

Abstract
Bcr‐abl1 oncogene causes a shift in the transcription start site of the SMS1 gene (SGMS1) encoding the sphingomyelin (SM) synthesizing enzyme, sphingomyelin synthase 1 (SMS1). This results in an mRNA with a significantly shorter 5′‐UTR, called 7‐SGMS1, which is translated more efficiently than another transcript (IIb‐SGMS1) with a longer 5′UTR in Bcr‐abl1‐positive cells. Here, we determine the effects of these alternative 5′UTRs on SMS1 translation and investigate the key features underlying such regulation. First, the presence of the longer IIb 5′UTR is sufficient to greatly impair translation of a reporter gene. Deletion of the upstream open reading frame (−164 nt) or of the predicted stem‐loops in the 5′UTR of IIb‐SGMS1 has minimal effects on SGMS1 translation. Conversely, deletion of nucleotides −310 to −132 enhanced transcription of IIb‐SGMS1 to reach that of 7‐SGMS1. We thus suggest that regulatory features within nucleotides −310 and −132 modulate IIb‐SGMS1 translation efficiency.
Keywords: 5′ UTR, mRNA secondary structures, SGMS1, sphingomyelin synthase 1, translational regulation

Abbreviations
AvGFP, aequorea victoria GFP
CML, chronic myelogenous leukemia
DAG, diacylglycerol
DMEM, Dulbecco's modified Eagle's medium
GCS, glucosylceramide synthase
MNE, mean of normalized expression
SM, sphingomyelin
SMS1, sphingomyelin synthase 1
TGN, trans‐Golgi network
Sphingomyelin synthase 1 (SMS1) is one of two isoenzymes responsible for the synthesis of sphingomyelin (SM), an important structural component of cell membranes in mammals [1, 2]. In the plasma membrane, SM is a key component of lipid microdomains, and, together with cholesterol, it enables the homeostasis and signaling functions of these microdomains. Several membrane receptors have been found to segregate and to be activated within these SM‐rich microdomains, and loss or downregulation of SMS1, via reduction in SM, impaired their activation. For instance, modulation of SMS1 has been shown to regulate CD95 activation in murine lymphoid cells [3], TCR in Jurkat cells [4], murine CD41 in T cells [5], and BCR activation in B cells [6]. Furthermore, SM produced by SMS1 is critical for the attachment and cell entry of the Japanese encephalitis virus (JEV) [7] in lymphoma cells and SMS1 deficiency decreased clathrin‐dependent endocytosis of transferrin–transferrin receptor complexes [8].
In the course of the biochemical reaction to produce SM, SMS1 utilizes ceramide and phosphatidylcholine as substrates and produces diacylglycerol (DAG) in addition to SM [9]. Importantly, ceramide and DAG are both bioactive lipids, ceramide mostly associated with negative effects on proliferation and survival and DAG with prosurvival functions and as a regulator of secretion via the trans‐Golgi Network (TGN). Thus, the biological importance of SMS1 also resides in its ability to modulate such signaling lipids. Indeed, in mouse neuroblastoma cells, upregulation of SMS1 attenuates the apoptotic response to oxidative stress by reducing ceramide levels [10], while in Jurkat cells, overexpression of SMS1 increases resistance to photodamage‐induced apoptosis by decreasing the accumulation of ceramide [11, 12]. In the case of DAG, regulation of SMS1 at the Golgi (where the enzyme resides) affects the local pool of DAG impacting translocation and activation of the DAG‐responsive protein kinase D and altering TGN‐mediated trafficking and secretion [13, 14].
Given SMS1 regulates critical lipids such as SM, ceramide, and DAG, it is not surprising that loss of SMS1 in mice causes several adverse effects, such as defective insulin secretion, hearing loss, reduced growth, infertility and loss of function of the white adipose tissue [15, 16, 17, 18, 19].
In spite of the biological importance of SMS1, only very limited knowledge of the molecular mechanisms that regulate SMS1 is available. At the protein level, the homodimerization of SMS1 via its carboxy termini has been proposed to play a role in SMS1 transport from the ER to its final destination at the Golgi [20]. Once in the Golgi, a study also found that SMS1 heterodimerizes with another enzyme of the sphingolipid pathway, glucosylceramide synthase (GCS) via its N‐terminal tail; this interaction seems to increase SMS1 activity while decreasing GCS activity [21]. Furthermore, CD95 engagement in Jurkat cells caused inactivation of SMS1 via caspase‐mediated cleavage and SMS1 release from the Golgi into the cytoplasm [22].
At the gene level, SMS1 has been proposed to be regulated transcriptionally and post‐transcriptionally. The SMS1 gene (SGMS1) is comprised of 11 exons and is located in human chromosome 10. Many alternative mRNA transcripts were found, each differing in its combination of exons, length of their 5′‐UTR, and length of their 3′‐UTR [23]. In addition, it has been observed that the expression of each alternative transcript is tissue‐specific. Alternate intron polyadenylation and alternative splicing have been implicated to produce truncated SMS1 mRNA transcripts which do not result in the formation of the full‐length SMS1 protein [24, 25]. Other studies have found circular noncoding RNA (circRNA) within human, rat, and mouse brain tissue which contain sequences of 5′‐UTR and/or exonic portions of the SGMS1 gene. These SMS1 circRNAs were found to have high probability of binding microRNAs and are suggested to play a role in the regulation of SMS1 expression [26].
Prior research in our laboratory has defined the first oncogenic‐driven molecular mechanism for the regulation of SMS1. Using a cell model for chronic myelogenous leukemia (CML), it was shown that the Bcr‐abl 1 oncogene, etiological agent for CML, promoted SGMS1 expression and activity which in turn sustained proliferation of Bcr‐abl 1‐positive leukemic cells [27]. A recent follow‐up study demonstrated that the regulation of SMS1 in CML was driven by a novel mechanism of oncogenic‐mediated protein upregulation promoted by the presence of Bcr‐abl 1 [28]. Bcr‐Abl 1 caused an increase in SGMS1 transcription from a novel start site (TSS‐7) which resulted in an SGMS1 transcript with a significantly shorter 5′‐UTR (7‐SGMS1). We showed that the 7‐SGMS1 transcript was translated 10‐ to 15‐fold more efficiently than another less abundant transcript found in CML cells and characterized by a significantly longer 5′UTR (IIb‐SGMS1), which approximates the one found on canonical SGMS1 mRNA. We proposed that the presence of specific RNA features on the long 5′UTR of IIb‐SGMS1 could negatively impact translation and that the shorter 5′UTR was responsible for the enhanced rate of SMS1 translation of 7‐SGMS1.
In this study, we set to determine the contribution of the two 5′UTRs to translation of SMS1 and to investigate in detail the regulatory features potentially involved in such regulation.
Materials and methods
Cell culture
HeLa cells were purchased from the American Type Culture Collection (Rockville, MD, USA) and used between passages 3 and 10. Cells were grown in Dulbecco's modified Eagle's medium (DMEM) high glucose (4500 mg·L−1 glucose; Gibco/Thermo Fisher Scientific, Waltham, MA, USA), 10% FBS (heat inactivated FBS; Gibco/Thermo Fisher Scientific), 100 U·mL−1 penicillin, and 100 μg·mL−1 streptomycin (Gibco/Thermo Fisher Scientific) and maintained at 37 °C and 5% CO2. When passing and seeding for experiments, cells were routinely stained with trypan blue to assess viability; viability was usually higher than 95%. Cells were also routinely tested for mycoplasma contamination using the mycoAlert detection PCR kit from Lonza (Morristown, NJ, USA), and no contamination was ever detected.
Generation of mammalian expression constructs
All constructs and their characteristics are listed in Tables 1 and 2. GFP transcripts carrying SGMS1 5′UTR IIb (IIb‐GFP) or 7 (7‐GFP) [28] were synthesized by GenScript USA Inc. (Piscataway, NJ, USA) and cloned into the pUC57 plasmid flanked by sequences for the restriction enzymes BamH1 (at 5′) and Xba1 (at 3′). The GFP sequence from Aequorea Victoria (Av, Jellyfish, Takara Bio, Mountain View, CA, USA, formerly Clontech) was utilized. The GenScript transcripts IIb‐GFP and 7‐GFP were subcloned into pEF6 for mammalian expression using BamH1 and Xba1.
Table 1.
Progressive deletions of SGMS1 5′UTR. The first coding nucleotide is conventionally given the number 1 while the nucleotides in the 5′UTR are assigned negative numbers starting with −1 for the nucleotide 5′ to the first coding nucleotide.
| Constructs |
Number of deleted nucleotides (while maintaining TSS‐AAA) |
Nucleotides in the 5’UTR (including TSS‐AAA) |
|---|---|---|
| Transcript IIb‐SGMS1 | None (wild‐type) | −670/−1 |
| Transcript IIb Δ1–3 SGMS1 | (80 from 5′ of wild‐type IIb) | −590/−1 |
| Transcript IIb Δ1–6 SGMS1 | (78 from 5′ of 1–3 SGMS1) | −512/−1 |
| Transcript IIb Δ1–9 SGMS1 | (82 from 5′ of Δ1–6 SGMS1) | −430/−1 |
| Transcript IIb Δ1–10 SGMS1 | (117 from 5′ of Δ1–9 SGMS1) | −313/−1 |
| Transcript IIb Δ1–11 SGMS1 | (178 from 5′ of Δ1–10 SGMS1) | −135/−1 |
| Transcript 7 |
None (wild‐type) Initial nucleotides, CTT |
−135(CTT)/ −1 |
Table 2.
Mutations and short deletions of SGMS1.
| Constructs | Type of change |
|---|---|
| Transcript IIb ΔATG SGMS1 | Deletion of canonical ATG |
| Transcript IIb ΔuORF SGMS1 | Deletion of predicted uORF (−164/−162 nt) |
| Transcript IIb(AAA → CTT)‐SGMS1 | Substitution of first 3nt AAA with CTT |
All SGMS1 deletion/mutation constructs were obtained by PCR using the Phusion High‐fidelity DNA polymerase (NEB Inc. Ipswich, MA, USA) according to manufacturer's instructions and, as template, the previously characterized wild‐type transcript IIb‐SGMS1 [28]. A list of all primers' combinations can be found in Table 3 while the sequences for IIb‐SGMS1 and 7‐SGMS1 can be found in Table 4. All PCR‐generated deletions were flanked by BamH1 (at 5′) and Xba1 (at 3′) restriction sites, and they included a Flag tag sequence at the 3′. Purified PCR products were digested with BamH1 and Xba1 and ligated with digested pEF6 plasmid. Ligated constructs were sequenced by the DNA Sequencing Facility at Stony Brook University. All plasmids ultimately used for transfection of HeLa cells were purified using the EndoFree Plasmid Maxi Kit from Qiagen (Waltham, MA, USA).
Table 3.
List of primers.
| Primers | Sequence |
|---|---|
| Transcript IIb ΔATG SGMS1 |
Forward: 5′‐CTGCTGTCTGCCAGTACAAAGGAAGTGGTTTATTGG‐3′ Reverse: 5′‐CCAATAAACCACTTCCTTTGTACTGGCAGACAGCAG‐3′ |
| Transcript IIb ΔuORF SGMS1 |
Forward: 5′‐GAACCCTGGACAGCTACAGGTGTTTAAAAACTGC Reverse: 5′‐GCAGTTTTTAAACACCTGTAGCTGTCCAGGGTTC |
| Transcript IIb (AAA → CTT)‐SGMS1 |
Forward: 5′‐CCGAGCTCGGATCCCTTGCAGGAAGATGGTG‐3′ Reverse: 5′‐CACCATCTTCCTGCAAGGGATCCGAGCTCGG‐3′ |
| Transcript IIb Δ1–3 SGMS1 |
Forward: 5′‐CAATGGATCCAAATAAAGCTTCAGCGACTGAAG‐3′ Reverse: 5′‐CACTCTAGATTACTTGTCGTCATCGTCTTTGTAGTCTGTGTCATTCACCAGCC‐3′ |
| Transcript IIb Δ1–6 SGMS1 |
Forward: 5′‐CAATGGATCCAAAAATAAAATACAGATTGGAAA‐3′ Reverse: 5′‐CACTCTAGATTACTTGTCGTCATCGTCTTTGTAGTCTGTGTCATTCACCAGCC‐3′ |
| Transcript IIb Δ1–9 SGMS1 |
Forward: 5′‐CAATGGATCCAAACGAAAGTGTCTGGTTGGGAA‐3′ Reverse: 5′‐CACTCTAGATTACTTGTCGTCATCGTCTTTGTAGTCTGTGTCATTCACCAGCC‐3′ |
| Transcript IIb Δ1–10 SGMS1 |
Forward: 5′‐CAATGGATCCAAAGATGGAACTGTACTGGAAC‐3′ Reverse: 5′‐CACTCTAGATTACTTGTCGTCATCGTCTTTGTAGTCTGTGTCATTCACCAGCC‐3′ |
| Transcript IIb Δ1–11 SGMS1 |
Forward: 5′‐CAATGGATCCAAAGCCAAACAAGTCTCTGCTC‐3′ Reverse: 5′‐CACTCTAGATTACTTGTCGTCATCGTCTTTGTAGTCTGTGTCATTCACCAGCC‐3′ |
|
GFP (Jellyfish) qRT–PCR primers |
Forward: 5′‐GGAGAGGGTGAAGGTGATGC‐3′ Reverse: 5′‐ACCATAAGAGAAAGTAGTGACAAGTG‐3′ |
|
Human SGMS1 qRT–PCR primers |
Forward: 5′‐GCCAGGACTTGATCAACCTAACC‐3′ Reverse: 5′‐CCATTGGCATGGCCGTTCTTG‐3′ |
|
Human β‐Actin qRT–PCR primers |
Forward: 5′‐ATTGGCAATGAGCGGTTCC‐3′ Reverse: 5′‐GGTAGTTTCGTGGATGCCACA‐3′ |
Table 4.
Nucleotide sequence of IIb‐SGMS1 and 7‐SGMS1.
| IIb‐SGMS1. The underlined sequence represents the predicted uORF; in bold is the canonical ATG. Italic sequence before stop codon is the Flag tag. |
| AAAGCAGGAAGATGGTGAACTTTATGCAATTCTGAGTTGACTCCAATCCAGATGTATTTTGGAACTAGGTAGAATATGACTGGTAAAGCTTCAGCGACTGAAGGAGTGTGGACTTTTTGAATTTCAAGAACAGTAAAGTTGGAACACAGCAGAGGGTGTGTAATAAAATACAGATTGGAAAGAAGACTGATGAAGAAAAATAACAGCATGATTCAGGCACACCATTGAGTTCTTTGGGATGCCCGAAAGTGTCTGGTTGGGAAACATGAAATAGAAGACGAGAACCTGCCTCAGCAAGCGTACCTGAGAATACTTTTAAAGGAACCTGTTGATAAATGCAAAACAGTGGCTGTTGCAGGTGATGGAACTGTACTGGAACAATGGGAGAAGCTGGTAGCTTATAGGAAGCCAAGATGACCAGAATGTTTTTAAATAATCCAAGGAAGAATCCTGCTCAAAAATGAGGTGAATTAATACTTGGGCGCTCAGGAACCCTGGACAGCTACATGAGGTGTTTAAAAACTGCCCTGACCATCTTGCCAAACAAGTCTCTGCTCATGAGGCCCCACAGGATGAACGAGCCGAGGCAGGACAGTGTCCTGCCCTGTCGGAACAGTGACTGCTGACCTGCCAAGAGAGAGCTGGGGACTGCCTGCTGTCTGCCAGTACAATGAAGGAAGTGGTTTATTGGTCACCCAAGAAGGTGGCAGACTGGCTGCTGGAGAATGCTATGCCAGAATACTGTGAGCCTCTGGAGCATTTCACAGGCCAGGACTTGATCAACCTAACCCAAGAGGATTTCAAAAAACCCCCCTTGTGCCGAGTCTCCTCTGACAATGGGCAGCGGCTCCTGGACATGATAGAAACCCTGAAAATGGAGCACCATTTGGAAGCACACAAGAACGGCCATGCCAATGGGCACCTCAACATTGGCGTAGACATCCCCACCCCCGACGGCAGCTTCAGCATCAAGATTAAACCCAACGGGATGCCAAATGGGTATAGGAAAGAGATGATAAAGATCCCCATGCCAGAACTGGAGCGCTCTCAGTACCCCATGGAGTGGGGCAAGACTTTTCTGGCCTTTCTTTATGCACTTTCCTGTTTCGTTCTCACCACAGTGATGATCTCGGTCGTCCACGAACGAGTACCTCCTAAGGAGGTGCAGCCTCCACTACCGGACACATTTTTTGACCATTTTAACCGGGTGCAGTGGGCCTTTTCTATTTGTGAAATTAATGGCATGATCCTTGTAGGACTCTGGTTAATTCAGTGGCTGCTCTTAAAATACAAGTCTATTATTAGCAGAAGATTTTTCTGCATAGTTGGCACGCTGTACCTGTATCGGTGTATTACAATGTATGTAACTACACTCCCAGTACCTGGTATGCATTTCAACTGTTCTCCGAAGCTTTTCGGAGACTGGGAAGCCCAACTGCGAAGAATAATGAAGCTCATTGCTGGAGGTGGCTTGTCTATCACTGGCTCTCACAACATGTGTGGGGACTATCTGTACAGCGGCCACACGGTCATGCTAACACTTACCTACTTATTTATCAAAGAGTATTCCCCTCGGCGACTCTGGTGGTATCACTGGATTTGCTGGCTTCTCAGCGTAGTTGGAATCTTCTGTATTCTCTTAGCGCATGACCACTACACTGTGGACGTGGTGGTGGCATATTACATCACCACGAGACTCTTCTGGTGGTATCACACTATGGCCAATCAGCAAGTGCTAAAGGAAGCTTCCCAGATGAACCTCCTGGCCAGGGTGTGGTGGTACAGGCCATTTCAGTACTTTGAAAAGAATGTCCAAGGAATTGTACCTCGATCTTACCATTGGCCTTTCCCCTGGCCAGTAGTCCACCTCAGTAGGCAAGTTAAATACAGCCGGCTGGTGAATGACACAGACTACAAAGACGATGACGACAAGTAA |
| 7‐SGMS1. In bold is the canonical ATG. Italic sequence before stop codon is the Flag tag. |
| CTTGCCAAACAAGTCTCTGCTCATGAGGCCCCACAGGATGAACGAGCCGAGGCAGGACAGTGTCCTGCCCTGTCGGAACAGTGACTGCTGACCTGCCAAGAGAGAGCTGGGGACTGCCTGCTGTCTGCCAGTACAATGAAGGAAGTGGTTTATTGGTCACCCAAGAAGGTGGCAGACTGGCTGCTGGAGAATGCTATGCCAGAATACTGTGAGCCTCTGGAGCATTTCACAGGCCAGGACTTGATCAACCTAACCCAAGAGGATTTCAAAAAACCCCCCTTGTGCCGAGTCTCCTCTGACAATGGGCAGCGGCTCCTGGACATGATAGAAACCCTGAAAATGGAGCACCATTTGGAAGCACACAAGAACGGCCATGCCAATGGGCACCTCAACATTGGCGTAGACATCCCCACCCCCGACGGCAGCTTCAGCATCAAGATTAAACCCAACGGGATGCCAAATGGGTATAGGAAAGAGATGATAAAGATCCCCATGCCAGAACTGGAGCGCTCTCAGTACCCCATGGAGTGGGGCAAGACTTTTCTGGCCTTTCTTTATGCACTTTCCTGTTTCGTTCTCACCACAGTGATGATCTCGGTCGTCCACGAACGAGTACCTCCTAAGGAGGTGCAGCCTCCACTACCGGACACATTTTTTGACCATTTTAACCGGGTGCAGTGGGCCTTTTCTATTTGTGAAATTAATGGCATGATCCTTGTAGGACTCTGGTTAATTCAGTGGCTGCTCTTAAAATACAAGTCTATTATTAGCAGAAGATTTTTCTGCATAGTTGGCACGCTGTACCTGTATCGGTGTATTACAATGTATGTAACTACACTCCCAGTACCTGGTATGCATTTCAACTGTTCTCCGAAGCTTTTCGGAGACTGGGAAGCCCAACTGCGAAGAATAATGAAGCTCATTGCTGGAGGTGGCTTGTCTATCACTGGCTCTCACAACATGTGTGGGGACTATCTGTACAGCGGCCACACGGTCATGCTAACACTTACCTACTTATTTATCAAAGAGTATTCCCCTCGGCGACTCTGGTGGTATCACTGGATTTGCTGGCTTCTCAGCGTAGTTGGAATCTTCTGTATTCTCTTAGCGCATGACCACTACACTGTGGACGTGGTGGTGGCATATTACATCACCACGAGACTCTTCTGGTGGTATCACACTATGGCCAATCAGCAAGTGCTAAAGGAAGCTTCCCAGATGAACCTCCTGGCCAGGGTGTGGTGGTACAGGCCATTTCAGTACTTTGAAAAGAATGTCCAAGGAATTGTACCTCGATCTTACCATTGGCCTTTCCCCTGGCCAGTAGTCCACCTCAGTAGGCAAGTTAAATACAGCCGGCTGGTGAATGACACAGACTACAAAGACGATGACGACAAGTAA |
Transfection of HeLa cells
On day 1, HeLa cells were plated in 6‐wells dishes at a concentration of 0.1 million cells per well in 3 mL of growth medium. On day 2, medium was changed with 2.8 mL per well of DMEM high glucose containing 10% FBS and no antibiotics, and cells in each well were transfected with a mix of 6 μL of X‐tremeGene 9 DNA transfection reagent (Roche/Sigma‐Aldrich , Indianapolis, IA, USA/St. Louis, MO, USA), 2 μg of plasmid DNA and OptiMEM (Gibco/Thermo Fisher Scientific) in a total volume of 200 μL. At the indicated times, cells were collected either for RNA isolation or protein analysis.
RNA isolation and cDNA synthesis
To analyze RNA expression, total RNA was first purified using the RNeasy mini kit from Qiagen. Briefly, the medium was removed, cells from one well were scraped with 350 μL of RLT buffer containing 2‐βmercaptoethanol (Sigma‐Aldrich), and total RNA was isolated using the RNeasy Mini Kit according to the manufacturer's protocol, including the on‐column DNase digestion (DNAse kit was from Qiagen). cDNA was prepared by using a two‐step method. First, an RNA mix was prepared with 1 μg of total RNA, 1 μL of Oligo (dT)20 primer (50 μm; Thermo Fisher Scientific), and 1 μL of 10 mm dNTP (Thermo Fisher Scientific) mixed to a final volume of 13 μL with sterile water and incubated at 65 °C for 5 min. Next, a second master mix of 4 μL of 5× first strand buffer, 2 μL of 0.1 m DTT, and 1 μL each of RNase Out and Superscript III (Thermo Fisher Scientific) was added to the RNA mix. The 20‐μL reaction was then incubated at 42 °C for 52 min and then at 70 °C for 15 min.
Quantitative RT–PCR (qRT–PCR)
To evaluate mRNA expression, we performed qRT–PCR using the SYBR green mixture (Bio‐Rad, Hercules, CA, USA) and primers probing either aequorea victoria GFP (AvGFP) or targeting exon 7 for SGMS1 and β‐actin (primer sequences are reported in Table 3). Measurements were performed on an Applied Biosystems 7500 Real‐time PCR system (Thermo Fisher Scientific). The following cycling conditions were used: one cycle of 3 min at 95 °C, 40 cycles of 10 s at 95 °C, and 45 s at 61 °C. Amplification efficiency for each primer pair was assessed as being between 90% and 100% across a range of cDNA concentrations. Results were normalized to the β‐actin internal control gene and amplification efficiency. qRT–PCR results were analyzed using q‐gene software [29] as the mean of normalized expression (MNE).
Western blotting
Cells from one well were scraped in 100 μL 0.75% SDS and tip‐sonicated for 20 s. An aliquot of the lysate was used for protein determination (BCA, Thermo Fisher Pierce Biotechnology, Waltham, MA, USA) while the rest was combined with 6X sample buffer and boiled for 7 min. Equal amount of proteins (10 μg) was run on an 10% SDS/PAGE under reducing conditions and transferred to nitrocellulose membranes (Bio‐Rad). Membranes were blocked with 5% milk in PBS‐Tween 0.1% for 1 h at room temperature. After washing, membranes were then incubated overnight at 4 °C in 2% BSA in PBS‐Tween 0.1% containing Sms1 antibodies raised against the full‐length protein (1 : 1000, rabbit polyclonal, Ex‐ alpha Biologics, Maynard, MA, USA) or antibodies against AvGFP (1 : 1000 dilution, D5.1 XP rabbit monoclonal; Cell Signaling Technology, Danvers, MA, USA) or β‐actin antibodies (I‐19, 1 : 2500 dilution; Santa Cruz Biotechnology, Dallas, TX, USA). After extensive washing, all membranes were incubated with peroxidase‐conjugated secondary antibodies in 5% milk PBS‐Tween 0.1% for 1 h at room temperature (for sms1 and AvGFP membranes: anti‐rabbit, 1 : 5000, Jackson ImmunoResearch Laboratories, Inc, West Grove, PA, USA; for actin membranes: anti‐goat, 1 : 5000, Jackson ImmunoResearch Laboratories, Inc). Signals were visualized using Super Signal (Pierce Chemical Co.) and exposure to Kodak BioMax MR Film (Eastman Kodak Co., Rochester, NY, USA).
Translation efficiency
Translation efficiency of SMS1 was calculated as previously reported [28] by dividing the normalized western blot intensity of SMS1 over MNE of SGMS1 mRNA. SMS1 western blot band intensities were normalized against corresponding β‐actin. Western blot band intensity was measured by using ImageJ (National Institutes of Health, Bethesda, MD, USA) [30]. Background intensity of SMS1 protein and mRNA expression in the empty pEF6 vector group was subtracted from all other experimental groups. For normalization across different experiments, western blotting for SMS1 and actin from all IIb samples (all experiments) were run in the same gel and SMS1/actin levels from each IIb sample were used as reference for quantitation of all other samples in the corresponding experiment.
Statistical analysis
Statistical analysis was performed using graphpad prism 8 software, San Diego, CA, USA. Translation efficiencies were compared to see whether there were statistically significant differences between groups as mentioned below. Figures 1 and 2 were analyzed using one‐tailed unpaired t‐test with Welch's correction. P values < 0.05 were considered significant. Figures 3 and 4 were analyzed using Brown–Forsythe and Welch's ANOVA tests with Dunnett T3 comparisons. P values < 0.05 were considered significant.
Fig. 1.
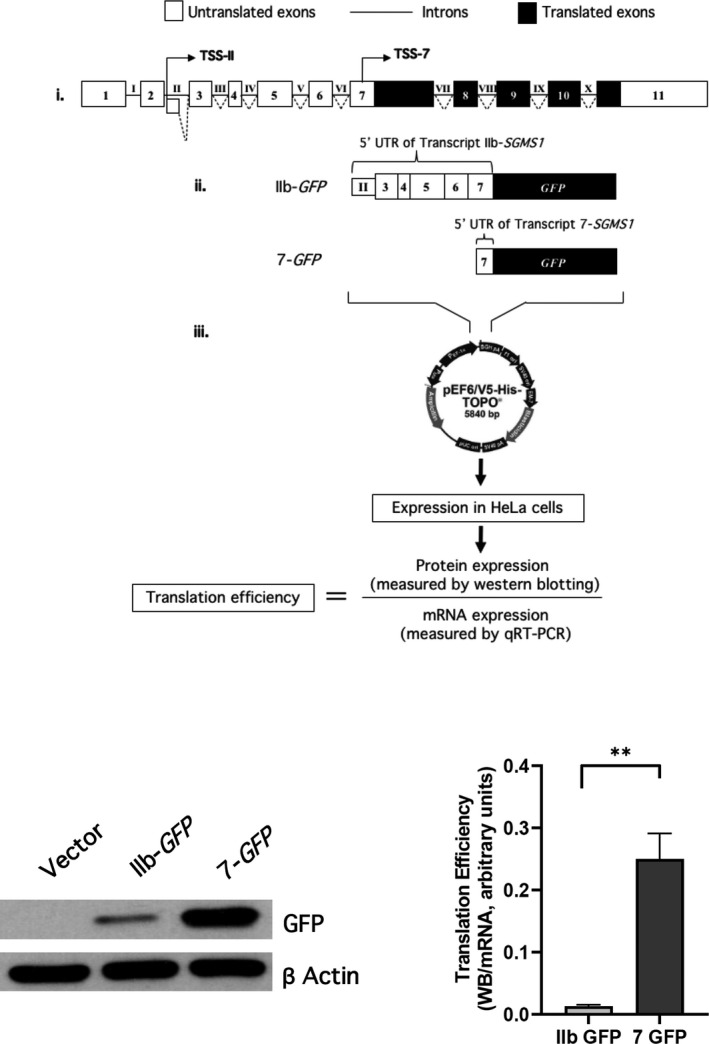
The 5′UTR of SM synthase 1 gene (SGMS1) regulates SMS1 translation. (A) Schematic of the experimental design. (i) Schematic representation of canonical SGMS1 locus (NM_147156.4). White boxes indicate untranslated exons; black boxes indicate translated exons; lines indicate introns. Two main TSSs have been confirmed in CLM cells, and they are in intron II (TSS‐II) and in exon 7 (TSS‐7), respectively [28]. (ii) Schematic representation of the GFP constructs carrying the 5′UTRs starting at TSS‐IIB and TSS‐7, respectively. The 5′UTR associated with TSS‐IIb includes a portion of intron II and annotated exons 3 to exon 7 and counts a total of 670 nucleotides. The 5′UTR associated with exon 7 counts 135 nucleotides upstream of the coding ATG. (iii) Calculation of translation efficiency. The two GFP clones were inserted in the pEF6/V5‐His‐TOPO mammalian expression plasmid and transfected into HeLa cells. Transfection conditions are described in the Materials and methods. Translation efficiency was calculated by quantification of the amount of produced SMS1 protein after 18 h of transfection, quantified by western blotting, and normalized against SGMS1 mRNA levels, as previously described [28]. (B, C) HeLa cells were plated and transfected as described in the Materials and methods. After 18 h from transfection, cells were collected with 1% SDS for western blotting or with RLT buffer for mRNA extraction. Samples were processed as indicated in the Materials and methods. (B) Effect of different 5′UTR on GFP protein levels. Representative western blot of GFP and β‐actin levels at 18 h of transfection of either pEF6 empty vector (vector), IIb GFP, or 7‐GFP in HeLa cells. Full images of western blots for GFP and actin are shown in Fig. S1A. (C) Translation efficiency of IIb‐GFP and 7‐GFP. Translation efficiency of the two constructs at 18 h after transfection was calculated as described above. The results represent the mean ± SD of four independent experiments; **P < 0.005. Quantification of normalized western blot and mRNA levels are provided in Fig. S1.
Fig. 2.
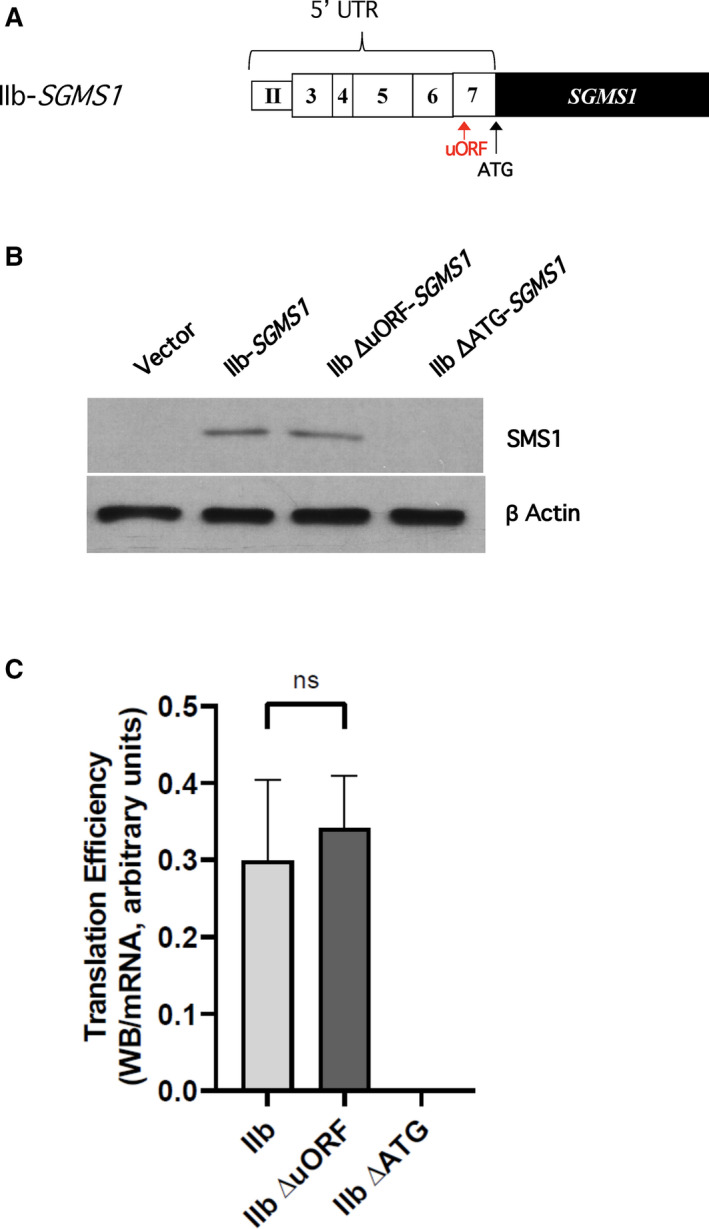
The uORF in the 5′UTR of IIb‐SGMS1 does not exert a significant role in inhibiting translation efficiency of SMS1. (A) Schematic representation to indicate the position of the predicted uORF and coding ATG. Computational analysis of the 5′UTR of IIb‐SGMS1 performed using uORFdb indicated the presence of a putative uORF at position −164 nt from the coding ATG [28]. (B, C) HeLa cells were plated and transfected as described in the Materials and methods. After 18 h from transfection, cells were collected with 1% SDS for western blotting or with RLT buffer for mRNA extraction. Samples were processed as indicated in the Materials and methods. (B) Effect of deletion of uORF or coding ATG on SMS1 levels. Representative results for western blot of SMS1 and actin levels in HeLa cells after 18 h of transfection with expression constructs pEF6 empty vector (vector), wild‐type IIb‐SGMS1, deleted uORF (IIb ΔuORF‐SGMS1), or deleted coding ATG (IIb ΔATG‐SGMS1). Full images of western blots for SMS1 and actin are shown in Fig. S2A. (C) SMS1 translation efficiency of constructs with deleted uORF or canonical ATG. Translation efficiency of SMS1 was expressed as the ratio of western blot normalized intensity of SMS1 over normalized SGMS1 mRNA expressed as MNE. The results represent the mean ± SD of 3–15 independent experiments; ns, not significant. Quantification of normalized western blot and mRNA levels are provided in Fig. S2.
Fig. 3.
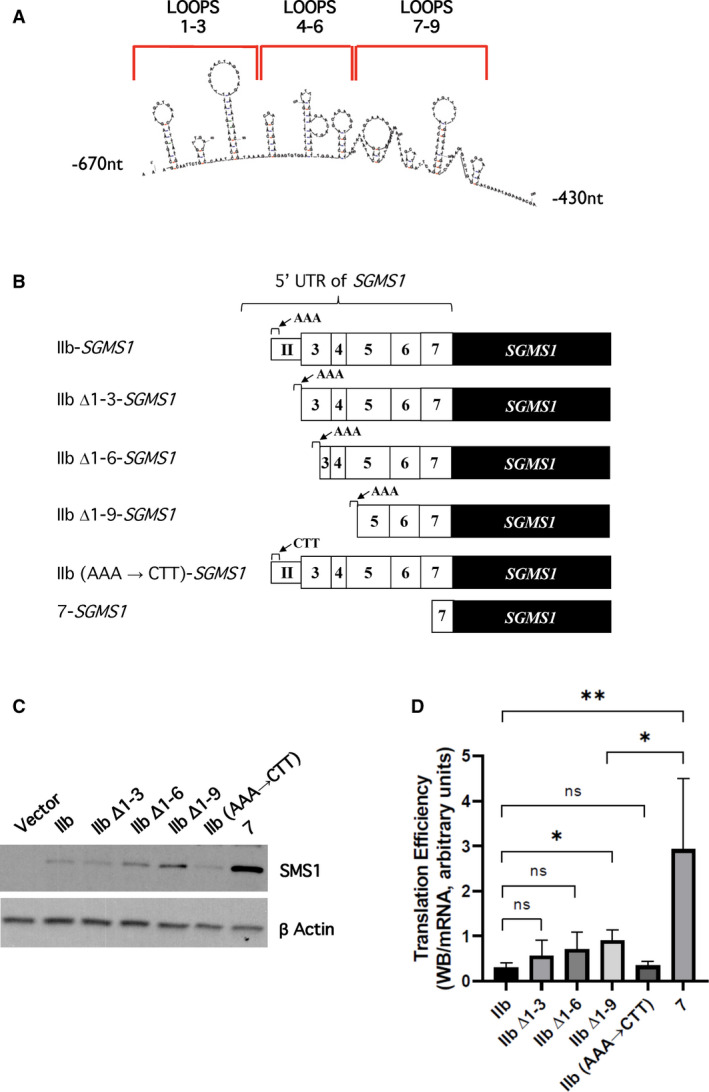
Contribution of putative stem‐loops in the first 80 nucleotides of IIb‐SGMS1 5′UTR to SMS1 translation efficiency. (A) Prediction of secondary structures. A maximum of nine hairpin loops were predicted by mFold in the mRNA stretch containing the first 83 nucleotides of the 5′UTR of IIb‐SGMS1 transcript. (B) Schematic representation of the IIb‐SGMS1 stem‐loop deletion constructs and mutants. A series of progressive deletions of the 5′UTR of IIb‐SGMS1 were obtained to assess the effect of the predicted stem‐loops on translation efficiency. A detailed summary of all deletions and mutations can be found in Tables 1 and 2. The predicted stem‐loops were progressively deleted in groups of 3, generating the constructs IIb Δ1–3‐SGMS1, IIb Δ1–6‐SGMS1, IIb Δ1–9‐SGMS1. In addition, a construct was generated in which the first 3 nucleotides at IIb TSS (AAA) were substituted with those found at TSS of 7 (CTT) generating the construct [IIb (AAA → CTT)‐SGMS1]. (C, D) HeLa cells were plated and transfected as described in the Materials and methods. After 18 h from transfection, cells were collected with 1% SDS for western blotting or with RLT buffer for mRNA extraction. Samples were processed as indicated in the Materials and methods. (C) Effect of stem‐loop deletions and mutations on SMS1 levels. Representative results from western blot of SMS1 and actin levels in HeLa cells after 18 h of transfection with expression constructs: pEF6 empty vector (vector), IIb‐SGMS1, IIb Δ1–3‐SGMS1, IIb Δ1–6‐SGMS1, IIb Δ1–9‐SGMS1, IIb (AAA → CTT)‐SGMS1, and 7‐SGMS1. Full images of western blots for SMS1 and actin are shown in Fig. S3A. D) Effect of stem‐loop deletions and mutations on SMS1 translation efficiency. Translation efficiency of SMS1 was expressed as the ratio of western blot normalized intensity of SMS1 over normalized SGMS1 mRNA expressed as MNE. The results represent the mean ± SD of 3–15 independent experiments; *P < 0.05, **P < 0.005, ns, not significant. Quantification of normalized western blot and mRNA levels is provided in Fig. S3.
Fig. 4.
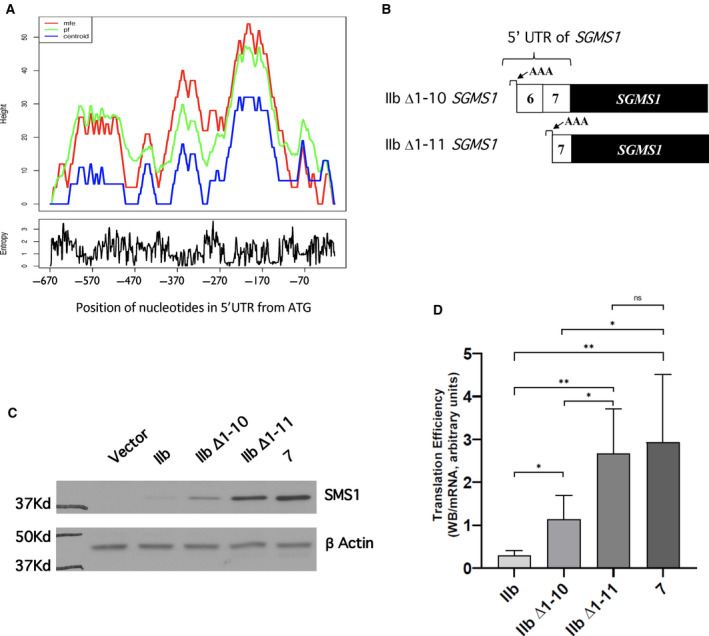
Contribution of additional areas of complexity of IIb‐SGMS1 5′UTR downstream of the stem‐loops to SMS1 translation efficiency. (A) Prediction of secondary structures by RNAFold. The full IIb‐SGMS1 5′UTR (−670 to −1) was analyzed with RNAFold (http://rna.tbi.univie.ac.at/cgi‐bin/RNAWebSuite/RNAfold.cgi). Predictions were generated using three different parameters: the mfe (red line), the centroid secondary structure (centroid, blue line), and the Pf (green line). Two areas of complexity downstream of the nine stem‐loops were predicted spanning nucleotides −400 to −310 and nucleotides −310 to −110, respectively. (B) Schematic representation of the deletion constructs based on RNAFold analysis. Two additional progressive deletions of the 5′UTR of IIb‐SGMS1 were generated to assess the effect of the predicted areas of complexity downstream of the nine stem‐loops on SMS1 translation efficiency (for details, see Table 1). The new deletion constructs were IIb Δ1–10‐SGMS1 (missing nucleotides up to −313) and IIb Δ1–11‐SGMS1 (missing nucleotides up to −135). (C, D) HeLa cells were plated and transfected as described in the Materials and methods. After 18 h from transfection, cells were collected with 1% SDS for western blotting or with RLT buffer for mRNA extraction. Samples were processed as indicated in the Materials and methods. (C) Effect of IIb Δ1–10‐SGMS1 and IIb Δ1–11‐SGMS1 deletions and mutations on SMS1 levels. Representative results from western blot of SMS1 and actin levels in HeLa cells after 18 h of transfection with expression constructs: pEF6 empty vector (vector), IIb‐SGMS1, IIb Δ1–10‐SGMS1, IIb Δ1–11‐SGMS1, and 7‐SGMS1. Full images of western blots for SMS1 and actin are shown in Fig. S4A. (D) Effect of IIb Δ1–10‐SGMS1 and IIb Δ1–11‐SGMS1 deletions on SMS1 translation efficiency. Translation efficiency of SMS1 was expressed as the ratio of western blot normalized intensity of SMS1 over normalized SGMS1 mRNA expressed as MNE. The results represent the mean ± SD of 9–15 independent experiments. *P < 0.05, **P < 0.005, ns, not significant. Quantification of normalized western blot and mRNA levels is provided in Fig. S4.
Results and Discussion
We previously showed that, in CML cells, SGMS1 is mostly represented by two different mRNA transcripts (IIb‐SGMS1 and 7‐SGMS1), associated with dramatically different translation efficiencies [28]. A main difference between the two transcripts is their 5′UTRs resulting from different transcription start sites (TSSs). IIb‐SGMS1 is transcribed from TSS‐II located within intron II of the annotated SGMS1 gene, and 7‐SGMS1 is transcribed from TSS‐7 located within exon 7 (Fig. 1A,i) [28]. IIb‐SGMS1 has a 5′UTR of 670 nt while 7‐SGMS1 has a 5′UTR of 135 nt (Fig. 1A,ii; Table 1). Here, we wanted to determine the extent of the contribution of the two 5′UTRs to the different translation efficiency. To this aim, we cloned each 5′UTR upstream of a GFP reporter gene and expressed the constructs into HeLa cells. As previously published [28], the efficiency of translation was calculated by the ratio of protein produced over the mRNA (Fig. 1A,iv). Preliminary experiments verified that at 18 h following transfection, the GFP translation rate was linear, and thus, 18 h were chosen as end point for calculations. Additionally, results from qRT–PCR showed similar level of expression of transfected plasmids between the two constructs, as comparable levels of mRNAs were measured (Fig. S1B).
As shown in Fig. 1B, there was a dramatic difference in translation depending on whether the GFP mRNA carried IIb or 7 at the 5′UTR. The difference in translation between the two constructs (Fig. 1C) recapitulated the 10‐ to 15‐fold higher efficiency of SGMS1 when transcribed from TSS‐7 and carrying the shorter 5′UTR [28]. These results indicate that the presence of the longer IIb 5′UTR is sufficient to greatly impair translation.
Next, we wanted to identify the molecular features responsible for such effect. Several features on an mRNA can reduce its translation efficiency [31]. The presence of upstream open reading frames (uORFs) is one of them [32]. uORFs are short, peptide‐coding sequences which contain start and stop codons in frame. They are generally thought to constitutively suppress or transiently hinder translation by halting the scanning of the mRNA by the 43S preinitiation complex [33, 34] or favoring ribosome dissociation from the mRNA [35, 36]. Additionally, they can position the scanning machine out‐of‐frame and thus cause it to bypass the translation start of the coding sequence [37]. However, in some instances, they have also been described to enhance translation possibly by favoring the retention of initiation complexes and accelerating translation of subsequent primary ORFs [33, 34]. We have previously predicted that the IIb‐SGMS1 transcript has one uORF in position −164 nt from the proposed canonical ATG [28]. Thus, we proceeded to delete the sequence directly in the IIb 5′UTR (Fig. 2A, Table 2) and assess the effect on SGMS1 translation. In this and all following experiments, we utilized SMS1 as read‐out (instead of GFP) to also include functionally relevant regions potentially present in the SGMS1 cDNA. As shown in Fig. 2B,C, deletion of the putative uORF did not exert any significant effect on the translation efficiency of SGMS1. Optimal experimental conditions to assess impact on translation efficiency (i.e., appropriate transfection conditions and time points) were previously determined [28] and followed in these experiments. As positive control, we deleted the ATG associated with the primary coding sequence and located within exon 7 (Fig. 2A, Table 2), and, as expected, a complete loss of translation was observed (Fig. 2B,C) [1]. All together these results indicate that the uORF (−164 nt) does not affect translation of SGMS1.
Next, the presence and effect of mRNA secondary structures on the IIb‐SGMS1 5′UTR was investigated. RNA molecules fold on themselves assuming complex secondary and tertiary structures composed by simple modules: basepairs and unpaired nucleotides, that, when combined, make stem‐loops. While specific RNA complex structures are sites of recognition for interacting partners (proteins, small molecules, or other RNA molecules), the actual binding mostly occurs at sites of low complexity where the RNA is not paired [38]. Thus, the identification of secondary structures is an important tool for understanding the regulation of specific RNA molecules. Importantly, secondary structures need to be unraveled before the mRNA can be translated. As the 43S preinitiation complex scans the RNA from the 5′CAP, the action of the RNA helicase eIF4 in the eIF4F complex unwinds secondary structures allowing the binding of the 60S large ribosomal subunits to optimal start codons. It is therefore generally accepted that the presence of stem‐loop (or hairpin) structures negatively affects translation by making this process heavily dependent on the eIF4. On the other hand, the presence of stem‐loop structures within the coding sequence might actually favor its translation [39].
Prediction of RNA secondary structures can be made computationally; however, prediction softwares are limited as they are based on specific criteria such as selective inclusion of canonical base pairs and of the G‐U wobble pair; also they do not consider the formation of pseudoknots in the RNA and they compute on the assumption that the more likely natural RNA structure is the one with the minimum free energy (mfe). Even with these limitations, they represent a useful tool to start dissecting structure–function relationships. The analysis of the 5′UTR of IIb‐SGMS1 using the mFold software generated different prediction models for the presence of stem‐loops in the first 200 nucleotides; the maximum number of predicted stem‐loops was 9 (Fig. 3A). Thus, we determined whether the sequences associated with these predicted loops contributed to the translation efficiency of the transcript. The loops were progressively deleted in groups of three from the 5′UTR (Δ1–3, Δ1–6, Δ1–9), and all the constructs retained the first 3 nucleotides of IIb‐SGMS1 (AAA; Fig. 3B and Table 1). As shown in Fig. 3C,D, while there is a tendency for each progressive deletion to increase translation efficiency, only the loss of all 9 loops shows a significant translation gain. It is to be noted that deleting all 9 loops increased overall translation only by about 2‐fold, suggesting that most of the translational regulation is still retained (Fig. 3C).
Additionally, we also tested whether the TSS of IIb‐SGMS1 (AAA) might play a role in inhibiting translation efficiency (Table 1); however, as shown in Fig. 3C,D, the substitution of these nucleotides with the TSS of the 7‐SGMS1 transcript (CTT) did not affect translation.
Altogether, these results indicate that mechanisms of regulation that affect translation of SGMS1 in the IIb‐SGMS1 for the most part are still retained in the 400 nucleotides closest to the coding ATG (−400 to −1nt).
Interestingly, analysis of the full IIb‐SGMS1 5′UTR with RNAFold (http://rna.tbi.univie.ac.at/cgi‐bin/RNAWebSuite/RNAfold.cgi; Fig. 4A) suggested the presence of two areas of complexity in the region, one spanning nucleotides −400 to −310 and one spanning nucleotides −310 to −110. Predictions were generated using three different parameters: the mfe, centroid secondary structure (centroid) and partition function (Pf). Mfe generated the red plot indicating the mRNA structure with the lowest free energy and therefore expected to be the most likely to occur in nature. The centroid secondary structure (blue line) indicates the structure with the minimum total basepair distance among the structures in the set. The Pf (green line) calculates the probability for a certain base pair to form. In the mountain plot in Fig. 4A, the height of each nucleotide indicates the number of bases enclosing that specific base; in other words, peaks indicate stem‐loops, plateaus indicate single strand portions or loops, and slopes indicate helices. The three plots showed a similar pattern, reinforcing the confidence of these general predictions.
Based on these predicted areas of complexity and considering that the 5′UTR of 7‐SGMS1 starts at −135 nt (Fig. 4B and Table 1), we generated two additional deletion constructs: one where the 5′UTR started at position −310 (Δ1–10) of IIb‐SGMS1 and one starting at −132nt (Δ1–11); both 5′UTRs contained the first three nucleotides (AAA) found in IIb‐SGMS1 (Fig. 4B and Table 1). Of note, deletion construct Δ11 has therefore the same 5′UTR of 7‐SGMS1 except for the initial AAA in place of CTT present in the 7‐SGMS1. As shown in Fig. 4C,D, Δ1–10 IIb‐SGMS1 showed a two‐fold gain of translation compared to full‐length IIb‐SGMS1, and thus was similar to Δ1–9 IIb‐SGMS1 (Fig. 3D). These results indicate that deletion of nucleotides −427 to −311 has no significant additional gain in translation efficiency. On the other hand, deletion of the next 178nt (Δ1–11 IIb‐SGMS1) significantly enhanced translation efficiency, reaching the levels observed with 7‐SGMS1. These results indicate that a major area of regulation resides is the 5′UTR sequence between nucleotides −310 and −132.
Interestingly, even though no distinctive cis‐regulatory sequences (such as Rfam, ERPIN, similar functional RNA sequences, 5′ terminal oligopyrimidine (TOP) motif; pyrimidine‐rich translational element, cytosine‐enriched regulator of translation, the G‐quadruplex structure, eukaryotic initiation factor 3‐binding stem‐loop structure) were identified on the full‐length 5′UTR of IIb‐SGMS1 by multiple prediction software's analysis using RegRNA2.0 (http://regrna2.mbc.nctu.edu.tw) revealed a putative binding sequence for miR‐4712‐5p at position −315 nt to −294 nt. miRs are thought to impact translation possibly by binding at or in the vicinity of the ribosome‐binding site, thereby directly competing with the 30S ribosomal subunit for binding to the mRNA. Interestingly, the putative miR binding sequence is absent in the Δ1–11 IIb‐SGMS1, where the translation efficiency is restored to the levels of 7‐SGMS1. Therefore, the presence of the miR‐4712‐5p binding sequence on the IIb‐SGMS1 and the gain in translation efficiency when the sequence containing this particular stretch of nucleotides is missing (Fig. 4D) raises the interesting possibility that the regulation of IIb‐SGMS1 translation could occur, at least in part, via this miR. Of note, another miR sequence (has‐miR‐1208) was predicted to bind from nucleotide −71 to −52, indicating an additional potential site of regulation within the 5′UTR of 7‐SGMS1. In agreement with the potential regulation of SGMS1 by miRs in this area, another study has found a particularly high number of potential miR binding sequences in exons 5, 6, and 7 corresponding to the sequence retained in the 5′UTR of Δ1–10 IIb‐SGMS1 (Fig. 4B) [26]. Additionally, we compared the sequence of IIb‐SGMS1 starting within exon 5 and spanning exon 6 and the first portion of exon 7 across different vertebrates (a total of 360 nt, from −404 to −44 of IIb‐SGMS1, which included the two predicted miR binding sites; Fig. S5). Interestingly, the alignment showed good homology among the different organisms considered (Homo sapiens, Macaca mulatta, Bos taurus, Rattus norvegicus, and Mus musculus) for both putative binding sequences (as well as the overall 360 nt sequence). This further supports the potential regulatory importance of this portion of SGMS1 across different species.
Human SGMS1 mRNA (NM_147156.4) annotated in the NCBI database carries a 5′UTR of 933 nt (Fig. 5A); however, as previously reported, this variant is not present in CML cells [28]. Interestingly, the prediction of secondary structures by RNAFold for this longer annotated SGMS1 variant yields a significantly different profile from IIb‐SGMS1 (Fig. 4A vs Fig. 5B). It is worth noting that, while the two SGMS1 transcripts share nucleotides −588 to −1, the plots of the predicted secondary structures for this region are quite different between the two variants. This indicates that the presence of annotated exon 1 and exon 2 in NM_147156.4 might have a significant impact on the complexity of the overall sequence and possibly on the regulation of translation.
Fig. 5.
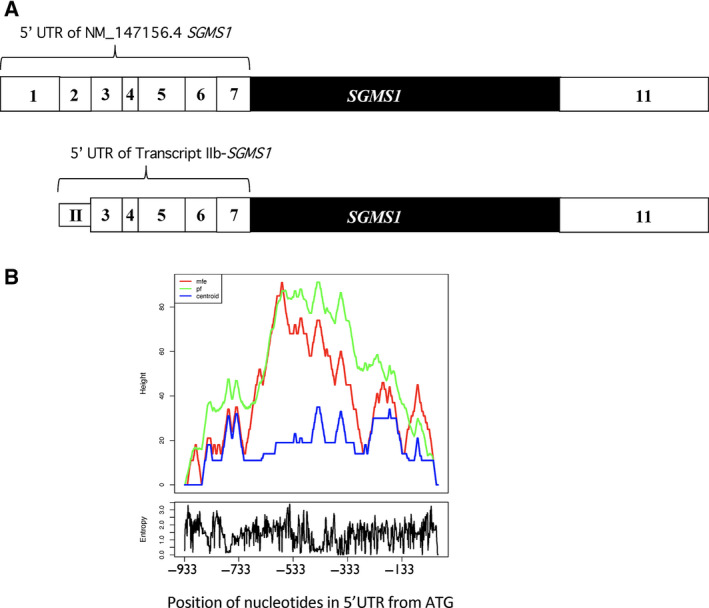
Prediction of secondary structure of the SGMS1 gene annotated in NCBI (NM_147156.4) by RNAFold. (A) Comparison of the schematic representation of canonical SGMS1 transcript (NM_147156.4) vs IIb‐SGMS1. White boxes indicate untranslated exons, and black boxes indicate translated exons. The 5′UTR of NM_147156.4 counts 933 nucleotides (−933 to −1) compared to 670 nt of IIb‐SGMS1. The two 5′UTRs share nucleotides −588 to −1, while they differ in their initial nucleotides. (B) Prediction of secondary structures of canonical SGMS1 by RNAFold. The 5′UTR of canonical SGMS1 was analyzed with RNAFold (http://rna.tbi.univie.ac.at/cgi‐bin/RNAWebSuite/RNAfold.cgi). Predictions were generated using the mfe (red line), centroid secondary structure (centroid, blue line), and Pf (green line).
Analysis of the overall distribution of the length of human 5′UTRs (cage ≥ 5) performed by UTRdb (http://www.ba.itb.cnr.it) reveals that the vast majority of 5′UTRs has a length spanning from 50 to 200 nt, with the highest representation having 5′UTRs of 100 to 150 nt. mRNAs with 5′UTRs of more than 600 nt are a small minority. Based on the CART model for the analysis of 5′UTRs [40], the length of the 5′UTR is a strong predictor of poor translation, together with the mFold prediction of secondary structure and the presence of 5′ TOP motifs. Importantly, regulatory proteins tend to have long and/or more structurally complex 5′UTRs, and 5′UTRs of 670 or 933 nt as those for IIb‐SGMS1 and canonical SGMS1, potentially place SGMS1 in this category. On the other hand, our mutational analysis for IIb‐SGMS1 indicates that the most consequential regulatory features of this transcript variant reside within the first 310 nt from the canonical ATG, further supporting the importance of potential secondary structures and other features that might modulate translation efficiency in addition to the length of the 5′UTR.
Author contributions
FD and NA performed experiments, analyzed data and contributed to portions of the manuscript; BSE designed and performed experiments and analyzed data; GQY and GL performed experiments; SM designed experiments and analyzed data; CL designed experiments, analyzed data and wrote the manuscript.
Supporting information
Fig. S1. Quantitation of GFP protein and mRNA levels in transfected HeLa cells.
Fig. S2. Quantitation of SMS1 protein and SGMS1 mRNA levels in HeLa cells transfected with SGMS1 deleted in the uORF or in the coding ATG.
Fig. S3. Quantitation of SMS1 protein and SGMS1 mRNA levels in HeLa cells transfected with SGMS1 lacking the first predicted stem loops.
Fig. S4. Quantitation of SMS1 protein and SGMS1 mRNA levels in HeLa cells transfected with different SGMS1 deletion mutants of nucleotides up to −135 of the IIb 5′UTR.
Fig. S5. Nucleotide alignment of the putative regulatory portion of IIb‐SGMS1 across different vertebrates.
Legend
Acknowledgements
The authors thank the Stony Brook DNA Sequencing facility for timely assistance. This work was supported by U.S. National Institutes of Health, National Cancer Institute Grant P01‐CA097132 (to CL for Project #4).
Edited by Giovanni D'Angelo
References
- 1. Huitema K, van den Dikkenberg J Brouwers JFHM and Holthuis JCM (2004) Identification of a family of animal sphingomyelin synthases. EMBO J 23, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamaoka S, Miyaji M, Kitano T, Umehara H and Okazaki T (2004) Expression cloning of a human cDNA restoring sphingomyelin synthesis and cell growth in sphingomyelin synthase‐defective lymphoid cells. J Biol Chem 279, 18688–18693. [DOI] [PubMed] [Google Scholar]
- 3. Miyaji M, Jin Z‐X, Yamaoka S, Amakawa R, Fukuhara S, Sato SB, Kobayashi T, Domae N, Mimori T, Bloom ET et al (2005) Role of membrane sphingomyelin and ceramide in platform formation for Fas‐mediated apoptosis. J Exp Med 202, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jin ZX, Huang C‐R, Dong L, Goda S, Kawanami T, Sawaki T, Sakai T, Tong X‐P, Masaki Y, Fukushima T et al (2008) Impaired TCR signaling through dysfunction of lipid rafts in sphingomyelin synthase 1 (SMS1)‐knockdown T cells. Int Immunol 20, 1427–1437. [DOI] [PubMed] [Google Scholar]
- 5. Dong L, Watanabe K, Itoh M, Huan C‐R, Tong X‐P, Nakamura T, Miki M, Iwao H, Nakajima A, Sakai T et al (2012) CD4+ T‐cell dysfunctions through the impaired lipid rafts ameliorate concanavalin A‐induced hepatitis in sphingomyelin synthase 1‐knockout mice. Int Immunol 24, 327–337. [DOI] [PubMed] [Google Scholar]
- 6. Wang C, Ming B, Wu X, Wu T, Cai S, Hu P, Tang J, Tan Z, Liu C, Zhong J et al (2019) Sphingomyelin synthase 1 enhances BCR signaling to promote lupus‐like autoimmune response. EBioMedicine 45, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taniguchi M, Tasaki T, Ninomiya H, Ueda Y, Kuremoto K‐I, Mitsutake S, Igarashi Y, Okazaki T and Takegami T (2016) Sphingomyelin generated by sphingomyelin synthase 1 is involved in attachment and infection with Japanese encephalitis virus. Sci Rep 6, 37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shakor AB, Taniguchi M, Kitatani K, Hashimoto M, Asano S, Hayashi A, Nomura K, Bielawski J, Bielawska A, Watanabe K et al (2011) Sphingomyelin synthase 1‐generated sphingomyelin plays an important role in transferrin trafficking and cell proliferation. J Biol Chem 286, 36053–36062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D'Angelo G, Moorthi S and Luberto C (2018) Role and function of sphingomyelin biosynthesis in the development of cancer. Adv Cancer Res 140, 61–96. [DOI] [PubMed] [Google Scholar]
- 10. Tu R, Yang W and Hu Z (2016) Inhibition of sphingomyelin synthase 1 affects ceramide accumulation and hydrogen peroxide‐induced apoptosis in Neuro‐2a cells. NeuroReport 27, 967–973. [DOI] [PubMed] [Google Scholar]
- 11. Separovic D, Hanada K, Awad Maitah MY, Nagy B, Hang I, Tainsky MA, Kraniak JM and Bielawski J (2007) Sphingomyelin synthase 1 suppresses ceramide production and apoptosis post‐photodamage. Biochem Biophys Res Commun 358, 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Separovic D, Semaan L, Tarca AL, Awad Maitah MY, Hanada K, Bielawski J, Villani M and Luberto C (2008) Suppression of sphingomyelin synthase 1 by small interference RNA is associated with enhanced ceramide production and apoptosis after photodamage. Exp Cell Res 314, 1860–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Villani M, Subathra M, Im Y‐B, Choi Y, Signorelli P, Del Poeta M and Luberto C (2008) Sphingomyelin synthases regulate production of diacylglycerol at the Golgi. Biochem J 414, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Subathra M, Qureshi A and Luberto C (2011) Sphingomyelin synthases regulate protein trafficking and secretion. PLoS One 6, e23644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yano M, Yamamoto T, Nishimura N, Gotoh T, Watanabe K, Ikeda K, Garan Y, Taguchi R, Node K, Okazaki T et al (2013) Increased oxidative stress impairs adipose tissue function in sphingomyelin synthase 1 null mice. PLoS One 8, e61380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wittmann A, Grimm MOW, Scherthan H, Horsch M, Beckers J, Fuchs H, Gailus‐Durner V, Hrabě de Angelis M, Ford SJ, Burton NC et al (2016) Sphingomyelin synthase 1 is essential for male fertility in mice. PLoS One 11, e0164298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yano M, Watanabe K, Yamamoto T, Ikeda K, Senokuchi T, Lu M, Kadomatsu T, Tsukano H, Ikawa M, Okabe M et al (2011) Mitochondrial dysfunction and increased reactive oxygen species impair insulin secretion in sphingomyelin synthase 1‐null mice. J Biol Chem 286, 3992–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu MH, Takemoto M, Watanabe K, Luo H, Nishimura M, Yano M, Tomimoto H, Okazaki T, Oike Y and Song W‐J (2012) Deficiency of sphingomyelin synthase‐1 but not sphingomyelin synthase‐2 causes hearing impairments in mice. J Physiol 590, 4029–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matsumoto G, Hashizume C, Watanabe K, Taniguchi Mand Okazaki T (2019) Deficiency of sphingomyelin synthase 1 but not sphingomyelin synthase 2 reduces bone formation due to impaired osteoblast differentiation. Mol Med 25, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hayashi Y, Nemoto‐Sasaki Y, Matsumoto N, Tanikawa T, Oka S, Tanaka Y, Arai S, Wada I, Sugiura T and Yamashita A (2017) Carboxyl‐terminal Tail‐mediated homodimerizations of sphingomyelin synthases are responsible for efficient export from the endoplasmic reticulum. J Biol Chem 292, 1122–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hayashi Y, Nemoto‐Sasaki Y, Matsumoto N, Hama K, Tanikawa T, Oka S, Saeki T, Kumasaka T, Koizumi T, Arai S et al (2018) Complex formation of sphingomyelin synthase 1 with glucosylceramide synthase increases sphingomyelin and decreases glucosylceramide levels. J Biol Chem 293, 17505–17522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lafont E, Milhas D, Carpentier S, Garcia V, Jin Z‐X, Umehara H, Okazaki T, Schulze‐Osthoff K, Levade T, Benoist H et al (2010) Caspase‐mediated inhibition of sphingomyelin synthesis is involved in FasL‐triggered cell death. Cell Death Differ 17, 642–654. [DOI] [PubMed] [Google Scholar]
- 23. Rozhkova AV, Dmitrieva VG, Zhapparova ON, Sudarkina OY, Nadezhdina ES, Limborska SA and Dergunova LV (2011) Human sphingomyelin synthase 1 gene (SMS1): organization, multiple mRNA splice variants and expression in adult tissues. Gene 481, 65–75. [DOI] [PubMed] [Google Scholar]
- 24. Dergunova LV, Rozhkova AV, Sudarkina OY and Limborska SA (2013) The use of alternative polyadenylation in the tissue‐specific regulation of human SMS1 gene expression. Mol Biol Rep 40, 6685–6690. [DOI] [PubMed] [Google Scholar]
- 25. Sudarkina OY, Filippenkov IB, Brodsky IB, Limborska SA and Dergunova LV (2015) Comparative analysis of sphingomyelin synthase 1 gene expression at the transcriptional and translational levels in human tissues. Mol Cell Biochem 406, 91–99. [DOI] [PubMed] [Google Scholar]
- 26. Filippenkov IB, Sudarkina OY, Limborska SA and Dergunova LV (2015) Circular RNA of the human sphingomyelin synthase 1 gene: Multiple splice variants, evolutionary conservatism and expression in different tissues. RNA Biol 12, 1030–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burns TA, Subathra M, Signorelli P, Choi Y, Yang X, Wang Y, Villani M, Bhalla K, Zhou D and Luberto C (2013) Sphingomyelin synthase 1 activity is regulated by the BCR‐ABL oncogene. J Lipid Res 54, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moorthi S, Burns TA, Yu G‐Q and Luberto C (2018) Bcr‐Abl regulation of sphingomyelin synthase 1 reveals a novel oncogenic‐driven mechanism of protein up‐regulation. FASEB J 32, 4270–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Simon P (2003) Q‐Gene: processing quantitative real‐time RT‐PCR data. Bioinformatics 19, 1439–1440. [DOI] [PubMed] [Google Scholar]
- 30. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hinnebusch AG, Ivanov IP and Sonenberg N (2016) Translational control by 5'‐untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Young SK and Wek RC (2016) Upstream open reading frames differentially regulate gene‐specific translation in the integrated stress response. J Biol Chem 291, 16927–16935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kozak M (2001) Constraints on reinitiation of translation in mammals. Nucleic Acids Res 29, 5226–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Szamecz B, Rutkai E, Cuchalova L, Munzarova V, Herrmannova A, Nielsen KH, Burela L, Hinnebusch AG and Valasek L (2008) eIF3a cooperates with sequences 5' of uORF1 to promote resumption of scanning by post‐termination ribosomes for reinitiation on GCN4 mRNA. Genes Dev 22, 2414–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abastado JP, Miller PF, Jackson BM and Hinnebusch AG (1991) Suppression of ribosomal reinitiation at upstream open reading frames in amino acid‐starved cells forms the basis for GCN4 translational control. Mol Cell Biol 11, 486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grant CM and Hinnebusch AG (1994) Effect of sequence context at stop codons on efficiency of reinitiation in GCN4 translational control. Mol Cell Biol 14, 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Young SK, Baird TD and Wek RC (2016) Translation regulation of the glutamyl‐prolyl‐tRNA synthetase gene EPRS through bypass of upstream open reading frames with noncanonical initiation codons. J Biol Chem 291, 10824–10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lorenz R, Wolfinger MT, Tanzer A and Hofacker IL (2016) Predicting RNA secondary structures from sequence and probing data. Methods 103, 86–98. [DOI] [PubMed] [Google Scholar]
- 39. Jagodnik J, Chiaruttini C and Guillier M (2017) Stem‐loop structures within mrna coding sequences activate translation initiation and mediate control by small regulatory RNAs. Mol Cell 68, 158–170.e3. [DOI] [PubMed] [Google Scholar]
- 40. Davuluri RV, Suzuki Y, Sugano S and Zhang MQ (2000) CART classification of human 5' UTR sequences. Genome Res 10, 1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Quantitation of GFP protein and mRNA levels in transfected HeLa cells.
Fig. S2. Quantitation of SMS1 protein and SGMS1 mRNA levels in HeLa cells transfected with SGMS1 deleted in the uORF or in the coding ATG.
Fig. S3. Quantitation of SMS1 protein and SGMS1 mRNA levels in HeLa cells transfected with SGMS1 lacking the first predicted stem loops.
Fig. S4. Quantitation of SMS1 protein and SGMS1 mRNA levels in HeLa cells transfected with different SGMS1 deletion mutants of nucleotides up to −135 of the IIb 5′UTR.
Fig. S5. Nucleotide alignment of the putative regulatory portion of IIb‐SGMS1 across different vertebrates.
Legend