

Abstract
A novel route for the production of the versatile chemical building block phthalide from biorenewable furfuryl alcohol and acrylate esters is presented. Two challenges that limit sustainable aromatics production via Diels–Alder (DA) aromatisation—an unfavourable equilibrium position and undesired regioselectivity when using asymmetric addends—were addressed using a dynamic kinetic trapping strategy. Activated acrylates were used to speed up the forward and reverse DA reactions, allowing for one of the four DA adducts to undergo a selective intramolecular lactonisation reaction in the presence of a weak base. The adduct is removed from the equilibrium pool, pulling the system completely to the product with a fixed, desired regiochemistry. A single 1,2‐regioisomeric lactone product was formed in up to 86 % yield and the acrylate activating agent liberated for reuse. The lactone was aromatised to give phthalide in almost quantitative yield in the presence of Ac2O and a catalytic amount of strong acid, or in 79 % using only catalytic acid.
Keywords: biomass, dehydration, Diels–Alder reactions, lactonisation, sustainable chemistry
An intramolecular trapping strategy allows inherent problems in the Diels–Alder reaction of biomass‐derived furfuryl alcohol, such as regioselectivity and unfavourable equilibrium yields, to be overcome on the way to biomass‐derived aromatics. This strategy provides a high yielding, redox and atom economical approach to produce the versatile phthalide synthon.
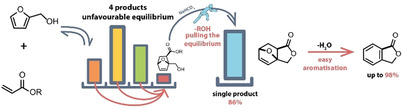
The synthesis of renewable “drop‐in” aromatics from biomass‐derived chemical building blocks is a rapidly emerging technology aimed at reducing the dependency of the chemical industry on fossil resources. [1] Additionally, the rise of shale gas and its potential impact on the production of certain key aromatic building blocks has increased interest in feedstock diversification in order to safeguard production. [2] One route to aromatics that has received significant attention of late combines a Diels–Alder (DA) reaction of biomass‐derived furanics with a second dehydration (aromatisation) step (Scheme 1 A). [1] Notable examples include the synthesis of: p‐xylene from dimethylfuran and ethylene,[ 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 ] acrylic acid[ 15 , 16 ] or acrolein; [17] benzene/toluene from furan and ethylene/propylene; [18] and (substituted) phthalic anhydride from (substituted) furan and maleic anhydride.[ 19 , 20 , 21 ] In general, there are two key considerations that determine how successful these reactions are: 1) the kinetic and thermodynamic efficiency of the DA reaction and 2) the ease with which the resulting DA cycloadduct can be aromatised. Either one can make or break the process and the outcome depends heavily on the diene/dienophile combination used.
Scheme 1.
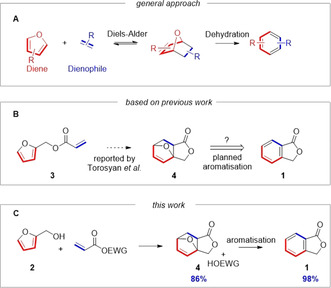
A) General scheme for the production of biomass‐derived aromatics from furanics via DA and dehydration reactions. B) Intramolecular DA reaction reported by Torosyan et al. [22] that could not be reproduced. C) This work: dynamic kinetic trapping of DA adducts for the efficient synthesis of phthalide.
As can be seen from the previously highlighted examples, biomass‐derived DA routes to aromatics typically make use of normal electron‐demand cycloadditions between electron‐rich (reduced and defunctionalized) furans and electron‐deficient or neutral dienophiles. Furthermore, it is common for either one or both addends to be symmetrical, thereby ensuring only one regioisomer is present in the final aromatised products. Unfortunately, these requirements lead to routes with poor atom and redox economy when considering the C5 or C6 carbohydrate feedstocks used to generate the biomass‐derived furanics. For example, the synthesis of furan, which is also used to synthesize maleic anhydride, requires decarbonylation of furfural, [23] while (di)methylfuran requires extensive reduction with hydrogen. From an efficiency point of view, it would clearly be advantageous to develop new routes utilizing feedstocks that are closer to those obtained directly from biomass. To this end, Davis et al. have reported the synthesis of terephthalic acid via DA reactions of ethylene and 5‐hydroxymethylfurfural (HMF) variants, with moderate yields and selectivities, [24] and Jerome et al. very recently demonstrated the synthesis of a mixture of ortho and meta substituted aromatics starting from activated furfural derivatives and acrylonitrile. [25] Here, we report on a new strategy for renewable aromatics production via DA aromatisation, that is, a new route to the versatile synthon phthalide (1). This 1,2‐substituted aromatic building block is synthesized from biobased furfuryl alcohol (2) and acrylic acid [23] derivatives which can be accessed in 100 % carbon yield from the C5 and C6 carbohydrate fractions, via furfural [26] and lactic acid, [23] respectively. Alternatively, lactic acid or directly acrylic acid are available via the chemocatalytic upgrading of bioglycerol.[ 27 , 28 ] By exploiting a dynamic kinetic trapping strategy, we overcome limitations concerning the regioselectivity and equilibrium position that are inherent to this particular DA reaction and prototypical of some of the general challenges faced in DA aromatisation as a method for sustainable aromatics production. In this way, 1 is obtained in an overall yield of 84 % from 2.
As 2 and acrylic acid are both asymmetric, their DA reaction naturally not only leads to a mixture of endo/exo stereoisomers, which are inconsequential in the preparation of aromatic compounds, but also to ortho/meta regioisomers, [29] with no apparent strong kinetic or thermodynamic preference for a single product. Upon dehydration this would generate mixtures of 1,2‐ and 1,3‐substituted aromatics. To overcome this regioselectivity issue, we originally pursued a strategy centred around an intramolecular DA reaction that would give complete control over the regio‐ and relative stereo‐chemical outcome of the DA reaction, thus ensuring the correct connectivity for the eventual formation of only 1,2‐substituted aromatic products. This was inspired by the work of Torosyan et al. who reported that the intramolecular DA reaction of furfuryl acrylate (3) proceeded to give cycloadduct 4 in a 50 % (isolated) yield, [22] which we expected could be readily aromatised to give 1 (Scheme 1 B).
In our hands, however, the reported procedure for the preparation of 4, which required 1 to be supported on diatomaceous earth and then heated at 60 °C for 60 h, gave no detectable intramolecular DA product. Indeed, no reaction was observed at all. Further, attempts to perform the reaction in solution in a range of solvents ([D6]DMSO, CDCl3, [D6]benzene, [D8]toluene, [D4]methanol, [D6]acetone, D2O), or in the presence of Lewis acids (e.g. ZnCl2, Sc(OTf)3, Hf(OTf)4) also failed to provide any of the expected product, as judged by 1H NMR. Given this disappointing result, and with reports of the successful intramolecular DA reaction of closely related furfuryl fumarates[ 30 , 31 ] and maleates in mind, [32] we decided to carry out a computational investigation into the feasibility of the target reaction using Density Functional Theory method. The computational results supported our experimental observations, suggesting the targeted intramolecular DA is not feasible (Figure 1). In line with previous suggestions from experimental studies on furfuryl fumarates, [30] we found that initial formation of the reactive conformation (rotamer) was associated with a relatively large increase in Gibbs free energy (ΔG) of approximately 10.0 or 10.8 kcal mol−1 for the endo or exo reaction pathways, respectively. Accessing this reactive conformer involves repositioning of the acrylate group in closer proximity to the diene and a change of the ester from a s‐cis to a s‐trans arrangement. This is associated with a large energy penalty of 10.0 to 10.8 kcal mol−1 due to the loss of the stabilising anomeric effect, increased steric strain and a larger overall dipole moment (0.9 D for the lowest energy conformer versus 4.5–4.8 D for the reactive rotamers), meaning the equilibrium population of either of the reactive conformers will be very low. Furthermore, examining the transition state energies shows that the endo pathway has a significantly higher activation energy than the exo one (30.1 vs. 39.8 kcal mol−1), but both are sufficiently high that the forward reaction would not be feasible. Additionally, the high relative energies of the products indicate a highly endergonic reaction, further ruling out the feasibility of this intramolecular reaction, in line with our experimental results.
Figure 1.
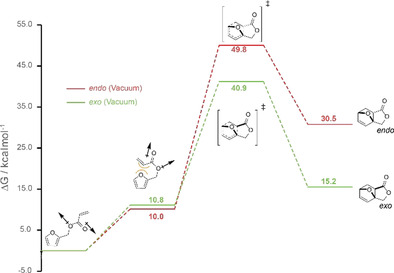
Gibbs free energy profiles of intramolecular DA reactions calculated in the gas phase at 50 °C using the B3LYPD3 XC functional together with the TZ2P basis set. The global minimum substrate conformer was set to zero and all other energies were calculated with respect to these zero points.
One additional important outcome of the computational analysis was that it allowed us to identify 4‐exo as a potentially stable compound, depending on synthesis conditions, with an activation energy for the retro‐DA reaction of 25.7 kcal mol−1, leading us to consider an alternative synthetic route. We decided to employ a dynamic kinetic trapping strategy (Scheme 1 C) to produce 4‐exo, making use of acyclic activated acrylates. Based on the principle of rate enhancement by the proximity effect, we anticipated to be able to trap, as the lactone, a single isomer from the mixture of isomers produced from the DA reaction of 2 and a suitably activated acrylate (Scheme 2). This approach is related to one previously utilised in the DA reaction of 2 and itaconic anhydride. [33] From surveying commercially available acrylates we identified hexafluoroisopropyl (HFIP) acrylate (5) as a potentially suitable dienophile where the electron withdrawing HFIP group serves a dual purpose to both activate the dienophile towards DA reactions and also the ester group towards lactonisation. Indeed, HFIP esters have been shown to be viable alternatives to other traditional activated esters in amide bond forming reactions, [34] although their use in DA reactions is surprisingly seldom reported.[ 35 , 36 ]
Scheme 2.
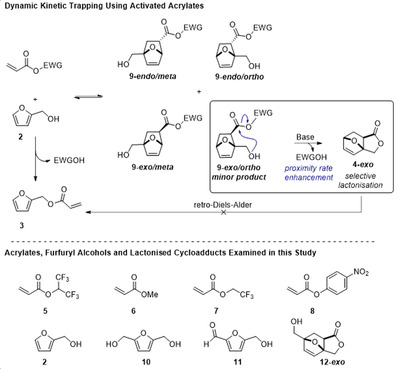
A new route to phthalide involving an intermolecular DA reaction of furfuryl alcohols with activated acrylates followed by selective dynamic kinetic trapping of only one isomer.
The reaction of 2 and 5 (1:1 molar ratio) neat at room temperature, proceeded smoothly to give, after 96 h, an equilibrium mixture of the four expected cycloadducts (9), totalling 66 %, and remaining 2 and 5. In contrast to anhydrides, [33] no spontaneously lactonized products could be detected under these conditions. Therefore, to promote this reaction, a catalytic amount of a weak base, NaHCO3 (20 mol %), was added to the crude mixture of cycloadducts. Gratifyingly, 1H NMR analysis (Supporting Information, Figure S2) revealed that one of the cycloadducts, the minor 9‐exo/ortho isomer, is selectively transformed into a new compound, assigned to 4‐exo based on conformational modelling, 2D NMR analysis and later confirmed via single crystal X‐ray structure determination (Figure S3). The observed stereochemistry is consistent with the above computational studies showing 4‐exo to be the thermodynamically favoured product. Notably, the analytical data obtained for 4‐exo is substantially different to that reported by Torosyan et al. (Supporting Information, Table S2), [22] and given that computational analysis indicates 4‐endo would be an unstable compound with respect to the retro‐DA reaction (Figure 1), it seems highly unlikely that the original assignment of 4 was correct.
Due to the low equilibrium yield of the key 9‐exo/ortho cycloadduct and the relatively slow kinetics of this DA reaction at room temperature, this did not represent a practical route to 4‐exo and so optimization studies were required (Table 1). Therefore, the reaction temperature was increased to 60 °C, allowing for relatively rapid equilibration of addends and adducts, thus allowing us to apply dynamic trapping of the exo/ortho cycloadduct, by irreversible lactonisation as 4‐exo, as an optimization strategy. At this temperature and 20 mol % NaHCO3 loading, 4‐exo was formed in 17 % yield but with a disappointing selectivity of just 20 % (Entry 1). Furfuryl acrylate (3) was identified as the major side product leading to the low selectivity. Two possible routes exist for the formation of 3: direct base‐catalysed transesterification of 5 by 2 or via a retro‐DA reaction from 4‐exo. Stability testing of the (isolated) lactone (Figure S4) suggested that 3 is formed via the former route, as 4‐exo showed a relatively high thermal stability up to 100 °C. Thus, optimization of the base loading and reaction temperature led to an improved yield of 64 %, with an 82 % selectivity (Entry 5). A small increase in yield was observed when using 1.5 equiv. of 5 (Entry 7) but changing the base or running the reaction in a solvent did not further improve the yield. Changing the acrylate to methyl acrylate (6) showed that activation of the ester was critical for the success of the reaction (Entry 14). Ultimately, the best yield of 4‐exo (86 % at 91 % selectivity), was obtained using 4‐nitrophenol acrylate (8) as the activated dienophile (Entry 16). For both 5 and 8 recovery of the activating group should be readily achievable, for example 1,1,1,3,3,3‐hexafluoroisopropyl alcohol can be recovered by distillation and 4‐nitrophenol by selective extraction with a mild base, allowing recycling of these groups. Furthermore, we found that the same trapping approach could be used with 2,5‐bis(hydroxymethyl)furan (10, Entry 17), but introduction of a deactivating electron withdrawing group on the furan ring, as in hydroxymethylfurfural (11), was not tolerated (Entry 18).
Table 1.
Optimization of the DA‐lactonisation reaction between furfuryl alcohols and activated acrylates. Yields after 22 h using 2 as the substrate, unless otherwise stated.
|
Entry |
Acrylate |
Base |
Amount mol % |
T [°C] |
Yield [%] |
Selectivity |
|---|---|---|---|---|---|---|
|
1 |
HFIP |
NaHCO3 |
20 |
60 |
17 |
20 |
|
2 |
|
|
2 |
60 |
34 |
43 |
|
3 |
|
|
20 |
80 |
26 |
28 |
|
4 |
|
|
2 |
80 |
54 |
66 |
|
5 |
|
|
1 |
80 |
64 |
82 |
|
6 |
|
|
0.5 |
80 |
38 |
70 |
|
7[b] |
|
|
1 |
80 |
68 |
88 |
|
8 |
|
CH3O2Na |
20 |
80 |
23 |
27 |
|
9 |
|
|
2 |
80 |
44 |
55 |
|
10 |
|
CHCl2CO2Na |
20 |
80 |
45 |
58 |
|
11 |
|
|
2 |
80 |
31 |
50 |
|
12 |
|
NEt3 |
2 |
80 |
30 |
34 |
|
13[a] |
|
NaHCO3 |
2 |
80 |
43 (68)[c] |
83 (74)[c] |
|
14 |
Me |
NaHCO3 |
20 |
80 |
2 |
8 |
|
15 |
TFE |
NaHCO3 |
20 |
80 |
33 |
39 |
|
16 |
4NP |
NaHCO3 |
2 |
80 |
86 |
91 |
|
17[d] |
HFIP |
|
1 |
80 |
61 |
66 |
|
18[e] |
HFIP |
|
1 |
80 |
0 |
0 |
[a] Reaction run in EtOAc. [b] 1.5 equiv. of HFIP acrylate. [c] Yield after 216 h. [d] Using 10 as substrate. [e] Using 11 as substrate.
With a reliable, high‐yielding route to 4‐exo in hand, our focus turned to the aromatisation reaction (Table 2). Using a mixture of methanesulfonic acid (MsOH) and Ac2O, as reported by Mahmoud et al.,[ 20 , 37 ] 4‐exo could be readily aromatised to 1 in excellent yield (Entry 1). Interestingly, in this case, we found that only catalytic amounts of MsOH are required (Entry 2–6), with lower loadings actually proving beneficial to the yield, which was near quantitative down to 2 mol % MsOH. Following this reaction at low temperature (0–4 °C) by NMR revealed the formation of several intermediates (Figure 2; Figure S10) which based on NMR analysis and literature precedence [20] we assigned to di‐ and mono‐acetates representing the different stages of dehydration in the presence of Ac2O.
Table 2.
Optimization of the aromatisation reaction to give phthalides. All reactions with 4‐exo, unless stated otherwise.
|
Entry |
Solvent |
Catalyst |
Loading [equiv] |
Ac2O |
T [°C][a] |
Yield [%][b] |
|---|---|---|---|---|---|---|
|
1 |
Neat |
MsOH |
13 |
20 vol % |
20 |
75 |
|
2 |
|
|
0.5 |
4 equiv |
20/80 |
97[c] |
|
3 |
|
|
0.5 |
4 equiv |
80 |
98 |
|
4 |
|
|
0.1 |
4 equiv |
80 |
95 |
|
5 |
|
|
0.02 |
4 equiv |
80 |
94 |
|
6 |
|
|
0.01 |
4 equiv |
80 |
39 |
|
7 |
|
TfOH |
0.01 |
4 equiv |
80 |
95 |
|
8 |
|
H2SO4 |
0.01 |
4 equiv |
80 |
80 |
|
9 |
|
Amberlyst 15 |
0.1 |
4 equiv |
80 |
82 |
|
10 |
EtOAc |
|
0.1 |
4 equiv |
80 |
78 |
|
11 |
|
|
0.1 |
2 equiv |
80 |
89 (79)[d] |
|
12[e] |
Neat |
MsOH |
0.5 |
8 equiv |
80 |
60[f] |
|
13 |
Neat |
MsOH |
13 |
0 |
20 |
66 |
|
14 |
d8‐Toluene |
MsOH |
0.1 |
0 |
80 |
66 |
|
15 |
|
Hf(OTf)4 |
0.1 |
0 |
80 |
60 |
|
16 |
|
TfOH |
0.1 |
0 |
80 |
63 |
|
17 |
|
TfOH |
0.01 |
0 |
80 |
63 |
|
18 |
|
TfOH[g] |
0.01 |
0 |
80 |
58 |
|
19 |
CDCl3 |
TfOH |
0.1 |
0 |
80 |
79 |
|
20 |
CH3COOH |
TfOH |
0.1 |
0 |
80 |
56 |
[a] Acid/Ac2O added at 0 °C and then warmed to 20 or 80 °C as specified. [b] Determined by quantitative 1H NMR. [c] Reaction mixture heated to 80 °C for 1 h after 22 h at 20 °C. [d] Yield using recycled Amberlyst catalyst. [e] Using 12‐exo as the substrate. [f] Isolated yield. [g] Silica‐supported TfOH acid. [38]
Figure 2.
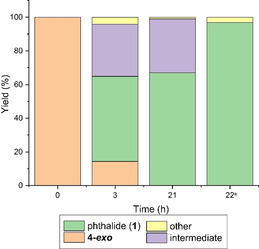
Time course for the aromatisation of 4‐exo corresponding to Table 2, entry 2. [a] Mixture heated at 80 °C for 1 h.
Further acid screening showed that trifluoromethanesulfonic acid (TfOH) (Entry 7), H2SO4 (Entry 8) and (recyclable) Amberlyst 15 resin (Entry 9) were all capable of catalysing the aromatisation reaction. With TfOH, the strongest acid tested, loadings as low as 1 mol % gave excellent yields of 1. The reaction could also be run in EtOAc as a solvent and with lower amounts of Ac2O without negative effects (Entry 10–11). 12‐exo could also be aromatised under these conditions (Entry 12), although the yield was lower than for 4‐exo. In the absence of a strong acid catalyst (i.e. neat Ac2O only) no conversion was observed. Interestingly, in the absence of Ac2O but presence of strong acid the dehydration proceeded, but with somewhat lower yields of 56–79 %, highlighting the important role of Ac2O in intercepting and stabilizing reactive intermediates during the aromatisation process and thereby limiting non‐productive polymerisation reactions. Notably, these yields are still much higher than those obtained previously in the synthesis of phthalic anhydride, [37] suggesting 4‐exo is particularly amenable to dehydration. Although rigorously dried solvents are not required for the aromatisation reaction, we did find that added water has a negative effect, significantly reducing both the conversion and selectivity (Table S1).
In conclusion, we have developed a new and high‐yielding route (84 % over two steps) to biomass‐derived phthalide (1) from furfuryl alcohol (2) and activated acrylates such as 1,1,1,3,3,3‐hexafluoroisopropyl acrylate (5) and 4‐nitrophenyl acrylate (8). The choice of these substrates provides excellent overall atom and redox efficiency considering the potential parent sugars and the opportunities to recycle the activating groups. The key step in this route involves dynamic kinetic trapping of only one of the four DA cycloadducts through selective, base‐assisted lactonisation. This overcomes the inherent thermodynamic and regioselectivity challenges of DA reactions using asymmetric addends, providing a high‐yield route to phthalide (1), which is itself a commodity chemical and a potential precursor to bulk chemicals such as phthalic anhydride. [39]
Conflict of interest
Folker, Lancefield, Crockatt and Bruijnincx are listed as inventors on a patent application filed by TNO related to the results described in the manuscript.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
R.B. acknowledges the Netherlands Organization for Scientific Research (NWO) (Vidi 723.012.104). C.S.L. thanks the Leverhulme Trust Early Career Fellowship (ECF‐2018‐480). We also acknowledge the NWO for funding the X‐ray diffractometer used in this study. K.S. acknowledges financial support from The Netherlands Centre for Multiscale Catalytic Energy Conversion (MCEC). The support of Prof. B. M. Weckhuysen is kindly acknowledged.
C. S. Lancefield, B. Fölker, R. C. Cioc, K. Stanciakova, R. E. Bulo, M. Lutz, M. Crockatt, P. C. A. Bruijnincx, Angew. Chem. Int. Ed. 2020, 59, 23480.
References
- 1. Settle A. E., Berstis L., Rorrer N. A., Roman-Leshkóv Y., Beckham G. T., Richards R. M., Vardon D. R., Green Chem. 2017, 19, 3468–3492. [Google Scholar]
- 2. Bruijnincx P. C. A., Weckhuysen B. M., Angew. Chem. Int. Ed. 2013, 52, 11980–11987; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12198–12206. [Google Scholar]
- 3. Wijaya Y. P., Suh D. J., Jae J., Catal. Commun. 2015, 70, 12–16. [Google Scholar]
- 4. Williams C. L., Chang C.-C., Do P., Nikbin N., Caratzoulas S., Vlachos D. G., Lobo R. F., Fan W., Dauenhauer P. J., ACS Catal. 2012, 2, 935–939. [Google Scholar]
- 5. Rohling R. Y., Uslamin E., Zijlstra B., Tranca I. C., Filot I. A. W., Hensen E. J. M., Pidko E. A., ACS Catal. 2018, 8, 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim J. C., Kim T. W., Kim Y., Ryoo R., Jeong S. Y., Kim C. U., Appl. Catal. B 2017, 206, 490–500. [Google Scholar]
- 7. Cho H. J., Ren L., Vattipalli V., Yeh Y.-H., Gould N., Xu B., Gorte R. J., Lobo R., Dauenhauer P. J., Tsapatsis M., et al., ChemCatChem 2017, 9, 398–402. [Google Scholar]
- 8. Yin J., Shen C., Feng X., Ji K., Du L., ACS Sustainable Chem. Eng. 2018, 6, 1891–1899. [Google Scholar]
- 9. Wijaya Y. P., Winoto H. P., Park Y. K., Suh D. J., Lee H., Ha J. M., Jae J., Catal. Today 2017, 293–294, 167–175. [Google Scholar]
- 10. Nikbin N., Feng S., Caratzoulas S., Vlachos D. G., J. Phys. Chem. C 2014, 118, 24415–24424. [Google Scholar]
- 11. Feng X., Cui Z., Ji K., Shen C., Tan T., Appl. Catal. B Environ. 2019, 259, 118108. [Google Scholar]
- 12. Chang C. C., Je Cho H., Yu J., Gorte R. J., Gulbinski J., Dauenhauer P., Fan W., Green Chem. 2016, 18, 1368–1376. [Google Scholar]
- 13. Kim T. W., Kim S. Y., Kim J. C., Kim Y., Ryoo R., Kim C. U., Appl. Catal. B 2016, 185, 100–109. [Google Scholar]
- 14. Feng X., Shen C., Tian C., Tan T., Ind. Eng. Chem. Res. 2017, 56, 5852–5859. [Google Scholar]
- 15. Ni L., Xin J., Dong H., Lu X., Liu X., Zhang S., ChemSusChem 2017, 10, 2394–2401. [DOI] [PubMed] [Google Scholar]
- 16. Ni L., Xin J., Jiang K., Chen L., Yan D., Lu X., Zhang S., ACS Sustainable Chem. Eng. 2018, 6, 2541–2551. [Google Scholar]
- 17. Shiramizu M., Toste F. D., Chem. Eur. J. 2011, 17, 12452–12457. [DOI] [PubMed] [Google Scholar]
- 18. Cheng Y.-T., Huber G. W., Green Chem. 2012, 14, 3114. [Google Scholar]
- 19. Thiyagarajan S., Genuino H. C., Śliwa M., Van Der Waal J. C., De Jong E., Van Haveren J., Weckhuysen B. M., Bruijnincx P. C. A., Van Es D. S., ChemSusChem 2015, 8, 3052–3056. [DOI] [PubMed] [Google Scholar]
- 20. Mahmoud E., Watson D. A., Lobo R. F., Green Chem. 2014, 16, 167–175. [Google Scholar]
- 21. Thiyagarajan S., Genuino H. C., Van Der Waal J. C., De Jong E., Weckhuysen B. M., Van Haveren J., Bruijnincx P. C. A., Van Es D. S., Angew. Chem. Int. Ed. 2016, 55, 1368–1371; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1390–1393. [Google Scholar]
- 22. Torosyan G. O., Akopyan A. A., Torosyan A. T., Babayan A. T., Chem. Heterocycl. Compd. 1990, 26, 390–392. [Google Scholar]
- 23. Katryniok B., Bonnotte T., Dumeignil F., Paul S., Chemicals and Fuels from Bio-Based Building Blocks, Wiley-VCH, Weinheim, 2016, pp. 217–244. [Google Scholar]
- 24. Pacheco J. J., Davis M. E., Proc. Natl. Acad. Sci. USA 2014, 111, 8363–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scodeller I., Mansouri S., Morvan D., Muller E., de Oliveira Vigier K., Wischert R., Jérôme F., Angew. Chem. Int. Ed. 2018, 57, 10510–10514; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10670–10674. [Google Scholar]
- 26. Hoydonckx H. E., Van Rhijn W. M., Van Rhijn W., De Vos D. E., Jacobs P. A., in Ullmann′S Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2007. [Google Scholar]
- 27. Dusselier M., Van Wouwe P., Dewaele A., Makshina E., Sels B. F., Energy Environ. Sci. 2013, 6, 1415–1442. [Google Scholar]
- 28. Kim M., Lee H., ACS Sustainable Chem. Eng. 2017, 5, 11371–11376. [Google Scholar]
- 29.王静刚, 马松琪, 刘小青, 朱锦, 那海宁, 倪金平, 一种4-羟甲基-7-氧杂二环[2.2.1]庚-5-烯单体及其制备方法, 2014, CN104193759A.
- 30. Jung M. E., Gervay J., Tetrahedron Lett. 1988, 29, 2429–2432. [Google Scholar]
- 31. Jung M. E., Kiankarimi M., J. Org. Chem. 1998, 63, 2968–2974. [Google Scholar]
- 32. Pelter A., Singaram B., J. Chem. Soc. Perkin Trans. 1 1983, 1383–1386. [Google Scholar]
- 33. Pehere A. D., Xu S., Thompson S. K., Hillmyer M. A., Hoye T. R., Org. Lett. 2016, 18, 2584–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kelly C. L., Leadbeater N. E., Int. J. Adv. Res. Chem. Sci. 2016, 3, 27–34. [Google Scholar]
- 35.“Aromatic Compounds From Furanics”: Crockatt M., Urbanus J. H., WO2017146581, 2017.
- 36. Artigas A., Vila J., Lledó A., Solà M., Pla-Quintana A., Roglans A., Org. Lett. 2019, 21, 6608–6613. [DOI] [PubMed] [Google Scholar]
- 37. Mahmoud E., Yu J., Gorte R. J., Lobo R. F., ACS Catal. 2015, 5, 6946–6955. [Google Scholar]
- 38. Liu P. N., Xia F., Wang Q. W., Ren Y. J., Chen J. Q., Green Chem. 2010, 12, 1049–1055. [Google Scholar]
- 39. Mahyari M., Laeini M. S., Shaabani A., Kazerooni H., Appl. Organomet. Chem. 2015, 29, 456–461. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary