

Abstract
The first adducts of NHCs (=N‐heterocyclic carbenes) with aromatic polyphosphorus complexes are reported. The reactions of [Cp*Fe(η5‐P5)] (1) (Cp*=pentamethyl‐cyclopentadienyl) with IMe (=1,3,4,5‐tetramethylimidazolin‐2‐ylidene), IMes (=1,3‐bis(2,4,6‐trimethylphenyl)‐imidazolin‐2‐ylidene) and IDipp (=1,3‐bis(2,6‐diisopropylphenyl)‐imidazolin‐2‐ylidene) led to the corresponding neutral adducts which can be isolated in the solid state. However, in solution, they quickly undergo a dissociative equilibrium between the adduct and 1 including the corresponding NHC. The equilibrium is influenced by the bulkiness of the NHC. [Cp′′Ta(CO)2(η4‐P4)] (Cp′′=1,3‐di‐tert‐butylcyclopentadienyl) reacts with IMe under P atom abstraction to give an unprecedented cyclo‐P3‐containing anionic tantalum complex. DFT calculations shed light onto the energetics of the reaction pathways.
Keywords: iron, N-heterocyclic carbenes, NMR spectroscopy, nucleophilic attack, phosphorus, tantalum
Access to polyphosphorus complexes: A dynamic behavior between [Cp*Fe(η5‐P5)] and NHCs in solution is reported. NMR investigations showed a bond formation and bond breaking process between [Cp*Fe(η5‐P5)] and the NHCs. Despite this dynamic behavior in solution, adducts of [Cp*Fe(η5‐P5)] and NHCs can be isolated as solids. The tantalum complex [Cp′′Ta(CO)2(η4‐P4)] reacts with the NHC via a phosphorus atom abstraction and a ring contraction to a novel anionic cyclo‐P3 complex.
Pentaphosphaferrocene [Cp*Fe(η5‐P5)] (1) (Cp*=pentamethyl‐cyclopentadienyl) was first synthesized by Scherer et al. [1] and its reactivity was intensively investigated. It was shown that 1 can coordinate to transition‐metal carbonyl species as the cyclo‐P5 unit acts as a nucleophile. As a result, triple‐decker complexes and other organometallic compounds containing distorted P5 units were obtained. [2] In the reaction with CuI halides, 1D and 2D polymers result, [3] as well as fullerene‐like superballs. [4] Furthermore, Winter and Geiger studied the redox properties of 1 by cyclovoltammetry and predicted a dimerization of the resulting monoionic species. [5] Later, it was possible to isolate and fully characterize these species, which are the dication [(Cp*Fe)2(μ,η4:4‐P10)]2+ and the dianion [(Cp*Fe)2(μ,η4:4‐P10)]2− as well as the monomeric dianion [(Cp*Fe(η4‐P5)]2−. [6] Compared to the starting material 1, the cyclo‐P5 unit in these ionic complexes loses its planarity and adopts an envelope‐like structure. This structural motif is also observed when 1 reacts with charged main group element nucleophiles that bind to one phosphorus atom. [7] For this type of reactions, only anionic nucleophiles have been used so far, which give strong adducts since ionic products are formed. However, the reactivity of 1 towards neutral nucleophiles was not yet investigated and, in case of success, neutral and thus moderately stable products are expected to form. One such neutral nucleophile could be the NHCs (=N‐heterocyclic carbenes). After the discovery of the first stable NHC, [8] many others were synthesized and characterized. [9] In fact, NHCs are strong σ donors, [10] which is why they are used as ligands for transition metals, [11] in homogenous catalysis [12] and for stabilizing small molecules. [13] In 2000 Nixon et al. reported of a NHC induced ring contraction of a triphosphabenzene, leading to the formation of a 1,2,4‐triphosphole. By treating the 1,2,4‐triphosphole with [PtCl2(PMe3)]2 this transformation can be reverse. [14] Our first use of NHCs in their reactions towards polyphosphorus complexes containing cyclo‐P4 and cyclo‐P6 ligands as end‐ and as middle deck, respectively, showed that P atom elimination reactions occur leading to ring contractions in which the NHCs act as strong nucleophiles and not as simple donor molecules. [15] Therefore, the question arises whether a ring contraction would occur also towards the cyclo‐P5 ring in 1 resulting in an anionic cyclo‐P4 ligand complex of Fe for which a precedent was recently reported ([CpArFe(η4‐P4)]−), [16] or whether unprecedented metastable neutral adducts would result, representing a novel class of compounds, which, for the first time, might reveal no static structures in solution. This question is of general interest, since NHCs are known to react in deprotonation and dehydrocoupling reactions, respectively, E−H bond activation or ring opening towards main‐group element compounds, depending on both the nature of the NHC and the feature (as for instance acidity) of the corresponding compound. [17] Reports on the reactivity of NHCs towards homoatomic aromates in general are unknown, because they act as nucleophiles. In this respect the behavior of NHCs towards non‐carbon containing aromates bound in the coordination sphere of transition metals is of general interest, paving the way for our understanding of bonding and interactions.
Herein, we report on the reactivity of NHCs towards the polyphosphorus ligand complexes [Cp*Fe(η5‐P5)] (1) and [Cp′′Ta(CO)2(η4‐P4)] (2) (Cp′′=1,3‐di‐tert‐butylcyclopentadienyl) in which the first neutral adducts of 1 could be isolated. Depending on the bulkiness and the nucleophilicity of the NHC, adducts of different stability were isolated whose dynamic behavior in solution was elucidated by VT NMR and EXSY experiments. Moreover, in comparison, the behavior of a cyclo‐P4 complex of tantalum towards NHC was investigated, showing a reaction pattern in which a novel contracted ring product was formed [Eq. 1].
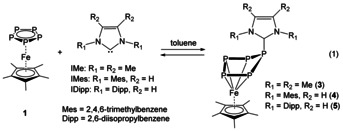
An equimolar mixture of 1 with each of these three different NHCs: IMe (=1,3,4,5‐tetramethylimidazolin‐2‐ylidene), IMes (=1,3‐bis(2,4,6‐trimethylphenyl)‐imidazolin‐2‐ylidene) and IDipp (=1,3‐bis(2,6‐diisopropylphenyl)‐imidazolin‐2‐ylidene) in toluene was stirred for 1 h at room temperature. After removing the volatiles, each reaction residue was dissolved in THF and layered with n‐hexane. Dark green crystals of [Cp*Fe(η4‐P5IMe)] (3), [Cp*Fe(η4‐P5IMes)] (4) and [Cp*Fe(η4‐P5IDipp)] (5) formed in moderate to good yields at 4 °C for 3, −30 °C for 4 and −78 °C for 5, respectively (Equation (1)).
The central structure motif of these neutral complexes in the solid state is a P5 ring, which loses its planarity and adopts an envelope‐like conformation with the NHC being bonded to one phosphorus atom (Figure 1). These structures are reminiscent of products of 1 with anionic nucleophiles, [7] with the difference that, for the first time, neutral adducts (3, 4 and 5) are now accessible. While in ionic derivatives coulomb forces contribute decisively to their stability, there are no such additional forces in the present case of neutral compounds. In comparison to [Cp*Fe(η4‐P5CH2SiMe3)]− (1 a), the P−P bond lengths (2.1342(11)–2.1535(9) Å) in 3 and 4 are similar (3: 2.1302(8)–2.1572(7) Å, and 4: 2.063(3)–2.184(5) Å). This indicates the multiple‐bond character for all P−P bonds. The P−C bonds of 3 and 4 (1.860(2) and 1.849(2) Å, respectively) are slightly longer than in 1 a (1.843(3) Å), indicating a weaker bonding in the case of the NHCs as nucleophiles. Despite numerous efforts, the obtained single crystals of 5 were of limited quality and therefore the bond features are not discussed in detail. [18]
Figure 1.
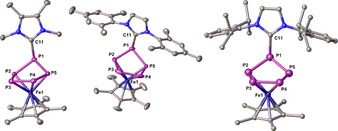
Molecular structure of 3 (left), 4 (middle) and 5 (right) in the solid state. H atoms are omitted for clarity. Selected distances [Å] and angles [°]: for 3: P1−P2 2.1572(7), P1−P5 2.1621(7), P2−P3 2.1552(8), P3−P4 2.1302(8), P4−P5 2.1575(7), P1−C11 1.860(2), Fe1−P2 2.2979(6), Fe1−P3 2.3415(6), Fe1−P4 2.3422(6), Fe1−P5 2.3077(5); P5‐P1‐P2 95.03(3), P3‐P2‐P1 107.62(3), P4‐P3‐P2 104.18(3), P3‐P4‐P5 104.19(3), P4‐P5‐P1 107.86(3). For 4: P1−P2 2.063(3), P1−P5 2.184(5), P2−P3 2.143(3), P3−P4 2.115(3), P4−P5 2.138(5), P1−C11 1.849(2), Fe1−P2 2.339(3), Fe1−P3 2.3531(19), Fe1−P4 2.303(2), Fe1−P5 2.263(5); P5‐P1‐P2 96.04(14), P3‐P2‐P1 107.57(11), P4‐P3‐P2 104.12(12), P3‐P4‐P5 104.08(16), P4‐P5‐P1 105.7(2). For 5: The Figure is drawn as a balls and sticks model (For more details, see the Supporting Information).
The 31P{1H} NMR spectra of a 1:1 mixture of 1 and IMe, IMes or IDipp (the same as if crystals of 3 or 4 were dissolved at low temperatures) showed broad signals over a wide range of temperatures, indicating a dynamic behavior of the adducts of 1 with these NHCs. Furthermore, the 31P{1H} NMR spectra of the 1:1 mixture of 1 and another NHC (ItBu=1,3‐di‐tert‐butyl‐imidazolin‐2‐ylidene) or an NHO (=N‐heterocyclic olefins), IDipp=CH2 (=(HCNDipp)2C=CH2, Dipp=2,6‐di‐isopropylphenyl) were also recorded at room temperature, because of the pronounced donor ability of ItBu and IDipp=CH2. [19] However, no broad signals or a broadening for 1 were detected in these 31P{1H} NMR spectra, not even at −80 °C.
To shed light onto this behavior, DFT calculations at the B3LYP/6‐31G* level of theory were performed for adducts of 1 with IMe, IMes and IDipp as well as with ItBu and IDipp=CH2 (Table 1). The complexation reactions with IMe, IMes and IDipp are exothermic, and therefore the interactions with 1 are energetically favorable. However, in the case of ItBu and IDipp=CH2, gas phase reactions with 1 are predicted to be energetically unfavorable. But, the absolute values of the standard enthalpies are small, and considering that the reaction is accompanied by a lowering of the entropy, the Gibbs energies for all gaseous reactions are positive (the complex formation is endergonic). Since, experimentally, the reaction proceeds in toluene solution, this indifferent solvent will only slightly affect the enthalpy of the reaction, but the entropy loss will be much less than in the gas phase. The estimation of the reaction entropy in solution according to the literature [20] leads to the values of the equilibrium constants at room temperature of 1.82×103, 0.37, and 1.1×10−4 for reactions of 1 with IMe, IMes and IDipp, respectively (see Table 1).
Table 1.
Standard enthalpies of complex formation between [Cp*Fe(η5‐P5)] (1) and NHCs/NHO (hereinafter referred to as LB): 1 + LB=1⋅LB. Reaction energies ΔE°0, standard enthalpies ΔH°298, Gibbs energies ΔG°298 (kJ mol−1) and standard entropies ΔS°298 (J mol−1 K−1) for the considered gas phase processes. B3LYP/6‐31G* level of theory.
|
LB |
ΔE°0 |
ΔH°298 |
ΔS°298(g) |
ΔG°298(g) |
ΔS°298(soln) |
ΔG°298(soln) |
K298(soln) |
T(K=1) [K] |
|---|---|---|---|---|---|---|---|---|
|
IMe |
−47.0 |
−38.6 |
−157.2 |
8.2 |
−67.2 |
−18.6 |
1.82×103 |
575 |
|
IMes |
−32.2 |
−28.1 |
−192.6 |
29.3 |
−102.6 |
2.5 |
0.37 |
274 |
|
IDipp |
−16.4 |
−9.8 |
−198.7 |
49.4 |
−108.7 |
22.6 |
1.1×10−14 |
90 |
|
ItBu |
41.6 |
46.8 |
−185.2 |
102.0 |
−95.2 |
75.2 |
6.6×10−14 |
– |
|
IDip=CH2 |
10.4 |
18.9 |
−179.8 |
72.6 |
−89.8 |
45.7 |
9.8×10−9 |
– |
The exothermic reactions are thermodynamically favorable at low temperatures. The estimated temperatures at which the equilibrium constant equals 1.0, are 575, 274, and 90 K for IMe, IMes and IDipp, respectively. Therefore, the reaction of 1 with IDipp is expected to occur at much lower temperatures than reactions with IMe and IMes. This reflects the preparative accessibility of the products as seen by the required crystallization temperature of the adducts. In contrast, in the case of ItBu and IDipp=CH2, reactions with 1 both in the gas phase and in solution are predicted to be highly endergonic and thermodynamically prohibited at any temperature.
To gain insight into the dynamic behavior of 1 and IMe in solution, 31P{1H} NMR spectra of a 1:1 mixture of 1 and IMe at variable temperatures were recorded (Figure 2). Two very broad signals were observed at room temperature, centered at δ 40 and −50 ppm. By lowering the temperature, the signals sharpen and a new signal at δ 150.2 appears which can be assigned to free 1. However, at 193 K, besides the signal for free 1, three signals at δ 34.7, 31.6 and −49.1 ppm in an integral ratio of 2:1:2 were detected. This NMR spectrum indicates the formation of a compound containing an envelope‐like P5 ring at low temperatures according to the molecular structure of 3. Additionally, these spectroscopic investigations show the occurrence of a highly dynamic system in solution. To explain this dynamic process between 1 and IMe, two mechanisms are conceivable: either a tumbling process where the IMe migrates around the P5 ring and interacts with more than one P atom at the same time or a P−C bond formation and breaking (dissociative/associative) process.
Figure 2.
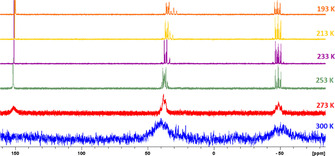
31P{1H} NMR spectra of 1 and IMe in [D8]toluene in the range of 300 to 193 K.
To clarify which of the two mechanisms does take place, a 1H EXSY spectrum of a [D8]toluene solution containing 1 and IMe at 263 K was recorded (Figure 3) which showed two cross peaks: one between the signals at δ 1.5 and 0.6 ppm and one between the signals at δ 3.5 and 2.8 ppm. The signals at δ 1.5 and 3.5 ppm are assigned to the C−CH3 and N−CH3 methyl groups of free IMe, respectively, whereas the signals at δ 0.6 and 2.8 ppm are assigned to the C−CH3 and N−CH3 methyl groups in the adduct [Cp*Fe(η4‐P5IMe)] (3). Thus, the 1H EXSY spectrum of 1 plus IMe revealed a strong exchange between free and P‐bonded IMe at 263 K, which is compatible with the dissociative/associative process.
Figure 3.
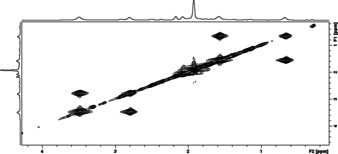
1H EXSY spectrum of 1 plus IMe in [D8]toluene at 263 K.
The definite proof that the dynamic process occurring for 1 and IMe in solution is the P−C bond formation and breaking process was derived from the 31P{1H} EXSY spectrum of a solution of 1 and IMe in [D8]toluene at 263 K (Figure 4). This spectrum showed cross peaks between each of the 31P signals of the diagonal (at δ 150.2 ppm for 1 and at δ 37 and −48.7 ppm for 3) and both other signals, indicating that the 31P nucleus of 1 interchanges with each of the 31P nuclei of 3 (and that the 31P nuclei of 3 exchange each other). Considering exclusively a tumbling process of 3 in solution, there would not be a cross peak between the signals for 1 and 3. Consequentially, the dynamic behavior of IMe and 1 at 263 K can be explained by a bond formation and bond breaking process.
Figure 4.
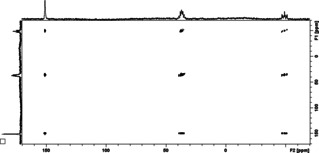
31P{1H} EXSY spectrum of 1 plus IMe in [D8]toluene at 263 K.
In addition to the NMR investigations in solution, a study of the solid‐state behavior by recording the 31P MAS NMR exemplified for 3 was executed (Figure 5). Two measurements with spinning frequencies of 14.5 and 12.5 kHz were carried out in order to determine the isotropic chemical shift of the 31P nuclei which were found at δ 123, 70, 50, −27 and −46 ppm, indicating that the five 31P nuclei in 3 are inequivalent, as expected on the basis of the X‐ray structure.
Figure 5.
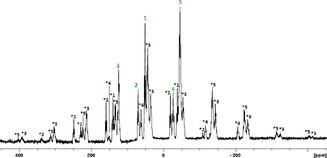
31P MAS NMR spectrum of 3 obtained at 298 K at a spinning rate of 14.5 kHz. The green numbers without asterisk denote the isotropic 31P chemical shifts.
The 31P{1H} NMR spectrum of a 1:1 mixture of 1 and IMes at room temperature showed a very broad signal at δ 49 which, upon cooling, shows coalescence at 273 K and, upon further cooling, decoalesces giving rise, below 253 K, to three signals at δ 44.0, 33.0 and −66.0 ppm with an integral ratio of 1:2:2 that can be assigned to 4, plus a singlet at δ 150.2 ppm, ascribable to 1 (Figure S3). This behavior is interpreted assuming that complex 4 is more labile in solution than 3 and that at room temperature only an averaged signal for the 31P nuclei of 1 and 4 (in fast equilibrium, see Eq. (1)) is detectable. The higher lability of 4 as opposed to 3 is in accordance with the DFT calculations (vide supra), which predict a minimum stability for the adduct 5 with respect to 4 and 3. And, in fact, a distinct interaction between 1 and IDipp was detected at much lower temperatures than with IMe or IMes. The 31P{1H} NMR spectrum of the 1:1 mixture of 1 and IDipp showed clear signals for 5 (δ 40.5, 35.7 and −62.1 ppm with an integral ratio of 1:2:2), along with 1, only at a temperature as low as 193 K (Figure S5). From 233 K upwards, the averaged signal for the 31P nuclei of 1 and 5 is detected with chemical shifts that move towards that of 1 when the temperature is raised. At room temperature, only the singlet at δ 150.2 assigned to 1 could be detected, indicating that at room temperature a negligible interaction between 1 and IDipp occurs. Isolated crystals of 5 are extremely temperature‐sensitive and can only be handled at temperatures below 195 K.
Once having ascertained that the cyclo‐P5 ring does form neutral adducts with NHCs, the question arises if such neutral adduct formation can be transferred also to cyclo‐P4 rings. To answer this question, we reacted [Cp′′Ta(CO)2(η4‐P4)] (2) [21] with IMe in toluene, obtaining, to our surprise, the unprecedented cyclo‐P3 complex [(IMe)2P][Cp′′Ta(CO)2(η3‐P3)] (6) in good yields (Equation 2). If less than two equivalents of IMe are used, the conversion is not complete and the 31P{1H} NMR spectrum shows unreacted 2 and 6. Thus, the tantalum species 2 loses one phosphorus atom to form an anionic complex with an η3‐P3 ring and the eliminated phosphorus atom is coordinated by two NHC molecules forming the [(IMe)2P]+ cation. The 31P{1H} NMR spectrum of 6 showed a singlet at δ −113.1 ppm for the [(IMe)2P]+ cation and a singlet at δ −421.9 ppm for the [(Cp′′Ta(CO)2(η3‐P3)]− anion in a 1:3 integral ratio. Red crystals of 6 were obtained in a saturated acetonitrile solution, which were submitted to XRD analysis. In the solid state, the anion of 6 shows an unprecedented cyclic P3 unit coordinated to the tantalum atom carrying additionally two carbonyl and the Cp′′ ligands (Figure 6). The distances between the tantalum and the phosphorus atoms differ. Two shorter bonds (2.563(2) and 2.568(2) Å) and one slightly elongated bond (2.614(2) Å) are found.
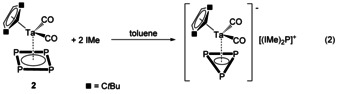
Figure 6.
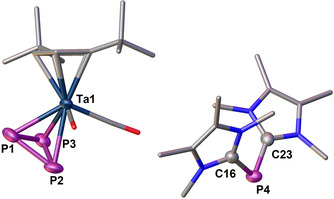
Molecular structure of 6 in the solid state. H atoms are omitted for clarity. Selected distances [Å] and angles [°]: P1−P2 2.169(4), P1−P3 2.186(3), P2−P3 2.184(3), P4−C16 1.797(8), P4−C23 1.796(8), Ta1−P1 2.568(2), Ta1−P2 2.614(2), Ta1−P3 2.563(2); P1‐P2‐P3 60.26(12), P2‐P3‐P1 59.53(13), P3‐P1‐P2 60.21(12), C16‐P4‐C23 97.9(3).
The calculated Wiberg bond indexes (WBIs) using the experimental solid‐state geometry of the anion at the B3LYP/def2‐SVPD level of theory (see Supporting Information) reveal for the shorter Ta−P distances values of 0.85 and 0.87 Å. The WBI for the elongated Ta−P distance is 0.76 Å. The P−P bond lengths within the P3 ring are 2.184(4), 2.186(3) and 2.169(4) Å with WBIs of 1.00, 1.00 and 1.01. The P−C distances of the cation are 1.797(8) and 1.796(8) Å and are therefore characteristic of P−C single bonds. The angle between the two NHC carbon atoms and the phosphorus atom (C23‐P4‐C16) in the cation is close to right (97.9(3)°).
In summary, we reported the synthesis of the first neutral iron complexes [Cp*Fe(η4‐P5NHC)] (NHC=IMe: 3, NHC=IMes: 4, NHC=IDipp: 5) in which the NHCs act as neutral donors and the cyclo‐P5 unit adopts an envelope‐like structure. VT NMR experiments elucidated their dynamic behavior in solution, consisting in C−P bond breaking and reformation. The stability of the adducts 3–5 decreases in the order 3>4>5, as indicated by DFT calculations and dynamic NMR studies. In contrast to the reaction of the NHCs with the cyclo‐P5 ring of 1, the cyclo‐P4 ring of [Cp′′Ta(CO)2(η4‐P4)] reacts with IMe with a phosphorus atom being abstracted to form an unprecedented anionic tantalum complex with a cyclo‐P3 unit and a cationic phosphorus atom stabilized by two NHC units. These results exhibit the high potential of NHCs in the chemistry of heteroaromates revealing adduct formations or elimination reactions, a topic which will further investigated in a broader scope.
Experimental Section
Crystallographic data
Deposition numbers 2015543, 2015544, 2015545, and 2015546 (3, 4, 5, and, 6) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the project Sche 384/38‐1. Open access funding enabled and organized by Projekt DEAL.
F. Riedlberger, S. Todisco, P. Mastrorilli, A. Y. Timoshkin, M. Seidl, M. Scheer, Chem. Eur. J. 2020, 26, 16251.
Dedicated to Professor H. W. Roesky on the occasion of his 85th birthday
Contributor Information
Dr. Felix Riedlberger, www.uni‐regensburg.de/chemie‐pharmazie/anorganische‐chemie‐scheer
Prof. Dr. Manfred Scheer, Email: manfred.scheer@ur.de.
References
- 1. Scherer O. J., Brück T., Angew. Chem. Int. Ed. Engl. 1987, 26, 59; [Google Scholar]; Angew. Chem. 1987, 99, 59. [Google Scholar]
- 2.
- 2a. Detzel M., Mohr T., Scherer O. J., Wolmershäuser G., Angew. Chem. Int. Ed. Engl. 1994, 33, 1110–1112; [Google Scholar]; Angew. Chem. 1994, 106, 1142–1144; [Google Scholar]
- 2b. Scherer O. J., Acc. Chem. Res. 1999, 32, 751–762. [Google Scholar]
- 3.
- 3a. Scheer M., Gregoriades L. J., Virovets A. V., Kunz W., Neueder R., Krossing I., Angew. Chem. Int. Ed. 2006, 45, 5689–5693; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5818–5822; [Google Scholar]
- 3b. Bai J., Virovets A. V., Scheer M., Angew. Chem. Int. Ed. 2002, 41, 1737–1740; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1808–1811. [Google Scholar]
- 4.
- 4a. Welsch S., Groger C., Sierka M., Scheer M., Angew. Chem. Int. Ed. 2011, 50, 1435–1438; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1471–1474; [Google Scholar]
- 4b. Li T., Wiecko J., Pushkarevsky N. A., Gamer M. T., Köppe R., Konchenko S. N., Scheer M., Roesky P. W., Angew. Chem. Int. Ed. 2011, 50, 9491–9495; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9663–9667; [Google Scholar]
- 4c. Scheer M., Schindler A., Bai J., Johnson B. P., Merkle R., Winter R., Virovets A. V., Peresypkina E. V., Blatov V. A., Sierka M., Eckert H., Chem. Eur. J. 2010, 16, 2092–2107; [DOI] [PubMed] [Google Scholar]
- 4d. Scheer M., Schindler A., Groger C., Virovets A. V., Peresypkina E. V., Angew. Chem. Int. Ed. 2009, 48, 5046–5049; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5148–5151; [Google Scholar]
- 4e. Scheer M., Schindler A., Merkle R., Johnson B. P., Linseis M., Winter R., Anson C. E., Virovets A. V., J. Am. Chem. Soc. 2007, 129, 13386–13387; [DOI] [PubMed] [Google Scholar]
- 4f. Scheer M., Bai J., Johnson B. P., Merkle R., Virovets A. V., Anson C. E., Eur. J. Inorg. Chem. 2005, 4023–4026; [Google Scholar]
- 4g. Bai J., Virovets A. V., Scheer M., Science 2003, 300, 781–783. [DOI] [PubMed] [Google Scholar]
- 5. Winter R. F., Geiger W. E., Organometallics 1999, 18, 1827–1833. [Google Scholar]
- 6. Butovskiy M. V., Balázs G., Bodensteiner M., Peresypkina E. V., Virovets A. V., Sutter J., Scheer M., Angew. Chem. Int. Ed. 2013, 52, 2972–2976; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3045–3049. [Google Scholar]
- 7. Mädl E., Butovskii M. V., Balázs G., Peresypkina E. V., Virovets A. V., Seidl M., Scheer M., Angew. Chem. Int. Ed. 2014, 53, 7643–7646; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7774–7777. [Google Scholar]
- 8. A. J. Arduengo III , Harlow R. L., Kline M., J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar]
- 9.
- 9a. Martin D., Melaimi M., Soleilhavoup M., Bertrand G., Organometallics 2011, 30, 5304–5313; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Melaimi M., Soleilhavoup M., Bertrand G., Angew. Chem. Int. Ed. Angew. Chem. Int. Ed. Engl. 2010, 49, 8810–8849; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8992–9032; [Google Scholar]
- 9c. Vignolle J., Cattoën X., Bourissou D., Chem. Rev. 2009, 109, 3333–3384; [DOI] [PubMed] [Google Scholar]
- 9d. Schuster O., Yang L., Raubenheimer H. G., Albrecht M., Chem. Rev. 2009, 109, 3445–3478; [DOI] [PubMed] [Google Scholar]
- 9e. Hahn F. E., Jahnke M. C., Angew. Chem. Int. Ed. 2008, 47, 3122–3172; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3166–3216. [Google Scholar]
- 10. Back O., Henry-Ellinger M., Martin C. D., Martin D., Bertrand G., Angew. Chem. Int. Ed. 2013, 52, 2939–2943; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3011–3015. [Google Scholar]
- 11. Herrmann W. A., Angew. Chem. Int. Ed. 2002, 41, 1290–1309; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1342–1363. [Google Scholar]
- 12.
- 12a. Vougioukalakis G. C., Grubbs R. H., Chem. Rev. 2010, 110, 1746–1787; [DOI] [PubMed] [Google Scholar]
- 12b. Díez-González S., Marion N., Nolan S. P., Chem. Rev. 2009, 109, 3612–3676. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Krachko T., Slootweg J. C., Eur. J. Inorg. Chem. 2018, 2734–2754; [Google Scholar]
- 13b. Nesterov V., Reiter D., Bag P., Frisch P., Holzner R., Porzelt A., Inoue S., Chem. Rev. 2018, 118, 9678–9842; [DOI] [PubMed] [Google Scholar]
- 13c. Back O., Donnadieu B., Parameswaran P., Frenking G., Bertrand G., Nat. Chem. 2010, 2, 369–373; [DOI] [PubMed] [Google Scholar]
- 13d. Wang Y., Xie Y., Wei P., King R. B., H. F. Schaefer III , von Schleyer P., Robinson G. H., Science 2008, 321, 1069–1071. [DOI] [PubMed] [Google Scholar]
- 14. Clendenning S. B., Hitchcock P. B., Nixon J. F., Nyulászi L., Chem. Commun. 2000, 1305–1306. [Google Scholar]
- 15. Piesch M., Reichl S., Seidl M., Balázs G., Scheer M., Angew. Chem. Int. Ed. 2019, 58, 16563–16568; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16716–16721. [Google Scholar]
- 16. Chakraborty U., Leitl J., Mühldorf B., Bodensteiner M., Pelties S., Wolf R., Dalton Trans. 2018, 47, 3693–3697. [DOI] [PubMed] [Google Scholar]
- 17. Würtemberger-Pietsch S., Schneider H., Marder T. B., Radius U., Chem. Eur. J. 2016, 22, 13032–13036. [DOI] [PubMed] [Google Scholar]
- 18.Due to the limited quality of single crystals of 5 and their extreme sensibility (they can only be handled at temperatures −70 °C, also in the solid state), the best X-ray structure analysis only gave a confident atom connectivity (for more details see the Supporting Information).
- 19. Roy M. M. D., Rivard E., Acc. Chem. Res. 2017, 50, 2017–2025. [DOI] [PubMed] [Google Scholar]
- 20. Lisovenko A. S., Timoshkin A. Y., Inorg. Chem. 2010, 49, 10357–10369. [DOI] [PubMed] [Google Scholar]
- 21. Scherer O. J., Winter R., Wolmershäuser G., Z. Anorg. Allg. Chem. 1993, 619, 827–835. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary