

Abstract
Cancer patients with liver metastasis demonstrate significantly worse outcomes than those without liver metastasis when treated with anti-PD-1 immunotherapy. The mechanism of liver metastases-induced reduction in systemic antitumor immunity is unclear. Using a dual-tumor immunocompetent mouse model, we found that the immune response to tumor antigen presence within the liver led to the systemic suppression of antitumor immunity. The immune suppression was antigen-specific and associated with the coordinated activation of regulatory T cells (Tregs) and modulation of intratumoral CD11b+ monocytes. The dysfunctional immune state could not be reversed by anti-PD-1 monotherapy unless Treg cells were depleted (anti-CTLA-4) or destabilized (EZH2 inhibitor). Thus, this study provides a mechanistic understanding and rationale for adding Treg and CD11b+ monocyte targeting agents in combination with anti-PD-1 to treat cancer patients with liver metastasis.
Introduction:
In many solid and liquid tumors, checkpoint inhibitor immunotherapies (CPIs) can reinvigorate preexisting antitumor immunity to achieve durable response rates. However, for melanoma, lung, kidney, and several other malignancies where CPIs have shown efficacy, accumulating evidence suggests that the presence of liver metastasis reduces response rate, progression-free and overall survival (1–7). For patients who have disease progression despite CPIs, there are limited salvage options. Since the liver is one of the most common sites of metastases of all malignancies, this problem poses a significant unmet challenge in the field of immuno-oncology (1, 4, 7–10).
Despite the accumulating clinical data, it remains unclear how liver metastasis modulates systemic antitumor immunity, and the mechanistic underpinnings behind the CPI resistance in these patients are not well understood. Our group has previously demonstrated in melanoma patients that the presence of liver metastases, as opposed to other metastatic sites, correlated with the reduced expression of activation and functional markers on CD8+ tumor-infiltrating lymphocytes (TILs) when pre-CPI treatment cutaneous tumor biopsies were analyzed (11, 12). This finding raises the possibility that liver-specific tolerance mechanisms could be triggered in the context of liver metastasis to suppress systemic antitumor T cell immunity and undermine current forms of cancer immunotherapy. Previous investigations of the tolerogenic properties of the liver either focused on settings outside of cancer (such as infectious disease, transplantation, and autoimmunity) or suggested that the premetastatic potential of the liver and cancer-related immunosuppression was based on local effects within the confines of/ the liver parenchyma (13–16). These explanations do not account for the potential impact of liver tolerance on systemic or distant antitumor immunity. To date, approaches to study the tumor immunotherapy resistance have focused on preclinical models that rely on the single subcutaneous (SQ) tumor because of its efficiency and convenience (17). However, those models rarely represent the most common clinical scenario where immunotherapy is deployed, when tumors are at multiple anatomical sites and often have distinct response patterns (18).
Here, we developed a preclinical model to explore if tumor presence within the liver influences antitumor immunity at a distant SQ site. The model addressed the potential tolerogenic mechanisms leading to CPI immunotherapy resistance. We show that the presence of tumor within the liver significantly reduced systemic tumor-specific immunity in an antigen-specific, PD-1-independent manner. This liver-specific regulatory process led to distinct changes in T effector CD4+ and CD8+ T cells at distant tumor sites. Immunosuppression was dependent on Tregs and associated with significant differential remodeling of tumor-infiltrating myeloid-derived monocytes. Finally, Treg-targeting combination immunotherapy strategies, including Treg-depleting anti-CTLA-4 antibodies or functional inhibition using an EZH2 inhibitor, restored systemic antitumor immunity and CPI-responsiveness. These results are consistent with the hypothesis that liver involvement in cancer patients could significantly compromise systemic antitumor immunity even at distant sites and may provide a mechanism of CPI resistance in the setting of liver metastasis.
Results:
Liver tumor reduces distant antitumor immunity and systemic response to anti-PD-1 immunotherapy
In the present study, we developed a mouse model to study the effects of the tumor within the liver on antitumor immunity at a distant site. As such, the model was based on a two-site tumor system in which tumor cells were injected simultaneously into a SQ site and the liver or other anatomic sites of a mouse. Specifically, immunocompetent C57BL/6 (B6) mice were injected subcutaneously with syngeneic MC38 tumor cells concurrently with the subcapsular injection of MC38 cells into the liver or intravenous delivery into the lung (19, 20) (Fig. 1A). Mice with tumors present within the liver exhibited significantly increased tumor growth of the distant SQ tumor as compared to a secondary tumor in the lungs (Fig. 1B). Previous studies have shown that high levels of expression of the activation markers, PD-1 and CTLA-4, on CD8+ tumor-infiltrating lymphocytes (TILs) correlate with tumor immunity and predict better responses to CPI treatment (12, 21–23). Therefore, the expression of surface PD-1 and intracellular CTLA-4 was analyzed on tumor-infiltrating CD8+ T cells at the SQ site of all three groups. Mice bearing liver tumors had a significantly lower percentage of PD-1 and CTLA-4 expression on their SQ CD8+ TILs, as compared to mice bearing lung tumors and control SQ tumor-only mice (Fig. 1C). To test the impact of anti-PD-1 immunotherapy treatment in this model, we injected anti-mouse PD-1 mAb to three groups of mice - those with SQ tumor only, and SQ plus experimental liver or lung metastasis. While anti-PD-1 treatment promoted tumor rejection and improved the survival in mice bearing SQ or SQ plus lung tumors, mice bearing SQ plus liver tumors were refractory to anti-PD-1 monotherapy, and 10 out of 10 mice required euthanizing by day 25 post tumor injection due to large tumor burden (Fig. 1, D and E). The relative expression of PD-1 and CTLA-4 by the SQ CD8+ TILs was compared between mice with experimental liver metastasis versus other tumor-bearing groups treated with anti-PD-1 monotherapy. There was a significant reduction in the frequency of PD-1 and CTLA-4 co-expressing cells and lower MFI of CTLA-4 and PD-1 on SQ CD8+ TILs isolated from liver tumor-bearing mice (Fig. 1F). The reduced survival and low anti-PD-1 response rate in mice with liver tumors were not related to hepatotoxicity or liver dysfunction. Standardized liver function tests performed at the experimental endpoint showed that liver tumor-bearing mice displayed similar results as mice without liver tumors and had serum values within the healthy control reference range (fig. S1). Taken together, these findings show that the presence of tumor in the liver reduced antitumor immunity at a distant tumor site and recapitulated clinical observations in melanoma patients with liver metastasis where lower co-expression of PD-1 and CTLA-4 on cutaneous CD8+ TILs correlated with lower survival and response rate to anti-PD-1 immunotherapy (1, 12, 21).
Fig. 1. Experimental liver metastasis reduces distant antitumor immunity and systemic response to anti-PD-1 immunotherapy.
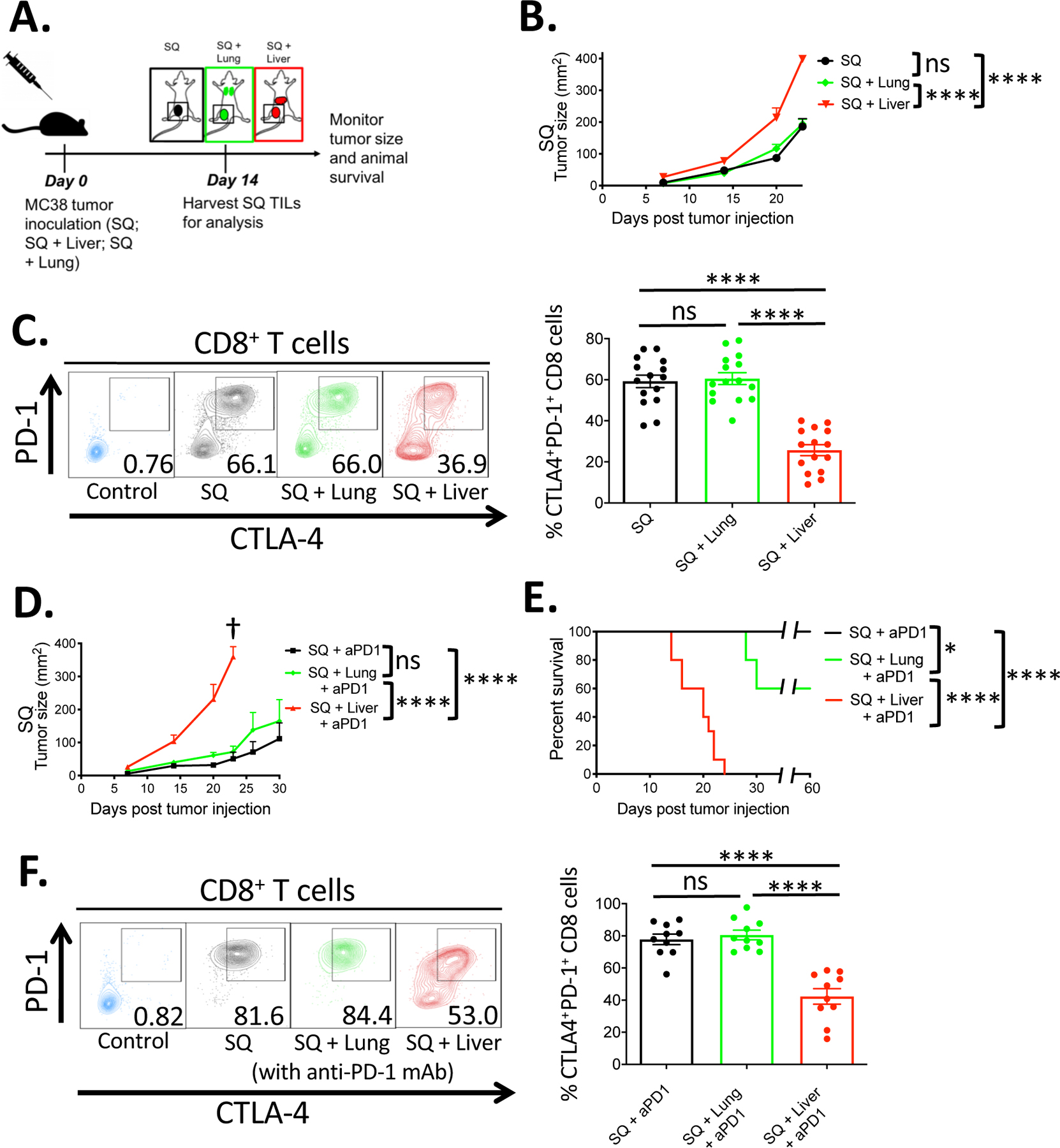
(A) Two-site tumor model schema. C57BL/6 mice were implanted with MC38 tumor cells (5 × 105) subcutaneously (black), subcutaneously and at the lungs (green), or subcutaneously and at the liver (red) and monitored for tumor growth and survival. SQ = Subcutaneous. (B) SQ Tumor growth curves of the three experimental groups (n = 15 mice per group). (C) Representative flow cytometric plots and percentage of surface PD-1 and intracellular CTLA-4 co-expression on CD8+ T cells sampled from the subcutaneous tumor of the indicated groups on day 14 post tumor implantation (n=15). (D) SQ Tumor growth curves of the indicated experimental groups treated with anti-PD1 antibody treatment (n = 10 mice per group). (E) Survival curves of the indicated experimental groups (n = 10 mice per group). (F) Representative flow cytometric plots and percentage of PD-1 and CTLA-4 co-expression on CD8+ T cells from the indicated anti-PD-1 antibody treated groups (n=10). (C, F) Representative staining controls (blue) of PD-1 and CTLA-4 on CD8+ T cells from splenocytes of naïve mice. (B, E) Asterisks indicating significance determined by Log-rank tests between groups are * p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001, data pooled from two or more experiments. (B, D) Data are shown as mean +/− s.e.m pooled from two or more experiments. Asterisks indicating significance until day 23 post tumor injection determined by two-way ANOVA with Sidak’s multiple comparisons test are * p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. † indicate that all mice reached experimental endpoint and were euthanized per institutional guidelines.
The liver facilitates distant tumor antigen-specific immunosuppression independent of tumor burden
Previous studies have suggested that higher overall tumor burden, as seen in the liver tumor-bearing mice, correlated with a poor response (24–26). To investigate whether an increased tumor burden was responsible for immune suppression, equal amounts of tumors were implanted at alternative host anatomical sites to examine the immunosuppressive effects on the distant SQ site. Unlike mice with a liver tumor, mice that had tumors implanted in their kidney or peritoneum did not show any significant change in tumor growth at their SQ site compared to SQ tumor-only mice (Fig. 2A). In many instances, the overall tumor burden was similar or significantly higher, as seen in the intraperitoneal tumor group (fig. S2A). There were no significant differences in SQ tumor growth in the immunodeficient NSG nor Rag-1−/− mice implanted with secondary liver tumor (fig. S2B), suggesting that the modulation of tumor growth at the distal SQ site was dependent on adaptive immunity. Notably, implantation of irradiated MC38 tumor cells or tumor lysate alone into the tumor was sufficient to mediate the effects. Five × 105 irradiated tumor cells, or lysates from 5 × 105 tumor cell equivalents, resulted in a significant increase in tumor growth as compared to mice animals implanted with MC38 tumor only in the SQ site (Fig. 2B). The degree of enhanced tumor growth was similar to that seen in animals transplanted with live tumors in the liver, supporting the conclusion that tumor burden was not associated with the systemic suppression in tumor immune responses. In fact, implantation of 10-fold less irradiated MC38 tumor cells showed similar results (Fig. 2C). Finally, liver-mediated immune suppression was tumor antigen-specific, as implantation of an unrelated B16F10 tumor into the liver did not promote tumor growth at the distant SQ site (Fig. 2D). Thus, liver tumor-dependent suppression in systemic antitumor immunity was not merely a consequence of large tumor burden but likely due to tumor-antigen and liver-specific tolerogenic processes that modulated systemic antitumor response.
Fig. 2. Liver-mediated suppression of antitumor immunity is tumor-antigen specific and independent of tumor burden.
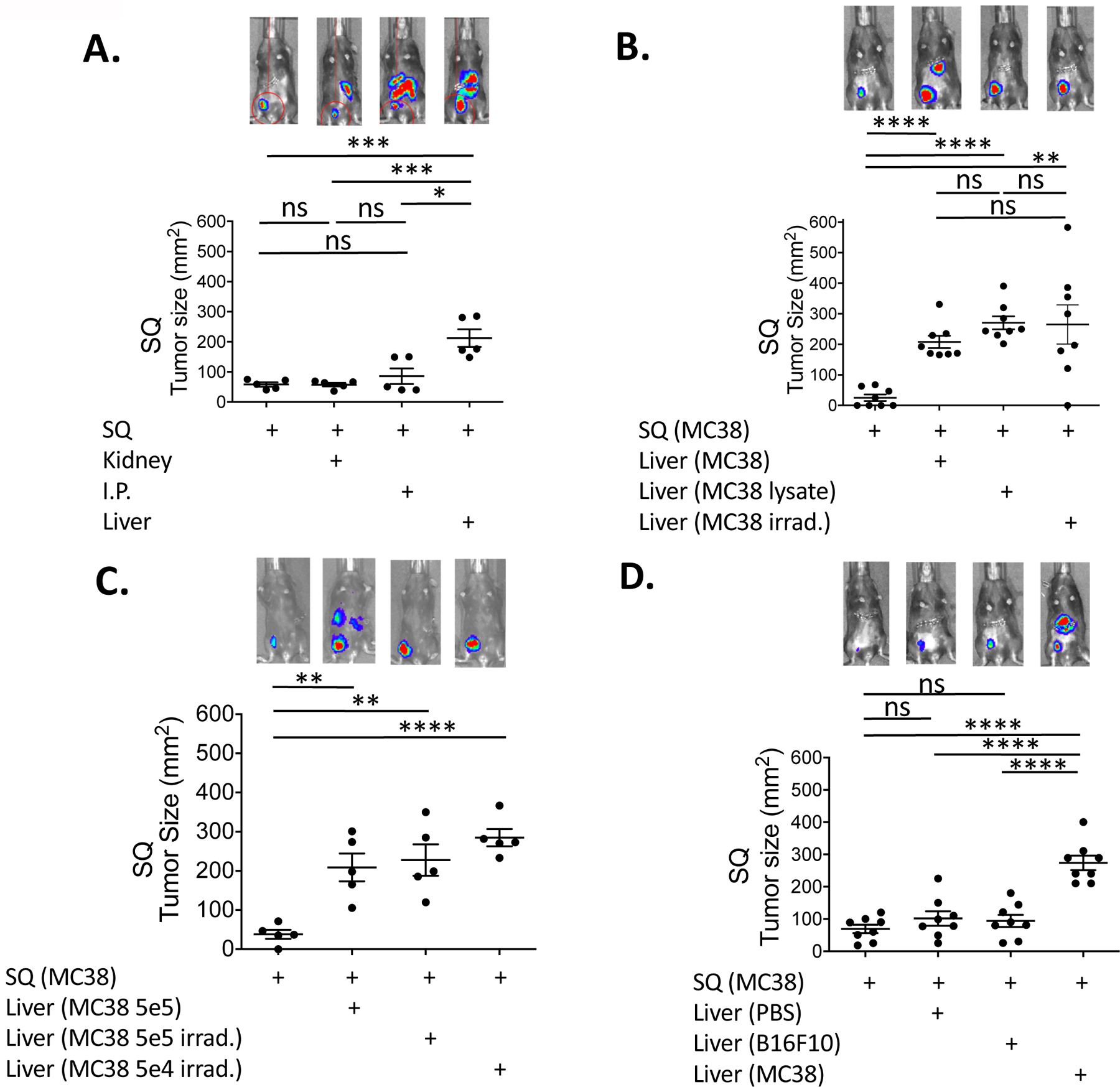
(A) Day 20 subcutaneous (SQ) tumor size from B6 mice that were implanted with MC38 tumor cells (5 × 105) subcutaneously alone as a control, subcutaneously and at the left kidney, subcutaneously and intra-peritoneum (I.P.), and subcutaneously and at the liver. (B) SQ tumor sizes in mice implanted with MC38 tumor cells subcutaneously, subcutaneously and at the liver, subcutaneously with irradiated (50 Gy) MC38 tumor cells (5 × 105) at the liver, and subcutaneously with MC38 tumor cell lysates (from 5 × 105 cells) at the liver. (C) SQ tumor sizes in mice implanted with MC38 tumor cells subcutaneously, subcutaneously and at the liver, subcutaneously with 5 × 105 irradiated MC38 tumors cells at the liver, and subcutaneously with 5 × 104 irradiated MC38 tumor cells at the liver. (D) SQ tumor sizes in mice implanted with MC38 tumor cells subcutaneously, subcutaneously with saline (PBS) at the liver, subcutaneously with heterologous B16F10 tumor cells (5 × 105) at the liver, and subcutaneously with homologous MC38 tumor cells at the liver. Representative whole mouse bioluminescent image of implanted MC38 tumors from each experimental group for day 20 are shown (A-D, upper row). Data are shown as mean +/− s.e.m. Asterisks indicating significance determined by unpaired t tests between groups are * p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. Representative data from one out of at least two independent experiments are shown.
Experimental liver metastasis is associated with phenotypic changes in distant tumor-infiltrating lymphocytes
The phenotype of CD8+ and CD4+ T cells within the SQ tumor in mice with and without a matched tumor within their livers was analyzed to investigate systemic changes to specific lymphocyte subsets in the context of the liver tumor (Fig. 3A). In addition to PD-1 and CTLA-4, presented above, other activation markers including ICOS and effector cytokines, previously shown to correlate with antitumor responses (12, 22), were altered due to the presence of liver tumors. Although the percentage of CD8+ TILs among CD45+ cells within the tumor remained similar between the two groups, CTLA-4, ICOS and IFNγ expression were significantly lower on the CD8+ T cells isolated from mice bearing liver tumors (Fig. 3, B and C). Moreover, the most highly activated T cell subsets, based on the co-expression of multiple activation markers, PD-1, CTLA-4, ICOS, and Ki-67, were the most significantly affected, with liver-tumor implanted mice showing less than 5% of CD8+ T cells co-expressing all four markers as compared to over 12% in SQ only tumor-bearing animals (fig. S3A). Conventional CD4+ TILs, within the SQ tumor of liver tumor-bearing mice, had lower ICOS expression, but higher expression of PD-1 (fig. S3B). These results were consistent with other studies showing that ICOS expression on conventional CD4+ TILs is linked to enhanced antitumor immunity, while high expression of PD-1 correlates with reduced antitumor responses (22, 27, 28). Thus, both CD8+ and CD4+ effector T cell subsets were compromised in the distant SQ tumor site in mice with a secondary tumor implant within the liver. In striking contrast, the CD4+ Foxp3+ Treg population within the SQ tumor in mice with secondary liver tumor implants showed a highly activated phenotype. Although the percentage of Foxp3+ Tregs (~35% of total CD4+ T cells) and absolute Treg number within the SQ tumors of the two groups were not significantly different (fig. S4, A and B), the expression of PD-1, CTLA-4, and ICOS, was significantly higher on Tregs from liver tumor-bearing mice (Fig. 3D). When multiple activation markers were combined in the analysis, we found that the two experimental groups’ phenotypic difference was even more pronounced (fig. S3C). Similar experiments were performed in a second tumor model, the B16F10 melanoma model, with similar results observed where there was both a significant enhancement of SQ tumor growth in mice co-injected with liver tumor and reduced expression of activation markers and cytokines in cytotoxic T cells isolated from the SQ tumors (fig. S5). These data are in line with the recent reports showing an increase in activated CTLA-4 and ICOS expressing Tregs in the blood and tumors of cancer-bearing hosts (29–31).
Fig 3. Liver tumor-associated distant CD8+ T cell dysfunction is antigen-specific and dependent on Tregs.
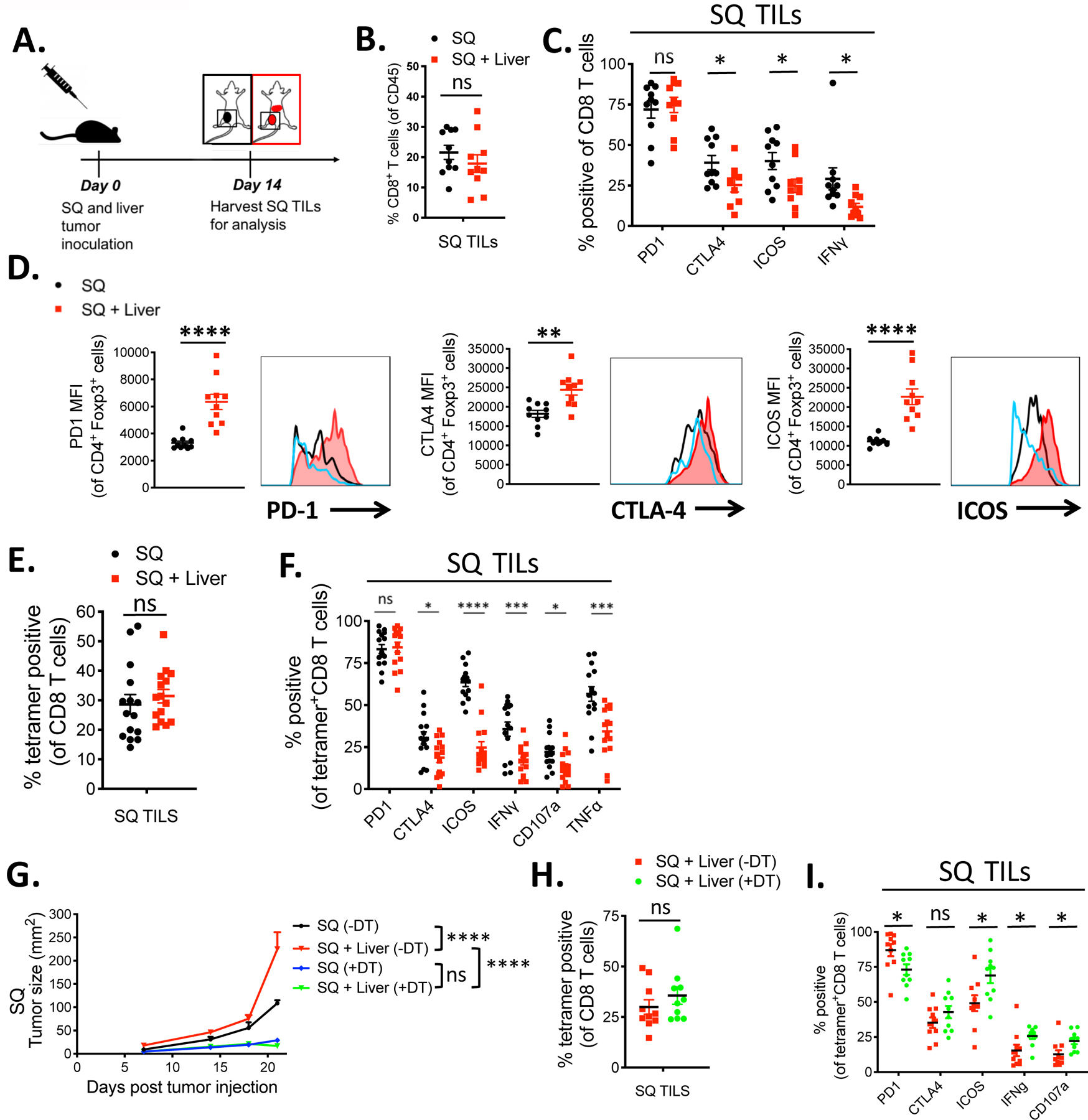
(A) Tumor model schema. C57BL/6 mice were implanted with MC38 tumor cells (5 × 105) subcutaneously alone (black) or subcutaneously plus in the liver (red) and TILs were harvested on day 14 post tumor implantation. (B) Percentage of CD8+ T cells of viable CD45+ immune cells and (C) percentage of CD8+ T cells that express PD-1, CTLA-4, ICOS, and IFNγ in the subcutaneous (SQ) tumor of mice with (red) or without (black) concurrent liver tumor by flow cytometry. (D) PD-1, CTLA-4, and ICOS MFIs from Foxp3+ CD4+ Tregs in the SQ tumor from mice with (red) and without (black) concurrent liver tumor and naïve CD4 T cells (blue). (E) Percentage of tetramer+ (KSPWFTTL) cells of total CD8 T cells in the SQ tumor of mice with or without concurrent liver tumor. (F) Percentage of tetramer+ CD8+ T cells positive for PD-1, CTLA-4, ICOS, IFNγ, CD107a, and TNFα. (Unpaired t tests, * p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001) (G) SQ tumor growth curves of mice with SQ tumor without DT (black ), SQ and liver tumor without DT (red ), SQ tumor with DT (blue), and SQ and liver tumor with DT (green). (H) Percentage of tetramer+ CD8+ T cells and (I) percentage of tetramer+ CD8+ T cells that are positive for PD-1, CTLA-4, ICOS, IFNγ, and CD107a in mice bearing liver tumor with (green) or without (red) DT administration. Data shown as mean +/− s.e.m., (n=15 for E, n=10 for all others). For tumor growth curves, asterisks indicating significance up till day 23 post tumor injection were determined by two-way ANOVA with Sidak’s multiple comparisons test are * p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. Data were pooled from 2 or more experiments.
The above findings suggested that the presence of tumor cells in the liver led to a significant decrease in the number and activation state of effector T cells at a distant site. This raised the possibility that tumor antigen within the liver either induced clonal deletion or altered the activation state of tumor antigen-specific T cells (32, 33). To directly monitor the tumor antigen-specific T cell response, we employed an MC38 tumor antigen-derived peptide KSPWFTTL-H-2Kb-tetramer (KSP) that reacts specifically with tumor-specific CD8+ in MC38 tumor-bearing mice (34, 35). The KSP tetramer bound to CD8+ T cells infiltrating the tumors and tumor-draining lymph nodes of mice bearing MC38 tumors, but not CD8+ T cells from mice bearing the unrelated B16F10 tumors. Additionally, the tetramer reactivity was confirmed to be highly specific when compared to an irrelevant control peptide tetramer (fig. S6, A, B and C). A comparison of tetramer+ CD8+ TILs at the SQ site showed that there was only a small, but not significant effect on the percentage or the absolute number of MC38-specific tetramer+ CD8+ T cells in the tumors between mice with and without concurrent liver tumors (Fig. 3E, fig. S4C). These results suggested that the generation and expansion of CD8+ T cells were not altered significantly by tumor presence at the liver site. Moreover, there was no evidence of sequestration or clonal deletion in the liver tumor site, as the absolute numbers of KSP tetramer+ CD8+ TILs were similar to the SQ sites (fig. S4C). However, there was a significant reduction in the expression of several functional markers on the tetramer+ CD8+ T cells within the SQ tumor of liver-tumor bearing mice (Fig. 3, E and F). Tetramer+ CD8+ T cells in the SQ tumor of mice with liver tumors expressed reduced cell surface levels of CTLA-4, ICOS, and CD107a as well as reduced production of IFNγ and TNFα (Fig. 3F). Finally, the inhibition on the CD8+ T cells was both liver- and antigen-specific, since the suppression of ICOS expression, as a surrogate marker of activation, did not occur in mice bearing lung tumors nor when an unrelated tumor (B16F10) was implanted in the liver (fig. S7, A and B). Together, these results indicate that the liver-mediated reduction of tumor-specific immunity at the distant SQ site was the result of a systemic dysfunction of tumor-specific CD8+ T cells. Importantly, this dysfunctional state was irreversible with anti-PD-1 treatment despite the high expression of PD-1 on the Tet+ CD8+ T cells, suggesting that T cell “exhaustion” through the PD-1 axis was unlikely to be the main mechanism involved in immune suppression of the antitumor response.
Tregs control the suppression of distant tumor immunity in the presence of liver tumors
Numerous lines of evidence suggest that Tregs play an essential role in hepatic immune tolerance and can suppress CD8+ T cell activation and cytotoxicity (13, 14, 32, 36–38). Therefore, the potential impact of Tregs in the systemic suppression was examined by selectively depleting Tregs using a mouse strain expressing the diphtheria toxin receptor under the control of Foxp3 (FP3-DTR) (fig. S8). FP3-DTR mice, implanted with both SQ and liver tumors, were treated with diphtheria toxin (DT) every other day for up to 24 days to eliminate Tregs systemically during the experiment (39, 40). In the untreated setting, ~30% of CD4 TILs were Tregs. DT treatment led to near-complete elimination of Foxp3+ Tregs in the circulation and the SQ tumor as compared to untreated mice (fig. S9). Treg depletion led to enhanced tumor rejection in both the SQ only and SQ plus liver tumor groups, suggesting that Tregs were involved in the liver tumor-specific suppression (Fig. 3G). Moreover, depletion of Tregs restored the activation and functional status of tumor specific CD8+ T cells, based on a significant increase in ICOS, IFNγ and CD107a expression (Fig. 3, H and I). PD-1 expression was lower in the DT treated group. This result was likely explained by the reduced tumor burden (caused by Treg depletion) as previously observed(41). Collectively, the results suggest that Tregs play a critical role in modulating the distant tumor-specific CD8+ T cell immunity in the presence of liver-resident tumors.
Treg depletion or inactivation reverses liver tumor-associated systemic immune suppression
The results above suggested that Tregs may be responsible for the inability of anti-PD-1 monotherapy to induce tumor regression in this tolerogenic model. Therefore, we tested two independent approaches to selectively compromise Tregs to see if it would synergize with anti-PD-1 treatment. First, we tested whether Treg depleting anti-CTLA-4 mAb, clone 9H10, in combination with anti-PD-1 mAb, enhanced tumor rejection in SQ and liver tumor-bearing mice. Treatment with three doses of clone 9H10 on days 7, 9, and 11 post tumor implantations led to the depletion of SQ tumor infiltrating Tregs and enhanced rejection of the tumors at both the SQ and liver sites in the dual tumor-bearing mice (Fig. 4, A and B; S10A). While treatment with 9H10 as monotherapy had a partial impact on tumor growth and survival, in combination with anti-PD-1 mAb, 9H10 treatment achieved 100% survival at 60 days (n=10) and resulted in the complete rejection of tumors at both sites (Fig. 4, B and C; S10A). The complete rejection of tumors at the liver site was confirmed by full-body bioluminescent visualization of the mice as well as necropsy (fig. S10, A and B). Finally, treatment with the anti-CTLA4, clone 9H10, was able to restore the activation and functional status of suppressed tumor antigen-specific tetramer+ CD8+ T cells, as demonstrated by their increase in ICOS, IFNγ, and TNFα relative to an isotype control (Fig. 4D).
Fig 4. Treg targeting immunotherapy enhances efficacy and reverses immune dysfunction in liver metastasis.
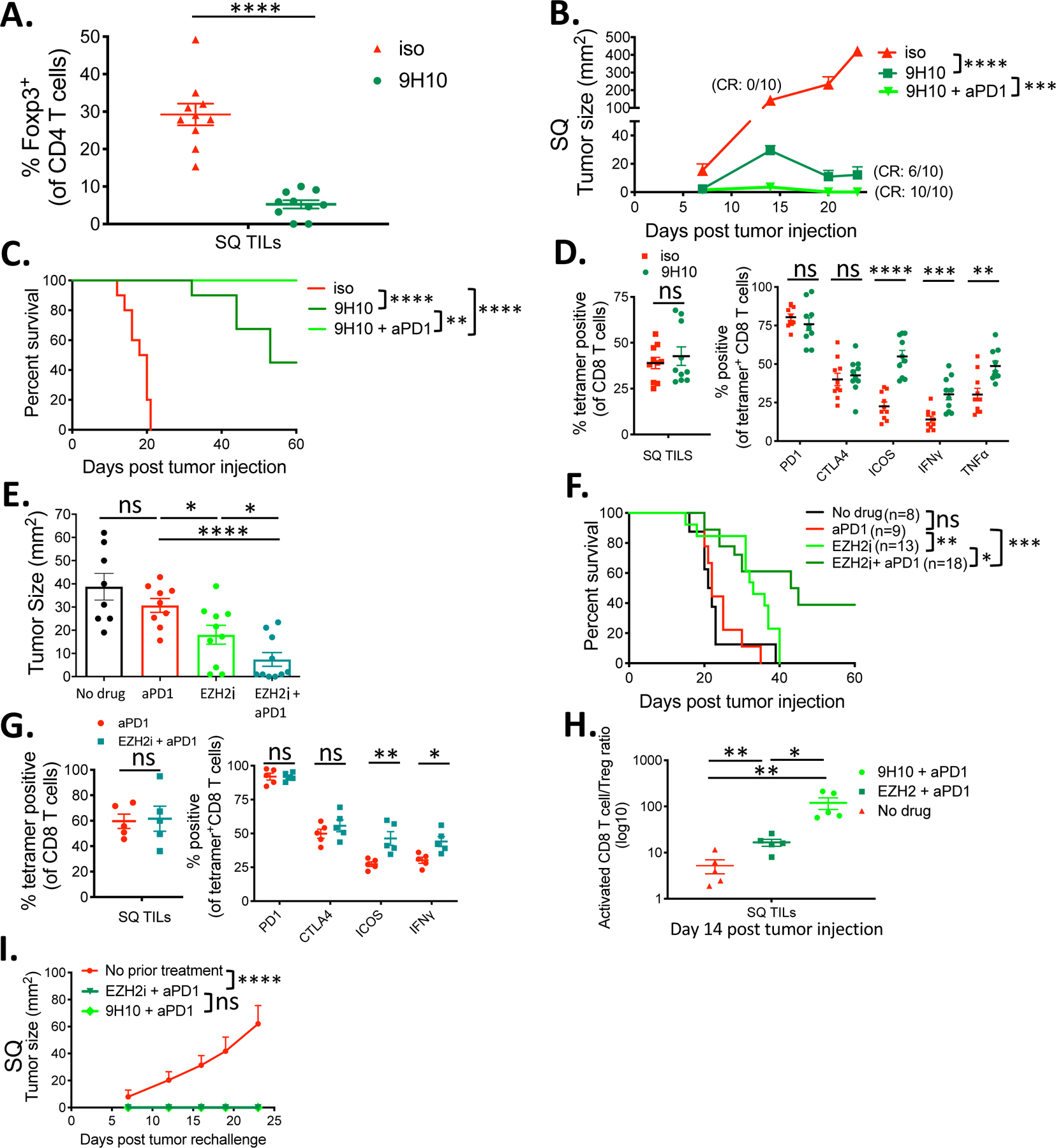
(A) Percentage of Foxp3+ CD4 Tregs within the indicated Subcutaneous (SQ) tumors. (B) SQ tumor growth curves of liver-tumor mice treated with anti-CTLA-4 antibody clone 9H10, 9H10 plus anti-PD-1 antibody, or isotype control. CR= complete rejection with no measurable SQ tumor at endpoint. (C) Survival curves of indicated groups. (D) Percentage of KSP tetramer+ CD8+ TILs and percentage that are positive for PD-1, CTLA-4, ICOS, IFNγ, and TNFα in mice treated with 9H10 versus control. (E) Day 14 SQ tumor sizes from liver-tumor bearing mice treated with EZH2 inhibitor CPI-1205(n=10), anti-PD-1 antibody (n=9), or a combination of both (n=10) compared to control (n=8). (F) Survival curves of indicated groups. (G) Percentage of KSP tetramer+ CD8+ TILs and percentage that are positive for PD-1, CTLA-4, ICOS, and IFNγ in mice treated with anti-PD-1 plus CPI-1205 versus anti-PD-1 alone. (H) Activated CD8+ T cell to Treg ratio within the SQ tumor sample of the indicated treatment groups. (I) SQ tumor growth curves from the MC38 tumor rechallenge experiment of the indicated groups. All data are shown as mean +/− s.e.m. All experiments besides E and F were n=5 or 10 and were representative of three or more independent experiments. Survival curves were analyzed by Log-rank tests, tumor growth curves were analyzed by two-way ANOVA with Sidak’s multiple comparisons, all others were analyzed by unpaired t tests. Asterisks indicating significance determined between groups are * p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
To test the hypothesis that Treg depletion was essential for the anti-CTLA-4 activity, we compared the antitumor effects of two different isotypes of the mouse anti-CTLA-4 antibody clone 9D9 that have been shown to block CTLA-4-B7 interactions but have varying abilities to deplete Tregs intratumorally. The anti-CTLA-4 IgG2a isotype has been shown to have high Treg-depleting capability intratumorally in contrast to the anti-CTLA-4 IgG2b isotype, which conferred minimal Treg-depletion (42). Similar to the anti-CTLA-4 9H10 antibody results, only the Treg-depleting IgG2a antibody was capable of achieving complete rejection of tumors at both sites in the majority of treated mice (fig. S16, A and B).
Systemic depletion of Tregs can trigger systemic autoimmune response (43). Therefore, as a second approach, we took advantage of previous studies showing that inhibitors of Enhancer of Zeste 2 (EZH2), an H3K27 methyltransferase induced upon Treg activation, destabilize and disrupt tumor-infiltrating Treg function resulting in enhanced antitumor immunity without a systemic effect (40, 44). Treatment of mice with the EZH2 inhibitor, CPI-1205 (Constellation Pharmaceuticals), in combination with anti-PD-1 mAb led to enhanced tumor rejection and improved survival (Fig. 4, E and F).
These combination treatments restored the activation and functional status of tetramer+ CD8+ TILs within the SQ tumor microenvironment of liver-tumor bearing mice (Fig. 4G). Moreover, analysis of the distant SQ TILs showed that either combination treatment modality augmented the activated tumor-specific CD8+ T cell (tetramer/PD-1/CTLA-4/ICOS+) to Treg ratio within the tumor microenvironment (Fig. 4H). Finally, 7 out of 18 mice that achieved 60-day tumor-free survival in the CPI-1205 + anti-PD1 group and 10 out 10 mice in the 9H10 + anti-PD1 group rejected MC38 tumors in subsequent rechallenge experiments confirming that the immune response was durable (Fig. 4I). These results suggest that combination strategies that include PD-1 checkpoint blockade and the depletion or disruption of Treg function can enhance durable antitumor immunity in the setting of liver tumor mediated immunosuppression where anti-PD-1 monotherapy is ineffective.
Phenotypic analyses and adoptive transfer of Tregs from liver tumor-bearing mice suppress tumor rejection
Given that Tregs play a significant role in the systemic immunosuppression in the liver tumor model, we sought to directly compare the in vivo suppressive capability of Tregs derived from mice with and without liver tumors to determine if there were functional differences in the Treg populations. For these studies, transgenic mice that co-express EGFP and Foxp3 under the control of the endogenous promoter were used to efficiently isolate Treg cells for adoptive transfer (45). Phenotypic analysis of Tregs isolated from SQ tumors in the absence or presence of liver tumor showed increased levels of PD-1 and CTLA-4 as seen previously. In addition, liver-tumor bearing mice derived Tregs expressed higher levels of Neuropilin-1 and Helios (Fig. 5A), which are known Treg activation markers previously correlated with their suppressive potency (46, 47). Moreover, the transfer of the Treg cells isolated from liver tumor-bearing mice significantly enhanced tumor growth when compared to the no Treg transfer or the transfer of Treg cells from SQ tumor-only bearing mice (Fig. 5B). Together, these results suggested that the presence of liver tumor induced a highly active population of Tregs that more effectively suppressed antitumor immunity on a per cell basis.
Fig 5. Presence of liver tumor changes phenotype and transcriptome of SQ tumor-infiltrating adaptive and innate immune cells.
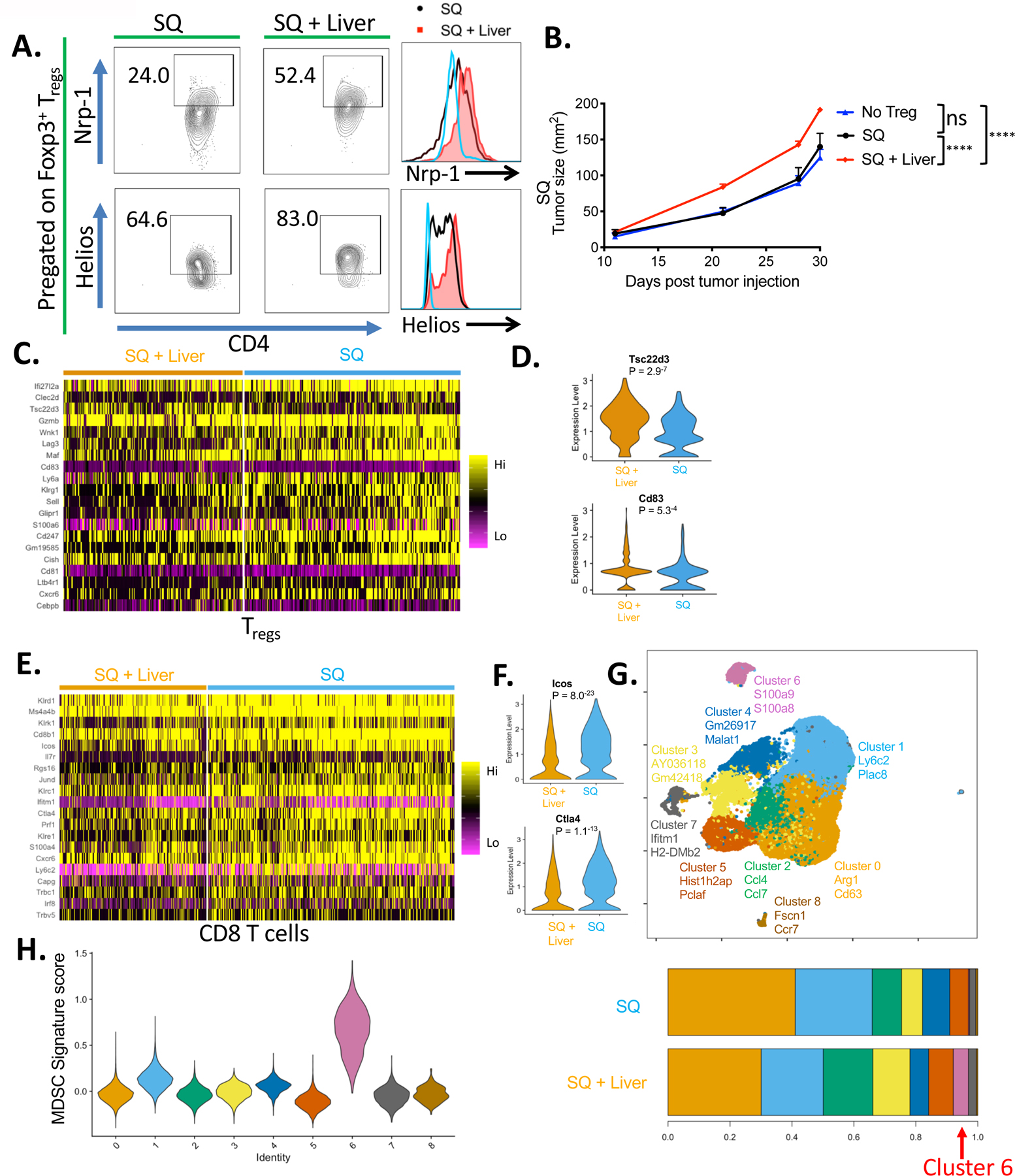
(A) Percentage of Foxp3+ Treg cells that express Neuropilin-1 and Helios in the subcutaneous (SQ) tumor of mice with and without concurrent liver tumor by flow cytometry, and their relative MFIs between the indicated groups. Naïve splenic Foxp3- CD4+ T cells (blue) were stained as control. (B) SQ tumor growth curves from mice that received adoptive transfer of Tregs isolated from mice with (red, n=6) or without liver tumor (black, n=5) versus no Treg control (blue, n=5). Data representative of two independent experiments. (C) Heatmap displaying the top 20 differentially expressed genes by SQ Tregs between the two groups via scRNAseq. (D) Violin plots showing the top 2 differentially upregulated genes by SQ Tregs from mice with liver tumor. (E) Heatmap displaying the top 20 differentially upregulated genes by SQ CD8+ T cells between the two groups. (F) Violin plots showing differential expression of ICOS and CTLA-4 by SQ CD8+ T cells between the two groups. (G) Unbiased reclustering (UMAP) and stacked bar graph of relative frequencies of monocyte/myeloid cells showing 9 distinct scRNAseq subclusters. (H) Violin plots showing relative MDSC score ordered by monocyte/myeloid cell subclusters. Tumor growth curves were analyzed by two-way ANOVA with Sidak’s multiple comparisons. Asterisks indicating significance determined between groups are * p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
High-dimensional gene expression profiling reveals distinct liver tumor-associated changes in Tregs and CD8+ T cells.
To more fully understand the differences between the Treg populations present in the SQ tumor site in mice where tumor was also present in the liver, single cell transcriptomic analysis (scRNAseq) was performed using the 10x Genomics Chromium platform. CD45+ SQ tumor-infiltrating cells, pooled from 6 mice per group from mice with and without concurrent liver tumors, were analyzed across two separate experiments. In total, 62,743 cells (SQ only; 35,589 and SQ & Liver; 27154) were analyzed. Informatics analysis was used to identify 14 unique clusters, including nine myeloid/monocyte cell, three T cell, one NK cell, one plasmacytoid dendritic cell, and one erythrocyte cluster (fig. S11A). Further analysis of the Treg cluster, based on the expression canonical lineage markers CD3, CD4, and FoxP3, identified 82 differentially expressed genes between the Tregs from the two experimental groups (61 upregulated and 11 down-regulated). The top-20 upregulated genes, shown in Fig. 5C, suggest that the Tregs present in the SQ tumor underwent significant transcriptomic changes as a result of liver tumor implantation. The top 2 genes differentially upregulated by Tregs from mice with liver tumor were Tsc22d3 (GILZ) and Cd83. Specifically, the violin plots suggested a significant number of Tregs upregulated these two genes as compared to Tregs isolated from the SQ only group (Fig. 5D). Previous studies have shown that these proteins are essential for Treg stability and suppressive function (48–51) consistent with the differential effect on tumor growth seen in the adoptive transfer experiments. Differential gene expression analysis on the CD8+ T cells revealed significant transcriptomic changes between the two groups as well. There was decreased expression in activation-associated genes expressed by CD8+ T cells isolated from mice with liver tumors, including Klrk1, Prf1, Cd28, Il7r, Ctla4, and Icos (Fig. 5E). The RNAseq findings that ICOS and CTLA-4 expression levels were significantly lower on the CD8+ T cells in the liver tumor group (Fig. 5F) were similar to the flow cytometry studies showing reduced ICOS and CTLA-4 expression on Tet+ CD8+ T cells from liver tumor bearing mice (Fig. 3F). These results suggest that CD8+ T cells in the distant tumor of liver-tumor bearing mice underwent significant transcriptomic changes consistent with the resulting dysfunction.
High-dimensional profiling of innate immune cells in the tumor microenvironment implicates changes in gene and protein expression in differential antitumor immunity
Previously, Schreiber and colleagues showed that treatment of mice with CPIs led to distinct changes in the macrophage phenotypes (52) and other studies showed that innate immune cell reprogramming in the tumor microenvironment is highly correlated with antitumor responses (53, 54). Thus, we analyzed the transcriptome of the cells of the innate immune compartment by scRNAseq. Among the 9 distinct myeloid/monocyte/macrophage clusters, cluster 6 was significantly increased in the SQ sites of mice bearing liver tumors (Fig. 5G, S11B). The cells in cluster 6 were identified as myeloid-derived suppressor cells (MDSCs) based on a previously described gene signature (55), and showed quantitative and qualitative differences between MDSCs present in the SQ tumor only versus SQ plus liver tumor conditions (Fig. 5, G and H). There were significant decreases in mRNA expression associated with immunosuppression in this monocyte subset, including Socs3, Xbp1, Rhoh, Wfdc17, GOs2, and Acod1 (fig. S12) suggesting that the liver tumor-induced immunosuppression might be a consequence of crosstalk between Treg and MDSCs (56).
Liver-tumor associated Treg control of distant antigen-specific immunity is mediated by tolerogenic CD11b+ MDSCs
Previous studies have suggested that Tregs can exert tumor antigen-specific suppression of effector T cells indirectly by altering antigen-presenting cells (APCs) (57, 58). Our results indicate that the presence of liver tumors can significantly impact the immune composition of a distant SQ tumor. These findings suggested that the systemic antigen-specific immunoregulatory mechanism might have been coordinated by Tregs and MDSCs. MDSCs are known to be potent suppressors of tumor-specific immunity within the tumor microenvironment (54), and the crosstalk between Tregs with MDSCs resulting in antigen-specific suppression within tumors has been previously described (59). To explore this possibility in our model and extend gene expression analysis studies, other cells of the myeloid/monocyte/macrophage lineage in the SQ tumor microenvironment were surveyed using a 14-color flow cytometry panel of well-established canonical immune cell subset markers to identify significant cellular changes in the SQ tumor-infiltrating APCs between mice bearing a SQ tumor only and those expressing SQ plus liver tumors. Unbiased t-distributed stochastic neighbor embedding (t-SNE) clustering revealed two innate immune cell populations that underwent extensive phenotypic changes. These changes were predominately seen as changes in cell surface expression of key cell subset markers on APCs isolated from the SQ site of the liver tumor-bearing group, including changes in the canonical myeloid-derived monocyte differentiation marker CD11b as well as other granulocytic and monocyte markers. For example, there were substantial increases in two clusters of cells in the liver tumor group, the CD11b+Ly6G+Ly6C− granulocytic myeloid-derived suppressor cells (G-MDSCs) and the CD11b+Ly6G−Ly6C+ monocytic myeloid-derived suppressor cells (M-MDSCs) (fig. S13A). Using the B16F10-OVA tumor cell line that expresses the model antigen chicken ovalbumin, we were able to reproduce the liver-tumor mediated suppression seen in the MC38 tumor model (fig. S13, B and C). Importantly, the increase in MDSCs within the SQ tumors of mice with matched liver tumors was also evident when comparing the MDSC subset expressing the OVA-peptide in the context of the H-2Kb class I MHC (fig. S13D). Only CD11b+ monocytes, not dendritic cells (CD11c+) or B cells (CD19+), showed a significant increase in percentage within the SQ tumors from the liver tumor-bearing group (Fig. 6A). Notably, the surface expression of CD80/86 on the CD45+ CD11b+ monocytes from the liver group was significantly reduced, suggesting an increase of potentially tolerogenic CD11b+ MDSCs in the tumor microenvironment, similar to what was found in the gene expression analysis (Fig. 6B). To determine if these changes were limited to the SQ site of mice bearing liver tumors as the second site, the SQ immune infiltrate was analyzed in mice with secondary tumors at various other tissue sites. The significant increase in CD11b+ MDSC infiltrates with lower surface CD80/86 expression occurred only in the presence of a concurrent liver tumor but not tumors at other anatomical sites (fig. S14, A and B).
Fig 6. Treg control of distant tumor-antigen specific immunity is mediated by tolerogenic MDSCs.
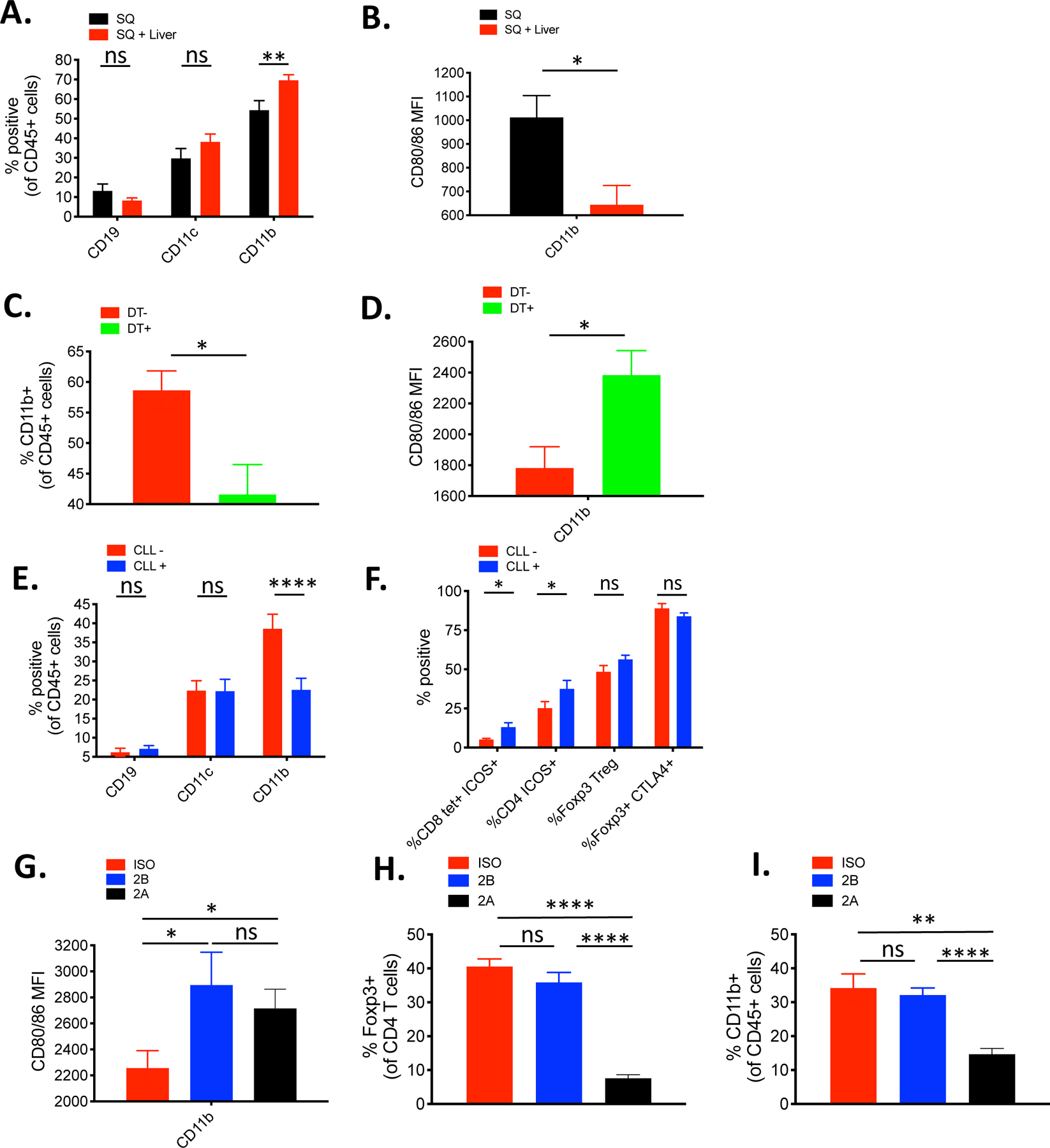
(A) Percentage of CD19, CD11b, and CD11c positive cells in the subcutaneous (SQ) tumor of mice with (red, n=10) or without (black, n=7) concurrent liver tumor by flow cytometry (B) MFI of CD80/86 from CD11b+ cells in the SQ tumor from mice with and without concurrent liver tumor by flow cytometry (each n=7). (C) Percentage of CD11b+ cells in the SQ tumor of Foxp3-DTR mice bearing liver tumor with (green) or without (red) DT administration (each n=8). (D) MFI of CD80/86 from CD11b+ cells in the SQ tumor of Foxp3-DTR mice bearing liver tumor with (green) or without (red) DT administration (each n=8). (E) Percentage of CD19, CD11b, and CD11c positive cells in the SQ tumor of liver-tumor bearing mice treated with liposomal clodronate (CLL) (blue, n=10) or vehicle control (red, n=8). (F) Comparative percentage of tetramer+ ICOS+ CD8+ T cells, ICOS+ CD4+ T cells, Tregs, and CTLA-4+ Tregs in the SQ tumor of mice bearing liver tumor treated with CLL (n=10) or vehicle control (n=8). (G) MFI of CD80/86 from CD45+ CD11b+ cells in the SQ tumor of mice bearing liver tumor treated with isotype control (red), 9D9 IgG2a (blue), or 9D9 IgG2b (black). (H) Percentage of Foxp3+ CD4 Tregs within the SQ tumor of the indicated groups. (I) Percentage of CD45+ CD11b+ cells within SQ tumor of the indicated groups. (G-I) N=6 for all groups. Data representative from at least 2 independent experiments. Data analyzed by unpaired t tests, and shown as mean +/− s.e.m. Asterisks indicating significance determined between groups are * p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Previous studies have shown that ICOS+, CTLA-4hiTregs can induce transendocytosis of costimulatory CD80/86 ligands from APCs through binding by CTLA-4 (58). Thus, the enhanced Tregs in this setting could coordinate distant antigen-specific effector T cell suppression by reducing costimulation signals on the tumor-antigen specific monocytes expanded in the distant tumor microenvironment in the presence of liver tumors. Moreover, these findings are consistent with the observation that the dysfunctional effector T cells in the liver tumor models show significantly decreased ICOS expression, which is known to be directly correlated with the strength of CD28-mediated costimulation available within the tumor microenvironment (60, 61). To examine whether the activated Tregs exert such influence on the MDSCs in this setting, we used the FoxP3-DTR mice with established SQ + liver tumors to evaluate their direct impact when the Tregs are depleted. Analysis of the distant SQ tumor showed that the depletion of Tregs led to a decrease in the percentage of infiltrating MDSCs (Fig. 6C). Moreover, the remaining CD11b+ monocytes in the tumor microenvironment expressed higher levels of CD80/86, suggesting their functionality within the tumor microenvironment may be reprogrammed to become APCs capable of providing costimulation to T cells (Fig. 6D), similar to findings previously described(62, 63).
The SQ tumor infiltrating CD45+CD11b+ monocytes were the only professional APCs, of those analyzed, which significantly changed in quantity in the context of liver tumor, supporting the hypothesis that Tregs may be responsible for their presence in the SQ tumor microenvironment. Clodronate liposomes (CLL) has been used to systemically deplete MDSCs in a variety of in vivo mouse models (64). Therefore, we examined what impact CLL treatment would have on the Tregs and vice versa. CLL administration reduced the percentage of intratumoral CD45+ CD11b+ MDSCs (Fig. 6E), increased the percentage of tumor-infiltrating activated ICOS+ tetramer+ CD8+ and ICOS+ CD4+ T cells (Fig. 6F), and significantly enhanced tumor rejection (fig. S15A). However, the percentage of total Foxp3+ Tregs in the tumor and CTLA-4+ expression in the Tregs did not change, suggesting that it is the Tregs that recruit MDSCs to the microenvironment and change their phenotype and not vice versa. Finally, to test our hypothesis that hepatic tumor priming increases activated CTLA-4+ Tregs at the distant tumor site, which leads to the recruitment of MDSCs and reduction of their CD80/86 expression, we compared two different isotypes of the mouse anti-CTLA-4 antibody clone 9D9. Notably, treatment with either the Treg depleting or non-depleting antibody led to an increase in CD80/86 expression on the CD45+ CD11b+ MDSCs, however, only the Treg depleting IgG2a antibody resulted in a reduction in the suppressive monocyte population (Fig. 6 G–I). Importantly, similar to the anti-CTLA-4 9H10 antibody, only the Treg-depleting IgG2a antibody treatment was capable of achieving complete rejection of tumors at both sites in the majority of treated mice when combined with anti-PD-1 antibody treatment (fig. S16, A and B). Thus, our data suggest that while CTLA-4 blockade can restore costimulatory surface CD80/86 expression on MDSCs, Treg-depletion may be necessary to further prevent any suppressive MDSCs’ recruitment to the tumor microenvironment and enable maximal systemic antitumor immunity in the presence of liver tumor.
Discussion
There is an urgent need to understand the mechanisms of CPI treatment failure and discover strategies that may augment therapeutic efficacy. In particular, liver metastasis continues as a significant unmet clinical challenge in immune-oncology. Our prior work revealed phenotypic changes to effector TILs at the distant biopsy sites in patients with liver metastasis (11, 12). This result raised the possibility that cancer cells invading the liver may trigger liver-specific tolerance mechanisms that reduce systemic antitumor immunity and the cancer immunotherapy efficacy. Here, we developed a preclinical model that allows us to study the influence of tumor engraftment at one anatomical site on the antitumor immune response at a distant site, and demonstrate that experimental liver metastasis can significantly reduce distant tumor-specific immunity as well as systemic anti-PD-1 treatment response in a manner mechanistically dependent on the crosstalk between Tregs and MDSCs. The selective anatomic location of the secondary tumor site in the liver resulted in significant suppression of tumor-specific immunity and anti-PD-1 immunotherapy response at the SQ site. Moreover, the data show that altering these suppressive pathways restores the favorable immune environment to promote tumor rejection by PD-1 blockade observed in the SQ tumor models (17). The findings argue against tumor-intrinsic adaptive or acquired mechanisms of resistance, as the same MC38 tumor cell line and even tumor lysates alone could achieve a similar effect in the liver but not when they are injected at other secondary tumor sites or with a different tumor line (B16F10). This latter point is critical as it rules out tumor burden but emphasizes the antigen-specificity of the effect. Thus, our data suggest a coordinated antigen-specific suppression reminiscent of multiple studies demonstrating the liver’s ability to facilitate systemic antigen-specific tolerance in the case of autoimmunity, liver organ transplantation and viral immunity (25, 65, 66).
To better understand the mechanism of liver tumor-mediated antitumor immune tolerance at the distant SQ site, we did extensive profiling of the immune infiltrates at the SQ tumor with and without the concurrent engraftment of a liver tumor. There was no evidence of clonal deletion of tumor-specific tetramer+ CD8+ T cells by the liver, in contrast to other studies suggesting FAS/FASL pathway-mediated effector T cell depletion as a major liver-associated tolerogenic mechanism (13). Moreover, treatment with PD-1 blockade could not enhance T cell function or tumor elimination in the SQ site of mice with liver tumor, suggesting an additional or alternative suppressive mechanism is involved systemically when the liver is engrafted with tumors. However, the tumor-specific CD8+ T cells in the SQ sites differed qualitatively between the two experimental groups, with cells from the liver tumor group having reduced expression of activation markers and effector cytokines. Notably, both effector CD4 T cells and tumor-specific CD8+ T cells showed significantly decreased ICOS expression in the SQ tumor microenvironment in the liver tumor-bearing mice. ICOS is an activation marker crucial in the function and identity of active antitumor T cells in the tumor microenvironment (22, 27) and is correlated with the strength of CD28-CD80/86 costimulation signals provided to T cells locally (60, 61). These results are consistent with the suggestion that tumor-specific T cell activation was deficient due to insufficient costimulation, which can be explained by the increased frequency of tumor-infiltrating MDSCs that expressed reduced levels of CD80/86 surface proteins. These results suggested that the MDSCs participate in systemic tumor-specific tolerance through the induction of clonal anergy in the tumor microenvironment (67).
The dynamic interaction between Tregs and MDSCs in the tumor microenvironment remains an area of active investigation (54, 59). Within the in vivo tumor microenvironment, CTLA-4 expressing Tregs downregulate or strip CD80/86 from APCs by transendocytosis preventing CD28-mediated costimulation of antigen-specific effector T cells (58). In the experimental liver metastasis model, the Tregs in the SQ site clearly had elevated expression of CTLA-4 as compared to groups without liver tumors. Our molecular analysis revealed that the genes encoding the glucocorticoid-induced leucine zipper and CD83, two proteins which are reported to be important in Treg suppressive function and ability to modulate APCs to promote tolerance, were upregulated in the Tregs from mice with liver tumors, suggesting that changes in Tregs may confer enhanced suppression (48–51). Additionally, there was decreased MDSC infiltration into the SQ tumor upon systemic depletion of Tregs, but no decrease in Tregs when MDSCs were depleted. This suggests that the Tregs were the driver of tolerance in this setting and supports the hypothesis that the liver tumor priming microenvironment alters Treg phenotype and function, promoting the circulation of the cells to the SQ site where they impact tumor-specific MDSC quantity, phenotype, and function leading to the inhibition of antitumor T cell activation and tumor rejection. Indeed, clinical tumor hyperprogression after PD-1 blockade treatment has been observed in the setting of an abundance of activated PD-1+ Tregs found in patients with liver metastasis, and in cases of hepatocellular carcinoma, there is evidence of higher frequencies of CTLA-4hi Tregs and MDSCs with associated systemic effector T-cell dysfunction (68, 69).
Finally, the seminal role of Tregs is supported by anti-CTLA-4 9D9 IgG2b blocking studies showing that the effects of CTLA-4hi Treg on MDSC function could be “reversed” without Treg depletion, resulting in an increased expression of CD80/86, an improved ability to provide costimulation to T cells, and somewhat restored antitumor immunity. However, these effects were insufficient to completely reverse the MDSC-mediated suppression to allow complete tumor regression when combined with anti-PD-1 treatment. In contrast, depletion of intratumoral CTLA-4hi Tregs by the anti-CTLA-4 antibody 9D9 IgG2a led to a significant reduction of the tolerogenic MDSCs within the tumor as well as superior tumor rejection in combination with PD-1 blockade. Similar results were observed by functional inactivation of intratumoral Tregs via the EZH2 inhibitor CPI-1205. Importantly, the site of most effective depletion by the anti-CTLA-4 mAb or functional inactivation by CPI-1205 is localized to intratumoral Tregs, which express higher levels of CTLA-4 and EZH2, respectively (40, 70). In this regard, clinical studies are currently underway testing Treg-depleting anti-CTLA-4 antibodies and EZH2 inhibitors and should be tried in the liver metastases setting which are often excluded from these trials.
Our data suggest that Tregs undergo specific priming in the presence of liver tumor and the enhanced Tregs can modify the tumor-antigen specific MDSCs that migrate to distant SQ sites, ultimately suppressing the activation of antigen-specific CD8+ T cells via clonal anergy. While the precise mechanism of intrahepatic priming of tumor-specific Tregs remains an area of active investigation, it is well-documented that suppressive hepatic antigenic priming of highly suppressive Tregs occurs by various tolerogenic liver-resident APCs including dendritic cells, Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells (LSECs), and even hepatocytes (25, 71, 72).
In summary, our findings complement the mounting clinical studies showing a reduced response to CPIs in cancer patients with liver metastases and implicate additional regulatory pathways that are engaged in the presence of liver metastases. The liver tumors compromise not only intrahepatic immunity but also impact distant antitumor immunity and may contribute to the reduced efficacy of systemic anti-PD-1-treatment seen in the clinical setting. Our observation further serves as a proof-of-concept that resistance patterns to current forms of immunotherapy could vary as a result of cancer invasion to distinct anatomic sites that promote additional, non-redundant, host tolerogenic mechanisms that PD-1 blockade alone cannot overcome. Importantly, our data support the rationale to more precisely deploy Treg-targeting combination immunotherapy in scenarios where Treg activity may be biologically enhanced.
Materials and Methods
Mice and cell lines
Six- to eight-week-old female and male C57BL/6 (B6) mice were purchased from The Jackson Laboratory. Foxp3DTR mice (FP3-DTR) express the human diphtheria toxin receptor and EGFP genes from the Foxp3 locus without disrupting expression of the endogenous Foxp3 gene were described elsewhere (39). Foxp3DTR mice were bred onto a B6 background a minimum of five generations. All mouse experiments (or cells from mice of given genotypes) used comparisons between littermates or age-matched control mice. All mice were maintained in an American Association for the Accreditation of Laboratory Animal Care–accredited barrier facility. All protocols were approved by the University of California, San Francisco, Institutional Animal Care and Use Committee. B6 derived MC38 colon adenocarcinoma cell line expressing luciferase was a gift from R. Daniel Beauchamp (Vanderbilt University). MC38 cell line was maintained in RPMI 1640 with Glutamax, penicillin, streptomycin, and 10% BSA. B16-F10 melanoma cells were purchased from ATCC and maintained in DMEM with Glutamax, penicillin, streptomycin, and 10% BSA. Cell lines were routinely tested for mycoplasma contamination.
In vivo tumor models
All tumor challenge was performed with the injection of 5 × 105 MC38 or B16F10 cells into syngeneic B6, B6 derived Rag-1−/− or immunocompromised NSG mice. Subcutaneous (SQ) tumor inoculations were performed in 50 μL of PBS in the right or left inguinal sites. Hepatic and kidney tumors were generated by subcapsular injection of tumor cells in 50 μL of PBS directly into the liver or left kidney. In some experiments, hepatic tumors were generated by injecting tumor cells into the upper hemispleen followed by ligation and resection of the hemispleen post injection Lung tumors (MC38) were generated via tail vein injection of tumor cells in 50 μL of PBS. Intraperitoneal tumors were generated via intraperitoneal injection of tumor cells. Kidney tumors were generated via direct injection of tumor cells into the left kidney capsule. Tumor measurements were performed by a single operator in two dimensions using calipers at a minimum of twice a week. Tumor lysates were generated by 10 cycles of freeze (liquid nitrogen) and thaw (37C water bath) of 5 × 105 whole MC38 cells. For experiments using irradiated MC38 cells, 50 Gy radiation was applied to the cells before injection of 5 × 105 cells into each animal as described above. For tumor rechallenge experiments, all mice treated with immunotherapy that survived past 60 days without any detectable tumor were re-injected SQ with 5 × 105 MC38 tumor cells and monitored for tumor growth compared to treatment naïve mouse injected with the same cell line. For adoptive Treg transfer experiments, live Foxp3+ Tregs were FACS-sorted from SQ MC38 TILs obtained from FoxP3-EGFP mice implanted with a secondary MC38 liver tumor versus non-liver tumor bearing mouse. The SQ tumors from each group were procured 14 days after inoculation. Equal numbers (5 × 104) of sorted Tregs from each group were co-injected SQ with 5 × 105 MC38 tumor cells to a matched cohort with just SQ tumor and monitored for tumor growth compared to mice injected with the same tumor cell line without any Tregs.
Diphtheria toxin administration
For FoxP3-DTR mouse studies, continuous systemic depletion of Tregs using FoxP3-DTR mice was achieved by administering of 0.04 mg/kg diphtheria toxin (DT) 1 day before tumor inoculation followed by every other day DT dosing for the entire duration of the experiment up to 24 days.
Checkpoint inhibitors administration
For checkpoint inhibitor studies, 250 μg of anti-mouse PD-1 (RMP1-14, BioXCell) and/or anti-mouse CTLA-4 (clone 9H10, BioXCell; clone 9D9 IgG2a/IgG2b, Bristol-Myers Squibb) antibody were injected intraperitoneally on days 7, 9, and 11 after tumor implantations.
EZH2 inhibitor administration
Mice were treated twice daily with CPI-1205 (Constellation Pharmaceuticals) by oral gavage with 300 mg/kg CPI-1205 made in a vehicle of 0.5% methylcellulose and 0.2% Tween 80 per the manufacturer’s instruction. The treatment regimen began one day after tumor inoculation and was continued throughout the experiment.
Lymphocyte isolation
For tumor-infiltrating lymphocytes, tumors were resected from the tissue of origin and were minced to 3–5mm3 fragments and digested in RPMI media supplemented with HEPES, 20 mg/ml DNase I (Roche), and 125 U/ml Collagenase D (Roche) using a GentleMACS™ tissue dissociator per manufacture’s protocol (Miltenyi). Liver tissues were processed by maceration through a 70 μm filter followed by obtaining the pellet of a 40% percent Percoll gradient separation. Cells from lymphoid organs were prepared by maceration through a 70 μm filter. The single-cell suspensions underwent ACK erythrocyte lysis and all suspensions were passaged over through 40 μm filters before cell staining or in vitro stimulation.
Flow Cytometry
Fresh or PMA/Ionomycin stimulated cells were stained with BV650 conjugated anti-mouse CD4 (RM4-5, BioLegend), PerCP-Cy5.5 conjugated anti-mouse CD8 (53-6.7, BD Biosciences), PE-Cy7 conjugated anti-mouse PD-1 (RMP1-30 or J43, BioLegend), PE conjugated anti-mouse CTLA-4 (intracellular staining, UC10-4F10-11, BD Biosciences), PE-CF594 conjugated ICOS (C398.4, BD Biosciences), BUV395 conjugated anti-mouse CD45 (30-F11, BD Biosciences), PE-Cy7 conjugated CD107a (1D4B, BioLegend), FITC conjugated anti-mouse Foxp3 (FJK-16s, eBioscience), PerCP-eFlour 710 conjugated SIINFEKL peptide/H-2Kb antibody (25-D1.16, ThermoFisher), BV650 conjugated anti-mouse CD80 (16-10A1, BioLegend), BV650 conjugated anti-mouse CD86 (GL-1, BioLegend), APC/Cy7 conjugated anti-mouse CD11b (M1/70, BioLegend), and LIVE/DEAD® Fixable Blue Dead Cell Stain (Invitrogen). Intracellular staining of cytokines was accomplished with 3 × 106 cells after 60-minute culture in RPMI media with Brefeldin A (eBioscience), followed by 180-minute in vitro stimulation with PMA (Sigma) and Ionomycin (Sigma). Fixation and permeabilization of cells were conducted for intracellular cytokine staining using BD Cytofix/Cytoperm (BD Biosciences), or transcription factor staining using eBioscience Foxp3 fixation/permeabilization kit (ThermoFisher) according to manufacturer’s protocol. PE conjugated anti-mouse TNFα (MP6-XT22, BioLegend), eFluor450 conjugated anti-mouse IFNγ (XMG1.2, eBioscience), and BV711 conjugated anti-mouse Ki-67 (B56, BD Biosciences) were used. Control staining for activation markers was performed on T cells from the splenocytes or lymph nodes of naïve mice. Flow cytometry was performed on an LSRII (BD Biosciences) and data sets were analyzed using FlowJo software (Tree Star, Inc).
Single-Cell RNA sequencing
FACS-sorted live CD45+ tumor infiltrating cells from the subcutaneous tumor were washed and resuspended in PBS with 0.04% BSA to a concentration of 1000 cell/μL, and loaded onto the 10x Genomics Chromium platform for droplet-based massively parallel single-cell RNA sequencing (scRNAseq) according to the manufacturer’s instructions. Libraries were prepared using 10x Genomics Chromium Single Cell 5’ Reagent (GEX + VDJ) Kit according to manufacturer’s protocol and sequenced using an Illumina HiSeq 2500 according to the manufacturer’s instructions (Illumina). FASTQ were aligned using the Cell Ranger pipeline (10x Genomics, Inc., version 3.0.2) using mouse genome reference dataset (GRCm38/Ensembl/10x, 3.0.2) or VDJ reference dataset (GRCm38_alts_ensembl).
Single-Cell RNA sequencing data processing
The Seurat pipeline was used to cluster and identify the cell subsets with the dataset (73, 74). Data was read into R (Version 4). Cells with low gene detection (<300 genes) as well as high mitochondrial gene content (>5%), were removed and SCTransform was used to perform normalization, variance stabilization, cluster identification, and feature selection based on a UMI-based gene expression matrix (75). Specific markers for each cluster identified by using the “FindAllMarkers” function within the Seurat pipeline. The top differential expressed genes were used to identify the cell types within the clusters. The monocyte/myeloid and T cell compartment was subset and re-clustered for analysis. For gene scoring analysis, “AddModuleScore” was used as previously published (55). For statistics, adjusted p-values where used from the “FindAllMarkers” in Seurat and unpaired Wilcoxon ran sum test with Bonferroni correction was used for the “AddModuleScore”.
Bioluminescent imaging
For in vivo imaging, MC38 colon adenocarcinoma cells modified to express firefly luciferase were inoculated into mice to establish a tumor model. Mice were intraperitoneally injected with 150 mg/kg of D-Luciferin (Gold Biotechnology) 7 minutes before imaging with the Xenogen IVIS Imaging System.
Tetramer staining
To identify MC38 tumor-specific CD8+ T cells, freshly isolated lymphocytes were stained with APC-conjugated H-2Kb restricted KSPWFTTL peptide-MHC tetramer or H-2Kb restricted irrelevant influenza peptide-MHC tetramer control for 30 minutes in 4C prior to surface or intracellular antibody staining as detailed above. All tetramers were made by the NIH Tetramer Core Facility at Emory University.
Statistical analysis
Prism software (GraphPad, version 8) was used for all statistical analysis. For all comparisons between two groups, unpaired two-tailed Student’s t test was used to assess P values, and data were displayed as means +/− SEMs. For survival analysis, Kaplan-Meier curves were used, and P values were assessed using the log-rank test. For tumor growth curves, two-way ANOVA was used with Sidak’s multiple comparisons test was performed on available time points. P values <0.05 were considered significant, and are denoted in the figures as * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
Supplementary Material
Fig. S1. Mice with experimental liver metastasis have a normal liver metabolic function.
Fig. S2. Liver-mediated suppression of distant antitumor immunity requires adaptive immunity but not high tumor burden.
Fig. S3. Activation markers are decreased on effector CD4+ and CD8+ T cells but increased on Tregs in mice with experimental liver metastasis.
Fig. S4. The percentage and number of Tregs and tetramer+ CD8+ T cells are similar in distant tumors.
Fig. S5. Experimental liver metastasis mediated suppression in the B16F10 tumor model.
Fig. S6. KSPWFTTL-H-2Kb (KSP) tetramer specifically stains MC38 tumor-targeted CD8+ T cells.
Fig. S7. Suppression of distant CD8+ T cells is liver-mediated and tumor-antigen specific.
Fig. S8. Foxp3-DTR mouse model schema.
Fig. S9. DT induces profound Treg depletion in the blood and the subcutaneous tumor of treated Foxp3-DTR mice.
Fig. S10. Combination therapy with Treg-depleting anti-CTLA-4 and anti-PD-1 antibodies overcomes experimental liver metastasis immune suppression.
Fig. S11. Clustering of tumor-infiltrating immune cell subsets using scRNAseq.
Fig. S12. Differential gene expression in distant MDSCs driven by presence of liver tumor.
Fig. S13. Liver-tumor mediated suppression is associated with distant increase in CD11b+ monocyte populations.
Fig. S14. Increase in distant tolerogenic MDSCs is anatomically unique to liver tumor.
Fig. S15. Treg or MDSC depletion can enhance tumor rejection in mice with experimental liver metastasis.
Fig. S16. Treg-depleting versus non-depleting anti-CTLA-4 antibody in combination with anti-PD-1 treatment in experimental liver metastasis.
Table S1. Raw data table (Excel spreadsheet)
Acknowledgements:
We thank Caroline Raffin and Karen Hammen for critically reviewing this manuscript; Michael Lee, Claudia Bispo, Vinh Nguyen, Ashley Carlos, the UCSF single cell analysis core facility, and UCSF Institute for Human Genetics Sequencing Core; R. Daniel Beauchamp (Vanderbilt University) for the MC38-luciferase tumor cell line; Jonathan Sockolosky (K. Christopher Garcia Lab, Stanford University) for the B16F10-OVA tumor cell line; NIH Tetramer Core Facility for the tetramers used in our experiments; Bristol Myers Squibb for the anti-CTLA-4 9D9 antibody, and Constellation Pharmaceuticals for providing the EZH2 inhibitor CPI-1205. A.D. and J.A.B. are members of the Parker Institute for Cancer Immunotherapy. J.A.B. is the A.W. and Mary Margaret Clausen Distinguished Professor in Metabolism and Endocrinology.
Funding: Support for this research came from Parker Institute for Cancer Immunotherapy. J.C.L. was supported by the A.P. Giannini Postdoctoral Research Fellowship Award and the NIH/NIAID (T32 5T32AI007334-28). A.Y. was supported by an NHMRC C.J. Martin Fellowship (1143981). J.S. was supported by the Larry L. Hillblom Foundation (2019-D-006-FEL). C.T.M. was supported by the UCSF ImmunoX Computational Immunology Fellowship and the NIH (T32GM007618).
Footnotes
Competing interests: J.A.B. is the founder of Sonoma BioTherapeutics (a Treg therapy company) and Celsius Therapeutics; a member of the board of directors of Rheos Medicines, Provention Bio, Sonoma BioTherapeutics, and the Parker Institute for Cancer Immunotherapy; and is a member of the scientific advisory board of Arcus Biosciences, Solid Biosciences, and Vir Biotechnology. A.D. discloses research funding from the Amoroso and Cook Fund, the Parker Institute for Cancer Immunotherapy, Novartis, Merck, Bristol Myers Squibb, Incyte, AbbVie, OncoSec, Xencor, Pfizer, Roche/Genentech, and Exelixis; participation in Advisory Boards with Amgen, Array BioPharma, and Roche/Genentech; and stock ownership in Trex Bio and Pionyr Immunotherapeutics. J.C.L. is on the Scientific Advisory Board of Chimera Bioengineering and has provide consulting services to OncoSec Medical Incorporated. The remaining authors declare they have no competing interests.
Data and materials availability: The anti-CTLA-4 9D9 antibody (Bristol Myers Squibb) and the EZH2 inhibitor CPI-1205 (Constellation Pharmaceuticals) are available through Material Transfer Agreements with the individual companies. The scRNAseq data is available in the NCBI GEO database through accession number GSE157653. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes:
- 1.Tumeh PC, Hellmann MD, Hamid O, Tsai KK, Loo KL, Gubens MA, Rosenblum M, Harview CL, Taube JM, Handley N, Khurana N, Nosrati A, Krummel MF, Tucker A, Sosa EV, Sanchez PJ, Banayan N, Osorio JC, Nguyen-Kim DL, Chang J, Shintaku IP, Boasberg PD, Taylor EJ, Munster PN, Algazi AP, Chmielowski B, Dummer R, Grogan TR, Elashoff D, Hwang J, Goldinger SM, Garon EB, Pierce RH, Daud A, Liver Metastasis and Treatment Outcome with Anti-PD-1 Monoclonal Antibody in Patients with Melanoma and NSCLC, Cancer Immunol. Res 5, 417–424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pires da Silva I, Lo S, Quek C, Gonzalez M, Carlino MS, Long GV, Menzies AM, Site-specific response patterns, pseudoprogression, and acquired resistance in patients with melanoma treated with ipilimumab combined with anti-PD-1 therapy, Cancer (2020), 10.1002/cncr.32522. [DOI] [PubMed]
- 3.Schmid S, Diem S, Li Q, Krapf M, Flatz L, Leschka S, Desbiolles L, Klingbiel D, Jochum W, Früh M, Organ-specific response to nivolumab in patients with non-small cell lung cancer (NSCLC), Cancer Immunol. Immunother 67, 1825–1832 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bilen MA, Shabto JM, Martini DJ, Liu Y, Lewis C, Collins HH, Akce M, Kissick H, Carthon BC, Shaib WL, Alese OB, Steuer CE, Wu C, Lawson DH, Kudchadkar R, Master V, El-Rayes B, Ramalingam SS, Owonikoko TK, Harvey RD, Sites of metastasis and association with clinical outcome (CO) in advanced stage cancer patients (pts) treated with immunotherapy (IO), BMC Cancer 19 (2019), 10.1093/annonc/mdy288.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sasaki A, Nakamura Y, Mishima S, Kawazoe A, Kuboki Y, Bando H, Kojima T, Doi T, Ohtsu A, Yoshino T, Kuwata T, Akimoto T, Shitara K, Predictive factors for hyperprogressive disease during nivolumab as anti-PD1 treatment in patients with advanced gastric cancer, Gastric Cancer (2019), 10.1007/s10120-018-00922-8. [DOI] [PubMed]
- 6.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Drilon A, Wolchok JD, Carvajal RD, McHenry MB, Hosein F, Harbison CT, Grosso JF, Sznol M, Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non-Small Cell Lung Cancer Treated With Nivolumab, JAMA Oncol (2019), 10.1001/jamaoncol.2019.2187. [DOI] [PMC free article] [PubMed]
- 7.Halabi S, Kelly WK, Ma H, Zhou H, Solomon NC, Fizazi K, Tangen CM, Rosenthal M, Petrylak DP, Hussain M, Vogelzang NJ, Thompson IM, Chi KN, De Bono J, Armstrong AJ, Eisenberger MA, Fandi A, Li S, Araujo JC, Logothetis CJ, Quinn DI, Morris MJ, Higano CS, Tannock IF, Small EJ, Meta-analysis evaluating the impact of site of metastasis on overall survival in men with castration-resistant prostate cancer, J. Clin. Oncol (2016), 10.1200/JCO.2015.65.7270. [DOI] [PMC free article] [PubMed]
- 8.Budczies J, von Winterfeld M, Klauschen F, Bockmayr M, Lennerz JK, Denkert C, Wolf T, Warth A, Dietel M, Anagnostopoulos I, Weichert W, Wittschieber D, Stenzinger A, The landscape of metastatic progression patterns across major human cancers, Oncotarget (2015), 10.18632/oncotarget.2677. [DOI] [PMC free article] [PubMed]
- 9.Schmid S, Diem S, Li Q, Krapf M, Flatz L, Leschka S, Desbiolles L, Klingbiel D, Jochum W, Früh M, Organ-specific response to nivolumab in patients with non-small cell lung cancer (NSCLC), Cancer Immunol. Immunother 67, 1825–1832 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pires da Silva I, Lo S, Quek C, Gonzalez M, Carlino MS, Long GV, Menzies AM, Site‐specific response patterns, pseudoprogression, and acquired resistance in patients with melanoma treated with ipilimumab combined with anti-PD-1 therapy, Cancer (2019), 10.1002/cncr.32522. [DOI] [PubMed]
- 11.Loo K, Tsai KK, Mahuron K, Liu J, Pauli ML, Sandoval PM, Nosrati A, Lee J, Chen L, Hwang J, Levine LS, Krummel MF, Algazi AP, Alvarado MD, Rosenblum MD, Daud AI, Partially exhausted tumor-infiltrating lymphocytes predict response to combination immunotherapy, JCI Insight 2 (2017), 10.1172/jci.insight.93433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daud AI, Loo K, Pauli ML, Sanchez-rodriguez R, Sandoval PM, Taravati K, Tsai K, Nosrati A, Nardo L, Alvarado MD, Algazi AP, Pampaloni MH, V Lobach I, Hwang J, Pierce RH, Gratz IK, Krummel MF, Rosenblum MD, Tumor immune profiling predicts response to anti – PD-1 therapy in human melanoma, J. Clin. Invest 126, 1–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crispe IN, Immune tolerance in liver disease, Hepatology 60, 2109–2117 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, Ritz T, Longerich T, Theriot CM, McCulloch JA, Roy S, Yuan W, Thovarai V, Sen SK, Ruchirawat M, Korangy F, Wang XW, Trinchieri G, Greten TF, Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells, Science (80-.) (2018), 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed]
- 15.Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, Liu Z, Dong M, Hu X, Ouyang W, Peng J, Zhang Z, Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing, Cell (2017), 10.1016/j.cell.2017.05.035. [DOI] [PubMed]
- 16.Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, Huang D, Li J, Li H, Chen F, Liu J, Xing Y, Chen X, Su S, Song E, DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25, Nature (2020), 10.1038/s41586-020-2394-6. [DOI] [PubMed]
- 17.Mosely SIS, Prime JE, Sainson RCA, Koopmann J-O, Wang DYQ, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L, Marcus D, Anderton J, Watkins A, Coates Ulrichsen J, Brohawn P, Higgs BW, McCourt M, Jones H, Harper JA, Morrow M, Valge-Archer V, Stewart R, Dovedi SJ, Wilkinson RW, Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery, Cancer Immunol. Res 5, 29–41 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Oliver AJ, Darcy PK, Trapani JA, Kershaw MH, Slaney CY, Cross‐talk between tumors at anatomically distinct sites, FEBS J (2020), 10.1111/FEBS.15316. [DOI] [PubMed]
- 19.Brown ZJ, Establishment of Orthotopic Liver Tumors by Surgical Intrahepatic Tumor Injection in Mice with Underlying Non-Alcoholic Fatty Liver Disease, 1–9 (2018). [DOI] [PMC free article] [PubMed]
- 20.Zaynagetdinov R, Sherrill TP, Gleaves LA, McLoed AG, Saxon JA, Habermann AC, Connelly L, Dulek D, Peebles RS, Fingleton B, Yull FE, Stathopoulos GT, Blackwell TS, Interleukin-5 facilitates lung metastasis by modulating the immune microenvironment, Cancer Res (2015), 10.1158/0008-5472.CAN-14-2379. [DOI] [PMC free article] [PubMed]
- 21.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, Adamow M, Kuk D, Panageas KS, Carrera C, Wong P, Quagliarello F, Wubbenhorst B, D’Andrea K, Pauken KE, Herati RS, Staupe RP, Schenkel JM, McGettigan S, Kothari S, George SM, Vonderheide RH, Amaravadi RK, Karakousis GC, Schuchter LM, Xu X, Nathanson KL, Wolchok JD, Gangadhar TC, Wherry EJ, T-cell invigoration to tumour burden ratio associated with anti-PD-1 response, Nature 545, 60–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, Allison JP, Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade, Cell 170, 1120–1133.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ribas A, Wolchok JD, Cancer immunotherapy using checkpoint blockade, Science (80-.) 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crispe IN, Hepatocytes as Immunological Agents., J. Immunol 196, 17–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomson AW, a Knolle P, Antigen-presenting cell function in the tolerogenic liver environment., Nat. Rev. Immunol 10, 753–766 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Joseph RW, Elassaiss-Schaap J, Kefford RF, Hwu W-J, Wolchok JD, Joshua AM, Ribas A, Hodi FS, Hamid O, Robert C, Daud AI, Dronca RS, Hersey P, Weber JS, Patnaik A, de Alwis DP, Perrone AM, Zhang J, Kang SP, Ebbinghaus SW, Anderson KM, Gangadhar T, Baseline Tumor Size Is an Independent Prognostic Factor for Overall Survival in Patients With Melanoma Treated With Pembrolizumab, Clin. Cancer Res (2018) (available at http://clincancerres.aacrjournals.org/content/early/2018/04/21/1078-0432.CCR-17-2386.abstract). [DOI] [PMC free article] [PubMed]
- 27.Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP, Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy, J. Exp. Med (2014), 10.1084/jem.20130590. [DOI] [PMC free article] [PubMed]
- 28.Zappasodi R, Budhu S, Hellmann MD, Postow MA, Senbabaoglu Y, Manne S, Gasmi B, Liu C, Zhong H, Li Y, Huang AC, Hirschhorn-Cymerman D, Panageas KS, Wherry EJ, Merghoub T, Wolchok JD, Non-conventional Inhibitory CD4+Foxp3−PD-1hiT Cells as a Biomarker of Immune Checkpoint Blockade Activity, Cancer Cell (2018), [DOI] [PMC free article] [PubMed]
- 29.Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanović S, Robbins PF, Rosenberg SA, Tumor-infiltrating human CD4+ regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity, Sci. Immunol 4 (2019), 10.1126/sciimmunol.aao4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vocanson M, Rozieres A, Hennino A, Poyet G, Gaillard V, Renaudineau S, Achachi A, Benetiere J, Kaiserlian D, Dubois B, Nicolas JF, Inducible costimulator (ICOS) is a marker for highly suppressive antigen-specific T cells sharing features of TH17/TH1 and regulatory T cells, J. Allergy Clin. Immunol 126 (2010), 10.1016/j.jaci.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 31.Tu J-F, Ding Y-H, Ying X-H, Wu F-Z, Zhou X-M, Zhang D-K, Zou H, Ji J-S, Regulatory T cells, especially ICOS+ FOXP3+ regulatory T cells, are increased in the hepatocellular carcinoma microenvironment and predict reduced survival, Sci. Rep 6, 35056 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crispe IN, Hepatic T cells and liver tolerance, Nat. Rev. Immunol 3, 51–62 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Horst AK, Neumann K, Diehl L, Tiegs G, Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells, Cell. Mol. Immunol 13, 277–292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, Hudson TE, Vu UT, Francica BJ, Banda T, Katibah GE, Kanne DB, Leong JJ, Metchette K, Bruml JR, Ndubaku CO, McKenna JM, Feng Y, Zheng L, Bender SL, Cho CY, Leong ML, van Elsas A, Dubensky TW, McWhirter SM, Magnitude of Therapeutic STING Activation Determines CD8 + T Cell-Mediated Anti-tumor Immunity, Cell Rep 25, 3074–3085.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Sánchez-Paulete AR, Cueto FJ, Martínez-López M, Labiano S, Morales-Kastresana A, Rodríguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI, Sancho D, Melero I, Cancer immunotherapy with immunomodulatory anti-CD137 and anti–PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells, Cancer Discov 6, 71–79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greten TF, Sangro B, Targets for immunotherapy of liver cancer J. Hepatol (2018), 10.1016/j.jhep.2017.09.007. [DOI] [PMC free article] [PubMed]
- 37.McNally A, Hill GR, Sparwasser T, Thomas R, Steptoe RJ, CD4+CD25+ regulatory T cells control CD8+ T-cell effector differentiation by modulating IL-2 homeostasis, Proc. Natl. Acad. Sci. U. S. A (2011), 10.1073/pnas.1103782108. [DOI] [PMC free article] [PubMed]
- 38.Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, Von Boehmer H, Khazaie K, Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo, Proc. Natl. Acad. Sci. U. S. A (2005), 10.1073/pnas.0408197102. [DOI] [PMC free article] [PubMed]
- 39.Kim JM, Rasmussen JP, Rudensky AY, Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice, Nat. Immunol 8, 191–197 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, Silveria S, Harwin T, Abnousian A, Pagani M, Rosenblum MD, Van Gool F, Fong L, Bluestone JA, DuPage M, Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity, Cell Rep 23, 3262–3274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, Adamow M, Kuk D, Panageas KS, Carrera C, Wong P, Quagliarello F, Wubbenhorst B, Schenkel JM, Mcgettigan S, Andrea KD, Pauken KE, Herati RS, Ryan P, Vonderheide RH, Amaravadi RK, Karakousis GC, Kothari S, George SM, Schuchter LM, Xu X, Nathanson KL, Wolchok JD, Gangadhar TC, T-cell invigoration to tumour burden ratio associated with anti-PD-1 response, Nature, 1–18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ, Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells, Cancer Immunol. Res 1, 32–42 (2013). [DOI] [PubMed] [Google Scholar]
- 43.June CH, Warshauer JT, Bluestone JA, Is autoimmunity the Achilles’ heel of cancer immunotherapy?, Nat. Med 23, 540–547 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Trojer P, Anandhan S, Goswami S, Zhang X, Xiong L, Skepner J, Zhang J, Apostolou I, Zhao H, Sharma P, Subudhi SK, Aparicio A, Allison JP, Modulation of EZH2 expression in T cells improves efficacy of anti–CTLA-4 therapy, J. Clin. Invest 128, 3813–3818 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, Chatila TA, Regulatory T cell development in the absence of functional Foxp3, Nat. Immunol (2007), 10.1038/ni1445. [DOI] [PubMed]
- 46.Tanaka A, Sakaguchi S, Regulatory T cells in cancer immunotherapy Cell Res (2017), 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed]
- 47.Raffin C, Vo LT, Bluestone JA, Treg cell-based therapies: challenges and perspectives Nat. Rev. Immunol (2019), 10.1038/s41577+019-0232-6. [DOI] [PMC free article] [PubMed]
- 48.Doebbeler M, Koenig C, Krzyzak L, Seitz C, Wild A, Ulas T, Baßler K, Kopelyanskiy D, Butterhof A, Kuhnt C, Kreiser S, Stich L, Zinser E, Knippertz I, Wirtz S, Riegel C, Hoffmann P, Edinger M, Nitschke L, Winkler T, Schultze JL, Steinkasserer A, Lechmann M, CD83 expression is essential for Treg cell differentiation and stability, JCI insight 3, e99712 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bereshchenko O, Coppo M, Bruscoli S, Biagioli M, Cimino M, Frammartino T, Sorcini D, Venanzi A, Di Sante M, Riccardi C, GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-β signaling., Cell Rep 7, 464–475 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Engler JB, Kursawe N, Solano ME, Patas K, Wehrmann S, Heckmann N, Lühder F, Reichardt HM, Arck PC, Gold SM, Friese MA, Glucocorticoid receptor in T cells mediates protection from autoimmunity in pregnancy, Proc. Natl. Acad. Sci 114, E181 LP-E190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reinwald S, Wiethe C, Westendorf AM, Breloer M, Probst-Kepper M, Fleischer B, Steinkasserer A, Buer J, Hansen W, CD83 Expression in CD4 + T Cells Modulates Inflammation and Autoimmunity, J. Immunol (2008), 10.4049/jimmunol.180.9.5890. [DOI] [PubMed]
- 52.Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, Noguchi T, Arthur CD, Meng W, Alspach E, Medrano RFV, Fronick C, Fehlings M, Newell EW, Fulton RS, Sheehan KCF, Oh ST, Schreiber RD, Artyomov MN, High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy, Cell (2018), 10.1016/j.cell.2018.09.030. [DOI] [PMC free article] [PubMed]
- 53.Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, Nelson AE, Loo K, Kumar R, Rosenblum MD, Alvarado MD, Wolf DM, Bogunovic D, Bhardwaj N, Daud AI, Ha PK, Ryan WR, Pollack JL, Samad B, Asthana S, Chan V, Krummel MF, A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments, Nat. Med 24, 1178–1191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V, Coordinated regulation of myeloid cells by tumours Nat. Rev. Immunol (2012), 10.1038/nri3175. [DOI] [PMC free article] [PubMed]
- 55.Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA, Nee K, Hernandez G, Evans K, Torosian L, Silva A, Walsh C, Kessenbrock K, Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics, Sci. Immunol (2020), 10.1126/sciimmunol.aay6017. [DOI] [PMC free article] [PubMed]
- 56.Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, Merghoub T, Wolchok JD, Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner, Cell Rep (2015), 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed]
- 57.Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, Kabat J, Matsumura R, Dorward DW, Glass DD, Shevach EM, Regulatory T cells mediate specific suppression by depleting peptide – MHC class II from dendritic cells, Nat. Immunol 20 (2019), 10.1038/s41590-018-0280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ovcinnikovs V, Ross EM, Petersone L, Edner NM, Heuts F, Ntavli E, Kogimtzis A, Kennedy A, Wang CJ, Bennett CL, Sansom DM, Walker LSK, CTLA-4-mediated transendocytosis of costimulatory molecules primarily targets migratory dendritic cells, Sci. Immunol (2019), 10.1126/sciimmunol.aaw0902. [DOI] [PMC free article] [PubMed]
- 59.Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, Merghoub T, Wolchok JD, Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner, Cell Rep 13, 412–424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McAdam AJ, Chang TT, Lumelsky AE, Greenfield EA, Boussiotis VA, Duke-Cohan JS, Chernova T, Malenkovich N, Jabs C, Kuchroo VK, Ling V, Collins M, Sharpe AH, Freeman GJ, Mouse Inducible Costimulatory Molecule (ICOS) Expression Is Enhanced by CD28 Costimulation and Regulates Differentiation of CD4 + T Cells, J. Immunol 165, 5035–5040 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Rudd CE, Schneider H, Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling, Nat. Rev. Immunol 3, 544–556 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Thaci B, Ahmed AU, Ulasov IV, Wainwright DA, Nigam P, Auffinger B, Tobias AL, Han Y, Zhang L, Moon KS, Lesniak MS, Depletion of myeloid-derived suppressor cells during interleukin-12 immunogene therapy does not confer a survival advantage in experimental malignant glioma, Cancer Gene Ther 21, 38–44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P, Identification of a CD11b+/Gr-1+/CD31+ myeloid progenitor capable of activating or suppressing CD8+ T cells, Blood 96, 3838–3846 (2000). [PMC free article] [PubMed] [Google Scholar]
- 64.Weiss JM, Ridnour LA, Back T, Hussain SP, He P, Maciag AE, Keefer LK, Murphy WJ, Harris CC, Wink DA, Wiltrout RH, Macrophage-dependent nitric oxide expression regulates tumor cell detachment and metastasis after IL-2/anti-CD40 immunotherapy, J. Exp. Med (2010), 10.1084/jem.20100670. [DOI] [PMC free article] [PubMed]
- 65.Umeshappa CS, Singha S, Blanco J, Shao K, Nanjundappa RH, Yamanouchi J, Parés A, Serra P, Yang Y, Santamaria P, Suppression of a broad spectrum of liver autoimmune pathologies by single peptide-MHC-based nanomedicines, Nat. Commun (2019), 10.1038/s41467-019-09893-5. [DOI] [PMC free article] [PubMed]
- 66.Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, Reimer R, Heine M, Huber S, Waurisch C, Eychmüller A, Wraith DC, Korn T, Nielsen P, Weller H, Schramm C, Lüth S, Lohse AW, Heeren J, Herkel J, Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice, J. Hepatol 62, 1349–1356 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Zheng M, Tian Z, Liver-Mediated Adaptive Immune ToleranceFront. Immunol (2019), 10.3389/fimmu.2019.02525. [DOI] [PMC free article] [PubMed]
- 68.Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y, Higher frequencies of GARP+CTLA-4+Foxp3+ t regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired t-cell functionality, Cancer Res (2013), 10.1158/0008-5472.CAN-12-3381. [DOI] [PMC free article] [PubMed]
- 69.Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, Sato E, Fukuoka S, Tada Y, Tanaka A, Morikawa H, Kawazoe A, Kinoshita T, Shitara K, Sakaguchi S, Nishikawa H, PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer, Proc. Natl. Acad. Sci. U. S. A (2019), 10.1073/pnas.1822001116. [DOI] [PMC free article] [PubMed]
- 70.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA, Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma, J. Exp. Med 210, 1695–1710 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grakoui A, Crispe IN, Presentation of hepatocellular antigens, Cell. Mol. Immunol 13, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Doherty DG, Immunity, tolerance and autoimmunity in the liver: A comprehensive review, J. Autoimmun 66, 60–75 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species, Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data., Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression, Genome Biol (2019), 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Mice with experimental liver metastasis have a normal liver metabolic function.
Fig. S2. Liver-mediated suppression of distant antitumor immunity requires adaptive immunity but not high tumor burden.
Fig. S3. Activation markers are decreased on effector CD4+ and CD8+ T cells but increased on Tregs in mice with experimental liver metastasis.
Fig. S4. The percentage and number of Tregs and tetramer+ CD8+ T cells are similar in distant tumors.
Fig. S5. Experimental liver metastasis mediated suppression in the B16F10 tumor model.
Fig. S6. KSPWFTTL-H-2Kb (KSP) tetramer specifically stains MC38 tumor-targeted CD8+ T cells.
Fig. S7. Suppression of distant CD8+ T cells is liver-mediated and tumor-antigen specific.
Fig. S8. Foxp3-DTR mouse model schema.
Fig. S9. DT induces profound Treg depletion in the blood and the subcutaneous tumor of treated Foxp3-DTR mice.
Fig. S10. Combination therapy with Treg-depleting anti-CTLA-4 and anti-PD-1 antibodies overcomes experimental liver metastasis immune suppression.
Fig. S11. Clustering of tumor-infiltrating immune cell subsets using scRNAseq.
Fig. S12. Differential gene expression in distant MDSCs driven by presence of liver tumor.
Fig. S13. Liver-tumor mediated suppression is associated with distant increase in CD11b+ monocyte populations.
Fig. S14. Increase in distant tolerogenic MDSCs is anatomically unique to liver tumor.
Fig. S15. Treg or MDSC depletion can enhance tumor rejection in mice with experimental liver metastasis.
Fig. S16. Treg-depleting versus non-depleting anti-CTLA-4 antibody in combination with anti-PD-1 treatment in experimental liver metastasis.
Table S1. Raw data table (Excel spreadsheet)