

Abstract
Objectives
To describe two patients with progressive external ophthalmoplegia (PEO) and mitochondrial myopathy associated with mutations in mitochondrial DNA, encoding the tRNAAsn gene (MT‐TN), which have not previously been published with clinical descriptions.
Materials & Methods
Two unrelated patients with PEO were clinically examined. Muscle biopsy was performed and investigated by exome sequencing, enzyme histochemistry, and immunohistochemistry. The level of heteroplasmy was investigated in single muscle fibers and in other tissues.
Results
Patient 1 was a 52‐year‐old man with ptosis, PEO, and exercise intolerance since childhood. Muscle biopsy demonstrated mitochondrial myopathy with frequent cytochrome c oxidase (COX)‐deficient fibers and a heteroplasmic mutation, m.5669G>A in the MT‐TN gene, resulting in a substitution of a highly conserved C to T in the T stem of tRNAAsn. Patient 2 was a 66‐year‐old woman with ptosis, PEO, and exercise intolerance since many years. Muscle biopsy demonstrated mitochondrial myopathy with frequent COX‐deficient fibers. She had a novel m.5702delA mutation in MT‐TN, resulting in loss of a highly conserved U in the anticodon stem of tRNAAsn. Single fiber analysis in both cases showed highly significant differences in mutation load between COX‐deficient and COX‐normal fibers and a high threshold level for COX deficiency. The mutations were not found in blood, urine sediment or buccal cells.
Conclusion
We describe two MT‐TN mutations associated with PEO and mitochondrial myopathy, and their pathogenicity was demonstrated. Together with previous reports, the results indicate that MT‐TN is a hot spot for mutations causing sporadic PEO.
1. INTRODUCTION
Progressive external ophthalmoplegia (PEO) with mitochondrial myopathy is usually associated with single or multiple mitochondrial DNA (mtDNA) deletions. 1 Single deletions are usually sporadic but may be maternally inherited in 5% of the cases. PEO associated with multiple deletions shows autosomal dominant or recessive inheritance. Point mutations in mtDNA are more rarely causing isolated PEO. In this study, we investigated two apparently sporadic patients presenting with PEO and mitochondrial myopathy. We identified two mtDNA mutations, m.5669G>A and m.5702delA, in the MT‐TN gene, encoding the tRNAAsn and demonstrated their pathogenicity. Nine probably pathogenic mutations have previously been described in the MT‐TN gene. 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 Five of these were associated with sporadic PEO, which together with the two mutations described here makes the MT‐TN gene a hot spot for sporadic PEO associated with mtDNA point mutations.
2. MATERIALS AND METHODS
2.1. Patients
The patients were two unrelated adults without any family history of PEO or other suspected mitochondrial disease. The study was conducted in accordance with the Helsinki declaration, and the patients gave their informed consent for the performed investigations and publication of data.
2.2. Histological and histochemical analyses
Open muscle biopsy investigations of the deltoid muscle in patient 1 and vastus lateralis in patient 2 were performed. Standard diagnostic morphological and enzyme histochemical analyses were performed on 8‐µm‐thick cryosections of fresh frozen skeletal muscle according to established protocols, 15 including hematoxylin and eosin (H&E) staining, oxidative enzyme histochemical analyses of cytochrome c oxidase (COX), succinate dehydrogenase (SDH), and sequential COX‐SDH activities. Immunohistochemical studies of each mitochondrial respiratory complex were carried out with peroxidase/diaminobenzidine as chromogen (EnVision FLEX DAB + Substrate Chromogen System, Dako Omnis, Denmark), and the following antibodies from Abcam, UK, anti‐NDUFB8 (ab110242, 1:100) for complex I, anti‐SDHB (ab14714, 1:500) for complex II, anti‐UQCRC2 (ab14745, 1:2000) for complex III, anti‐MTCO1 (ab14705, 1:2000) for complex IV, anti‐ATPB (ab14730, 1:500) for complex V, and an antibody to VDAC1 (ab14734, 1:2000) served as a mitochondrial marker.
2.3. Molecular genetic analyses
Exome sequencing target enrichment was performed on genomic DNA extracted from skeletal muscle using the Sure SelectXT Human All Exon Kit version 6 (Agilent Technologies) and sequenced with 60× coverage on the HiSeq4000 platform (Illumina). The 150‐bp paired‐end reads were aligned to the reference genome (hg19) using the CLC Biomedical Genomics Workbench (Qiagen). Data were analyzed using Ingenuity Variant Analysis (IVA) (www.ingenuity.com/products/variant-analysis). We searched for candidate genes potentially associated with mitochondrial disease and included all 1158 genes listed in MitoCarta2.0, 16 and the entire mtDNA. Further filtering was done for high‐quality variants classified as deleterious (missense, nonsense, indel, and splice‐site variants ± 10 bp around exon boundaries) and rare (<0.5% minor allele frequency in the ExAC Browser, gnomAD or 1000 Genomes). Variants of interest were further evaluated using prediction tools: PhyloP, SIFT, PolyPhen‐2, and MutationTaster. Sanger sequencing was used for confirmation and segregation analysis in additional tissues.
To further study the mutations in muscle, we isolated single muscle fibers with a tungsten needle from a 16‐μm‐thick section of the fresh frozen biopsy that was subjected to double COX/SDH staining as described previously 17 ; the brown fibers were regarded as COX‐normal, the blue fibers were regarded as COX‐deficient, and no intermediate fibers were investigated. In total, over 50 fibers from each patient were dissected and analyzed.
3. RESULTS
3.1. Patients
Patient 1 was a 52‐year‐old man without a family history (his sister and her daughter, his mother and her three siblings) of suspected mitochondrial disease. He had had limited ability to look in different directions without turning his head and also droopy eyelids as long as he could remember. Last 2 years, he had experienced short episodes of double vision. He was seeing an ophthalmologist who referred him to our neuromuscular center for evaluation of PEO. He had never been able to run more than 50 m because of exercise intolerance with pronounced muscle fatigue, but did not complain of muscle weakness or other neurological symptoms. A clinical examination revealed subtotal external ophthalmoplegia and ptosis. Upward gaze was completely lost, and horizontal gaze was limited to 10 degrees. He also had a weakness of eye closure with a remaining gap of one mm. He had no impairment of vision or hearing, and no other muscle weakness or signs of nervous system disease. Electrocardiogram and echocardiography were normal. Creatine kinase (CK) level in blood was at the upper limit of the normal range, whereas blood glucose and kidney function tests were normal.
Patient 2 was a 66‐year‐old woman without a family history of the suspected mitochondrial disease, but her mother, who died at age 73 years, had “muscular rheumatism”. None of the patient's three siblings, her two children, and three grandchildren had similar symptoms. The patient experienced symptoms of exercise intolerance in the past 40 years expressed as pain and fatigue in her legs when climbing upstairs. She needed frequent rests when walking on flat ground. For many years, she had problems with droopy eyelids and had been operated twice on both sides for ptosis. She also had PEO since many years. She had in periods experienced memory deficit for 20 years, but she had no other neurological complaints except for her muscle problems. Clinical examination revealed complete ophthalmoplegia, ptosis, and slight muscle weakness in the elbow and hip flexion. Echocardiography, resting electrocardiogram, and 24‐hour electrocardiogram were all normal. Recent serum CK analyses, blood glucose, and serum creatinine in blood were normal.
3.2. Muscle histopathology
The muscle biopsy from both patients 1 and 2 demonstrated mitochondrial myopathy with ragged red fibers and frequent COX‐deficient fibers in a mosaic pattern (Figure 1). Analyses of the respiratory chain enzyme complexes showed fibers with reduced expression of complex I subunit NDUFB8 and complex IV subunit CO1 by immunohistochemistry, and reduced cytochrome c oxidase (complex IV) activity by enzyme histochemistry. There were no necrotic fibers and no increase in interstitial connective tissue.
Figure 1.
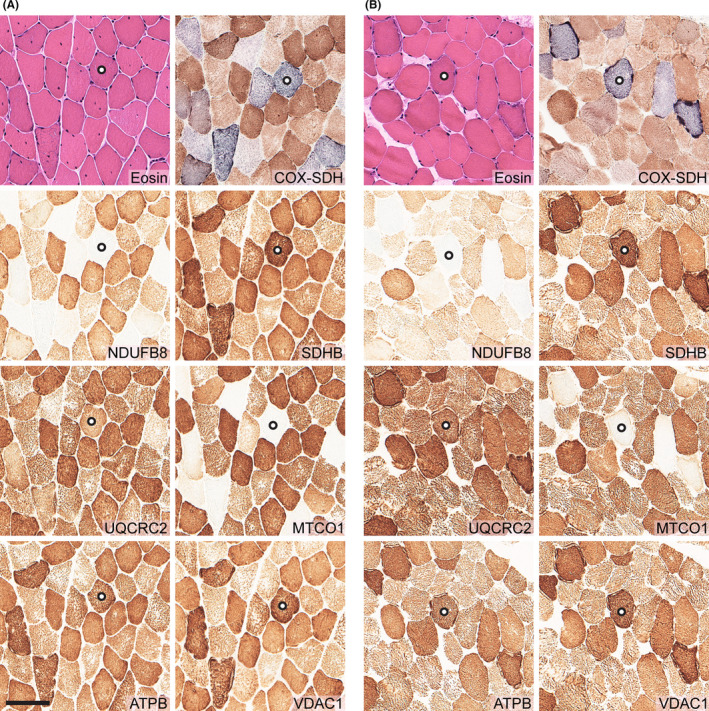
Morphological, immunohistochemical and enzyme histochemical analyses of skeletal muscle biopsy in patient 1 (A), and patient 2 (B). Each panel consists of hematoxylin and eosin (H&E) staining, cytochrome c oxidase (COX) and succinate dehydrogenase (SDH) double enzyme histochemistry, and immunohistochemistry of mitochondrial respiratory complex I subunit NDUFB8, complex II subunit SDHB, complex III subunit UQCRC2, complex IV subunit MTCO1, complex V subunit ATPB, and the mitochondrial marker VDAC1. Both patients showed mitochondrial myopathy with frequent COX‐deficient (blue) fibers, which were also deficient of complex I subunit NDUFB8, and complex IV subunit MTCO1. Serial sectioning was performed, and for orientation, one fiber in each case was marked with a circle. Scale bar represents 100 μm
3.3. Molecular genetic analyses
Large‐scale, single or multiple mtDNA deletions in muscle were excluded by LX‐PCR analyses. Exome sequencing revealed heteroplasmic mutations in the MT‐TN gene in muscle DNA: m.5669G>A in patient 1 and m.5702delA in patient 2. The mutations were both affecting evolutionary highly conserved bases of the T and anticodon stems, respectively, within the cloverleaf model of the human mt‐tRNAAsn (m.5657‐5729) (Figure 2). Sanger sequencing of mtDNA also confirmed the heteroplasmic levels of m.5669G>A in patient 1 and m.5702delA in patient 2 with 52% and 63% heteroplasmy, respectively. The mutations were only identified in mtDNA derived from muscle, but not from blood, buccal mucosa, and urinary epithelial cells in both patients (Figure 3A).
Figure 2.
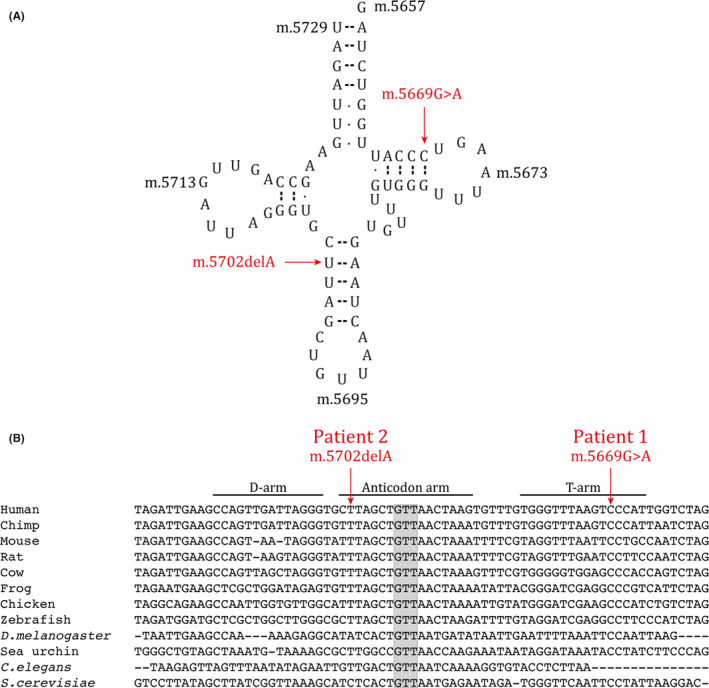
Mitochondrial tRNAAsn. The two novel mutations reported in this study are indicated with arrows. (A) Cloverleaf model of the human mt‐tRNAAsn (m.5657‐5729). The m.5669G>A mutation changes a highly conserved C to U in the T stem and the m.5702delA mutation results in loss of a highly conserved U in the anticodon stem. (B) Phylogenetic conservation of the MT‐TN gene. Anticodon bases are highlighted with gray background
Figure 3.
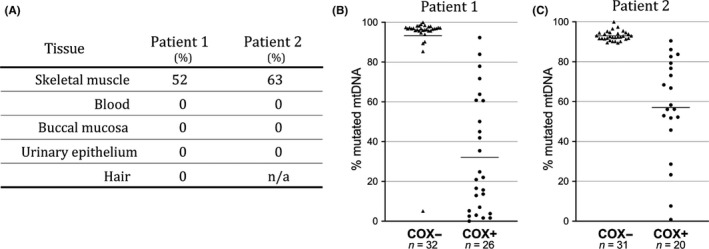
Mitochondrial DNA mutation load. (A) Mutation load in different tissues: skeletal muscle, blood, buccal mucosa, urinary epithelial cells, and hair shows that the mutations are only found in skeletal muscle in both patients. n/a indicates data are not available. (B and C) Distribution of the mutation load measured in single COX‐normal (COX+) and COX‐deficient (COX−) muscle fibers. Horizontal bars represent the calculated means. The results from patient 1 (m.5669G>A) and patient 2 (m.5702delA) demonstrate that a high level of mutation load correlates with respiratory chain deficiency
Single fiber PCR analyses showed significantly higher levels of both mutations in COX‐deficient fibers (two‐tailed unpaired t test) (Figure 3B and C). The mean levels of the m.5669G>A variant in patient 1 were 93% (range 5–100%) in COX‐deficient fibers, which was significantly higher (P < .0001) compared to COX‐normal fibers (32%, range 0–92%) (Figure 3B). In patient 2, the mean level of the m.5702delA variant was 93% (range 90–100%) in COX‐deficient fibers, which was significantly higher (P < .0001) compared to COX‐normal fibers (57%, range 1–91%) (Figure 3C). The threshold levels of mutation load for COX deficiency were above 80% in both patients (Figure 3B,C).
4. DISCUSSION
In this study, we describe two unrelated patients with mitochondrial myopathy and PEO associated with two mtDNA mutations, m.5669G>A and m.5702delA, in MT‐TN. The pathogenicity of the identified MT‐TN mutations was strongly supported by a clear correlation between mutation load in single muscle fibers and reduced expression of mtDNA‐encoded subunit MTCO1 in complex IV and a corresponding reduced cytochrome c oxidase activity in the same fibers. These fibers also showed reduced expression of complex I, which contains several essential subunits encoded by mtDNA. There was no reduction in either the entirely nuclear‐encoded complex II or the mitochondrial membrane protein porin (VDAC1). The identified MT‐TN mutations were only found in muscle tissue, which was the only tissue with a clear manifestation of mitochondrial disease. Therefore, the mutation load correlated with disease manifestations both at tissue and cell levels. Additional support for their pathogenicity is that the mutations affect evolutionarily conserved bases and they are predicted to disrupt essential base‐pairing which may severely affect the normal cloverleaf structure of the tRNAAsn molecule. The m.5702delA variant has not been identified as single nucleotide polymorphisms in MITOMAP, and m.5669G>A variant is reported as a likely pathogenic variant in one case. 18 The identified threshold levels for COX deficiency in single muscle fibers above 80% were also in accordance with what is usually found in myopathy due to pathogenic mtDNA point mutations. In patient 1, there was one COX‐deficient fiber with only 5% mutated mtDNA. This outlier was probably due to age‐related accumulation of another pathogenic mutation which occurs as a normal phenomenon in single fibers during aging. 19
From the clinical history, it appeared that both cases were sporadic and not inherited, although we could not completely rule out maternal transmission since such material was not available. Furthermore, both mutations were not identified in blood, which is a frequent finding in sporadic cases, and compatible with the idea that high risk for transmission of the mutation is associated with its presence in rapidly replicating tissues such as blood. 20
Nine different point mutations in MT‐TN have previously been reported to be associated with mitochondrial disease. Five of these appeared as isolated PEO with mitochondrial myopathy without demonstrated inheritance. 2 , 3 , 4 , 7 , 8 , 9 , 10 , 13 Together with the two mutations described here, seven out of eleven mutations show this phenotype, which is otherwise associated mainly with large‐scale deletions of mtDNA. Although sporadic PEO with mitochondrial myopathy has also been described to be related to other mtDNA tRNA genes, MT‐TN seems to be a hot spot for this particular phenotype together with MT‐TL2. 18 There is no clear genotype–phenotype correlation with regard to the different tRNA genes although some features may typically be associated with a particular tRNA gene, for example mitochondrial encephalomyopathy, lactic acidosis, and stroke‐like episodes (MELAS) is associated with several different MT‐L1 mutations. 18 The explanation for the apparent association of sporadic PEO with MT‐TN mutations remains unclear.
In conclusion, we describe two MT‐TN mutations associated with sporadic PEO and mitochondrial myopathy, adding to only a few previously reported mutations in this gene and indicating that MT‐TN is a hotspot for mutations causing sporadic PEO.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
ACKNOWLEDGEMENTS
The study was supported by a grant from the Swedish Research Council (Project no. 2018‐02821).
Visuttijai K, Hedberg‐Oldfors C, Lindgren U, et al. Progressive external ophthalmoplegia associated with novel MT‐TN mutations. Acta Neurol Scand. 2020;103–108. 10.1111/ane.13339
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017;139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pinos T, Melia MJ, Ortiz N, et al. Identification of the novel mutation m.5658T>C in the mitochondrial tRNA(Asn) gene in a patient with myopathy, bilateral ptosis and ophthalmoparesis. Neuromuscul Disord. 2013;23(4):330-336. [DOI] [PubMed] [Google Scholar]
- 3. Moraes CT, Ciacci F, Bonilla E, et al. Two novel pathogenic mitochondrial DNA mutations affecting organelle number and protein synthesis. Is the tRNA(Leu(UUR)) gene an etiologic hot spot? J Clin Invest. 1993;92(6):2906-2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blakely EL, Yarham JW, Alston CL, et al. Pathogenic mitochondrial tRNA point mutations: nine novel mutations affirm their importance as a cause of mitochondrial disease. Hum Mutat. 2013;34(9):1260-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meulemans A, Seneca S, Lagae L, et al. A novel mitochondrial transfer RNA(Asn) mutation causing multiorgan failure. Arch Neurol. 2006;63(8):1194-1198. [DOI] [PubMed] [Google Scholar]
- 6. Tarnopolsky M, Brady L, MacNeil L. Myasthenia graves-like symptoms associated with rare mitochondrial mutation (m.5728T>C). Mitochondrion. 2019;47:139-140. [DOI] [PubMed] [Google Scholar]
- 7. Vives‐Bauza C, Del Toro M, Solano A, Montoya J, Andreu AL, Roig M. Genotype‐phenotype correlation in the 5703G>A mutation in the tRNA(ASN) gene of mitochondrial DNA. J Inherit Metab Dis. 2003;26(5):507‐508. [DOI] [PubMed] [Google Scholar]
- 8. Nishigaki Y, Ueno H, Coku J, et al. Extensive screening system using suspension array technology to detect mitochondrial DNA point mutations. Mitochondrion. 2010;10(3):300‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Valente L, Piga D, Lamantea E, et al. Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim Biophys Acta. 2009;1787(5):491‐501. [DOI] [PubMed] [Google Scholar]
- 10. Sternberg D, Chatzoglou E, Laforet P, et al. Mitochondrial DNA transfer RNA gene sequence variations in patients with mitochondrial disorders. Brain. 2001;124(Pt 5):984‐994. [DOI] [PubMed] [Google Scholar]
- 11. Coulbault L, Herlicoviez D, Chapon F, et al. A novel mutation in the mitochondrial tRNA Asn gene associated with a lethal disease. Biochem Biophys Res Commun. 2005;329(3):1152‐1154. [DOI] [PubMed] [Google Scholar]
- 12. Bannwarth S, Procaccio V, Lebre AS, et al. Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J Med Genet. 2013;50(10):704‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ronchi D, Sciacco M, Bordoni A, et al. The novel mitochondrial tRNAAsn gene mutation m.5709T>C produces ophthalmoparesis and respiratory impairment. Eur J Hum Genet. 2012;20(3):357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seibel P, Lauber J, Klopstock T, Marsac C, Kadenbach B, Reichmann H. Chronic progressive external ophthalmoplegia is associated with a novel mutation in the mitochondrial tRNA(Asn) gene. Biochem Biophys Res Commun. 1994;204(2):482‐489. [DOI] [PubMed] [Google Scholar]
- 15. Dubowitz V, Sewry C, Oldfors A. Muscle Biopsy. A Practical Approach, 5th edn. Amsterdam, The Netherlands: Elsevier; 2020. [Google Scholar]
- 16. Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44(D1):D1251‐D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oldfors A, Moslemi AR, Fyhr IM, Holme E, Larsson NG, Lindberg C. Mitochondrial DNA deletions in muscle fibers in inclusion body myositis. J Neuropathol Exp Neurol. 1995;54(4):581‐587. [DOI] [PubMed] [Google Scholar]
- 18. MITOMAP . A Human Mitochondrial Genome Database. http://www.mitomap.org. 2020. [DOI] [PMC free article] [PubMed]
- 19. Fayet G, Jansson M, Sternberg D, et al. Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord. 2002;12(5):484‐493. [DOI] [PubMed] [Google Scholar]
- 20. Elson JL, Swalwell H, Blakely EL, McFarland R, Taylor RW, Turnbull DM. Pathogenic mitochondrial tRNA mutations–which mutations are inherited and why? Hum Mutat. 2009;30(11):E984‐E992. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.