

Abstract
The level of fetal hemoglobin (HbF) is an important disease modifier for β‐thalassemia and sickle cell disease patients. Indeed, genetic tinkering with the HbF repression machinery has demonstrated great potential for disease mitigation. Such genetic treatments are costly and the high incidence of β‐hemoglobinopathies in low‐income countries, therefore, calls for the development of affordable, off‐the‐shelf, oral treatments. The use of PROTAC (PRoteolysis TArgeting Chimeras) technology to influence the epigenetic mechanisms involved in HbF suppression may provide a solution. In this minireview, we briefly explain the HbF repression network highlighting the epigenetic factors that could be targeted for degradation by PROTACs. We hope that this review will inspire clinicians, molecular and chemical biologists to collaborate and contribute to this fascinating field, which should ultimately deliver drugs that reactivate HbF expression with high specificity and low toxicity.
Keywords: Drug discovery, Gene expression, Epigenetics, Targeted protein degradation, β-Hemoglobinopathies, Hemoglobin switching
Closing the gap! The level of fetal hemoglobin (HbF) is an important disease modifier for β‐thalassemia and sickle cell disease patients. Indeed, genetic tinkering with the HbF repression machinery has demonstrated great potential for disease mitigation. This minireview provides an overview of the epigenetic crosstalk involved in fetal HbF repression, discussing the possibility of using targeted protein degradation for β‐hemoglobinopathy treatments.
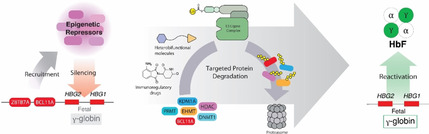
1. The urgent need to develop novel drugs for β‐hemoglobinopathy patients
To transport oxygen and carbon dioxide between the lungs and all other organs, red blood cells rely on adult hemoglobin (HbA), a tetrameric protein containing two α‐globin and two β‐globin subunits. Patients with dysfunctional or an insufficient amount of β‐globin suffer from the most common life‐threatening genetic diseases in the world, collectively referred to as β‐hemoglobinopathies. The molecular pathophysiology of these diseases, such as sickle cell disease, has been known for years, yet therapeutic options remain very limited. [1] Sickle cell disease is caused by a point mutation in the β‐globin encoding gene HBB that causes the affected sickle hemoglobin (HbS) to polymerize under low‐oxygen conditions. As a result, red blood cells become sickle‐shaped and tend to block capillaries, which causes the accumulation of ischemic damage throughout the body. Gene therapy holds great promise for curative treatment, [2] however, with the majority of patients living in low‐ and middle‐income countries, [3] most patients have no access to the healthcare infrastructure that gene therapy requires. A treatment in the form of an orally available drug is desperately needed. Before birth, red blood cells express fetal hemoglobin (HbF) that contains two α‐globin and two γ‐globin subunits. After birth, the switch from γ‐globin to β‐globin expression marks the onset of symptoms in β‐hemoglobinopathy patients. Conversely, patients with Hereditary Persistence of Fetal Hemoglobin (HPFH) show a milder phenotype, [4] hence drug development focusses on the reactivation of the γ‐globin encoding genes HBG1 and HBG2. [5] Hydroxyurea, a ribonucleotide reductase inhibitor, was the first FDA approved drug to elevate HbF levels in sickle cell patients [6] and is still considered the primary disease‐modifying compound with oral availability. [7] The level of HbF induction, however, varies among patients and thus the search continues for more potent and more specific compounds that derepress HbF. Repression of the HBG1/2 genes is well‐studied, and many transcriptional regulators of the switch from HbF to HbA have been identified (see for instance supplementary Table 1 in Houwing et al. [1] ). Epigenetic regulation facilitates and maintains the hemoglobin switch after birth, and provides avenues for drug development. This minireview briefly discusses targeted protein degradation as an avenue for drug development for β‐hemoglobinopathies. With the development of bi‐functional, proximity‐inducing small molecules [8] it has become possible to design drugs that target regulators of the hemoglobin switch. Since epigenetics concerns reversible changes in phenotype without dependency on the DNA sequence, using small molecules to interfere with the epigenetic regulators would bypass the need for costly gene therapy.[9] Oral compounds might be applied to deplete red blood cell progenitors of HBG1/2 repressors, thereby inducing HbF and ameliorating the symptoms of β‐hemoglobinopathy patients.
2. Targeted protein degradation
The molecular network in charge of balancing the entire protein abundance — proteostasisis — vital for almost every biological process since proteins play a pivotal role in the regulation of cellular functions. [10] Thus, protein turnover allows cells to respond to environmental and intracellular cues rapidly. In addition, the balance between protein synthesis and degradation enables different cellular identities. [11] Importantly, disturbed proteostasis is implicated in numerous human diseases [12] such as cancer, immune‐ and neurological disorders. The major pathway of protein degradation is the ubiquitin‐proteasome system, in which a bridging E3 ubiquitin ligase is capable of modifying a lysine side chain of the substrate protein via forming an isopeptide bond with the carboxy terminus of ubiquitin. This post‐translational modification is responsible for the identification of proteins that should be degraded. Around 20 years ago the first proof‐of‐principle for exploiting the canonical ubiquitin‐proteasome system as on‐demand degradation machinery was established. [13] The so‐called PROTACs (PROteolysis TArgeting Chimeras; Figure 1) promote cellular protein degradation by inducing proximity of a ubiquitin ligase to the protein substrate. However, only recently the development of low‐molecular weight ligands that bind to ubiquitin ligases with high affinity and specificity have brought the full potential of this target‐cleavage methodology to light. [14] PROTACs have been reviewed elsewhere recently. [15] Fewer than a dozen molecules have been used as recruitment elements for a handful of E3 ligases within the heterobifunctional chimeras. Interestingly, immunomodulatory drugs (IMiDs) such as thalidomide derivatives can directly induce degradation [16] of diverse zinc finger transcription factors [17] and some other proteins [18] via a unique structural arrangement of a conserved glycine‐containing β‐hairpin of the protein of interest and the phthalimide group of the IMiD, which is bound to cereblon (CRBN) ligase. PROTACs offer several advantages in comparison with occupancy‐based inhibitors. Unlike conventional inhibitors, PROTACs can function regardless of where they bind on the target protein. Consequently, they are not restricted to active sites and may target any accessible region, modulating previously “undruggable targets” such as transcription factors. In addition, PROTACs act sub‐stoichiometrically through a catalytic mechanism. Therefore, compared to conventional inhibitors the concentration required for activity tends to be lower for PROTACs. This leads to fewer off‐target effects and toxicity in a therapeutic setting and a more selective chemical intervention on the desired target. However, we still lack general principles for PROTAC design [19] as well as standards for degrader validation. [20] Currently, the field relies on trial‐and‐error efforts of large synthetically complex compounds that do not follow Lipinski's “rule‐of‐5”. [21] Consequently, the discovery and successful development of functional ternary complexes requires substantial empirical testing where factors including stability, re‐synthesis rate of the target, linker composition, ligase profile expression and cellular uptake all need to be considered. [22] Furthermore, the typical activity decrease of PROTACs at high concentration — hook effect — [23] together with potentially adverse effects of complete protein depletion may bring challenges to the actual drug development of PROTACs. Poor pharmacokinetic and pharmacodynamic profiles, off‐target effects and adverse immunological reactions are other potential drawbacks of PROTACs that will have to be addressed on the road to drug development. Nevertheless, many active PROTACs have recently been designed in the field of epigenetics,[ 9 , 24 ] and new approaches such as light‐activated degraders are now available. [25] A fascinating new era of drug development is on the horizon with the entry of PROTACs in clinical trials. [26] In addition to good oral bioavailability,[ 15d , 27 ] the fact that PROTAC effects can be limited to specific organs, or even to cellular compartments, [28] by targeting tissue‐specific E3 ubiquitin ligases [15a] opens up new avenues for precision medicine available. [25]
Figure 1.
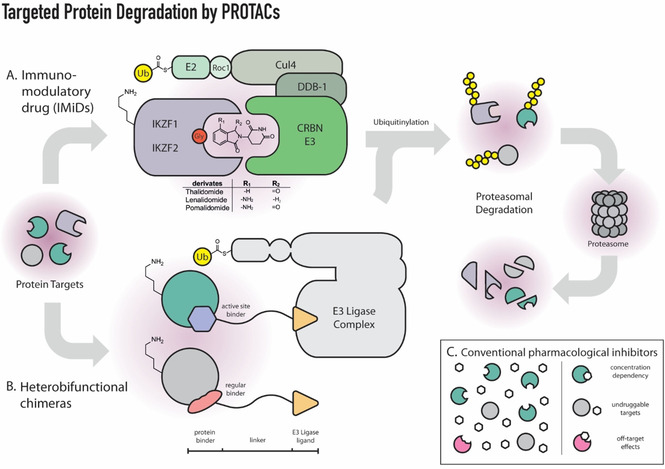
Schematic representation of the different types of substrate recruitment in PROTAC technology: A. Immunomodulatory drugs (IMiDs) illustrated for the degradation of the zinc finger proteins IKZF1 and IKZF2 via CRL4CRBN complex; B. Heterofunctional chimeras consisting of two protein‐binding molecules connected by a linker. Insert (C) highlights the concentration dependency and potential off‐target interactions of conventional pharmacological inhibitors.
3. Molecular regulation of HbF repression
After birth, hematopoiesis moves from the fetal liver to the bone marrow where adult erythrocytes are produced. PROTACs that aim to reverse the hemoglobin switch and instigate HbF expression should target the cellular factors that initiate and preserve repression of the HBG1/2 genes. Terminally differentiated erythrocytes are highly specialized small enucleated cells that contain a maximum amount of hemoglobin molecules to meet the respiratory needs of tissues throughout the body. To acquire this unique and highly specialized transcriptional program, the differentiating erythrocyte needs an equally unique epigenetic profile of DNA accessibility and DNA methylation. [29] An intricate network of transcription factors and chromatin remodeling complexes accounts for the differences in globin expression between fetal and adult erythrocytes. In adult erythrocytes, the zinc finger transcription factors BCL11A and ZBTB7A (also known as LRF) bind directly to the proximal promoters of the fetal hemoglobin genes HBG1 and HBG2. [30] The high‐level expression of genes in the HBB gene cluster is regulated by an upstream locus control region (LCR). [31] The LCR consists of five highly accessible chromatin sites that interact with transcription factors and RNA polymerase II. [32] Chromatin looping, facilitated by the lineage‐specific LDB1 protein, allows the LCR to activate the promoters of the cis‐linked β‐like globin genes (Figure 2). [33] In adult cells, BCL11A appears to prevent the formation of looped LCR‐promoter interactions at the fetal HBG1/2 genes. Consequently, the LCR loops further downstream to activate the adult HBB gene. Mutations that disrupt the BCL11A binding site in the HBG1/2 promoters result in higher γ‐globin expression. [34] Together with another key repressor, ZBTB7A, BCL11A recruits the Nucleosome Remodeling and Deacetylase (NuRD) silencing complex (Figure 3). [35] The NuRD complex represses the HBG1/2 genes with its epigenetic subunits such as HDAC2 which deacetylates lysine residues of histones. This results in a closed chromatin configuration and represses transcription. These epigenetic co‐regulators could be druggable targets, as illustrated by HDAC inhibitors that increase fetal hemoglobin levels. [36] Not only histone deacetylation but also histone demethylation regulates the hemoglobin switch. Inhibition of lysine‐specific histone demethylase KDM1 A (also known as LSD1) has shown therapeutic potential in experiments with both erythroid cells and mice. [17a] Treatment of baboons with LSD1 inhibitor RN‐1 also induced γ‐globin expression. [37] At the HBG1/2 promoters, RN‐1 treatment induced increased levels of H3K4me2 and H3K4me3, which are epigenetic histone modifications associated with increased chromatin accessibility and active transcription. [37] In addition to HDAC2, other subunits of the NuRD complex, are essential for full repression of fetal hemoglobin. These factors, such as nucleosome remodeler CHD4, structural subunits GATAD2A and MTA2, and MBD2, which binds methylated CpG residues in the DNA, would all be suitable targets for PROTAC‐mediated proteolysis. [38] A CRISPR‐Cas9‐mediated knock‐out of MBD2 in adult erythroid progenitor cells resulted in ±40 % fetal hemoglobin, [38] illustrating that depleting one NuRD component might be sufficient for successful treatment. A coiled‐coil domain of MBD2 interacts with GATAD2A to recruit the NuRD complex, and a knock‐down of GATAD2A results in a similar γ‐globin induction as was observed upon MBD2 knock‐down. [39] Furthermore, the administration of a GATAD2A‐binding peptide that competes with MBD2 increased γ‐globin expression levels. [39]
Figure 2.
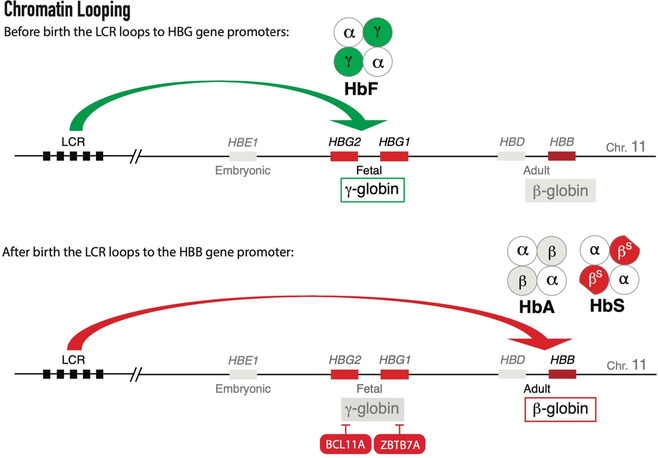
Chromatin looping activates genes in the human HBB locus: Before birth the locus control region loops to the fetal genes HBG1 and HBG2 activating γ‐globin expression; After birth, the fetal genes are silenced and the LCR loops further downstream to activate the adult gene HBB giving rise to adult (HbF) or sickling (HbS) hemoglobin tetramers.
Figure 3.
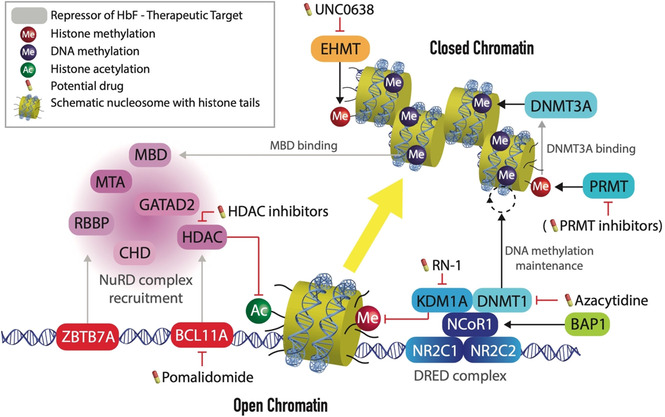
Therapeutic targets to raise HbF levels are the DNA‐binding and epigenetic factors that silence HbF. Silencing is initiated by key repressors BCL11A and ZBTB7A, that recruit the silencing NuRD complex. Together with the DRED complex, NuRD components instigate a closed chromatin conformation at the fetal genes stabilizing γ‐globin repression.
In addition to DNA accessibility, DNA methylation is a potentially important epigenetic determinant of γ‐globin expression. Active transcription in fetal cells is associated with hypomethylation of the HBG1/2 promoters. These promoters contain multiple CpG residues that — upon methylation — may enable DNA binding of MBD2, a component of the silencing NuRD complex. In mice, the Hbb genes are regulated by the interplay of H3K9 methyltransferase EHMT2 and H3K4 demethylase KDM5A. [40] In human cells, EHMT2 forms stable heterodimers with EHMT1 and both proteins are crucial for H3K9 methylation, a mark of heterochromatin. [41] These methyltransferases can be inhibited by the small molecule UNC0638 [42] and treatment of adult erythroid cells boosts HbF levels. [43] DNA methyltransferase DNMT1 is important for the maintenance of DNA methylation in the HBG1/2 promoters, and pharmacological inhibitors of this enzyme increase fetal hemoglobin levels. [16a] Hypomethylation of the promoters could explain the induction of fetal hemoglobin in patients treated with DNA hypomethylating agents such as azacytidine that was reported already in 1982. [44] More recently, a phase I clinical trial provided new evidence that by targeting DNMT1, HbF levels can be elevated in adult red blood cells. [45] The γ‐globin inducing potential of 5‐azacytidine is not attributed to global hypomethylation – including HBG1/2 promoters — but rather to a more focused (epi‐)genetic or post‐transcriptional mechanism. [46] DNMT1 is recruited to the HBG1/2 promoters as part of the DRED complex, together with KDM1A. Nuclear receptor corepressor‐1 (NCoR1) acts as a scaffold for these epigenetic enzymes and interacts with a heterodimer of DNA binding nuclear receptors NR2C1:NR2C2 (also known as TR2:TR4). [47] Full HbF repression requires sufficient amounts of NCoR1, which is regulated by the deubiquitinase BRCA1‐associated protein‐1 (BAP1). [34] Knock‐down of BAP1 was shown to increase NCoR1 ubiquitination and induce HbF expression unless it was combined with proteasomal inhibitor. [34] These experiments beautifully illustrate the therapeutic potential of hijacking the proteasomal machinery to target epigenetic repressors of HbF. While DNMT1 is responsible for maintenance of the DNA methylation, DNMT3 A is required for the initial methylation and is recruited by protein arginine methyltransferase PRMT5 which introduces the H4R3me2 repressive mark at the HBG1/2 promoters. [48] In this way, PRMT5 links histone methylation to DNA methylation. [48] In addition to PRMT5, PRMT1 and its chromatin target CHTOP provide additional therapeutic targets for γ‐globin induction. [49] Multiple small molecule inhibitors of PRMTs have been patented and, interestingly, the first three PRMT inhibitors that entered clinical trials target PRMT5 (GSK3326595 and JNJ‐64619178) or PRMT1 (GSK3368715). [50] Although these clinical trials are conducted in the context of malignancies, they merit further investigation of their use as HbF‐inducing agents.
Pomalidomide, a thalidomide derivative, was shown to increase HbF in erythroid cells [51] and mice. [52] Sievers et al. defined the C2H2 zinc finger degrome targeted by thalidomide derivatives. Their work suggests that the transcription factors BCL11A and ZBTB7A could be depleted with a specialized thalidomide derivative. [17c] Dulmovits et al., however, show that pomalidomide lowers cellular BCL11 A levels even in the presence of proteasomal inhibitors, indicating targeted proteasomal degradation cannot be the only mechanistic explanation for increased HbF levels. [53] PROTAC targeting of the epigenetic factors reviewed here might help destabilize the suppressed state of the HBG1/2 genes. Combinatorial treatments could increase the effect on γ‐globin expression while mitigating the risk of adverse effects. In addition to the now extensive list of druggable targets, researchers can choose from over 600 E3 ubiquitin ligases, of which until now only a small minority have been exploited for targeted protein degradation. Recently, SPOP, one of the substrate adapters of the CUL3 ubiquitin ligase complex, was shown to be important for HbF repression. [1] Genetic knock‐out of SPOP or CUL3 increased γ‐globin levels and slightly lowered BCL11A levels. [1]
4. Concluding remarks
The field of targeted protein degradation provides valuable research tools (reviewed by Wu et al. [54] ) and novel pharmacological modalities (reviewed by Bruslem and Crews [15d] ). It is rapidly evolving and is likely to expand significantly in the coming years. Kostic and Jones provided a tentative roadmap for further development of targeted protein degradation. [21] The field of drug development for β‐hemoglobinopathies should catch the wave of these newly developed tools. The current understanding of the fetal hemoglobin repression mechanism, [55] the availability of small compounds specifically targeting some of the key factors involved (Figure 3), and the demonstration that fetal hemoglobin expression can in principle be reactivated by peptides [39] collectively make a strong case for development of PROTACs in this field. To this end, this minireview combines a brief introduction to targeted protein degradation and an overview of the most tangible target proteins for epigenetic upregulation of fetal hemoglobin.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Sjaak Philipsen is a molecular biologist who studied at the University of Groningen (NL). For his postdoc he went to the National Institute for Medical Research (London, UK) where he performed functional dissection of the hypersensitive sites of the β‐globin Locus Control Region in transgenic mice. Currently, his laboratory in Rotterdam aims to understand the molecular control of erythropoiesis and to fully characterize the mechanisms that regulate the gamma‐globin suppression pathways.

Biographical Information
Thijs Verheul received his Bachelor of Science degree in Medicine from the Erasmus University in Rotterdam (NL). Before completing the clinical phase of the Master degree in Medicine, he interrupted his medical training for a Research Master in Molecular Medicine and is currently a PhD student in the Philipsen research group at the Erasmus MC in Rotterdam.

Biographical Information
Olalla Vázquez is currently enjoying an Assistant Professorship in Chemical Biology at Philipps‐Universität Marburg (DE). Her independent work focuses on the design of novel tools to understand and manipulate the biological processes behind relevant diseases. Optochemical tools and epigenetic chemical probes are the core of her research.

Biographical Information
Van Tuan Trinh received his Bachelor and Master degree in Chemistry from the Humboldt Universität zu Berlin (DE). Afterward, he joined the chemistry‐for‐healthcare enterprise Belyntic GmbH as a research scientist for six months. He is currently a PhD student in the Vázquez research group at Philipps‐Universität Marburg.

Acknowledgements
The laboratory of S.P. was supported by the Landsteiner Foundation for Blood Transfusion Research (1627), the Netherlands Scientific Organization ZonMw (40‐00812‐98‐12128), Erasmus MC Human Disease Model Award, and the Friends of Sophia Children's Hospital Foundation WAR20‐21. The laboratory of O.V. was supported by the DFG program TRR81: Chromatin Changes in Differentiation and Malignancies (TRR81/3 Z04). Open access funding enabled and organized by Projekt DEAL.
T. C. J. Verheul, V. T. Trinh, O. Vázquez, S. Philipsen, ChemMedChem 2020, 15, 2436.
Contributor Information
Prof. Dr. Olalla Vázquez, Email: olalla.vazquez@staff.uni-marburg.de.
Prof. Dr. Sjaak Philipsen, Email: j.philipsen@erasmusmc.nl.
References
- 1. Houwing M. E., de Pagter P. J., van Beers E. J., Biemond B. J., Rettenbacher E., Rijneveld A. W., Schols E. M., Philipsen J. N. J., Tamminga R. Y. J., Fijnvandraat K., Nur E., Cnossen M. H., Consortium S., Blood Rev 2019, 37, 100580. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Lidonnici M. R., Ferrari G., Blood Cells, Molecules, and Diseases 2018, 70, 87–101; [DOI] [PubMed] [Google Scholar]
- 2b. Bourzac K., Nature 2017, 549, S28-S30; [DOI] [PubMed] [Google Scholar]
- 2c. Zeng J., Wu Y., Ren C., Bonanno J., Shen A. H., Shea D., Gehrke J. M., Clement K., Luk K., Yao Q., Kim R., Wolfe S. A., Manis J. P., Pinello L., Joung J. K., Bauer D. E., Nat. Med. 2020, 26, 535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Kato G. J., Piel F. B., Reid C. D., Gaston M. H., Ohene-Frempong K., Krishnamurti L., Smith W. R., Panepinto J. A., Weatherall D. J., Costa F. F., Vichinsky E. P., Nature Reviews Disease Primers 2018, 4, 18010; [DOI] [PubMed] [Google Scholar]
- 3b. De Sanctis V., Med. J. Hematol. Inf. Dis. 2017, 9, e2017018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Ngo D. A., Aygun B., Akinsheye I., Hankins J. S., Bhan I., Luo H. Y., Steinberg M. H., Chui D. H. K., Br. J. Haematol. 2012, 156, 259–264; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Steinberg M. H., Chui D. H. K., Dover G. J., Sebastiani P., Alsultan A., Blood 2014, 123, 481–485. [DOI] [PubMed] [Google Scholar]
- 5. Lettre G., Bauer D. E., The Lancet 2016, 387, 2554–2564. [DOI] [PubMed] [Google Scholar]
- 6. Charache S., Terrin M. L., Moore R. D., Dover G. J., Barton F. B., Eckert S. V., McMahon R. P., Bonds D. R., N. Engl. J. Med. 1995, 332, 1317–1322. [DOI] [PubMed] [Google Scholar]
- 7. McGann P. T., Ware R. E., Expert Opin. Drug Saf. 2015, 14, 1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gerry C. J., Schreiber S. L., Nat. Chem. Biol. 2020, 16, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vogelmann A., Robaa D., Sippl W., Jung M., Curr. Opin. Chem. Biol. 2020, 57, 8–16. [DOI] [PubMed] [Google Scholar]
- 10. Ciechanover A., Biochim. Biophys. Acta 2012, 1824, 3–13. [DOI] [PubMed] [Google Scholar]
- 11. Bludau I., Aebersold R., Nat. Rev. Mol. Cell Biol. 2020, 21, 327–340. [DOI] [PubMed] [Google Scholar]
- 12. Labbadia J., Morimoto R. I., Annu. Rev. Biochem. 2015, 84, 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sakamoto K. M., Kim K. B., Kumagai A., Mercurio F., Crews C. M., Deshaies R. J., Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Toure M., Crews C. M., Angew. Chem. Int. Ed. Engl. 2016, 55, 1966–1973; [DOI] [PubMed] [Google Scholar]
- 14b. Raina K., Crews C. M., Curr. Opin. Chem. Biol. 2017, 39, 46–53; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Testa A., Hughes S. J., Lucas X., Wright J. E., Ciulli A., Angew. Chem. Int. Ed. Engl. 2020, 59, 1727–1734; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14d. An S., Fu L., EBioMedicine 2018, 36, 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Schapira M., Calabrese M. F., Bullock A. N., Crews C. M., Nat. Rev. Drug Discovery 2019, 18, 949–963; [DOI] [PubMed] [Google Scholar]
- 15b. Churcher I., J. Med. Chem. 2018, 61, 444–452; [DOI] [PubMed] [Google Scholar]
- 15c. Naito M., Ohoka N., Shibata N., Tsukumo Y., Front. Chem. 2019, 7, 849; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15d. Burslem G. M., Crews C. M., Cell 2020, 181, 102–114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15e. Gao H., Sun X., Rao Y., ACS Med. Chem. Lett. 2020, 11, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Banzon V., Ibanez V., Vaitkus K., Ruiz M. A., Peterson K., Desimone J., Lavelle D., Experimental Hematology 2011, 39, 26–36.e21; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Gandhi A. K., Kang J., Havens C. G., Conklin T., Ning Y., Wu L., Ito T., Ando H., Waldman M. F., Thakurta A., Klippel A., Handa H., Daniel T. O., Schafer P. H., Chopra R., Br. J. Haematol. 2014, 164, 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Kronke J., Udeshi N. D., Narla A., Grauman P., Hurst S. N., McConkey M., Svinkina T., Heckl D., Comer E., Li X., Ciarlo C., Hartman E., Munshi N., Schenone M., Schreiber S. L., Carr S. A., Ebert B. L., Science 2014, 343, 301–305; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Lu G., Middleton R. E., Sun H., Naniong M., Ott C. J., Mitsiades C. S., Wong K. K., Bradner J. E., W. G. Kaelin, Jr. , Science 2014, 343, 305–309; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c. Sievers Q. L., Petzold G., Bunker R. D., Renneville A., Slabicki M., Liddicoat B. J., Abdulrahman W., Mikkelsen T., Ebert B. L., Thoma N. H., Science 2018, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Petzold G., Fischer E. S., Thoma N. H., Nature 2016, 532, 127–130; [DOI] [PubMed] [Google Scholar]
- 18b. Matyskiela M. E., Lu G., Ito T., Pagarigan B., Lu C. C., Miller K., Fang W., Wang N. Y., Nguyen D., Houston J., Carmel G., Tran T., Riley M., Nosaka L., Lander G. C., Gaidarova S., Xu S., Ruchelman A. L., Handa H., Carmichael J., Daniel T. O., Cathers B. E., Lopez-Girona A., Chamberlain P. P., Nature 2016, 535, 252–257. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Nowak R. P., DeAngelo S. L., Buckley D., He Z., Donovan K. A., An J., Safaee N., Jedrychowski M. P., Ponthier C. M., Ishoey M., Zhang T., Mancias J. D., Gray N. S., Bradner J. E., Fischer E. S., Nat. Chem. Biol. 2018, 14, 706–714; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Cyrus K., Wehenkel M., Choi E. Y., Han H. J., Lee H., Swanson H., Kim K. B., Mol. BioSyst. 2011, 7, 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jaeger M. G., Winter G. E., Cell Chem. Biol. 2020, 27, 14–16. [DOI] [PubMed] [Google Scholar]
- 21. Kostic M., Jones L. H., Trends Pharmacol. Sci. 2020, 41, 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maniaci C., Ciulli A., Curr. Opin. Chem. Biol. 2019, 52, 145–156. [DOI] [PubMed] [Google Scholar]
- 23. Bett J. S., Essays Biochem. 2016, 60, 143–151. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a.K. Cermakova, H. C. Hodges, Molecules 2018, 28; [DOI] [PMC free article] [PubMed]
- 24b.M. Schiedel, M. Moroglu, D. M. H. Ascough, A. E. R. Chamberlain, J. Kamps, A. R. Sekirnik, S. J. Conway, Angew. Chem. Int. Ed. Engl 2019, 58, 17930–17952; [DOI] [PubMed]
- 24c.Y. Itoh, Chem. Rec 2018, 18, 1681–1700. [DOI] [PubMed]
- 25.
- 25a. Xue G., Wang K., Zhou D., Zhong H., Pan Z., J. Am. Chem. Soc. 2019, 141, 18370–18374; [DOI] [PubMed] [Google Scholar]
- 25b. Pfaff P., Samarasinghe K. T. G., Crews C. M., Carreira E. M., ACS Cent. Sci. 2019, 5, 1682–1690; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25c. Naro Y., Darrah K., Deiters A., J. Am. Chem. Soc. 2020, 142, 2193–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mullard A., Nat. Rev. Drug Discovery 2019. [Google Scholar]
- 27. Sun X., Wang J., Yao X., Zheng W., Mao Y., Lan T., Wang L., Sun Y., Zhang X., Zhao Q., Zhao J., Xiao R. P., Zhang X., Ji G., Rao Y., Cell Discov. 2019, 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Y., Zhang S., Zhang J., Hu Z., Xiao Y., Huang J., Dong C., Huang S., Zhou H. B., Eur. J. Med. Chem. 2019, 172, 48–61. [DOI] [PubMed] [Google Scholar]
- 29. Schulz V. P., Yan H., Lezon-Geyda K., An X., Hale J., Hillyer C. D., Mohandas N., Gallagher P. G., Cell Rep. 2019, 28, 2996–3009 e2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martyn G. E., Wienert B., Yang L., Shah M., Norton L. J., Burdach J., Kurita R., Nakamura Y., Pearson R. C. M., Funnell A. P. W., Quinlan K. G. R., Crossley M., Nat. Genet. 2018, 50, 498–503. [DOI] [PubMed] [Google Scholar]
- 31. Liang S., Moghimi B., Yang T. P., Strouboulis J., Bungert J., J. Cell. Biochem. 2008, 105, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson K. D., Grass J. A., Park C., Im H., Choi K., Bresnick E. H., Mol. Cell. Biol. 2003, 23, 6484–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krivega I., Dean A., Nucleic Acids Res. 2017, 45, 8255–8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu N., Hargreaves V. V., Zhu Q., Kurland J. V., Hong J., Kim W., Sher F., Macias-Trevino C., Rogers J. M., Kurita R., Nakamura Y., Yuan G.-C., Bauer D. E., Xu J., Bulyk M. L., Orkin S. H., Cell 2018, 173, 430–442.e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Masuda T., Wang X., Maeda M., Canver M. C., Sher F., Funnell A. P. W., Fisher C., Suciu M., Martyn G. E., Norton L. J., Zhu C., Kurita R., Nakamura Y., Xu J., Higgs D. R., Crossley M., Bauer D. E., Orkin S. H., Kharchenko P. V., Maeda T., Science 2016, 351, 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Esrick E. B., McConkey M., Lin K., Frisbee A., Ebert B. L., Am. J. Hematol. 2015, 90, 624–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rivers A., Vaitkus K., Ibanez V., Ruiz M. A., Jagadeeswaran R., Saunthararajah Y., Cui S., Engel J. D., Desimone J., Lavelle D., Haematologica 2016, 101, 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sher F., Hossain M., Seruggia D., Schoonenberg V. A. C., Yao Q., Cifani P., Dassama L. M. K., Cole M. A., Ren C., Vinjamur D. S., Macias-Trevino C., Luk K., McGuckin C., Schupp P. G., Canver M. C., Kurita R., Nakamura Y., Fujiwara Y., Wolfe S. A., Pinello L., Maeda T., Kentsis A., Orkin S. H., Bauer D. E., Nat. Genet. 2019, 51, 1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gnanapragasam M. N., Scarsdale J. N., Amaya M. L., Webb H. D., Desai M. A., Walavalkar N. M., Wang S. Z., Zu Zhu S., Ginder G. D., Williams D. C., Proc. Natl. Acad. Sci. USA 2011, 108, 7487–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chaturvedi C. P., Somasundaram B., Singh K., Carpenedo R. L., Stanford W. L., Dilworth F. J., Brand M., Proc. Natl. Acad. Sci. USA 2012, 109, 18845–18850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tachibana M., Ueda J., Fukuda M., Takeda N., Ohta T., Iwanari H., Sakihama T., Kodama T., Hamakubo T., Shinkai Y., Genes Dev. 2005, 19, 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vedadi M., Barsyte-Lovejoy D., Liu F., Rival-Gervier S., Allali-Hassani A., Labrie V., Wigle T. J., Dimaggio P. A., Wasney G. A., Siarheyeva A., Dong A., Tempel W., Wang S. C., Chen X., Chau I., Mangano T. J., Huang X. P., Simpson C. D., Pattenden S. G., Norris J. L., Kireev D. B., Tripathy A., Edwards A., Roth B. L., Janzen W. P., Garcia B. A., Petronis A., Ellis J., Brown P. J., Frye S. V., Arrowsmith C. H., Jin J., Nat. Chem. Biol. 2011, 7, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.
- 43a. Krivega I., Byrnes C., de Vasconcellos J. F., Lee Y. T., Kaushal M., Dean A., Miller J. L., Blood 2015, 126, 665–672; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43b. Renneville A., Van Galen P., Canver M. C., McConkey M., Krill-Burger J. M., Dorfman D. M., Holson E. B., Bernstein B. E., Orkin S. H., Bauer D. E., Ebert B. L., Blood 2015, 126, 1930–1939; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43c. Nualkaew T., Khamphikham P., Pongpaksupasin P., Kaewsakulthong W., Songdej D., Paiboonsukwong K., Sripichai O., Engel J. D., Hongeng S., Fucharoen S., Jearawiriyapaisarn N., Ann. Hematol. 2020, 99, 2027–2036. [DOI] [PubMed] [Google Scholar]
- 44. Ley T. J., DeSimone J., Anagnou N. P., Keller G. H., Humphries R. K., Turner P. H., Young N. S., Keller P., Nienhuis A. W., N. Engl. J. Med. 1982, 307, 1469–1475. [DOI] [PubMed] [Google Scholar]
- 45. Molokie R., Lavelle D., Gowhari M., Pacini M., Krauz L., Hassan J., Ibanez V., Ruiz M. A., Ng K. P., Woost P., Radivoyevitch T., Pacelli D., Fada S., Rump M., Hsieh M., Tisdale J. F., Jacobberger J., Phelps M., Engel J. D., Saraf S., Hsu L. L., Gordeuk V., Desimone J., Saunthararajah Y., PLoS Med. 2017, 14, e1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mabaera R., Greene M. R., Richardson C. A., Conine S. J., Kozul C. D., Lowrey C. H., Blood 2008, 111, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cui S., Kolodziej K. E., Obara N., Amaral-Psarris A., Demmers J., Shi L., Engel J. D., Grosveld F., Strouboulis J., Tanabe O., Mol. Cell. Biol. 2011, 31, 3298–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rank G., Cerruti L., Simpson R. J., Moritz R. L., Jane S. M., Zhao Q., Blood 2010, 116, 1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. van Dijk T. B., Gillemans N., Pourfarzad F., van Lom K., von Lindern M., Grosveld F., Philipsen S., Blood 2010, 116, 4349–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li X., Wang C., Jiang H., Luo C., Expert Opin. Ther. Pat. 2019, 29, 97–114. [DOI] [PubMed] [Google Scholar]
- 51. Moutouh-de Parseval L. A., Verhelle D., Glezer E., Jensen-Pergakes K., Ferguson G. D., Corral L. G., Morris C. L., Muller G., Brady H., Chan K., J. Clin. Invest. 2008, 118, 248–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meiler S. E., Wade M., Kutlar F., Yerigenahally S. D., Xue Y., Moutouh-de Parseval L. A., Corral L. G., Swerdlow P. S., Kutlar A., Blood 2011, 118, 1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dulmovits B. M., Appiah-Kubi A. O., Papoin J., Hale J., He M., Al-Abed Y., Didier S., Gould M., Husain-Krautter S., Singh S. A., Chan K. W., Vlachos A., Allen S. L., Taylor N., Marambaud P., An X., Gallagher P. G., Mohandas N., Lipton J. M., Liu J. M., Blanc L., Blood 2016, 127, 1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu T., Yoon H., Xiong Y., Dixon-Clarke S. E., Nowak R. P., Fischer E. S., Nat. Struct. Mol. Biol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vinjamur D. S., Bauer D. E., Orkin S. H., Br. J. Haematol. 2018, 180, 630–643. [DOI] [PubMed] [Google Scholar]