

Abstract
Genetic signal detection in genome‐wide association studies (GWAS) is enhanced by pooling small signals from multiple Single Nucleotide Polymorphism (SNP), for example, across genes and pathways. Because genes are believed to influence traits via gene expression, it is of interest to combine information from expression Quantitative Trait Loci (eQTLs) in a gene or genes in the same pathway. Such methods, widely referred to as transcriptomic wide association studies (TWAS), already exist for gene analysis. Due to the possibility of eliminating most of the confounding effects of linkage disequilibrium (LD) from TWAS gene statistics, pathway TWAS methods would be very useful in uncovering the true molecular basis of psychiatric disorders. However, such methods are not yet available for arbitrarily large pathways/gene sets. This is possibly due to the quadratic (as a function of the number of SNPs) computational burden for computing LD across large chromosomal regions. To overcome this obstacle, we propose JEPEGMIX2‐P, a novel TWAS pathway method that (a) has a linear computational burden, (b) uses a large and diverse reference panel (33 K subjects), (c) is competitive (adjusts for background enrichment in gene TWAS statistics), and (d) is applicable as‐is to ethnically mixed‐cohorts. To underline its potential for increasing the power to uncover genetic signals over the commonly used nontranscriptomics methods, for example, MAGMA, we applied JEPEGMIX2‐P to summary statistics of most large meta‐analyses from Psychiatric Genetics Consortium (PGC). While our work is just the very first step toward clinical translation of psychiatric disorders, PGC anorexia results suggest a possible avenue for treatment.
Keywords: gene expression, genetics, GWAS, pathway, TWAS
1. INTRODUCTION
Genome‐wide association studies (GWAS) have been very successful in identifying disease loci using single‐marker‐based association tests (Bush & Moore, 2012). Unfortunately, such methods have had limited power to identify causal genes or pathways (Wang, Li, & Hakonarson, 2010). For most complex traits, genetic risks are likely the result of the joint effect of multiple genes located in causal pathways (Ramanan, Shen, Moore, & Saykin, 2012). Consequently, pooling information across genes in a pathway is likely to greatly improve signal detection.
Given that gene expression (GE) is widely posited to be the critical causal mechanism linking variant to phenotype (Emilsson et al., 2008), the pooling of information across multiple variants should be mediated by GE. Gene expression informed methods, known as transcriptome wide association studies (TWAS), exist for gene‐level inference (Chatzinakos et al., 2018; Gamazon et al., 2015; Gusev et al., 2016). They combine summary statistics of those expression Quantitative Traits Loci (eQTL) known to best predict the expression of each gene to infer the association between trait and GE for the gene under investigation. While TWAS pathway methods do not currently exist, such methods would help tremendously the translation of psychiatric disorders by (a) eliminating the confounding effect of linkage disequilibrium (LD) on TWAS gene statistics and (b) increasing the power by directly modeling the LD between TWAS summary statistics. The dearth of such methods is likely due to the large storage/computational burden associated with computing the LD between the numerous SNPs, possibly, involved in computing TWAS statistics for numerous genes in a large region, for example, Major Histocompatibility (MHC) region from chromosome 6p (~25–35 Mbp) or even entire chromosome arms. The large computational burden is the result of using the estimated pairwise LD for all eQTL SNPs in order to assess the variance of the linear combinations for TWAS statistics (Jin et al., 2014), which, for m variants, imposes a heavy O(m2) computational burden. Currently, pathway analysis methods are nontranscriptomic, that is, they disregard the effect of SNPs as eQTLs of GE and in addition they disregard the LD among TWAS gene statistics, which inflates the type I error. Instead, current pathway methods search for “agnostic” (i.e., not GE mediated) signal enrichment in a pathway/gene set. Among existing pathway methods we mention ALIGATOR (Holmans et al., 2009), GSEA (Subramanian et al., 2005), DAPPPLE (Rossin et al., 2011), as MAGENTA (Segre et al., 2010), INRICH (Lee, O'Dushlaine, Thomas, & Purcell, 2012) and MAGMA (de Leeuw, Mooij, Heskes, & Posthuma, 2015), as well as online tools: GeneGo/MetaCore (www.genego.com), Ingenuity Pathway Analysis (www.ingenuity.com), PANTHER (www.pantherdb.org), WebGestalt (bioinfo.vanderbilt.edu/webgestalt), DAVID (david.abcc.ncifcrf.gov), and Pathway Painter (pathway.painter.gsa-online.de). While not designed for pathway analyses, LDpred (Bulik‐Sullivan et al., 2015; Finucane et al., 2015) can also be adapted to test whether pathways are enriched above the polygenic background while adjusting for genomic covariates. Given that even such GE‐“naïve” pathway analyses methodologies have shown promise, we believe that quantitative GE‐informed pathway analyses can greatly complement the “agnostic” findings of all these tools, mirroring the contribution of modern gene‐level TWAS analyses to the prior agnostic gene annotation studies.
To advance the translation of psychiatric disorders, we propose JEPEGMIX2 Pathway (JEPEGMIX2‐P) that extends the reach of TWAS methods to pathway‐level. It promises to increase power by directly modeling the LD between TWAS gene statistics. JEPEGMIX2‐P (a) uses a very large and diverse reference panel consisting of 33 K subjects (including >10 K Han Chinese), (b) automatically estimates ethnic composition of cohort, (c) uses these weights to compute LD for gene statistics via a linear running time procedure, (d) uses LD and GWAS summary statistics to rapidly test for the association between trait and expression of genes even in the largest pathways, (e) is competitive, that is, adjusts for the background enrichment of TWAS gene statistics, and (f) provides the option of a conditional analysis which can effectively moderate the effect of the SNPs in LD with significant SNP signals to avoid a “carrying” effect, that is, a large signal in a SNP inducing significant signals in all small pathways that include it and the SNPs in LD with it. Compared to MAGMA, our analyses of PGC GWAS data with JEPEGMIX2‐P yield a markedly increased number of significant signals.
2. METHODS
2.1. TWAS pathway analysis
Naïve application of many analysis methods comparing the statistic with the default null hypothesis (H0), when applied for genes/pathways with numerous SNPs/genes might yield large signals merely by accumulating “average” polygenic signals from well‐powered studies. This comparison to the default null is also known as uncompetitive statistic, as it does not take into account the average enrichment of the genome. To avoid such an accumulation of average polygenic information in the uncompetitive statistic, we use competitive tests that adjust the SNP and gene level χ2 statistics for the background enrichment of genome wide SNPs and TWAS gene statistics, respectively. We achieve this simply by adjusting gene statistics for average noncentrality (Text S1 and S2 of Supporting Information). Subsequently, as detailed in Text S3 and S4, we use the GWAS summary statistics (a) to estimate the ethnic composition of the study cohort and (b) use the estimated ethnic weights to build a pathway statistic that has a manageable O(m) computational burden.
2.2. Computation of pathway statistic
Generic TWAS methods, including our JEPEGMIX2, output Z‐score statistics by gene. Thus, if the correlation between gene statistics is available, for example, by using the O(m) method described above, these statistics can be combined using a Mahalanobis χ2 statistics with the number of degrees of freedom (df) equal to the number of genes. Unfortunately, this can quickly become very involved if we need to compute the LD between statistics of all ~20,000 genes. However, due to the (a) to the random orientation of homologous chromosome pairs during meiosis and (b) large number of recombination in the centromere, the genotypes of variants in different chromosomes and chromosome arms are practically independent. Consequently, as Z‐scores are functions of genotypes, it follows that, near the null hypothesis we use to build tests, Z‐scores for genes on different chromosome arms are independent (Lee, Bigdeli, Riley, Fanous, & Bacanu, 2013). Thus, the Mahalanobis type statistic can be computed more easily by (a) computing chromosome arm χ2 statistics and, subsequently, and (b) combining the resulting chromosome arm statistic in a χ2 pathway statistic (Figure 1). Similarly, the df for the χ2 pathway statistic equals the sum of the dfs for chromosome arm statistics.
FIGURE 1.

Computation of pathway statistics [Color figure can be viewed at wileyonlinelibrary.com]
2.3. Conditional analysis
For a pathway signal to be credible, it is useful to be based on having suggestive signals in multiple genes, not a significant signal in a single one. Intuitively, if only one gene in a pathway yields a signal this might be due to the passenger effect—that is, SNPs used to predict the respective gene expression might be just in LD with a large SNP signal from another gene. Thus, to avoid the passenger gene effects on pathway statistics, we also offer the option to eliminate the effect of SNPs with statistically significant signals, by applying a novel conditional analysis procedure (Text S5 in Supporting Information) to summary statistics before their use in our TWAS pathway tool.
2.4. Annotation panel for gene and pathway statistics
The annotation file for JEPEGMIX2‐P now includes an R‐like formula for the expression of each gene as a function of its eQTL genotypes and of the content for each pathway as a function of the names its constituting genes. More specifically, this updated annotation gene file includes cis‐eQTL for all tissues available in the v0.7 version of PredictDB (http://predictdb.hakyimlab.org/). These tissue specific‐models currently contain only cis‐eQTL and, currently, do not include trans‐eQTL effects, gene–gene or protein–protein interaction across the genome. To avoid making inference about genes poorly predicted by SNPs, for the 48 available tissues (Text S6 and Table S1 of Supporting Information), we retain only genes for which the expression is predicted from its eQTLs with reasonable accuracy (i.e., when the multiple testing adjustment of gene expression predictions using False Discovery Rate [FDR] yield q‐value <.05). Finally, for pathway database we used MSigDB (Liberzon, 2014; Liberzon et al., 2011; Liberzon et al., 2015), which is well maintained and widely used by researchers.
2.5. Reference panel for LD calculations
The current version uses the 32,953 subjects (~33 K) as a reference panel. It consists of 20,281 Europeans, 10,800 East Asians (from CONVERGE study, Text S5 of Supporting Information), 522 South Asians, 817 Africans and 533 Natives of Americas (Text S6 and Table S2 of Supporting Information).
2.6. Traits of interest
We applied JEPEGMIX2‐P to summary statistics coming from Psychiatric Genetics Consortium (PGC‐ http://www.med.unc.edu/pgc/) datasets, that is, Schizophrenia (SCZ), Attention Deficit Hyperactivity Disorder (ADHD), Autism (AUT), Bipolar (BIP), Eating Disorders (Anorexia) (ED), Major depression disorder (MDD), and Post traumatic stress disorder (PTSD) (Table 1). To limit the increase in Type I error rates of JEPEGMIX2‐P, we deem as significantly associated only those pathways that yield an FDR‐adjusted p‐value (q‐value)<.05q) < .05. Due to C4 explaining most of Major Histocompatibility (MHC) (chr6:25–33 Mb; McCarthy et al., 2016), gene/signals for SCZ, for this trait, we omit non‐C4 genes in this region. Moreover, due to the high correlation between SNPs in MHC (chr6:25–33 Mb), we also omit genes in this region for MDD, which also showed MHC signals (Wray et al., 2018).
TABLE 1.
Description of GWAS studies and traits that were analyses
| Trait | Trait abbreviation | Dataset description |
|---|---|---|
| Schizophrenia | SCZ | PGC2 SCZ (Schizophrenia Working Group of the Psychiatric Genomics, 2014) |
| Attention deficit hyperactivity disorder | ADHD | PGC ADHD (Demontis et al., 2019) |
| Autism | AUT | PGC AUT (Autism Spectrum Disorders Working Group of The Psychiatric Genomics, 2017) |
| Bipolar | BIP | PGC BIP (Stahl et al., 2019) |
| Eating disorders (anorexia) | ED | PGC EAT (Duncan et al., 2017) |
| Major depression disorder | MDD | PGC MDD (Wray et al., 2018) |
| Post‐traumatic stress disorder | PTSD | PGC PTSD (Nievergelt et al., 2019) |
2.7. MAGMA
MAGMA is one of the most used pathway analysis methods. Consequently, we compare the results obtained from our method with those obtained by this state‐of‐the‐art method. The original MAGMA software however cannot estimate tissue‐gene statistics, therefore to compare JEPEGMIX2‐P with MAGMA, we provided as input to MAGMA our JEPEGMIX2‐P TWAS results.
2.8. Across tissue analysis
To summarize the top signal across tissues (i.e., regardless of the tissue in which they had signal), we (a) FDR adjusted by‐tissue p‐values for each gene/pathway and (b) FDR adjusted the overall gene/pathway p‐values for multiple testing of genes/pathways. Moreover, to compare across signals across tissue types, we divided the 48 available tissue into 4 major categories: brain (13 tissues), peripheral (27 tissues), sex (7 tissues), and blood (1 tissue). In order to control the results for the widely variable number of tissues across tissue types, we first summarize their findings according to the above FDR procedure for each category.
2.9. Simulations
To estimate the false positive rates of JEPEGMIX2‐P, for five different cosmopolitan studies scenarios, we simulated (under H0) 100 cosmopolitan cohorts of 10,000 subjects for Ilumina 1 M autosomal SNPs using 1KG haplotype patterns (Lee et al., 2015) (Text S9 and Table S3 of Supporting Information). The subject phenotypes were simulated independent of genotypes as a random Gaussian sample. SNP phenotype–genotype association summary statistics were computed from a correlation test. For each cohort, we obtained JEPEGMIX2‐P statistics, for the two “null” enrichment scenarios (a) under null (H0), of no enrichment, and (b) polygenic null (Hp), that is, when enrichment is uniform over the entire genome regardless of functionality of individual genomic regions. For the JEPEGMIX2‐P analyses of the resulting data we used (a) prespecified (PRE) and (b) automatically estimated ethnic weights (EST). Given that (a) subjects were re‐assigned to subpopulations in the new panel and (b) the populations labels in the new panel do not correspond to the ones from 100 Genomes, this induced possible mismatches that might result in increased false positive rates. To avoid this, a second version of the PRE‐approach provides the published weights to continental superpopulations, that is, EUR, ASN, SAS, AFR, and AMR.
During our initial simulations, we observed that pathways with name lengths ≤8, for example, pathways denoting chromosome bands like chr3p21, ch6p21, and so on, have increased false positive rates due to having numerous genes in high LD because of their proximity. For that reason, we also estimated the size of the test for all cohort scenarios just for these high LD pathways.
Finally, given that the simulated cohorts might not reflect “real data,” we create “nullified” data sets from GWAS data sets. These nullified data sets are based on 20‐real GWAS (of mostly Caucasian cohorts) as: Schizophrenia (SCZ), attention deficit hyperactive disorder (ADHD), autism (AUT), major depressive disorder MDD (see Table 1), and a further (mainly European) 16 data sets that are not yet publicly available. This approximation for null data is obtained by substituting the expected quantile of the Gaussian distribution for the (ordered) Z‐score (see also Text S1, nonparametric robust estimation of weights section, of the Supporting Information) after eliminating SNPs with significant association p‐values in the original GWAS. However, one side effect of this approach consists of statistics within/near the peak signals in original GWAS, which might be still slightly more concentrated into the tails of the distribution compared to perfect “null data.” This can result in a slight increase in false positive rate, especially when applied to the nullified version of a GWAS with a lot of signals (e.g., PGC2 SCZ; Table 1). However, most of the data sets used in “nullification” were not highly enriched in association signals.
3. RESULTS
3.1. JEPEGMIX2‐P false positives rates results
JEPEGMIX2‐P using our proposed automatic weight detection procedure (see Methods), controlled the false positive rates at or below the nominal threshold, even when this threshold was 10−6, under both null (H0) and “polygenic null” scenario (Hp—enrichment in association signals is uniform over the entire genome, in keeping with the polygenicity of most traits). When the method used narrowly prespecified subpopulation weights (e.g., using the closest subpopulations from the reference sample, that is, as derived from the study description), the false positives rates were increased, especially for lower nominal rates, by up to ~220–450 (Text S9 and Figures S1–S5 in Supporting Information). However, JEPEGMIX2‐P with pre‐estimated weights based on super populations (i.e., European, East Asian, African, etc.) had a much lower inflation of false positive rates; only for 10−6 threshold the false positive rate was increased by ~2–4 times, under both H0 and Hp scenarios (Text S9 and Figure S6 in Supporting Information).
For high‐LD pathways, for example, those defined by single chromosome bands in MSigDB (Liberzon, 2014; Liberzon et al., 2011; Liberzon et al., 2015), the behavior of JEPEGMIX2‐P with automatically estimated weights is similar to the one for the whole set of MSigDB pathways. However, false positive rates increase by ~300–1,200 for the narrowly prespecified subpopulation weights (Text S9 and Figures S7–S11 in Supporting Information), while when using super population‐based weights, it remained practically unchanged from the 2–4X increase derived for all pathways (Text S9 and Figure S12 in Supporting Information).
3.2. JEPEGMIX2‐P gene statistics results
We applied JEPEGMIX2‐P to the PGC traits (Table 1). As conditional analyses (Section 2.3) are relevant mostly for the pathway analyses, all gene‐level results are based on the unconditional analysis. JEPEGMIX2‐P successfully identified several significant genes after adjusting for multiple testing using FDR (Table 2, Supplementary Excel file 1 and PDF S1–S4 with the Manhattan plots, https://cutt.ly/ypFpWdh). The results showed signals for multiple gene‐tissue pairs (Text S10 and Figures S13–S17 in Supporting Information). The top FDR‐significant signal for BIP was YJEFN3 in the Breast Mammary tissue (p = 7.3e‐09), for ED SUOX in Pituitary tissue (p = 4.5e‐09), for MDD NKAPL in the Esophagus Mucosa tissue (p = 9.6e‐10) and for SCZ RP5‐874C20.3 in the Breast Mammary tissue (p = 9.5e‐23), while ADHD, AUT and PTSD analyses did not yield any FDR‐significant gene signals. For BIP most of the signals were found in the sex specific tissues for ED at brain, for MDD in peripheral and sex and for SCZ in peripheral and sex (Text S10 and Figures S13–S16 in Supporting Information).
TABLE 2.
Numbers of genes signals found by JEPEGMIX2‐P
| Trait | JEPEGMIX2‐P analysis | |
|---|---|---|
| FDR | Holm | |
| ADHD | — | — |
| AUT | — | — |
| BIP | 452 | 21 |
| ED | 78 | 33 |
| MDD | 221 | 33 |
| SCZ | 4,207 | 936 |
| PTSD | — | — |
Moreover, the overlap of the gene‐tissue signals was higher for SCZ and BIP (238) compared to SCZ and MDD (168) while the ED signals (78) had no overlap with the other traits (Text S10 and Figure S17 in Supporting Information). Finally, two genes were common between SCZ, MDD and BIP (PRSS16 in the tissue Muscle Skeletal and in the tissue Skin Sun Exposed Lower Leg.
3.3. JEPEGMIX2‐P pathway statistics results
Using the gene statistics of both unconditional and the conservative conditional JEPEGMIX2‐P analyses, we uncovered numerous significant pathway signals. The most significant signals for each trait and their respective effect sizes are presented in PDF S5–S14 (https://cutt.ly/ypFpWdh). We summarized (see Section 2.8) top across‐tissues pathway signals (Table 3) and we hierarchically clustered the top 50 signals across all the traits (Figure 2). Additionally, we constructed an overall heatmap that includes the top 10 hits for each trait (Figure 3) and more detailed heatmaps (Text S10 and Figures S32–S40 in Supporting Information). We also include all signals in extended tables (Supplementary Excel file 2, https://cutt.ly/ypFpWdh).
TABLE 3.
Numbers of across‐tissues pathway signals found by JEPEGMIX2‐P
| Trait | JEPEGMIX2‐P without conditional analysis | JEPEGMIX2‐P conditional analysis | ||
|---|---|---|---|---|
| FDR | Holm | FDR | Holm | |
| ADHD | — | — | — | — |
| AUT | 2 | 2 | 2 | 2 |
| BIP | 92 | 6 | 35 | 2 |
| ED | 186 | 6 | 1 | 1 |
| MDD | 300 | 10 | 3 | 2 |
| SCZ | 886 | 117 | 15 | — |
| PTSD | — | — | — | — |
FIGURE 2.
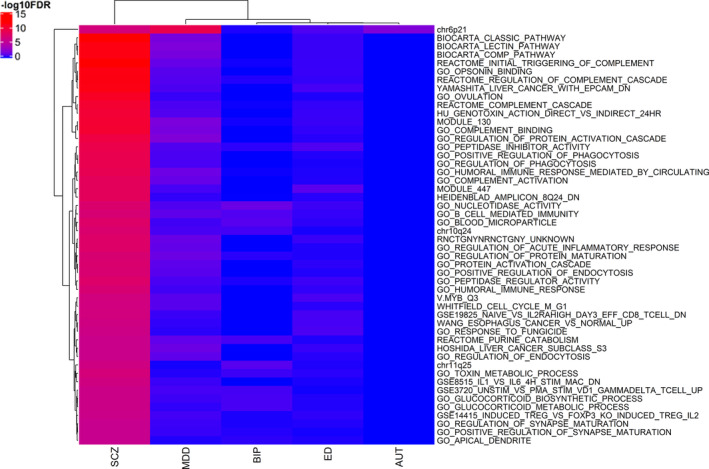
Top 50 pathway signals heatmap (unconditional analysis) across all traits. At the x‐axis are the hierarchical clustered Traits and at the y‐axis are the hierarchical clustered pathways according to the ‑log10FDR values [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 3.
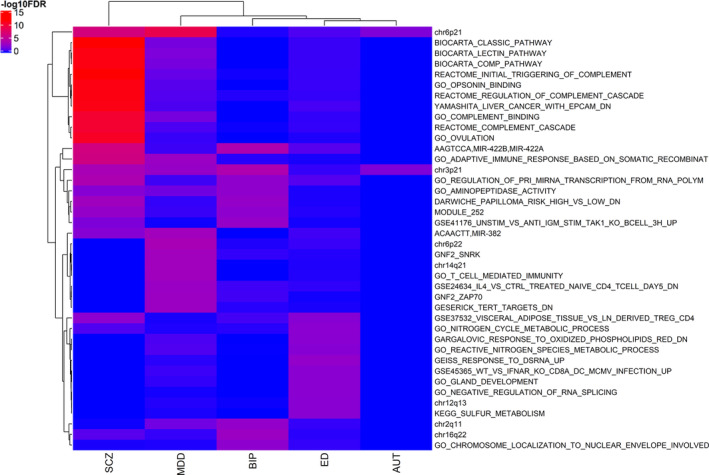
Pathway signals heatmap (unconditional analysis) for the 10 top pathways from each trait. At the x‐axis are the hierarchical clustered Traits and at the y‐axis are the hierarchical clustered pathways according to the ‑log10FDR values [Color figure can be viewed at wileyonlinelibrary.com]
JEPEGMIX2‐P pathway signals were computed for all pathway‐tissue pairs. As expected, due to eliminating large signals, the FDR signals in the conditional analysis were significantly less numerous than those in the unconditional analysis and, for this reason, we will focus mostly on the unconditional findings. Pathway chr3p21 was the top FDR for signal for both AUT (p = 2.3e‐08) and BIP (p = 1.4e‐10), in Skin Not Sun Exposed Suprapubic and Artery Tibial tissues. Pathway GEISS_RESPONSE_TO_DSRNA_UP in Pituitary tissue was the top FDR signal for ED (p = 3.5e‐09), and pathway chr6p21 in the Adrenal Gland tissue (p = 3.4e‐09) was the top FDR signal for MDD. Lastly, for SCZ the top pathway signal was found in REACTOME_INITIAL_TRIGGERING_OF_COMPLEMENT in Skeletal Muscle tissue (p = 4.4e‐44).
For all the traits (AUT, BIP, ED, MDD, and SCZ), most of the signals were found in the sex specific tissues (Text S10 and Figures S18–S28 in Supporting Information). While initially surprising, this finding is concordant with gender differences in the prevalence of psychiatric disorders, this finding might be driven mostly by the large number of genes having good gene expression prediction in testis (9,061) despite the relatively low number of subjects (157).
Common pathway‐tissue signals were higher for SCZ and MDD (41) compared to SCZ and BIP (31) while the ED signals (589) were unique except for one common with SCZ (GSE37532_VISCERAL_ADIPOSE_TISSUE_VS_LN_DERIVED_TREG_CD4_TCELL_DN in Breast Mammary tissue). In the unconditional analysis one chromosomal pathway, chr3p21, was common between SCZ, BIP, and MDD in seven different tissues, while in the conditional analysis this common pathway survives only for BIP and MDD (Text S10 and Figure S28 in Supporting Information).
3.4. JEPEGMIX2‐P comparison with MAGMA
Comparing our method with MAGMA, most likely due to not modeling the LD between gene statistics, MAGMA finds fewer signals when it was applied to the TWAS gene statistics from JEPEGMIX2 (Table 4). JEPEGMIX2‐P and MAGMA found 1 common specific tissue‐pathway for BIP (GO_REGULATION_OF_PROTEIN_SUMOYLATION in the Brain Cerebellar Hemisphere tissue, with/without conditional analyses) (Text S10 and Figure S29 in Supporting Information), 1 for MDD (Text S10 and Figure S30 in Supporting Information) only for the unconditional analysis (GNF2_ZAP70 for the Pancreas tissue) and 16 for SCZ (Text S10 and Figure S31 in Supporting Information) only for the unconditional analysis (e.g., GO_COMPLEMENT_BINDING, REACTOME_COMPLEMENT_CASCADE and REACTOME_REGULATION_OF_COMPLEMENT_CASCADE for the Brain Cerebellum tissue).
TABLE 4.
Numbers of pathway signals found by JEPEGMIX2‐P and MAGMA
| Trait | JEPEGMIX2‐P without conditional analysis | JEPEGMIX2‐P conditional analysis | MAGMA unconditional analysis | |||
|---|---|---|---|---|---|---|
| FDR | Holm | FDR | Holm | FDR | Holm | |
| ADHD | — | — | — | — | — | — |
| AUT | 2 | 2 | 2 | 2 | — | — |
| BIP | 149 | 14 | 88 | 16 | 3 | 3 |
| ED | 590 | 6 | 1 | 1 | ‐ | ‐ |
| MDD | 607 | 31 | 7 | 2 | 2 | 1 |
| SCZ | 3,342 | 824 | 27 | — | 113 | 16 |
| PTSD | — | — | — | — | — | — |
4. DISCUSSION
The discovery of biological pathways implicated in diseases is the target for any genetic analysis. Despite the numerous methods available for pathway analyses, none of these methods rely solely on eQTLs to infer the association between expression of genes in pathway and trait, which is widely posited to be the critical causal mechanism. To overcome these two main limitations, we propose JEPEGMIX2‐P method for testing the association between pathway expression and trait. Even for pathways enriched GWAS and in high LD, JEPEGMIX2‐P with its automatic weights fully controls the false positive rates at or below nominal levels.
While the method is a novel addition to the pathway tools repertoire, it still has major limitations when attempting to use it for assigning “causal” tissues/cell types. Firstly, due to the rather small sample sizes of existing GE experiments ~80% of genes do not have good GE prediction from cis‐SNPs. Secondly, there is a large difference between the sample sizes available for different tissues; generally, tissues that are more accessible (peripheral tissues and blood) have larger sample sizes, and thus have more accurate predictions for more genes. Given the large sample size discrepancies, from the applied TWAS analyses probably the most important findings are the pathways and genes that are associated with the trait, not necessarily the tissue/single cell where they are (the most) significant. This argument is supported by recent research (Hekselman & Yeger‐Lotem, 2020); it shows that while mis‐regulation of gene is causal in one/few tissues, these genes might be mis‐regulated in many other tissues. Consequently, even if we do not assay the gene/pathway in the causal cell types directly, we can find the mis‐regulated genes/pathways by detecting signals in some other tissues and cell types. Unfortunately, until we have similarly large sample sizes for all tissues, the causal tissue discovery will require additional validation effort involving wet‐lab experiments (Chatzinakos, Georgiadis, & Daskalakis, 2020).
After applying our method broadly to neuropsychiatric GWAS, our pathway results bring forward two main questions: i) why there are so many pathway signals and ii) how these signals can be used in practice. There are many signals for some diseases, for example, SCZ, mainly due (a) passenger effect (in unconditional analyses) for pathway having just one/a few genes near large signals, (b) the same pathway yielding significant findings in multiple tissues, and (c) some pathways in MSigDB database sharing many genes. The number of signals is greatly reduced by summarizing pathway p‐value across all tissues. While having numerous signals might be overwhelming for some researchers, they can be very useful for fine‐mapping gene signals. Intuitively, pathways with significant signals are more likely to include many genes that are causal, not just LD passengers alongside true signals, and that genes in these pathways have a larger probability to be causal. Consequently, an heuristic approach to gene fine‐mapping might consist of i) counting how many times each gene (with significant signals) is found in significant pathways and ii) revealing clusters of signals coming from genes in LD. The gene(s) with the highest number of counts of significant pathways including it (them) is (are) the most likely candidate(s) to be causal.
By applying JEPEGMIX2‐P to psychiatric phenotypes, we uncovered numerous genes and pathways with significant signals for SCZ, AUT, BIP, ED, and MDD. Here we provide a viewpoint on the overall picture of the pathway analyses results. The major finding is that the signals fall, in the order of their strength, roughly into three main named categories: (a) immune related, (b) cell cycle/RNA transcription, and (c) synapse/dendrite regulation. While it might appear counterintuitive that general biological processes (i.e., immunity and cell cycle) to be more heavily involved in psychiatric disorders than the neuron specific ones, it is confirmed by well‐known prior results: (a) most significant signals among the first batch of replicated psychiatric disorder signals were in the MHC region on chromosome 6 (Stefansson et al., 2009), and (b) immune pathways were already implicated in psychiatric diseases like SCZ, likely due to be being involved in synapse pruning/maintenance (Sekar et al., 2016). We note that “REACTOME_COMPLEMENT_CASCADE” pathway is the nonchromosomal band pathway for SCZ with the most significant signals, under both unconditional (MHC and C4 excluded) and conditional analyses (Figures 2 and 3); thus, our finding aligns well with the involvement of the complement pathway in SCZ pathogenesis (Sekar et al., 2016).
The fact that general biological processes seem more consequential rather than strictly neuronal might also indirectly explain the fact that the drug development for psychiatric disorders is an exceedingly low yield affair (Hyman, 2013), presumably due pharmaceutical companies focusing exclusively on neuronal specific pathways. These prior drug development failures in the last 15 years led GlaxoSmithKline to exit the field, and Pfizer and AstraZeneca to drastically reduce internal drug development for psychiatric disorders (Hyman, 2013). Nonetheless, the prominent involvement of the immune pathways suggests that it is reasonable to envision drug repurposing of common immune modulatory drugs in psychiatry.
Interpreting and validating all pathway signals require substantially more work. However, JEPEGMIX2‐P provides carefully vetted targets for wet‐lab validation (see Supplementary Excel 2, https://cutt.ly/ypFpWdh) as our findings allow to generate somewhat narrower hypotheses for future testing (Chatzinakos et al., 2020). For instance, in ED results, the most significant signal corresponds to GEISS_RESPONSE_TO_DSRNA_UP pathway (Supplementary Excel file 2 https://cutt.ly/ypFpWdh), which is a pathway that is involved in response to virus infections. The immune system was already strongly suspected to have a role in ED etiology, for example, a large study of 0.5 million Danish girls showed that a severe childhood infection greatly increases the chances of developing ED at puberty (Breithaupt et al., 2019). Thus, similar to other psychiatric disorder, our signals support previous findings that the immune system is implicated in ED. However, the dsRNA pathway ED signal suggests that a narrower focus could be accomplished, that is, pointing to the subset of the immune system responses dealing with virus infections could be the most relevant to ED etiology. This finding suggests that for ED clinicians might want to pay special attention to active virus infections in ED patients.
Concluding, JEPEGMIX2‐P is a novel transcriptomic method/software for pathway associations with complex traits. Unlike existing methods, JEPEGMIX2‐P has a linear computational burden when computing internally the LD correlation between SNPs and genes using a large and diverse reference panel (33 K). Moreover, it fully controls the false positive rates at or below nominal levels by performing a competitive test, that is, adjusting for the background enrichment of TWAS gene statistics. Being written in C ++, JEPEGMIX2‐P is very fast while future versions of the software will use (a) cis‐eQTL for all the available tissues in the v0.8 version of PredictDB (http://predictdb.hakyimlab.org/) and (b) larger reference panel (e.g., 10,000 more African American samples). Due to its internal computation of the correlations between gene/pathway TWAS signals, the software is very well positioned to have its functionality extended to gene level fine mapping.
CONFLICT OF INTEREST
Nikolaos P. Daskalakis has held a part‐time paid position at Cohen Veteran Biosciences, has served as a paid consultant for Sunovion Pharmaceuticals and is on the scientific advisory board for Sentio Solutions, Inc. for unrelated work. The remaining authors have nothing to disclose.
AUTHORS' CONTRIBUTIONS
Chris Chatzinakos and Silviu‐Alin Bacanu conceived and designed the method, simulations and applications. Chris Chatzinakos designed the code and performed all computations, conducted all simulations and produced the application results, created all visualizations and packaged/validated/maintained the software. Silviu‐Alin Bacanu supervised the work. Foivos Georgiadis, Vladimir I. Vladimirov, Anna Docherty, Bradley T. Webb and Brien P. Riley and Nikolaos P. Daskalakis contributed to the interpretation of the simulation experiments and application results. Donghyung Lee and Na Cai contributed to the data preparation. Jonathan Flint and Kenneth S. Kendler gave inputs regarding the overall interpretation of the method and results. Chris Chatzinakos and Silviu‐Alin Bacanu wrote the first draft and revisions of the manuscript. Foivos Georgiadis and Nikolaos P. Daskalakis gave input in the presentation of results and writing of the manuscript. All authors commented and edited all the version of the manuscript.
5.
Supporting information
Appendix S1. Supporting Information
ACKNOWLEDGMENTS
This study was supported by the 2019 Seed Grant from Silvio O. Conte Center for Stress Peptide Advanced Research, Education, & Dissemination (NIMH P50MH115874) to Chris Chatzinakos, an appointed KL2 award from Harvard Catalyst|The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences KL2TR002542, UL1TR002541) to Nikolaos P. Daskalakis, and NIMH (R21MH121909) to Nikolaos P. Daskalakis.
Chatzinakos C, Georgiadis F, Lee D, et al. TWAS pathway method greatly enhances the number of leads for uncovering the molecular underpinnings of psychiatric disorders. Am J Med Genet Part B. 2020;183B:454–463. 10.1002/ajmg.b.32823
Funding information National Center for Advancing Translational Sciences, Grant/Award Numbers: KL2TR002542, UL1TR002541; National Institute of Mental Health, Grant/Award Numbers: P50MH115874, R21MH121909
5.1. DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in https://www.med.unc.edu/pgc/download‐results/
REFERENCES
- Autism Spectrum Disorders Working Group of The Psychiatric Genomics, C . (2017). Meta‐analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Molecular Autism, 8, 21 10.1186/s13229-017-0137-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breithaupt, L. , Köhler‐Forsberg, O. , Larsen, J. T. , Benros, M. E. , Thornton, L. M. , Bulik, C. M. , & Petersen, L. (2019). Association of exposure to infections in childhood with risk of eating disorders in adolescent girls. JAMA Psychiatry, 76(8), 800–809. 10.1001/jamapsychiatry.2019.0297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik‐Sullivan, B. K. , Loh, P. R. , Finucane, H. K. , Ripke, S. , Yang, J. , Patterson, N. , … Neale, B. M. (2015). LD score regression distinguishes confounding from polygenicity in genome‐wide association studies. Nature Genetics, 47(3), 291–295. 10.1038/ng.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush, W. S. , & Moore, J. H. (2012). Chapter 11: Genome‐wide association studies. PLoS Computational Biology, 8(12), e1002822 10.1371/journal.pcbi.1002822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzinakos, C. , Georgiadis, F. , & Daskalakis, N. P. (2020). GWAS meets transcriptomics: From genetic letters to transcriptomic words of neuropsychiatric risk. Neuropsychopharmacology. 10.1038/s41386-020-00835-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzinakos, C. , Lee, D. , Webb, B. T. , Vladimirov, V. I. , Kendler, K. S. , & Bacanu, S. A. (2018). JEPEGMIX2: Improved gene‐level joint analysis of eQTLs in cosmopolitan cohorts. Bioinformatics, 34(2), 286–288. 10.1093/bioinformatics/btx509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw, C. A. , Mooij, J. M. , Heskes, T. , & Posthuma, D. (2015). MAGMA: Generalized gene‐set analysis of GWAS data. PLoS Computational Biology, 11(4), e1004219 10.1371/journal.pcbi.1004219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis, D. , Walters, R. K. , Martin, J. , Mattheisen, M. , Als, T. D. , Agerbo, E. , … Neale, B. M. (2019). Discovery of the first genome‐wide significant risk loci for ADHD. Nature Genetics, 51, 63–75. 10.1101/145581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan, L. , Yilmaz, Z. , Gaspar, H. , Walters, R. , Goldstein, J. , Anttila, V. , … Grp, E. D. W. (2017). Significant locus and metabolic genetic correlations revealed in genome‐wide association study of anorexia nervosa. American Journal of Psychiatry, 174(9), 850–858. doi: 10.1176/appi.ajp.2017.16121402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson, V. , Thorleifsson, G. , Zhang, B. , Leonardson, A. S. , Zink, F. , Zhu, J. , … Stefansson, K. (2008). Genetics of gene expression and its effect on disease. Nature, 452(7186), 423–428. doi: 10.1038/nature06758 [DOI] [PubMed] [Google Scholar]
- Finucane, H. K. , Bulik‐Sullivan, B. , Gusev, A. , Trynka, G. , Reshef, Y. , Loh, P. R. , … Price, A. L. (2015). Partitioning heritability by functional annotation using genome‐wide association summary statistics. Nature Genetics, 47(11), 1228–1235. 10.1038/ng.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon, E. R. , Wheeler, H. E. , Shah, K. P. , Mozaffari, S. V. , Aquino‐Michaels, K. , Carroll, R. J. , … Im, H. K. (2015). A gene‐based association method for mapping traits using reference transcriptome data. Nature Genetics, 47(9), 1091–1098. doi: 10.1038/ng.3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev, A. , Ko, A. , Shi, H. , Bhatia, G. , Chung, W. , Penninx, B. W. , … Pasaniuc, B. (2016). Integrative approaches for large‐scale transcriptome‐wide association studies. Nature Genetics, 48(3), 245–252. doi: 10.1038/ng.3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekselman, I. , & Yeger‐Lotem, E. (2020). Mechanisms of tissue and cell‐type specificity in heritable traits and diseases. Nature Reviews Genetics, 21, 137–150. 10.1038/s41576-019-0200-9 [DOI] [PubMed] [Google Scholar]
- Holmans, P. , Green, E. K. , Pahwa, J. S. , Ferreira, M. A. , Purcell, S. M. , Sklar, P. , … Craddock, N. (2009). Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. American Journal of Human Genetics, 85(1), 13–24. 10.1016/j.ajhg.2009.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman, S. E. (2013). Psychiatric drug development: Diagnosing a crisis. Cerebrum: The Dana Forum on Brain Science, 2013, 5–5. Retrieved from. https://pubmed.ncbi.nlm.nih.gov/23720708, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3662213/ [PMC free article] [PubMed] [Google Scholar]
- Jin, L. , Zuo, X. Y. , Su, W. Y. , Zhao, X. L. , Yuan, M. Q. , Han, L. Z. , … Rao, S. Q. (2014). Pathway‐based analysis tools for complex diseases: A review. Genomics, Proteomics & Bioinformatics, 12(5), 210–220. doi: 10.1016/j.gpb.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D. , Bigdeli, T. B. , Riley, B. P. , Fanous, A. H. , & Bacanu, S. A. (2013). DIST: Direct imputation of summary statistics for unmeasured SNPs. Bioinformatics, 29(22), 2925–2927. 10.1093/bioinformatics/btt500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D. , Bigdeli, T. B. , Williamson, V. S. , Vladimirov, V. I. , Riley, B. P. , Fanous, A. H. , & Bacanu, S. A. (2015). DISTMIX: Direct imputation of summary statistics for unmeasured SNPs from mixed ethnicity cohorts. Bioinformatics, 29(22), 2925–2927. 10.1093/bioinformatics/btv348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, P. H. , O'Dushlaine, C. , Thomas, B. , & Purcell, S. M. (2012). INRICH: Interval‐based enrichment analysis for genome‐wide association studies. Bioinformatics, 28(13), 1797–1799. 10.1093/bioinformatics/bts191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon, A. (2014). A description of the molecular signatures database (MSigDB) web site. Methods in Molecular Biology, 1150, 153–160. 10.1007/978-1-4939-0512-6_9 [DOI] [PubMed] [Google Scholar]
- Liberzon, A. , Birger, C. , Thorvaldsdottir, H. , Ghandi, M. , Mesirov, J. P. , & Tamayo, P. (2015). The molecular signatures database (MSigDB) hallmark gene set collection. Cell Systems, 1(6), 417–425. 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon, A. , Subramanian, A. , Pinchback, R. , Thorvaldsdottir, H. , Tamayo, P. , & Mesirov, J. P. (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics, 27(12), 1739–1740. 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, S. , Das, S. , Kretzschmar, W. , Delaneau, O. , Wood, A. R. , Teumer, A. , … Haplotype Reference, C. (2016). A reference panel of 64,976 haplotypes for genotype imputation. Nature Genetics, 48(10), 1279–1283. doi: 10.1038/ng.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nievergelt, C. M. , Maihofer, A. X. , Klengel, T. , Atkinson, E. G. , Chen, C. Y. , Choi, K. W. , … Koenen, K. C. (2019). International meta‐analysis of PTSD genome‐wide association studies identifies sex‐ and ancestry‐specific genetic risk loci. Nature Communications, 10(1), 4558. doi: 10.1038/s41467-019-12576-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan, V. K. , Shen, L. , Moore, J. H. , & Saykin, A. J. (2012). Pathway analysis of genomic data: Concepts, methods, and prospects for future development. Trends in Genetics, 28(7), 323–332. 10.1016/j.tig.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin, E. J. , Lage, K. , Raychaudhuri, S. , Xavier, R. J. , Tatar, D. , Benita, Y. , … Daly, M. J. (2011). Proteins encoded in genomic regions associated with immune‐mediated disease physically interact and suggest underlying biology. PLoS Genetics, 7(1), e1001273. doi: 10.1371/journal.pgen.1001273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics, C . (2014). Biological insights from 108 schizophrenia‐associated genetic loci. Nature, 511(7510), 421–427. 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segre, A. V. , Groop, L. , Mootha, V. K. , Daly, M. J. , Altshuler, D. , Consortium, D. , & Investigators, M . (2010). Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genetics, 6(8), e1001058 10.1371/journal.pgen.1001058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar, A. , Bialas, A. R. , de Rivera, H. , Davis, A. , Hammond, T. R. , Kamitaki, N. , … McCarroll, S. A. (2016). Schizophrenia risk from complex variation of complement component 4. Nature, 530(7589), 177–183. doi: 10.1038/nature16549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl, E. A. , Breen, G. , Forstner, A. J. , McQuillin, A. , Ripke, S. , Trubetskoy, V ., … Bipolar Disorder Working Group of the Psychiatric Genomics, C . (2019). Genome‐wide association study identifies 30 loci associated with bipolar disorder. Nature Genetics, 51(5), 793–803. doi: 10.1038/s41588-019-0397-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson, H. , Ophoff, R. A. , Steinberg, S. , Andreassen, O. A. , Cichon, S. , Rujescu, D. , … Collier, D. A. (2009). Common variants conferring risk of schizophrenia. Nature, 460(7256), 744–747. doi: 10.1038/nature08186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian, A. , Tamayo, P. , Mootha, V. K. , Mukherjee, S. , Ebert, B. L. , Gillette, M. A. , … Mesirov, J. P. (2005). Gene set enrichment analysis: A knowledge‐based approach for interpreting genome‐wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America, 102(43), 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). Analysing biological pathways in genome‐wide association studies. Nature Reviews Genetics, 11(12), 843–854. 10.1038/nrg2884 [DOI] [PubMed] [Google Scholar]
- Wray, N. R. , Ripke, S. , Mattheisen, M. , Trzaskowski, M. , Byrne, E. M. , Abdellaoui, A ., … Major Depressive Disorder Working Group of the Psychiatric Genomics, C . (2018). Genome‐wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nature Genetics, 50(5), 668–681. doi: 10.1038/s41588-018-0090-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information
Data Availability Statement
The data that support the findings of this study are openly available in https://www.med.unc.edu/pgc/download‐results/