

Abstract
In living organisms, protein folding and function take place in an inhomogeneous, highly crowded environment possessing a concentration of diverse macromolecules of up to 400 g/L. It has been shown that the intracellular environment has a pronounced effect on the stability, dynamics and function of the protein under study, and has for this reason to be considered. However, most protein studies neglect the presence of these macromolecules. Consequently, we probe here the overall thermodynamic stability of cold shock protein B from Bacillus subtilis (BsCspB) in cell lysate. We found that an increase in cell lysate concentration causes a monotonic increase in the thermodynamic stability of BsCspB. This result strongly underlines the importance of considering the biological environment when inherent protein parameters are quantitatively determined. Moreover, we demonstrate that targeted application of 19F NMR spectroscopy operates as an ideal tool for protein studies performed in complex cellular surroundings.
Keywords: biophysics, NMR spectroscopy, protein folding
The importance of biological environment: Fluorine NMR spectroscopy was applied to determine the thermodynamic stability, ΔG 0, of 19F‐labeled cold shock protein B, BsCspB, during fully reversible folding‐to‐unfolding transitions in cell lysate at different cell pellet concentrations.
Introduction
Most biophysical studies investigating the inherent structural, dynamic and functional characteristics of proteins apply molecular concentrations of less than 10 g/L [1] (so‐called dilute conditions). Under these conditions, solely intermolecular interactions with water molecules exist while the protein of interest is analyzed. [2] However, the intracellular environment is largely inhomogeneous and highly crowded (Figure 1) due to the presence of diverse macromolecules which occupy up to 40 % of the volume of the cell thus reaching an intracellular concentration of up to 400 g/L. [3] In this respect, numerous studies have shown that such pronounced macromolecular concentrations may have a major effect on proteins overall stability, dynamics and the ability for interactions to other molecules. [4] Such presence of a large macromolecular concentration leads to crowding and confinement effects resulting in hard and soft interactions among the agents used for macromolecular crowding (MC) and the analyzed protein. [5] Hard interactions are based on volume exclusion by MC agents yielding to a reduced space which is accessible for the protein under study. Since the unfolded protein ensemble has a lager hydrodynamic radius compared to the native state, volume exclusion is capable to increase the overall thermodynamic protein stability due to a shift in the equilibrium between native and unfolding protein conformations. [6] In contrast, soft interactions are based on stabilizing as well as destabilizing effects macromolecules have on adjacent proteins.[ 2 , 7 ]
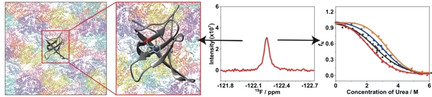
Figure 1.

Cartoon representing the fluorinated variant of cold shock protein B from Bacillus subtilis, 4‐19F−Trp−BsCspB (cartoon mode, colored in gray, PDB ID: 6SZZ [18b] ) present in a highly crowded environment comprising diverse macromolecules found in cell lysate. The main‐ and side‐chain atoms of 4‐19F−tryptophan are highlighted in blue, except for side chain fluorine at ring position four (colored in red).
It has been shown that adding MC agents to the protein solution of interest represents one way (“bottom‐up approach”) to mimic the intracellular environment in vitro.[ 2 , 4a , 4k , 7a , 8 ] Thus, comparable results, for example, for the determination of the overall thermodynamic stability of a protein present in vitro compared to in vivo have been obtained. [9] On the other hand, it should be noted that in vitro measurements using MC agents may also differ from experimental results obtained in vivo [10] or give quantitative results that precisely depend on MC agents that were used. [11] Reasons for such observed differences in experimental results comparing an in vitro with an in vivo setup may potentially be based on the fact that solutions containing MC agents are homogeneous in nature and that MC agents possess no or modified soft interactions with the protein compared to the in vivo scenario.[ 2 , 4e , 12 ] However, performing experiments directly in the cell or in a cell‐like environment provides several technical challenges to reliably observing the protein of interest in the presence of the variety of cellular constituents or the large quantity of (diverse) MC agents, as shown by probing protein stability, [13] diffusion [11b] or side chain dynamics. [14] In this respect, 19F NMR spectroscopy offers one promising approach to study proteins in such complex environments as fluorine is not naturally found in proteins.[ 8 , 15 ] Moreover, 19F nuclei possess a natural abundance of 100 % and a high sensitivity (83 % compared to 1H) that allows NMR spectroscopic measurements at a relatively low range of concentrations and inherently provides a large range of chemical shifts. This property provides an ideal prerequisite for the detection of changes in the chemical environment of the 19F nucleus under study.[ 8 , 16 ] Thus, NMR spectroscopic studies of proteins significantly larger than 25 kDa become feasible by selective labeling schemes used for fluorine. [17] As another advantage, it has been shown that the site‐directed incorporation of fluorinated tryptophan or phenylalanine residues into a target protein exhibits no or only mild effects on the overall thermodynamic stability and folding kinetics of the modified protein compared to the wild type form. [18]
In previous work we presented a robust method to incorporate fluorinated tryptophan or phenylalanine into the cold shock protein from Bacillus subtilis, BsCspB, which inherently contains seven phenylalanine (F9, F15, F17, F27, F30, F38, and F49) and one tryptophan residue (W8). Consequently, 5‐19F−Trp−BsCspB comprises a single fluorinated site whereas 4‐19F−Phe−BsCspB comprises seven fluorinated sites. We demonstrated the high purity of the protein variants with high‐resolution 1H and 19F NMR spectroscopy as well as mass spectrometry. [18a] Moreover, it could be shown by applying two dimensional heteronuclear 1H,15N HSQC NMR spectroscopy and X‐ray crystallography that the fluorine label has no significant impact on the three‐dimensional structure of BsCspB even when the orientation of fluorinated side chains is analyzed. [18b] BsCspB is a relatively small protein (M W=7.3 kDa) constituting a β‐barrel fold and interacting to singly stranded DNA and RNA molecules. [19] This protein has been extensively studied in terms of structural, dynamical and functional features under both dilute [20] and MC conditions [4a] using a broad range of diverse biophysical techniques before and makes it therefore to a perfect candidate for experimental investigations performed in cell lysate. BsCspB has been characterized as a two‐state folding protein and possesses an overall thermodynamic stability of about ΔG 0=10 kJ/mol at T=298 K under dilute conditions.[ 4a , 20a , 20c ]
Here, we use 4‐19F−Phe−BsCspB and 5‐19F−Trp−BsCspB as two protein variants enabling to monitor the overall thermodynamic stability of BsCspB in cell lysate in a cell lysate‐concentration dependent manner. The folding‐to‐unfolding transition of BsCspB was chemically induced by a stepwise increase in urea concentration and followed by one‐dimensional 19F NMR spectroscopy quantifying resonance signals representing native and unfolding protein. Our experimental results indicate that the overall thermodynamic stability of BsCspB is substantially increased in cell lysate compared to measurements conducted under dilute conditions. Moreover, this increase in thermodynamic stability can be precisely regulated by the concentration of the cell lysate applied, thus giving indispensable insights into the biological environment in which proteins operate.
Results and Discussion
Thermodynamic stability of fluorine labelled BsCspB in vitro
High‐resolution NMR spectroscopy enables to monitor the chemically induced folding‐to‐unfolding transition of a protein in equilibrium by a stepwise increase in urea concentration to obtain its overall thermodynamic stability, ΔG 0. [21] Urea is known to be able to directly interact with the protein under study [22] and to potentially change the water structure finally leading to protein denaturation. [23] First, one‐dimensional proton NMR spectra differing in urea concentration were acquired for both 5‐19F−Trp−BsCspB and 4‐19F−Phe−BsCspB under dilute (in vitro) conditions (Figure S1 in the Supporting Information). The subsequent acquisition of one‐dimensional fluorine NMR spectra of the same samples (Figure S2A, B) allows a direct comparison of the thermodynamic stability of 5‐19F−Trp−BsCspB and 4‐19F−Phe−BsCspB based on either 1H or 19F NMR spectra (Figure 2). Of note, the course of the folding‐to‐unfolding transitions obtained for both protein variants using 1H or 19F resonance signals are comparable (Figure 2A, B). Specifically, ΔG 0 was determined for 5‐19F−Trp−BsCspB and 4‐19F−Phe−BsCspB to ΔG 0=7.2±0.2 and 8.5±0.2 kJ/mol (1H dimension) and to ΔG 0=7.7±0.2 and 8.7±0.2 kJ/mol (19F dimension), respectively. These numbers are in good accordance to ΔG 0 values determined for fluorinated variants of BsCspB by fluorescence spectroscopy before using also chemical denaturation. [18b]
Figure 2.
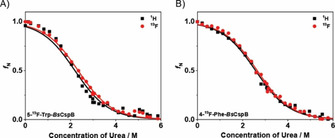
Folding‐to‐unfolding transitions determined for A) 5‐19F−Trp−BsCspB and B) 19F−Phe−BsCspB induced by an increasing concentration of urea under dilute conditions acquired at T=298 K. The folding‐to‐unfolding transitions were monitored using both one‐dimensional proton (•) and fluorine NMR spectroscopy (▪). Fitting Equation (S1) to the experimentally obtained data yields to an overall thermodynamic stability of ΔG 0=(7.2±0.2) kJ/mol (proton data set) and ΔG 0=(7.7±0.2) kJ/mol (fluorine data set) characterizing 5‐19F−Trp−BsCspB. The overall thermodynamic stability of 4‐19F−Phe−BsCspB was determined as ΔG 0=(8.5±0.2) kJ/mol (proton data set) and ΔG 0=(8.7±0.2) kJ/mol (fluorine data set). The fitting function is shown as a continuous line. The folding cooperativity, m, was used as a global parameter in the fitting process and was determined to be m=−3.3±0.1 kJ/(molM).
Thermodynamic stability of fluorine labelled BsCspB in cell lysate
As a next step, we have proceeded to the determination of ΔG 0 values of fluorine labelled BsCspB variants present in cell lysate. Before quantifying folding‐to‐unfolding transitions of BsCspB in cell lysate, we have acquired two‐dimensional 1H,15N HSQC NMR spectra of 5‐19F−Trp−BsCspB both in vitro and in cell lysate. The spectral comparison between these two experimental conditions indicates that 1H and 15N chemical shifts of cross signals comprising 5‐19F−Trp−BsCspB observed in vitro are conserved in the experiment conducted in cell lysate for almost all residues (Figure 3A). Moreover, the course of signal intensities of cross peaks monitored in 2D 1H,15N HSQC spectra is comparable under both conditions (Figure 3B). These observations indicate that the structural characteristics of 5‐19F−Trp−BsCspB are largely similar when experiments performed in vitro are compared with cell lysate condition. Confirming the overall structural characteristics of BsCspB in cell lysates with known features found in vitro brings us into a position to reliably compare the overall thermodynamic stability of this protein present either in vitro or in cell lysate applying 19F NMR spectroscopy. Note that fluorine NMR spectroscopy presents indeed a reliable method reporting on thermodynamic parameters of this protein as experiments monitoring protein unfolding observed under in vitro conditions have already indicated (Figure 2A, B). In this respect, one‐dimensional 1H NMR spectra acquired in cell lysate cannot be used to reliably quantify thermodynamic parameters of BsCspB due to the broadening of resonance signals and contributions of resonance signals arising from cell lysate in the spectral range used for data analysis (Figure S3A−C). Consequently, the chemically induced folding‐to‐unfolding transitions of 5‐19F−Trp−BsCspB and 4‐19F−Phe−BsCspB were followed in cell lysate with different concentrations of urea (Figures S4A−C and S5A, B). Analyzing the folding‐to‐unfolding transitions illuminates a significant increase in the thermodynamic stability of BsCspB comparing cell lysate with in vitro conditions (Figure 4). This increase in ΔG 0 is seen for both 5‐19F−Trp−BsCspB, and 4‐19F−Phe−BsCspB. Quantitatively, a cell pellet fraction of 120 g/L (holds for 5‐19F−Trp−BsCspB) or 150 g/L (holds for 4‐19F−Phe−BsCspB) is capable to increase ΔG 0 value by about 1 kJ/mol whereas the general two‐state folding character of BsCspB as observed under in vitro condition is fully conserved in cell lysate (Figure 4A, B). Importantly, the experimental setup chosen here applying chemical denaturation to probe thermodynamic stability illustrates the applicability to a complex biological environment as cell lysate by showing a full reversibility of the folding‐to‐unfolding transition of BsCspB. But how much does the concentration of macromolecules present in cell lysate impacts the thermodynamic stability of BsCspB? Addressing this question is of particular interest as several studies conducted on different proteins have shown that an increasing concentration of MC agents results in an increasing thermodynamic stability of the protein being probed.[ 4a , 4k , 5d , 6a ] We are now in a position to prove or disprove – at least for the case of BsCspB – the assumption that working with MC is indeed a reliable approach to imitate an environment of the protein under study which is found inside a cell. Thus chemically induced folding‐to‐unfolding transitions of 5‐19F−Trp−BsCspB and 4‐19F−Phe−BsCspB were measured at various cell pellet concentrations probing the impact of the concentration of dissolved macromolecules present in cell lysate on the thermodynamic stability of BsCspB. For 5‐19F−Trp−BsCspB, increasing the amount of cell pellet in a fixed volume of lysis buffer results in a monotonic increase of the overall thermodynamic stability (Figure 4C). Note that the thermodynamic stability of 5‐19F−Trp−BsCspB increases from ΔG 0=7.7±0.2 kJ/mol (in vitro condition) to finally ΔG 0=11.2±0.2 kJ/mol when 450 g/L cell pellet concentration is present (Figure 4C). To compare, probing ΔG 0 of wild type BsCspB in presence of 200 g/L PEG1, PEG8, or Dex20 used as MC agents in fluorescence spectroscopic experiments leads to an increase in thermodynamic stability of about 5 to 6 kJ/mol compared to dilute conditions. [4a] In the present study, an amount of 260 g cell pellet per liter lysis buffer generates ΔG 0=(10.0±0.2) kJ/mol for 5‐19F−Trp−BsCspB representing an increase of about 2.3 kJ/mol compared to in vitro condition (Figure 4C). Probing the impact of 450 g/L cell pellet concentration on the thermodynamic stability of 4‐19F−Phe−BsCspB leads to ΔG 0=(11.3±0.2) kJ/mol compared to ΔG 0=(8.7±0.2) kJ/mol obtained for in vitro condition (Figure 4C). At this point it can therefore be concluded that an increase in cell pellet concentration results in parallel in a monotonic increase of the overall thermodynamic stability of BsCspB. Additionally, we have analyzed the potential impact of varying cell pellet concentration on the spectral features of 5‐19F−Trp−BsCspB and 4‐19F−Phe−BsCspB present at native or unfolding conditions (Figure S6). As a result, the unfolded protein ensemble of 5‐19F−Trp−BsCspB or 4‐19F−Phe−BsCspB is not modified when cell pellet concentration changes from 150 g/L to 450 g/L (Figure S6B, D). Qualitatively, the same observation can be made when the native state of both proteins is analyzed (Figure S6A, C). Solely a slight change in line width of fluorine resonance signal arising from 5‐19F−Trp−BsCspB can be monitored which presumably results from an increase in sample viscosity comparing 150 g/L with 450 g/L cell pellet concentration (Figure S6A). Note that even a high concentration of cell pellet does not modify the resonance signal originating from urea in one‐dimensional 1H NMR spectra (Figure S7A). Also, the proton chemical shift observed for urea shows a monotonic and similar increase for both dilute and cell lysate conditions with increasing urea concentration (Figure S7B–D). The courses of the proton chemical shift of urea which is present in 120 or 450 g/L cell lysate are identical (Figure S7C, D).
Figure 3.
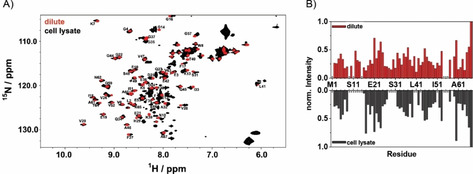
A) Two‐dimensional 1H,15N HSQC NMR spectroscopy on 5‐19F−Trp−BsCspB. The NMR spectra were acquired under both dilute conditions (red) and in cell lysate containing 250 g cell pellet per liter lysis buffer (black). The assignment of backbone resonances of 5‐19F−Trp−BsCspB is shown by using the one letter code for amino acids followed by the position in the primary sequence. Additional cross signals identified in cell lysate are caused by other common compounds in the cytosol of E. coli. [24] B) Cross signals assigned in two‐dimensional 1H,15N HSQC NMR spectra were also analyzed for their intensity. Signal intensities acquired under dilute conditions are shown at the top (red); signal intensities acquired in cell lysate are shown at the bottom (black). Signal intensities were normalized using the maximum found for C‐terminal A67. 2D 1H,15N HSQC NMR spectra were acquired at T=298 K and B 0=18.8 T (cell lysate) or 14.1 T (dilute). Stars on the x‐axis indicate no unambiguously assigned residues.
Figure 4.
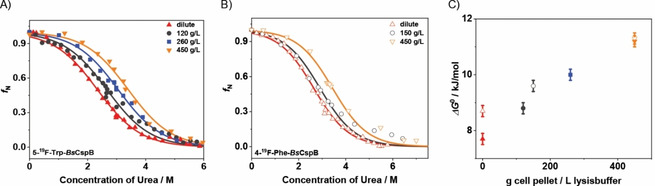
Chemically induced folding‐to‐unfolding transitions of 5‐19F−Trp−BsCspB and 4‐19F−Phe−BsCspB under dilute conditions and in cell lysate as probed by one‐dimensional 19F NMR spectroscopy at T=298 K. The overall thermodynamic stability, ΔG 0, increases for A) 5‐19F−Trp−BsCspB and B) 4‐19F−Phe−BsCspB with increasing mass of the cell pellet in the lysis buffer. The following experimental conditions for cell pellet concentration are shown: 0 g/L (▴, ▵), 120 g/L (•), 150 g/L (○), 260 g/L (▪), and 450 g/L (▾, ∇). The straight lines represent fitting of Equation (S1) to experimental data using a global value for folding cooperativity yielding m=−3.3±0.1 kJ/(molM). The fitting function is shown as a continuous line. C) Results for the determination of ΔG 0 with filled symbols for 5‐19F−Trp−BsCspB and open symbols for 4‐19F−Phe−BsCspB. Numerical values obtained for 5‐19F−Trp−BsCspB are: ΔG 0=7.7±0.2 (0 g/L), 8.8±0.2 (120 g/L), 10.0±0.2 (260 g/L), and 11.2±0.2 kJ/mol (450 g/L). For 4‐19F−Phe−BsCspB: ΔG 0=8.7±0.2 (0 g/L), 9.6±0.2 (150 g/L), and 11.3±0.2 kJ/mol (450 g/L).
As cell pellet concentration increases, the concentration of 5‐19F−Trp−BsCspB and 4‐19F−Phe−BsCspB will increase, too, and may potentially cause differences in the overall thermodynamic stability which has been observed. Consequently, we probed the overall thermodynamic stability of 5‐19F−Trp−BsCspB at two very different protein concentrations. Chemically induced folding‐to‐unfolding transitions of 5‐19F−Trp−BsCspB were monitored by applying one‐dimensional 1H NMR spectroscopy applying in vitro conditions using c BsCspB=8 and 200 μM (Figures 2A and S8). This setup covers a range of concentrations of BsCspB which can be expected to be present in cell lysate as used in this study (Tables S1 and S2). The overall thermodynamic stability for c BsCspB=8 μM was determined to be ΔG 0=(7.8±0.5) kJ/mol (Figure S8), which is in total agreement with ΔG 0=(7.2±0.2) kJ/mol as determined for c BsCspB=200 μM (Figure 2A). Thus we conclude that the increase in the overall thermodynamic stability as presented here for BsCspB in cell lysate compared to dilute conditions is not caused by a potential dependence on the protein concentration that was used.
As an important note, a reliable acquisition and analysis of a temperature‐induced folding‐to‐unfolding transition of 5‐19F−Trp−BsCspB present in cell lysate is not accessible (Figure S9). Even the unfolding process of 5‐19F−Trp−BsCspB appears to occur in cell lysate at a higher temperature compared to in vitro measurements, a quantitative assessment of the process of protein denaturation is not accomplishable. The temperature‐induced folding‐to‐unfolding transition of 5‐19F−Trp−BsCspB present in cell lysate is not a reversible process which prevents a subsequent quantitative thermodynamic analysis.
Estimation of concentration of macromolecules present in cell lysate
Controlling the weight of the cell pellet used for resuspension in a specific volume of lysis buffer made it possible to estimate the concentration of macromolecules which are present in cell lysate. Note, this numeric value indicates cell wet weight. Recently, Glazyrina et al. have shown that 1.70 g/L cell wet weight corresponds to 0.39 g/L cell dry weight in Escherichia coli cells by generating the same value in optical density at a wavelength of 600 nm. [25] Applying this relationship, the cell dry weight of samples used was determined (Table S3). Using numerical values of dry weight of samples strongly underlines the pronounced effect of an increasing overall thermodynamic stability of BsCspB when cell lysate instead of dilute conditions are experimentally applied. In detail, presence of 100 g dry cell pellet per liter lysis buffer is capable to increase ΔG 0 of BsCspB by about 3 kJ/mol. Notably, this number is in perfect agreement with the increase in ΔG 0 of BsCspB seen for the presence of 100 g/L PEG1, PEG8, or Dex20 in the test tube. [4a]
Conclusion
We have presented a reliable experimental method operating in a biological context of cell lysate that avoids handling of macromolecular crowding agents used for the imitation of the cellular environment. We applied 19F NMR spectroscopy to monitor fully reversible, folding‐to‐unfolding transitions of 4‐19F−Phe−BsCspB and 5‐19F−Trp−BsCspB at different concentrations of cell lysate. In this way increasing the concentration of cell lysate causes a monotonic increase in overall thermodynamic stability of BsCspB. Thus, we speculate that the quantity of macromolecules encompassing a protein intracellularly precisely controls its inherent thermodynamic features resulting in an adaptable functionality.
Experimental Section
Experimental Details are outlined in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Ulrich Weininger for critical reading of the manuscript. Financial support from the Juniorprofessurenprogramm handled by the Baden‐Württemberg Stiftung (grant ID: 647/16) and from the Deutsche Forschungsgemeinschaft (KO4687/4‐1) is gratefully acknowledged. Open access funding enabled and organized by Projekt DEAL.
H. Welte, M. Kovermann, ChemBioChem 2020, 21, 3575.
References
- 1. Barnes C. O., Pielak G. J., Proteins Struct. Funct. Bioinf. 2011, 79, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sarkar M., Li C., Pielak G. J., Biophys. Rev. Lett. 2013, 5, 187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Fulton A. B., Cell 1982, 30, 345–347; [DOI] [PubMed] [Google Scholar]
- 3b. Zimmerman S. B., Trach S. O., J. Mol. Biol. 1991, 222, 599–620. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Köhn B., Kovermann M., ChemBioChem 2019, 20, 759–763; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Olsen S. N., Thermochim. Acta 2006, 448, 12–18; [Google Scholar]
- 4c. Homchaudhuri L., Sarma N., Swaminathan R., Biopolymers 2006, 83, 477–486; [DOI] [PubMed] [Google Scholar]
- 4d. Ellis R. J., Trends Biochem. Sci. 2001, 26, 597–604; [DOI] [PubMed] [Google Scholar]
- 4e. Gierasch L. M., Gershenson A., Nat. Chem. Biol. 2009, 5, 774–777; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4f. Zhou H. X., Arch. Biochem. Biophys. 2008, 469, 76–82; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4g. Fodeke A. A., Minton A. P., J. Phys. Chem. B 2011, 115, 11261–11268; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4h. Schnell S., Turner T. E., Prog. Biophys. Mol. Biol. 2004, 85, 235–260; [DOI] [PubMed] [Google Scholar]
- 4i. van den Berg B., Ellis R. J., Dobson C. M., EMBO J. 1999, 18, 6927–6933; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4j. van den Berg B., Wain R., Dobson C. M., Ellis R. J., EMBO J. 2000, 19, 3870–3875; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4k. Charlton L. M., Barnes C. O., Li C. G., Orans J., Young G. B., Pielak G. J., J. Am. Chem. Soc. 2008, 130, 6826–6830; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4l. Ai X., Zhou Z., Bai Y., Choy W.-Y., J. Am. Chem. Soc. 2006, 128, 3916–3917; [DOI] [PubMed] [Google Scholar]
- 4m. Elcock A. H., Curr. Opin. Struct. Biol. 2010, 20, 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Zhou H. X., Rivas G. N., Minton A. P., Annu. Rev. Biophys. 2008, 37, 375–397; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Minton A. P., Biopolymers 2013, 99, 239–244; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Miklos A. C., Li C. G., Sharaf N. G., Pielak G. J., Biochemistry 2010, 49, 6984–6991; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Wang Y. Q., Sarkar M., Smith A. E., Krois A. S., Pielak G. J., J. Am. Chem. Soc. 2012, 134, 16614–16618; [DOI] [PubMed] [Google Scholar]
- 5e. Miklos A. C., Sarkar M., Wang Y. Q., Pieak G. J., J. Am. Chem. Soc. 2011, 133, 7116–7120. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Minton A. P., Biophys. J. 1980, 32, 77–79; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Minton A. P., Biopolymers 1981, 20, 2093–2120. [Google Scholar]
- 7.
- 7a. Sarkar M., Lu J., Pielak G. J., Biochemistry 2014, 53, 1601–1606; [DOI] [PubMed] [Google Scholar]
- 7b. Mcconkey E. H., Proc. Natl Acad Sci-Biol 1982, 79, 3236–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li C. G., Wang G. F., Wang Y. Q., Creager-Allen R., Lutz E. A., Scronce H., Slade K. M., Ruf R. A. S., Mehl R. A., Pielak G. J., J. Am. Chem. Soc. 2010, 132, 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghaemmaghami S., Oas T. G., Nat. Struct. Biol. 2001, 8, 879–882. [DOI] [PubMed] [Google Scholar]
- 10. Ignatova Z., Krishnan B., Bombardier J. P., Marcelino A. M. C., Hong J., Gierasch L. M., Biopolymers 2007, 88, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Bai J., Liu M. L., Pielak G. J., Li C. G., ChemPhysChem 2017, 18, 55–58; [DOI] [PubMed] [Google Scholar]
- 11b. Wang Y. Q., Li C. G., Pielak G. J., J. Am. Chem. Soc. 2010, 132, 9392–9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Theillet F. X., Binolfi A., Frembgen-Kesner T., Hingorani K., Sarkar M., Kyne C., Li C. G., Crowley P. B., Gierasch L., Pielak G. J., Elcock A. H., Gershenson A., Selenko P., Chem. Rev. 2014, 114, 6661–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Smith A. E., Sarkar M., Young G. B., Pielak G. J., Protein Sci. 2013, 22, 1313–1319; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Sarkar M., Smith A. E., Pielak G. J., Proc. Natl. Acad. Sci. USA 2013, 110, 19342–19347; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Kim M. S., Song J., Park C., Protein Sci. 2009, 18, 1051–1059; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Minde D. P., Maurice M. M., Rudiger S. G. D., PLoS One 2012, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Latham M. P., Kay L. E., PLoS One 2012, 7. [Google Scholar]
- 15. Li H. L., Frieden C., Proc. Natl. Acad. Sci. USA 2007, 104, 11993–11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crowley P. B., Kyne C., Monteith W. B., Chem. Commun. 2012, 48, 10681–10683. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Kim H. W., Perez J. A., Ferguson S. J., Campbell I. D., FEBS Lett. 1990, 272, 34–36; [DOI] [PubMed] [Google Scholar]
- 17b. Huestis W. H., Raftery M., Biochemistry 1972, 11, 1648–1654; [DOI] [PubMed] [Google Scholar]
- 17c. Sykes B. D., Weingarten H. I., Schlesinger M. J., Proc. Natl. Acad. Sci. USA 1974, 71, 469–473; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17d. Kimber B., Griffiths D., Birdsall B., King R., Scudder P., Feeney J., Roberts G., Burgen A., Biochemistry 1977, 16, 3492–3500. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Welte H., Kovermann M., Protein Expression Purif. 2019, 157, 86–91; [DOI] [PubMed] [Google Scholar]
- 18b. Welte H., Zhou T., Mihajlenko X., Mayans O., Kovermann M., Sci. Rep. 2020, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Sachs R., Max K. E., Heinemann U., Balbach J., RNA 2012, 18, 65–76; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Zeeb M., Max K. E., Weininger U., Löw C., Sticht H., Balbach J., Nucleic Acids Res. 2006, 34, 4561–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Schindler T., Perl D., Graumann P., Sieber V., Marahiel M. A., Schmid F. X., Proteins 1998, 30, 401–406; [DOI] [PubMed] [Google Scholar]
- 20b. Schindler T., Herrler M., Marahiel M. A., Schmid F. X., Nat. Struct. Biol. 1995, 2, 663–673; [DOI] [PubMed] [Google Scholar]
- 20c. Schindelin H., Marahiel M. A., Heinemann U., Nature 1993, 364, 164–168; [DOI] [PubMed] [Google Scholar]
- 20d. Schindelin H., Herrler M., Willimsky G., Marahiel M. A., Heinemann U., Proteins 1992, 14, 120–124; [DOI] [PubMed] [Google Scholar]
- 20e. Schnuchel A., Wiltscheck R., Czisch M., Herrler M., Willimsky G., Graumann P., Marahiel M. A., Holak T. A., Nature 1993, 364, 169–171. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Pace C. N., Methods Enzymol. 1986, 131, 266–280; [DOI] [PubMed] [Google Scholar]
- 21b. Greene R. F., Pace C. N., J. Biol. Chem. 1974, 249, 5388–5393; [PubMed] [Google Scholar]
- 21c. Monera O. D., Kay C. M., Hodges R. S., Protein Sci. 1994, 3, 1984–1991; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21d. Santoro M. M., Bolen D. W., Biochemistry 1988, 27, 8063–8068. [DOI] [PubMed] [Google Scholar]
- 22. Åstrand P.-O., Wallqvist A., Karlström G., J. Phys. Chem. 1994, 98, 8224–8233. [Google Scholar]
- 23. Frank H. S., Franks F., J. Chem. Phys. 1968, 48, 4746–4757. [Google Scholar]
- 24. Bryant J. E., Lecomte J. T. J., Lee A. L., Young G. B., Pielak G. J., Biochemistry 2007, 46, 8206–8206.17567051 [Google Scholar]
- 25. Glazyrina J., Materne E.-M., Dreher T., Storm D., Junne S., Adams T., Greller G., Neubauer P., Microb. Cell Fact. 2010, 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary