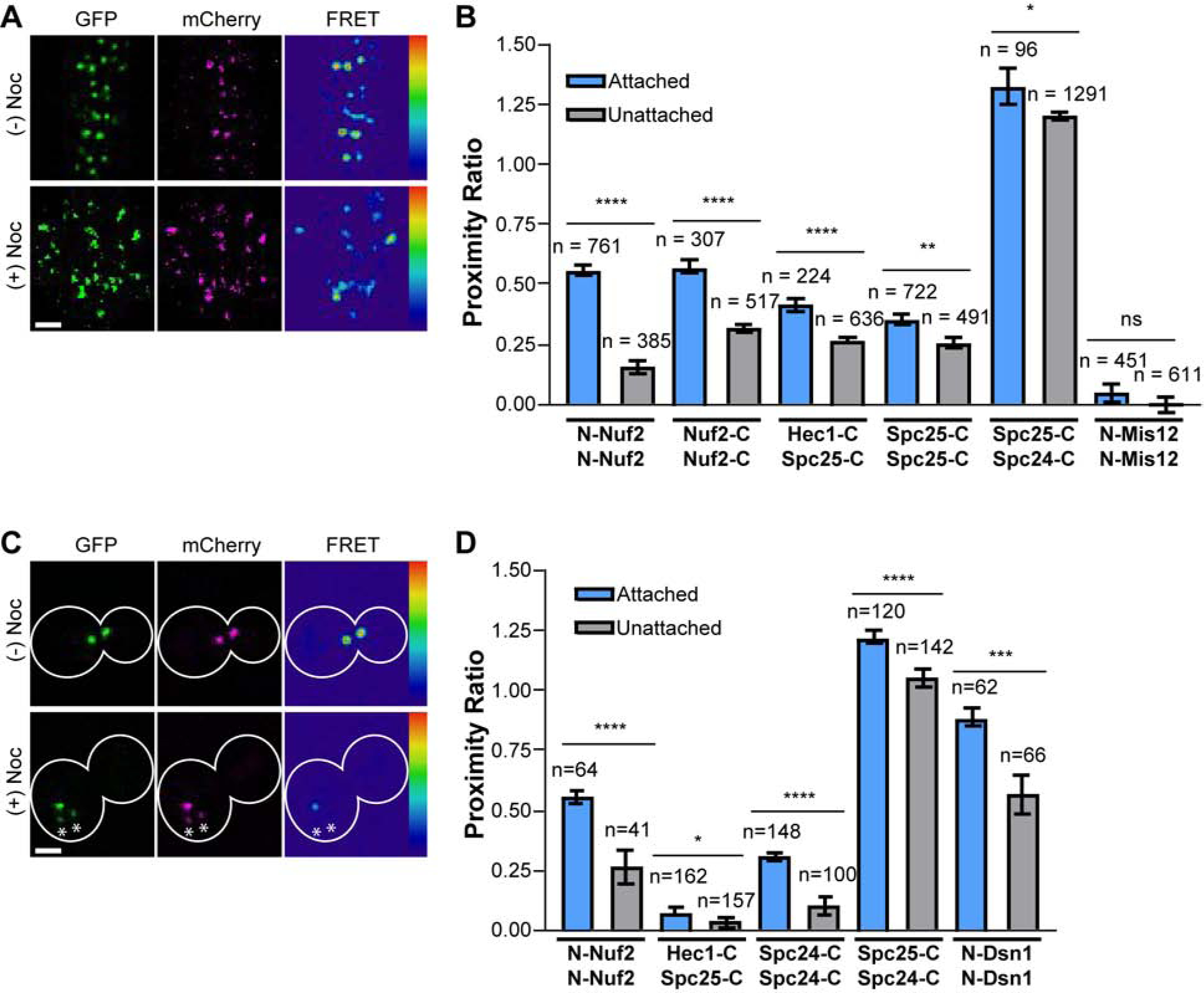
A) Micrographs of mitotic HeLa cells expressing N-Nuf2/N-Nuf2 with and without nocodazole (Noc) treatment. B) Nocodazole treatment reduces FRET between Ndc80C subunits. Measurements are from metaphase (blue) and nocodazole-treated (gray). C) Micrographs of budding yeast metaphase cells expressing N-Nuf2/N-Nuf2, with or without nocodazole. Asterisks highlight clusters of unattached kinetochores. D) Same as in B) but for budding yeast kinetochores. For A) and C), FRET micrographs are scaled equivalently; GFP and mCherry micrographs are scaled for ease of viewing. Scale bar, 2 μm. For D) and B), bars are average proximity ratio ± SEM. The number of measurements is indicated above the bars. All data are from N ≥ 3 experiments. Statistical significance was evaluated using the Mann Whitney test, ns = not significant; * = p<0.05; ** = p<0.01; *** = p<0.001; **** = p<0.0001. See also Figure S5 and Table S1.
Interestingly, the proximity ratio at the centromeric end of Ndc80C was only modestly reduced in unattached kinetochores. This observation suggests that the multivalent recruitment of Ndc80C by CenpT is responsible for Ndc80C centromeric clustering [37]. Moreover, Hec1-C/Spc25-C inter-complex FRET was also detectable in unattached kinetochores (Figure 4B). Therefore, Ndc80C staggering may also result from the multivalence of CenpT.
To understand the influence of multivalent CenpT interactions with Ndc80C in organizing the kinetochore, we adopted a comparative approach. In budding yeast, each centromeric linkage recruits only one Ndc80C [43–45]. The budding yeast CenpT homolog does not bind Ndc80C prior to anaphase [45–48]. Therefore, we expected that the yeast and human kinetochore architectures may respond differently to the loss of microtubule attachment. Accordingly, the centromeric ends of Ndc80C molecules were clustered during attachment in both yeast and human kinetochores. This clustering vanished in unattached yeast kinetochores (compare Spc25-C/Spc25-C in human kinetochores with Spc24-C/Spc24-C in yeast; Figures 4B & D). Furthermore, Hec1-C/Spc25-C FRET was undetectable in yeast kinetochores, consistent with a lack of Ndc80C staggering. The centromeric end of Mis12C (marked by N-Mis12 in the human kinetochore and N-Dsn1 in the yeast kinetochore; [3, 40, 43]) showed a significant degree of clustering in budding yeast kinetochores, but not in human kinetochores. Interestingly, the degree of clustering at the Ndc80C microtubule-binding ends was similar in both kinetochores, implying similar distribution relative to the plus-end.
These data reveal the influence of centromere organization on kinetochore architecture. They strengthen our proposal that the multivalent CenpT linkage is the main source of Ndc80C’s clustered centromeric ends and its longitudinal staggering in human kinetochores. Importantly, despite these differences both kinetochores adopt similar organization at the microtubule-binding ends of Ndc80C.
Centromeric tension and microtubule dynamics promote Ndc80C clustering
The sensitivity of Ndc80C clustering to microtubule attachment prompted us to study whether Ndc80C architecture is also sensitive to centromeric tension. Centromeric tension arises from the opposing forces generated by bioriented sister kinetochores. To reveal the relationship between Ndc80C clustering and centromeric tension, we plotted inter-complex FRET between Ndc80C molecules at their microtubule binding ends (N-Nuf2/N-Nuf2) and at their centromeric ends (Spc25-C/Spc25-C) against the sister kinetochore separation, a proxy for the centromeric tension (referred to as the K-K distance, Figure 5A). The proximity ratio in both cases showed a weak positive correlation, in part because of measurement noise and a smaller number of observations at high K-K distance values (Pearson’s correlation coefficients of 0.17 for N-Nuf2/N-Nuf2 and 0.09 for Spc25-C/Spc25-C, see STAR Methods). To expose the relationship between the proximity ratio and K-K distance, we binned the dataset according to the K-K distance, revealing positive linear correlations at both ends of Ndc80C (Figure 5A). Thus, the proximity between Ndc80C molecules increases with centromeric tension at both the microtubule-binding and centromere-anchored ends of Ndc80C.
Ndc80C clustering at the microtubule-binding ends can increase in response to tension because the number of microtubule-bound molecules increases, the spacing between bound molecules decreases, or both (Figure 5B). These two parameters can change due to Aurora B kinase mediated phosphoregulation of Ndc80C molecules [49], via Ndc80C’s numerous protein-protein interactions (e.g., oligomerization, accessory microtubule-binding proteins, etc.), and by changes in the available microtubule binding surface [50–54].
To understand the role of phosphoregulation on Ndc80C clustering, we treated HeLa cells with ZM447439, a small molecule inhibitor of the Aurora B kinase. ZM447439 treatment increased inter-complex FRET at both N-Nuf2 and Spc25-C such that the average value of the proximity ratio was equivalent to its value in kinetochores under the highest centromeric tension (Figure 5C). ZM447439 treatment did not affect the range of K-K distances as compared to untreated cells, eliminating any potential role of tension in this experiment (Figure S6; [49]). Thus, an increase in the number of microtubule-bound molecules results in more Ndc80C clustering at the plus-end.
To reveal how microtubule plus-end dynamics affects Ndc80C clustering, we treated cells with Taxol. Taxol stabilizes kinetochore-bound plus-ends by dampening tubulin polymerization dynamics, causing an increase in the number of kinetochore-bound microtubules [51, 55, 56]. Therefore, Ndc80C molecules will have a larger microtubule surface area for binding. Accordingly, we measured a small decrease in inter-complex FRET at N-Nuf2 in Taxol-treated cells as compared to untreated metaphase cells (Figure 5C). Interestingly, FRET at Spc25-C did not change with Taxol treatment, showing that plus-end stabilization has little effect on the organization of Ndc80C’s centromere-anchored ends. As an aside, we note that Taxol also lowers the turn-over rate of kinetochore-bound microtubules (i.e., the k-fiber; [51]) Therefore, k-fiber stabilization may play a role in Ndc80C clustering. To test this, we measured N-Nuf2/N-Nuf2 FRET in anaphase cells. During anaphase, the turn-over rate of kinetochore-microtubule attachments is reduced [57]. However, N-Nuf2/N-Nuf2 FRET did not change in anaphase kinetochores as compared to metaphase kinetochores (proximity ratio = 0.58 ± 0.05 versus 0.55 ± 0.02, respectively), suggesting that k-fiber stabilization plays an insignificant role in the organization of Ndc80C’s microtubule-binding ends.
Finally, we simultaneously treated cells with Taxol and ZM447439 to study the distribution of maximally bound Ndc80C molecules. Under this condition, N-Nuf2/N-Nuf2 FRET was intermediate between what we measured with either Taxol or ZM447439 alone (Figure 5C). However, Spc25-C/Spc25-C FRET was unchanged from ZM447439 treatment alone, consistent with the observation that Taxol does not influence Ndc80C centromeric clustering. Overall, these observations show that the number of microtubule-bound Ndc80C molecules and microtubule dynamics influence the relationship between centromeric tension and Ndc80C clustering.
Kinetochores depleted of Ndc80C recruitment linkages maintain Ndc80C clustering and form load-bearing microtubule attachments
Our data demonstrate that Ndc80C clustering occurs despite a ≥10 nm separation between its centromeric receptors (Figure 3C). To determine each receptor’s contribution to Ndc80C clustering, we used RNAi of either CenpT, CenpC, or Mis12C. Knock-down of these proteins reduces the number of Ndc80C molecules per kinetochore by 60–70% (Figure S6; [11, 39]). The lower copy number should lower the centromeric surface density of Ndc80C (Figure 5D). Additionally, because CenpT recruits multiple Ndc80C molecules, these experiment should also reveal the contribution of CenpT in clustering Ndc80C molecules (Figure 2A; [37, 39, 58]).
RNAi treatments caused minor perturbations in chromosome alignment and cell cycle timing. However, most sister kinetochores aligned at the metaphase plate and produced K-K distances similar to untreated cells (Figure S6). We only analyzed aligned kinetochores. We first measured Spc25-C/Spc25-C inter-complex FRET. CenpC and Mis12C siRNA treatments did not significantly influence centromeric clustering (Figure 5E). However, CenpT depletion caused a modest decrease consistent with its multivalent Ndc80C recruitment domain [37]. Although CenpC depletion reduces CenpT by ~40%, the remaining CenpT molecules should still recruit multiple Ndc80C molecules, explaining why CenpC RNAi may have little effect on Ndc80C’s centromeric clustering [11]. Finally, CenpT depletion does not completely eliminate centromeric clustering, suggesting that under this condition Ndc80C molecules recruited by CenpC come within 10 nm of each other.
We next assessed Ndc80C clustering at its microtubule-binding ends by quantifying N-Nuf2/N-Nuf2 FRET. In addition to lowering Ndc80C’s centromeric surface density, reduced numbers of Ndc80C also decreases the number of microtubules per kinetochore [11]. Therefore, RNAi-mediated knockdown of Ndc80C should reduce Ndc80C clustering at its microtubule-binding end (Figure 5D). Alternatively, the fraction of microtubule-bound Ndc80C molecules and/or the proximity between them may increase to compensate for the lower number of Ndc80C molecules (Figures 5D & S6D). Surprisingly, N-Nuf2/N-Nuf2 FRET was either unchanged or increased significantly (Figures 5E & S6B). Thus, Ndc80C clustering at the plus-end is either unchanged or increased during knockdowns of its recruitment linkages. This feature may explain how these kinetochores effectively formed load-bearing attachments despite reduced Ndc80C copy numbers (Figure 5D).
Discussion
Our analysis adds a new dimension to the emerging model of human kinetochore architecture by defining the distribution of key proteins around the plus-end and along the longitudinal axis of attached microtubules. We synthesized this information with protein structures and interactions to construct a model of the organization of human kinetochore-microtubule attachments (Figure 6A). In synthesizing this model, we considered the structure and interactions of the human kinetochore’s repeating ~26-subunit core seeded by the centromeric CenpA nucleosome [35, 36, 38, 42, 59]. The number and centromeric distribution of CenpA nucleosomes dictates CenpC, CenpT, and Ndc80C distribution within the kinetochore. Current estimates suggest that ~44 CenpA nucleosomes participate directly in nucleating the human kinetochore [19, 60]. Our quantitation of CenpHIK (Figure 3D) is consistent with this: one CenpA nucleosome recruits two copies of the CCAN, hence ~44 CenpA nucleosomes will recruit ~88 CCAN subunits [36].
Figure 6. Architectural models of human and budding yeast kinetochore microtubule attachments.
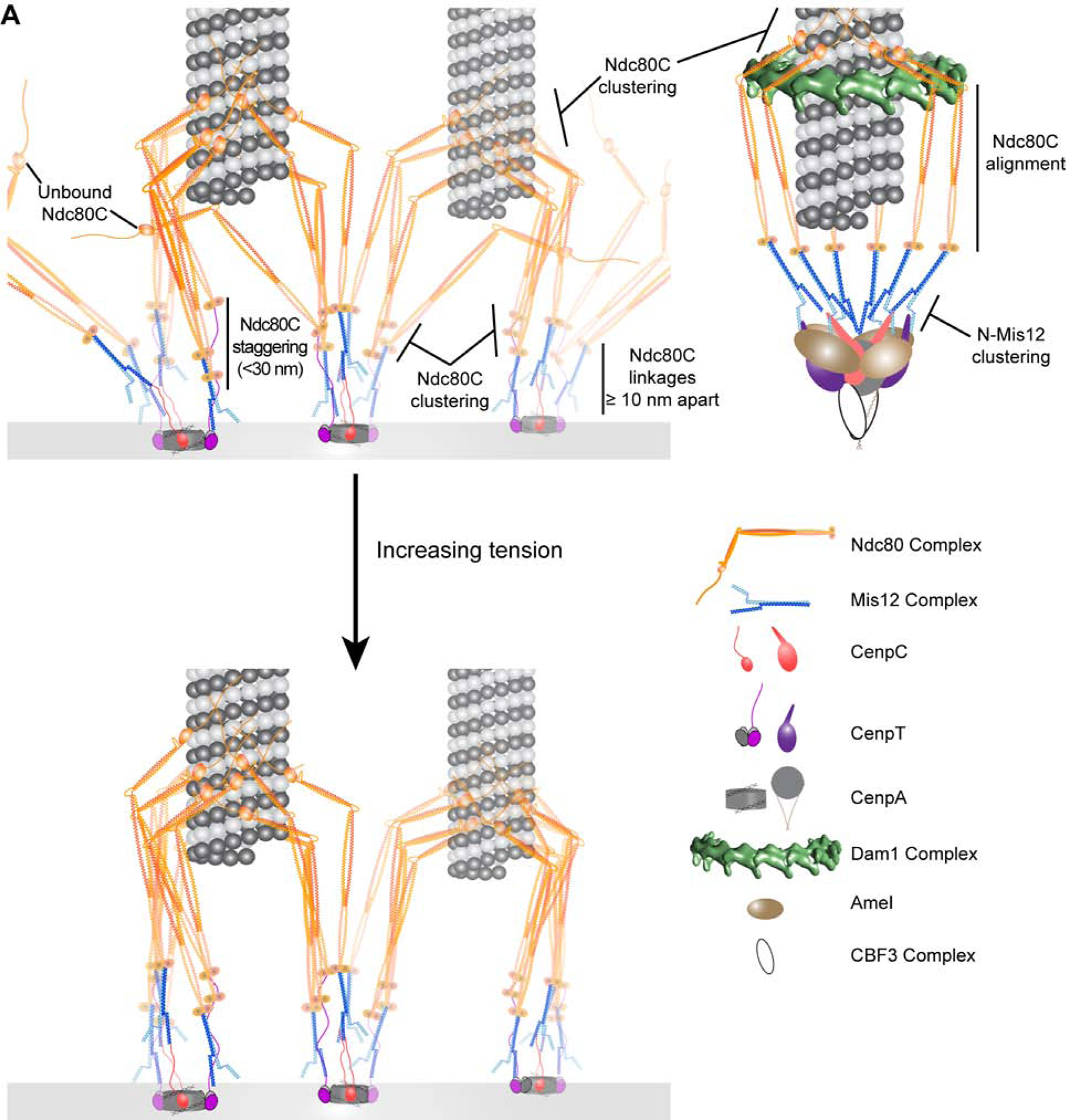
A) The protein organization of human kinetochore-microtubule attachment sites (left) is responsive to physical attachment to the microtubule lattice and to centromeric tension, both of which act to increase the density of microtubule-bound Ndc80C molecules. For comparison, we include a model of the budding yeast kinetochore (upper right). The legend (bottom right) identifies proteins for both models. Key architectural details are emphasized. See text for further details.
The human kinetochore binds 17–20 microtubules on average. Therefore, there are at least two CenpA-nucleated kinetochore subunits for every microtubule attachment. Whether the Ndc80C molecules recruited by a single CenpA-nucleated subunit interact exclusively with one microtubule plus-end like yeast kinetochores is unknown [61]. Our measurements of Ndc80C clustering upon depletion of its centromeric linkages suggest this is not the case (Figures 5D & E). These RNAi treatments reduce the number, and hence the surface density, of Ndc80C molecules per kinetochore by up to 60% [11]. Accordingly, each CenpA-nucleated subunit will recruit as few as 2 Ndc80C molecules, and the number of microtubules per kinetochore will see a proportionate decrease. Nevertheless, inter-complex FRET between Ndc80C’s microtubule-binding ends persists or even increases indicating that Ndc80C’s reduced surface density does not hinder its microtubule binding activity. Conceivably, the loss of centromeric structural integrity that accompanies CenpC and CenpT depletion may affect kinetochore/spindle interactions [6]. However, this concern does not apply to Mis12C RNAi. Mis12C does not bind the centromere and yet its depletion results in the highest clustering of Ndc80C molecules. These observations support the model that Ndc80C molecules in the human kinetochore operate as a “lawn”, allowing several neighboring CenpA-nucleated kinetochore subunits to cooperate in the formation of microtubule attachments despite their sparse centromeric distribution [62–64] (Figure 6).
The sensitivity of Ndc80C FRET to microtubule attachment reveals the adaptability of kinetochore architecture to its mechanical state. Upon attachment, Ndc80C molecules become clustered (Figures 2 and 4). This behavior resembles a recent model wherein Ndc80C molecules align along the spindle axis upon microtubule attachment [27]. Ndc80C clustering also increases proportionally with centromeric tension, suggesting that centromeric tension and the number of microtubule-bound Ndc80C molecules are correlated (Figure 5A). This hypothesis is supported by the significant increase in Ndc80C clustering upon Aurora B kinase inhibition, which promotes maximal Ndc80C binding (Figure 5C; [49]). The correlation between the number of microtubule-bound Ndc80C molecules and centromeric tension may also play a role in the persistent clustering of Ndc80C observed in our RNAi experiments. In these experiments, the force per Ndc80C molecule increases because Ndc80C numbers reduce without a change in the range of K-K distances (Figure S6). The higher force per Ndc80C may promote binding and clustering despite their lower centromeric surface density. Our studies also reveal that microtubule plus-end dynamics play a role in Ndc80C clustering. This is most clearly seen when Aurora B activity and plus-end dynamics are inhibited simultaneously: Ndc80C clustering decreases at its microtubule-binding domains compared to Aurora B inhibition alone (Figure 5C). How plus-end dynamics affects Ndc80C clustering is unclear. However, it is possible that dynamicity limits Ndc80C distribution at the plus-end [65]. Aurora B inhibition may also affect the function of key microtubule-binding proteins (e.g. the Astrin-SKAP complex) and indirectly affect Ndc80C architecture [66–68].
Finally, our study highlights the similarities and differences in kinetochore architectures built upon point centromeres (budding yeast) and regional centromeres (humans). Unlike the human kinetochore, the yeast kinetochore is nucleated by just one CenpA nucleosome, forming a persistent attachment with only one microtubule (Figure 6A; [61]). Therefore, all Ndc80C molecules interact with the same microtubule plus-end in budding yeast kinetochores. Consistent with this picture, both the Ndc80C microtubule-binding domains and the Mis12C centromere-binding ends cluster together in the yeast kinetochore (Figure 4D; [3, 43]). In contrast, only Ndc80C molecules cluster in the human kinetochore; Mis12C molecules do not (Figure 4B). Furthermore, Ndc80C molecules are aligned with one another in yeast kinetochores, but stagger along the microtubule axis in human kinetochores. For yeast, Ndc80C alignment is likely enforced by the point centromere and the Dam1 ring-like structure [69]. In humans, the staggered organization of Ndc80C arises because of the multivalence of CenpT and the flexibility of the centromeric linkages. We estimate that Ndc80C staggering is no greater than 30 nm, although we cannot rule out the possibility that non-adjacent Ndc80C molecules are staggered by even larger distances. The staggered arrangement of Ndc80C molecules will enhance the attachment persistence and tip-tracking ability of human kinetochores [17]. Given the significant differences in the organization of the human and yeast centromeres, it is remarkable that both kinetochores achieve similar degrees of Ndc80C clustering. This similarity in the kinetochore-microtubule interfaces of yeast and humans may represent a generally conserved feature of kinetochore architecture.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ajit P. Joglekar (ajitj@umich.edu).
Materials Availability
Plasmids and cell lines generated for this study are available upon request.
Data and Code Availability
The datasets supporting the current study are not publicly available but will be provided upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Culture conditions for HeLa cells
The parental HeLa A12 cell line was maintained in DMEM media (Gibco) supplemented with 10% FBS (Corning), and 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco). Parental cells stably-integrated with plasmids for the dual expression of fluorophore-tagged kinetochore proteins were maintained in the same media supplemented with 1 μg/mL puromycin. Cells were grown in a 37 °C/5% CO2 incubator. All plasmids integrated into the parental cell line were verified via DNA sequencing, and stably-integrated cells were authenticated by selection for puromycin resistance and subsequent fluorescence microscopy analysis for the co-localization of GFP- and mCherry-tagged proteins.
METHOD DETAILS
Construction of HeLa cells lines
For the co-expression of fluorophore-tagged kinetochore proteins, HeLa cell lines were generated containing a stable chromosomal insertion of a dual-expression vector. The HeLa A12 cell line (gift from the Lampson lab) contains a lentiviral-based chromosomal insertion of a pair of incompatible Cre/Lox sites in front of the human EF-1α promoter (see [18] for details). Using standard molecular cloning, we created cassettes capable of Cre recombinase-mediated integration at this chromosomal locus that were based on the pERB131 plasmid backbone (gift from the Lampson lab). Briefly, the pERB131 backbone contains two open-reading frames (ORFs), one that becomes under the control of the constitutive EF-1α promoter upon successful integration (ORF1) and a second (ORF2) which is controlled by a tetracycline responsive promoter (Tet-ON). All proteins examined in this study were cloned into one of these two ORFs. The cassette also contains a gene for puromycin resistance which aided in the selection of HeLa cells with successful integration. All HeLa cell lines generated for this study are listed in Table S2.
Integration was performed using the Lipofectamine 3000 Reagent kit (Thermo Fisher Scientific) to co-transfect cells with the pERB131 cassette of interest and a Cre-expression plasmid (gift from the Lampson lab). Two days post-transfection, 2 μg/mL puromycin was added to the cell media for selection over the course of two weeks. Successful transformants were then maintained in media containing 1 μg/mL puromycin.
Owing to the large number of cell lines generated for this study, we did not conduct a detailed analysis of cell cycle duration or mitotic defects. Uninduced cell lines were maintained at 37 °C/5% CO2 for 1 – 2 weeks without any obvious increases in cell death or mitotic index. Cells induced for dual protein expression by doxycycline were maintained for, at most, 3 days and under these conditions we also did not observe obvious increases in cell death or mitotic index. Additionally, the average sister kinetochore distances were in the normal range for all FRET pairs (see Table S1).
Fluorescence microscopy
All fluorescence and FRET imaging was performed on a Nikon Ti-E inverted microscope with a 1.4 NA, 100x, oil immersion objective. A Lumencor LED light engine (472/20 nm GFP excitation, 543/20 nm mCherry excitation) served as the laser power source. All filters are from Chroma and included: 1) a dual-band excitation filter ET/GFP-mCherry (59002x); 2) an excitation dichroic (89019bs); 3) an emission-side dichroic (T560lpxr); 4) and emission filters ET525/50m and ET595/50m. Images were acquired on an Andor iXon3 EMCCD camera (pixel size = 160 nm, 16-bit A/D converter). Cell images were either 20 or 10 plane z-stack image series for HeLa and budding yeast cells, respectively. The step size between planes was 0.25 μm. For most experiments, the acquisition rate for GFP and mCherry was set at 400 ms. Occasionally, when the copy number of fluorophore-tagged proteins was low (e.g., CCAN proteins or during siRNA mediated knockdowns) the acquisition rate was increased to obtain higher fluorescence signal. A simple linear correction was applied to normalize fluorescence intensity values to a 400 ms acquisition rate.
To account for fluctuations in laser power and other artifacts in our microscopy setup, we collected images of ~ 20 anaphase budding yeast cells expressing Ndc80-GFP and Spc25-mCherry before all experiments. Since budding yeast incorporate a stable number of proteins per kinetochore, any changes in GFP and mCherry brightness in these cells should be a result of instrument-derived fluctuations. In this way, ratiometric correction factors were derived for each day of imaging to normalize all FRET measurements throughout the course of this study.
For HeLa, cells were plated in multi-chamber glass-bottomed dishes (Lab-Tek®II) in DMEM media (Gibco) supplemented with 10% FBS (Corning), and 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco). Cells were treated with 1 – 2 μg/mL doxycycline for 48 hrs to induce the expression of ORF2 proteins. Treatments with siRNA were performed using the Lipofectamine RNAiMAX kit (Invitrogen), using 30 pmol of each protein-specific siRNA and an incubation period of at least 48 hrs (siRNAs listed in Table S3). During imaging, cell media was changed to DMEM without any phenol red and supplemented with 10% FBS and 100 U/mL penicillin and 100 μg/mL streptomycin. During imaging, the microscope stage is fitted with a heated chamber with CO2 respirator and objective warmer (Live Cell Instrument). For several experiments, we employed double-thymidine synchronization with 2.5 mM thymidine. For imaging unattached kinetochores, cells were treated with 100 ng/mL nocodazole and incubated at least 30 min before imaging. For imaging attached, tensionless kinetochores, cells were treated with 10 μM Taxol and incubated at least 10 min before imaging. For experiments with the Aurora B inhibitor, ZM447439, we added 10 μM MG132 and incubated 5 min before adding 3 μM of the ZM447439 drug. Cells were incubated an additional 10 min before imaging. For imaging with both ZM447429 and Taxol, the same procedure was followed as above, adding MG132 and ZM447439 first, incubating 10 min, then adding Taxol. Attached kinetochores were distinguished from unattached kinetochores by their positioning at the spindle mid-zone. Unattached kinetochores were often dispersed through the cell body with random orientations with respect to the spindle and greatly reduced (~ 800 nm) sister kinetochore separation.
For budding yeast, cells were grown at 30 °C to mid-log phase in yeast peptone (YP) media supplemented with 2% glucose. For strains with galactose-inducible promoters, the YP media was supplemented with 2% raffinose and varying concentrations of galactose. The appropriate galactose concentration was determined as that which produced average fluorescence signals at kinetochores that were equal to the fluorescence signal in strains without inducible-promoters. Prior to imaging, cells were rinsed and concentrated in synthetic drop-out media. For imaging of unattached kinetochores, mid-log phase cells were treated with 15 μg/mL nocodazole for 1.5 hrs before rinsing and concentrating cells in synthetic media supplemented with 15 μg/mL nocodazole. Metaphase kinetochore clusters were designated by sister pairs with a separation of ~ 0.8 to 1 μm. For nocodazole-treated cells, unattached kinetochores were identified as the dimmer fluorescent puncta separate from the brighter, spindle-localized attached kinetochores. Cells were imaged on 22×22 mm glass coverslips. All yeast strains used in this study are from [3].
Intensity-based FRET quantification
To measure FRET, a semi-automated graphical user interface written in MATLAB was used to analyze cell images. The implementation of this program is described in [25]. The raw FRET intensity, measured as the fluorescence intensity observed in the mCherry channel upon excitation with the GFP-specific laser, contains contaminating signal from GFP bleed-through and mCherry cross-excitation. The contribution of these signals was measured in HeLa cells expressing either Spc25-GFP or Spc25-mCherry alone (Figure S2; GFP bleed-through = 5.79 ± 0.17%, mCherry cross-excitation = 6.64 ± 0.18%). Subtracting these values from the raw FRET intensity yields the sensitized emission due to FRET. Given the variable number and stoichiometry of kinetochore protein subunits, the sensitized emission was further normalized by the sum of the GFP bleed-through and mCherry cross-excitation. Since these values are proportional to the number of fluorophore-tagged molecules, this normalization essentially yields the sensitized emission/molecule, a metric we refer to as the proximity ratio:
Filtering for kinetochore protein occupancy
To accurately measure FRET at HeLa kinetochores, we needed to ensure that our datasets contained only those kinetochores that are maximally occupied by donor- and acceptor-labeled proteins. To meet this requirement, we first defined the single molecule brightness values of GFP and mCherry. To do this, we measured the average background subtracted fluorescence signals for HeLa cells that co-expressed Spc25-GFP and Spc25-mCherry. For these measurements, HeLa cells were treated with Spc25 siRNA which specifically targets endogenous, unlabeled Spc25 but not the fluorophore-tagged versions of Spc25. The data set of fluorescence signal per kinetochore from these cells were first binned by their mCherry:GFP ratios. This binning suppresses the effect of variations in kinetochore size, which was noted in previous reports [19–24]. A plot of the bin average Spc25-mCherry v. Spc25-GFP fluorescence signals was fit by a linear regression, yielding the linear equation y = −0.4418x + 11010. The x- and y-intercepts of this equation predict the fluorescence intensity corresponding to kinetochores fully occupied by GFP or mCherry labeled Spc25 molecules, respectively (x-intercept = 24,921 a.u.; y-intercept = 11,010 a.u.; the data in Figure 1C are normalized by these values). These intensity values reflect the average number of Spc25 molecules per kinetochore. A prior study by Suzuki et al. found that there are 244 molecules of the Ndc80C per kinetochore (Spc25 is a subunit of the Ndc80 complex) [11]. Using this information, we determined the single molecule brightness of GFP and mCherry to be 102.1 ± 5.0 and 45.1 ± 0.9 a.u.
Using these single molecule brightness values, we converted all subsequent fluorescence signals per kinetochore into a molecular count. Only these brightness values that reflected the appropriate molecule counts for a given kinetochore subunit were retained. The following table defines the filtering bounds we used for each of the kinetochore protein complexes measured in this study:
| Protein Complex | Measurement Type | Filtering Bounds (# of molecules) | Reference |
|---|---|---|---|
| The Ndc80 complex | Untreated/Metaphase | 212 – 276 | [11] |
| Nocodazole | >212 | [11] | |
| Taxol | 212 – 488 | [11] | |
| ZM447439 | 212 – 276 | [11] | |
| ZM447439 + Taxol | 212 – 488 | [11] | |
| CenpT siRNA | 73 – 107 | [11] | |
| CenpC siRNA | 83 – 117 | [11] | |
| Mis12C siRNA | 40 – 146 | [11] | |
| The Mis12 complex | Untreated/Metaphase | 130 – 172 | [11] |
| Nocodazole | >130 | [11] | |
| CenpT | Untreated/Metaphase | 64 – 110 | This study & [11] |
| Nocodazole | >64 | This study & [11] | |
| CenpHIKM | Untreated/Metaphase | 64 – 110 | This study |
| Nocodazole | >64 | This study |
For nocodazole measurements, we adhered to the lower limits determined from [11], but did not place an upper limit on these values for two reasons: 1) it has been previously noted elsewhere and in our studies that nocodazole-treated, unattached kinetochores recruit greater numbers of molecules than attached, metaphase kinetochores (Figure S6C and [5, 7, 70, 71]); and 2) the disorganized spindles and reduced sister kinetochore separation made it difficult to measure single kinetochores accurately (Figure S6D and Table S1). Similarly, due to the reduced sister kinetochore separation upon Taxol treatment, we filtered Taxol measurements between 212–488 molecules (the upper-limit being twice the average number of molecules at a single kinetochore). Therefore, a small fraction of nocodazole and Taxol measurements may represent more than one kinetochore. For the purposes of quantifying FRET, however, this is not a problem as the proximity ratio is normalized by the total number of molecules (see “FRET quantification and image analysis”).
The number of Ndc80 complex molecules after siRNA mediated knockdown of CenpT or CenpC is documented [11]. For siRNA mediated knockdown of the Mis12 complex, however, we have defined our filter bounds for the Ndc80 complex indirectly by assuming that every Mis12 complex recruits exactly one Ndc80 complex [11, 37, 39, 40]. Thus, Mis12 complex knockdown should reduce the total number of Ndc80 complexes by 130 – 172 molecules.
The filter bounds for members of the CenpHIKM complex and CenpT were, in part, defined from the average of the unfiltered intensity values of CenpH, CenpI, CenpK, and CenpT (~87 molecules/kinetochore; Figure 3D). Since the CenpHIKM complex aids in the recruitment of CenpT, we set the lower limit of CenpHIKM molecules the same as for CenpT (i.e., 64 molecules/kinetochore; [11]). The upper limit was then set at 110 molecules/kinetochore to place the average value as the midpoint of these extremes.
An additional filter was used when two versions of the same protein compete for binding to the kinetochore (e.g. Spc25-C/Spc25-C FRET). In addition to filtering for full kinetochore occupancy, we further eliminated kinetochores with acceptor-to-donor ratios outside of the range of 0.2 – 5.0. Such filtering removes kinetochores that are saturated with only donor-labeled or acceptor-labeled molecules (i.e., incapable of FRET). As discussed in the main text and as demonstrated in Table S1, changing the bounds on this acceptor-to-donor ratio filter only mildly affects the value of the proximity ratio and the overall trends between different FRET pairs does not change.
In budding yeast measurements, nocodazole treatment creates unattached kinetochore clusters of variable size. Therefore, for consistency between metaphase attached and the unattached nocodazole-treated kinetochores, all measurements were filtered to contain only data points with an mCh:GFP ratio of 0.5 – 2.
Fluorescence lifetime imaging microscopy
Fluorescence lifetime imaging (FLIM) data were collected on an ISS ALBA time-resolved laser-scanning confocal system. This setup consists of: 1) an Olympus IX-81 microscope with a U-Plan S-APO 60X 1.2 NA water immerision objective; 2) an SPC-830 time-correlated single photon counting (TCSPC) board (Becker & Hickl); 3) an SC-400–6-PP supercontinuum laser (Fianium); 4) and two photomuliplier tubes (PMT) detectors (Hamamatsu H7422P-40). During data collection, the objective was also equipped with a 37 °C temperature-controlled sleeve. HeLa cells were plated in 35 mm glass-bottomed dishes (MatTek) and imaged in DMEM media without any phenol red supplemented with 10% FBS and 100 U/mL penicillin and 100 μg/mL streptomycin. GFP and mCherry excitation were performed with 488 nm and 561 nm lasers, interleaved with 20 MHz frequency and 256 ADC resolution. The pixel-dwell time was 0.2 ms and laser power was adjusted to keep photon counts between 500,000 – 1,000,000 per pixel.
FLIM data were analyzed using the VistaVision software analysis program (ISS). To distinguish between cytosolic versus kinetochore-localized GFP, we employed intensity threshold masks. This method proved effective since kinetochore-localized GFP always provided higher counts/pixel than cytosolic GFP. Additionally, kinetochore pixels were further isolated by cropping the images to contain only the cellular region corresponding to the metaphase plate. At minimum, cytosolic GFP had a lower threshold of 30 photon counts/pixel to distinguish from background. We also maintained a buffer of at least 25 photon counts/pixel between the upper threshold for the cytosolic GFP and the lower threshold for the kinetochore-localized GFP to prevent cross-contamination of signals. After appropriate thresholding, photon counts from all pixels were summed. As the GFP excitation laser was pulsed first during the interleaved excitation, only photons collected between the first 6.6 – 24.8 ns of each pulse were included. GFP lifetimes were estimated by fitting the histograms of the photon arrival times to single-component exponential decays, using a software generated instrument response function (IRF). The FRET efficiency was calculated by comparing the difference between the GFP lifetime in the absence and presence of an mCherry acceptor . We note that the GFP lifetime in the absence of an mCherry acceptor was highly dependent on the protein subunit to which it was attached and on temperature. Therefore, for all FRET pairs the GFP lifetimes without an mCherry acceptor were measured independently.
Western blot analysis
HeLa cell lysates were collected from cultures grown in 6-well plates (Corning), seeding at a density of ~ 100K cells/well. Cells were grown for 3 days with the appropriate drug and siRNA treatments applied, after which cells were rinsed and aspirated. Lysates were collected in 200 μL SDS-PAGE buffer containing 2-mercaptoethanol using a cell scarper. Lysates were denatured at 95 °C, vortexed and centrifuged before loading onto a 4% stacking/10% resolving SDS-PAGE gel. After running, PAGE gels were transferred to PVDF membranes (pre-activated by soaking in methanol) via electrophoresis in transfer buffer (1.4% glycine, 0.3% Tris-base in H2O). Blots were blocked with 5% milk in tris-buffered saline (TBS) for 30 min. and then incubated with primary antibody overnight at 4 °C with shaking (primary antibodies prepared in either 5% BSA or 5% milk in TBS + 0.1% Triton X100). After rinsing, blots were incubated with secondary antibody prepared in 5% BSA in TBS-T for 30 min. After rinsing, blots were developed via chemiluminescence (Immobilon Western reagent from Millipore) and imaged with an Azure c600 gel imager (Azure Biosystems). All antibodies used in this study are provided in Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-β-tubulin | Sigma-Aldrich | Cat# T7816; RRID: AB_261770 |
| Mouse monoclonal anti-DsRed2 | Santa Cruz Biotechnology | Cat# sc-101526; RRID: AB_1562589 |
| Rabbit polyclonal anti-Spc25 | Atlas Antibodies | Cat# HPA047144; RRID: AB_2679952 |
| Rabbit polyclonal anti-Nuf2 | Bethyl | Cat# A304–319A; RRID: AB_2620515 |
| Mouse monoclonal anti-GFP | Takara Bio | Cat# 632381; RRID: AB_2313808 |
| Goat monoclonal anti-mouse, horseradish peroxidase conjugated | Sigma-Aldrich | Cat# A4416; RRID: AB_258167 |
| Goat monoclonal anti-rabbit, horseradish peroxidase conjugated | Sigma-Aldrich | Cat# A4914; RRID: AB_258207 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nocodazole | Fisher | Cat# AC358240100; CAS: 31430-18-9 |
| Taxol | Fisher | Cat# NC9507351; CAS: 33069-62-4 |
| ZM447439 | Fisher | Cat# 508279 |
| Doxycycline | Fisher | Cat# BP26531; CAS: 10592-13-9 |
| Thymidine | Millipore | Cat# 6060; CAS: 50-89-5 |
| Puromycin | Fisher | Cat# ICN19453910 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| HeLa A12 | [18] | N/A |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: Strain background: YEF473 | ATCC | ATTC: |
| Oligonucleotides | ||
| For a list of all siRNAs used in this study, see Supplementary Table 3 | ||
| Recombinant DNA | ||
| pEM784 – pCAGGS-nls-Cre | [18] | N/A |
| pERB131 – Mis12-GFP-FKBPx3; inducible: mCh-Mps1 | Lampson lab | N/A |
| For plasmids for the expression of FRET pairs in HeLa A12 cells, see Supplementary Table 2 | This study | N/A |
| For plasmids for the expression of FRET pairs in S. cerevisiae, please see the accompanying reference | [3] | N/A |
| Software and Algorithms | ||
| Prism | Graphpad | Ver. 8 |
| MATLAB | Mathworks | Ver. 2017b |
| VistaVision | ISS | Ver. 4.0 |
| Other | ||
| DMEM | Thermo Fisher | N/A |
| Lipofectamine 3000 | Life Technologies | Cat# L3000008 |
| Lipofectamine RNAiMAX | Life Technologies | Cat# 13778075 |
QUANTIFICATION AND STATISTICAL ANALYSIS
Intensity-based FRET fluorescence microscopy images were measured using a semi-automated graphical user interface in MATLAB [25]. Fluorescence lifetime imaging microscopy data were analyzed using the VistaVision software analysis program (ISS). Statistical analyses were performed in GraphPad Prism. Details of statistical analyses performed in this study are provided in the figure legends and in Table S1.
Supplementary Material
Table S1. List of proximity ratio measurements for FRET pairs in this study. Related to Figures 1, 2, 3, 4, 5, S4, S5, and S6. FRET pairs are grouped by the complexes to which they belong. “Measurement Type” indicates treatments applied to cells before measurements (“Metaphase” indicates untreated cells). For all measurement types except Nocodazole, only bioriented kinetochores were selected for analysis. For all measurements, a one sample t-test was used to evaluate if the proximity ratio was statistically different from zero (n.s. = not significant, with p-values listed in parentheses; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001). aAverage of all filtered measurements, where filtering discards kinetochores that are not fully occupied by the fluorophore-tagged proteins. bThe Max Proximity Ratio indicates the average proximity ratio for all filtered measurements within an acceptor to donor ratio of 1 – 2. This is only done for FRET pairs that compete for binding to the kinetochore (i.e., acceptor and donor fluorophores on the same protein subunit).
Highlights:
Ndc80 complex molecules cluster and stagger relative to the microtubule plus-end
The centromeric receptors of Ndc80, CenpT and CenpC, are spaced ≥10 nm apart
Ndc80 clustering is sensitive to microtubule attachment and centromeric tension
Ndc80 molecules are organized as a flexible ‘lawn’ in the human kinetochore
Acknowledgements
This work was supported by R35 GM126983 to APJ. The authors also thank the Single Molecule Analysis in Real-Time (SMART) Center of the University of Michigan, seeded by NSF MRI-R2-ID award DBI-0959823 to Nils G. Walter, Damon Hoff for training, technical advice and use of the ALBA time-resolved confocal microscope, and Shih-Chu “Jeff” Liao for his help in debugging and running the VistaVision software.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests
References
- 1.McIntosh JR (1991). Structural and mechanical control of mitotic progression. Cold Spring Harb Symp Quant Biol 56, 613–619. [DOI] [PubMed] [Google Scholar]
- 2.Wan X, O’Quinn RP, Pierce HL, Joglekar AP, Gall WE, DeLuca JG, Carroll CW, Liu ST, Yen TJ, McEwen BF, et al. (2009). Protein architecture of the human kinetochore microtubule attachment site. Cell 137, 672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aravamudhan P, Felzer-Kim I, Gurunathan K, and Joglekar AP (2014). Assembling the protein architecture of the budding yeast kinetochore-microtubule attachment using FRET. Curr Biol 24, 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aravamudhan P, Goldfarb AA, and Joglekar AP (2015). The kinetochore encodes a mechanical switch to disrupt spindle assembly checkpoint signalling. Nat Cell Biol 17, 868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magidson V, He J, Ault JG, O’Connell CB, Yang N, Tikhonenko I, McEwen BF, Sui H, and Khodjakov A (2016). Unattached kinetochores rather than intrakinetochore tension arrest mitosis in taxol-treated cells. J Cell Biol 212, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki A, Badger BL, Wan X, DeLuca JG, and Salmon ED (2014). The architecture of CCAN proteins creates a structural integrity to resist spindle forces and achieve proper Intrakinetochore stretch. Dev Cell 30, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magidson V, Paul R, Yang N, Ault JG, O’Connell CB, Tikhonenko I, McEwen BF, Mogilner A, and Khodjakov A (2015). Adaptive changes in the kinetochore architecture facilitate proper spindle assembly. Nat Cell Biol 17, 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musacchio A, and Desai A (2017). A Molecular View of Kinetochore Assembly and Function. Biology 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinshaw SM, and Harrison SC (2018). Kinetochore Function from the Bottom Up. Trends Cell Biol 28, 22–33. [DOI] [PubMed] [Google Scholar]
- 10.Joglekar AP, and Kukreja AA (2017). How Kinetochore Architecture Shapes the Mechanisms of Its Function. Curr Biol 27, R816–R824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki A, Badger BL, and Salmon ED (2015). A quantitative description of Ndc80 complex linkage to human kinetochores. Nat Commun 6, 8161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith CA, McAinsh AD, and Burroughs NJ (2016). Human kinetochores are swivel joints that mediate microtubule attachments. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki A, Long SK, and Salmon ED (2018). An optimized method for 3D fluorescence co-localization applied to human kinetochore protein architecture. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeLuca JG, Gall WE, Ciferri C, Cimini D, Musacchio A, and Salmon ED (2006). Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127, 969–982. [DOI] [PubMed] [Google Scholar]
- 15.Ciferri C, Pasqualato S, Screpanti E, Varetti G, Santaguida S, Dos Reis G, Maiolica A, Polka J, De Luca JG, De Wulf P, et al. (2008). Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell 133, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wendell KL, Wilson L, and Jordan MA (1993). Mitotic block in HeLa cells by vinblastine: ultrastructural changes in kinetochore-microtubule attachment and in centrosomes. J Cell Sci 104 (Pt 2), 261–274. [DOI] [PubMed] [Google Scholar]
- 17.Hill TL (1985). Theoretical problems related to the attachment of microtubules to kinetochores. Proc Natl Acad Sci USA 82, 4404–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khandelia P, Yap K, and Makeyev EV (2011). Streamlined platform for short hairpin RNA interference and transgenesis in cultured mammalian cells. Proc Natl Acad Sci USA 108, 12799–12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bodor DL, Mata JF, Sergeev M, David AF, Salimian KJ, Panchenko T, Cleveland DW, Black BE, Shah JV, and Jansen LE (2014). The quantitative architecture of centromeric chromatin. Elife 3, e02137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherry LM, and Johnston DA (1987). Size variation in kinetochores of human chromosomes. Hum Genet 75, 155–158. [DOI] [PubMed] [Google Scholar]
- 21.Cherry LM, Faulkner AJ, Grossberg LA, and Balczon R (1989). Kinetochore size variation in mammalian chromosomes: an image analysis study with evolutionary implications. J Cell Sci 92 (Pt 2), 281–289. [DOI] [PubMed] [Google Scholar]
- 22.McEwen BF, Ding Y, and Heagle AB (1998). Relevance of kinetochore size and microtubule-binding capacity for stable chromosome attachment during mitosis in PtK1 cells. Chromosome Res 6, 123–132. [DOI] [PubMed] [Google Scholar]
- 23.Drpic D, Almeida AC, Aguiar P, Renda F, Damas J, Lewin HA, Larkin DM, Khodjakov A, and Maiato H (2018). Chromosome Segregation Is Biased by Kinetochore Size. Curr Biol 28, 1344–1356 e1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dumont M, Gamba R, Gestraud P, Klaasen S, Worrall JT, De Vries SG, Boudreau V, Salinas-Luypaert C, Maddox PS, Lens SM, et al. (2020). Human chromosome-specific aneuploidy is influenced by DNA-dependent centromeric features. EMBO J 39, e102924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joglekar A, Chen R, and Lawrimore J (2013). A Sensitized Emission Based Calibration of FRET Efficiency for Probing the Architecture of Macromolecular Machines. Cell Mol Bioeng 6, 369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller EG, Snydsman BE, Novik I, Hailey DW, Gestaut DR, Niemann CA, O’Toole ET, Giddings TH Jr., Sundin BA, and Davis TN (2005). The organization of the core proteins of the yeast spindle pole body. Mol Biol Cell 16, 3341–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roscioli E, Germanova TE, Smith CA, Embacher PA, Erent M, Thompson AI, Burroughs NJ, and McAinsh AD (2020). Ensemble-Level Organization of Human Kinetochores and Evidence for Distinct Tension and Attachment Sensors. Cell Rep 31, 107535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattiuzzo M, Vargiu G, Totta P, Fiore M, Ciferri C, Musacchio A, and Degrassi F (2011). Abnormal kinetochore-generated pulling forces from expressing a N-terminally modified Hec1. PLoS One 6, e16307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei RR, Sorger PK, and Harrison SC (2005). Molecular organization of the Ndc80 complex, an essential kinetochore component. Proc Natl Acad Sci USA 102, 5363–5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheeseman IM, Chappie JS, Wilson-Kubalek EM, and Desai A (2006). The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127, 983–997. [DOI] [PubMed] [Google Scholar]
- 31.Wang HW, Long S, Ciferri C, Westermann S, Drubin D, Barnes G, and Nogales E (2008). Architecture and flexibility of the yeast Ndc80 kinetochore complex. J Mol Biol 383, 894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Becker W (2012). Fluorescence lifetime imaging--techniques and applications. J Microsc 247, 119–136. [DOI] [PubMed] [Google Scholar]
- 33.Nishino T, Rago F, Hori T, Tomii K, Cheeseman IM, and Fukagawa T (2013). CENP-T provides a structural platform for outer kinetochore assembly. EMBO J 32, 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Screpanti E, De Antoni A, Alushin GM, Petrovic A, Melis T, Nogales E, and Musacchio A (2011). Direct binding of Cenp-C to the Mis12 complex joins the inner and outer kinetochore. Curr Biol 21, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klare K, Weir JR, Basilico F, Zimniak T, Massimiliano L, Ludwigs N, Herzog F, and Musacchio A (2015). CENP-C is a blueprint for constitutive centromere-associated network assembly within human kinetochores. J Cell Biol 210, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weir JR, Faesen AC, Klare K, Petrovic A, Basilico F, Fischbock J, Pentakota S, Keller J, Pesenti ME, Pan D, et al. (2016). Insights from biochemical reconstitution into the architecture of human kinetochores. Nature 537, 249–253. [DOI] [PubMed] [Google Scholar]
- 37.Huis In ‘t Veld PJ, Jeganathan S, Petrovic, Singh, John, Krenn, Weissmann, Bange, and Musacchio (2016). Molecular basis of outer kinetochore assembly on CENP-T. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gascoigne KE, Takeuchi K, Suzuki A, Hori T, Fukagawa T, and Cheeseman IM (2011). Induced ectopic kinetochore assembly bypasses the requirement for CENP-A nucleosomes. Cell 145, 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rago F, Gascoigne KE, and Cheeseman IM (2015). Distinct organization and regulation of the outer kinetochore KMN network downstream of CENP-C and CENP-T. Curr Biol 25, 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrovic A, Keller J, Liu Y, Overlack K, John J, Dimitrova YN, Jenni S, van Gerwen S, Stege P, Wohlgemuth S, et al. (2016). Structure of the MIS12 Complex and Molecular Basis of Its Interaction with CENP-C at Human Kinetochores. Cell 167, 1028–1040 e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basilico F, Maffini S, Weir JR, Prumbaum D, Rojas AM, Zimniak T, De Antoni A, Jeganathan S, Voss B, van Gerwen S, et al. (2014). The pseudo GTPase CENP-M drives human kinetochore assembly. Elife 3, e02978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKinley KL, Sekulic N, Guo LY, Tsinman T, Black BE, and Cheeseman IM (2015). The CENP-L-N Complex Forms a Critical Node in an Integrated Meshwork of Interactions at the Centromere-Kinetochore Interface. Mol Cell 60, 886–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dimitrova YN, Jenni S, Valverde R, Khin Y, and Harrison SC (2016). Structure of the MIND Complex Defines a Regulatory Focus for Yeast Kinetochore Assembly. Cell 167, 1014–1027 e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hornung P, Troc P, Malvezzi F, Maier M, Demianova Z, Zimniak T, Litos G, Lampert F, Schleiffer A, Brunner M, et al. (2014). A cooperative mechanism drives budding yeast kinetochore assembly downstream of CENP-A. J Cell Biol 206, 509–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malvezzi F, Litos G, Schleiffer A, Heuck A, Mechtler K, Clausen T, and Westermann S (2013). A structural basis for kinetochore recruitment of the Ndc80 complex via two distinct centromere receptors. EMBO J 32, 409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bock LJ, Pagliuca C, Kobayashi N, Grove RA, Oku Y, Shrestha K, Alfieri C, Golfieri C, Oldani A, Dal Maschio M, et al. (2012). Cnn1 inhibits the interactions between the KMN complexes of the yeast kinetochore. Nat Cell Biol 14, 614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dhatchinamoorthy K, Shivaraju M, Lange JJ, Rubinstein B, Unruh JR, Slaughter BD, and Gerton JL (2017). Structural plasticity of the living kinetochore. J Cell Biol 216, 3551–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schleiffer A, Maier M, Litos G, Lampert F, Hornung P, Mechtler K, and Westermann S (2012). CENP-T proteins are conserved centromere receptors of the Ndc80 complex. Nat Cell Biol 14, 604–613. [DOI] [PubMed] [Google Scholar]
- 49.Yoo TY, Choi JM, Conway W, Yu CH, Pappu RV, and Needleman DJ (2018). Measuring NDC80 binding reveals the molecular basis of tension-dependent kinetochore-microtubule attachments. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Helgeson LA, Zelter A, Riffle M, MacCoss MJ, Asbury CL, and Davis TN (2018). Human Ska complex and Ndc80 complex interact to form a load-bearing assembly that strengthens kinetochore-microtubule attachments. Proc Natl Acad Sci U S A 115, 2740–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McEwen BF, Heagle AB, Cassels GO, Buttle KF, and Rieder CL (1997). Kinetochore fiber maturation in PtK1 cells and its implications for the mechanisms of chromosome congression and anaphase onset. J Cell Biol 137, 1567–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alushin GM, Ramey VH, Pasqualato S, Ball DA, Grigorieff N, Musacchio A, and Nogales E (2010). The Ndc80 kinetochore complex forms oligomeric arrays along microtubules. Nature 467, 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alushin GM, Musinipally V, Matson D, Tooley J, Stukenberg PT, and Nogales E (2012). Multimodal microtubule binding by the Ndc80 kinetochore complex. Nat Struct Mol Biol 19, 1161–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janczyk PL, Skorupka KA, Tooley JG, Matson DR, Kestner CA, West T, Pornillos O, and Stukenberg PT (2017). Mechanism of Ska Recruitment by Ndc80 Complexes to Kinetochores. Dev Cell 41, 438–449 e434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar N (1981). Taxol-induced polymerization of purified tubulin. Mechanism of action. J Biol Chem 256, 10435–10441. [PubMed] [Google Scholar]
- 56.Fanara P, Turner S, Busch R, Killion S, Awada M, Turner H, Mahsut A, Laprade KL, Stark JM, and Hellerstein MK (2004). In vivo measurement of microtubule dynamics using stable isotope labeling with heavy water. Effect of taxanes. J Biol Chem 279, 49940–49947. [DOI] [PubMed] [Google Scholar]
- 57.Zhai Y, Kronebusch PJ, and Borisy GG (1995). Kinetochore microtubule dynamics and the metaphase-anaphase transition. J Cell Biol 131, 721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Volkov VA, Huis In ‘t Veld PJ, Dogterom M, and Musacchio A (2018). Multivalency of NDC80 in the outer kinetochore is essential to track shortening microtubules and generate forces. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pesenti ME, Prumbaum D, Auckland P, Smith CM, Faesen AC, Petrovic A, Erent M, Maffini S, Pentakota S, Weir JR, et al. (2018). Reconstitution of a 26-Subunit Human Kinetochore Reveals Cooperative Microtubule Binding by CENP-OPQUR and NDC80. Mol Cell 71, 923–939 e910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Logsdon GA, Barrey EJ, Bassett EA, DeNizio JE, Guo LY, Panchenko T, Dawicki-McKenna JM, Heun P, and Black BE (2015). Both tails and the centromere targeting domain of CENP-A are required for centromere establishment. J Cell Biol 208, 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McIntosh JR, O’Toole E, Zhudenkov K, Morphew M, Schwartz C, Ataullakhanov FI, and Grishchuk EL (2013). Conserved and divergent features of kinetochores and spindle microtubule ends from five species. J Cell Biol 200, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zaytsev AV, Sundin LJ, DeLuca KF, Grishchuk EL, and DeLuca JG (2014). Accurate phosphoregulation of kinetochore-microtubule affinity requires unconstrained molecular interactions. J Cell Biol 206, 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zaytsev AV, Mick JE, Maslennikov E, Nikashin B, DeLuca JG, and Grishchuk EL (2015). Multisite phosphorylation of the NDC80 complex gradually tunes its microtubule-binding affinity. Mol Biol Cell 26, 1829–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dong Y, Vanden Beldt KJ, Meng X, Khodjakov A, and McEwen BF (2007). The outer plate in vertebrate kinetochores is a flexible network with multiple microtubule interactions. Nat Cell Biol 9, 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Long AF, Udy DB, and Dumont S (2017). Hec1 Tail Phosphorylation Differentially Regulates Mammalian Kinetochore Coupling to Polymerizing and Depolymerizing Microtubules. Curr Biol 27, 1692–1699 e1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manning AL, Bakhoum SF, Maffini S, Correia-Melo C, Maiato H, and Compton DA (2010). CLASP1, astrin and Kif2b form a molecular switch that regulates kinetochore-microtubule dynamics to promote mitotic progression and fidelity. EMBO J 29, 3531–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt JC, Kiyomitsu T, Hori T, Backer CB, Fukagawa T, and Cheeseman IM (2010). Aurora B kinase controls the targeting of the Astrin-SKAP complex to bioriented kinetochores. J Cell Biol 191, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Redli PM, Gasic I, Meraldi P, Nigg EA, and Santamaria A (2016). The Ska complex promotes Aurora B activity to ensure chromosome biorientation. J Cell Biol 215, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ng CT, Deng L, Chen C, Lim HH, Shi J, Surana U, and Gan L (2019). Electron cryotomography analysis of Dam1C/DASH at the kinetochore-spindle interface in situ. J Cell Biol 218, 455–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wynne DJ, and Funabiki H (2015). Kinetochore function is controlled by a phosphor-dependent coexpansion of inner and outer components. J Cell Biol 210, 899–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wynne DJ, and Funabiki H (2016). Heterogeneous architecture of vertebrate kinetochores revealed by three-dimensional superresolution fluorescence microscopy. Mol Biol Cell 27, 3395–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of proximity ratio measurements for FRET pairs in this study. Related to Figures 1, 2, 3, 4, 5, S4, S5, and S6. FRET pairs are grouped by the complexes to which they belong. “Measurement Type” indicates treatments applied to cells before measurements (“Metaphase” indicates untreated cells). For all measurement types except Nocodazole, only bioriented kinetochores were selected for analysis. For all measurements, a one sample t-test was used to evaluate if the proximity ratio was statistically different from zero (n.s. = not significant, with p-values listed in parentheses; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001). aAverage of all filtered measurements, where filtering discards kinetochores that are not fully occupied by the fluorophore-tagged proteins. bThe Max Proximity Ratio indicates the average proximity ratio for all filtered measurements within an acceptor to donor ratio of 1 – 2. This is only done for FRET pairs that compete for binding to the kinetochore (i.e., acceptor and donor fluorophores on the same protein subunit).
Data Availability Statement
The datasets supporting the current study are not publicly available but will be provided upon request.