

Abstract
In this study we report the identification of a novel class of ligands for palladium‐catalyzed C(sp3)−H activation that enables the direct alkynylation of free carboxylic acid substrates. In contrast to previous synthetic methods, no introduction/removal of an exogenous directing group is required. A broad scope of acids including both α‐quaternary and challenging α‐non‐quaternary can be used as substrates. Additionally, the alkynylation in the distal γ‐position is reported. Finally, this study encompasses preliminary findings on an enantioselective variant of the title transformation as well as synthetic applications of the products obtained.
Keywords: Alkynylation, Carboxylic acids, C−H activation, Ligand-enabled catalysis, Palladium
No detours—The direct C(sp3)−H alkynylation of free carboxylic acids in the β‐ and γ‐position is achieved using a Pd‐catalyst with a newly discovered ligand class. The synthetic method features a broad scope including preliminary findings on an enantioselective variant and can be conducted on a preparatively useful scale.

The alkyne moiety is widely used in medicinal chemistry, material science, and synthetic chemistry. It often serves as synthetic linchpin, due to its tremendous synthetic versatility, which enables the synthesis of new materials and drugs, through subsequent transformations. [1] Thus, efficient methods for the introduction of alkyne moieties into a given organic molecule are required. In this context, the direct alkynylation of C−H bonds has received significant attention. [2]
The metal‐catalyzed C(sp3)−H alkynylation of carboxylic acids constitutes a particularly attractive goal, since many carboxylic acids are cheap, easily available, and abundant starting materials and the resulting products contain two highly versatile functional groups. [3] In this regard, the direct use of free, unfunctionalized carboxylic acids as substrates to achieve a C(sp3)−H activation/alkynylation can be considered the ideal approach (Scheme 1, Path A). However, the development of such methods is hampered by several key challenges inherent to the carboxylic acid moiety as directing group, including, among other reasons, the low directing ability of the carboxylate unit and the existence of competing coordination modes between palladium and the carboxylate moiety. [4] These challenges have traditionally been addressed through the introduction of specialized exogenous directing groups (DGs) that enable the key C−H alkynylation, but add synthetic steps to the overall sequence (Scheme 1, Path B). [5] The first palladium‐catalyzed alkynylation of non‐activated C(sp3)−H bonds in aliphatic carboxylic acid derivatives was described by Chatani et al. in 2011 using a bidentate auxiliary. [6] Later, Yu et al. developed a β‐methyl C(sp3)−H alkynylation, using aliphatic amides bearing a perfluorinated arene auxiliary on the nitrogen. [7] In 2017 the same group reported the first example of an enantioselective C(sp3)−H alkynylation, [8] as well as the first C(sp3)−H alkynylation of oligopeptides. [9] In parallel, other research groups such as the groups of Chen, Shi, Colobert, and Verho developed new catalytic systems and directing groups to achieve C(sp3)−H alkynylation. [10] Additionally to the development of palladium‐catalyzed alkynylation, cobalt‐ and nickel‐catalyzed reactions using a bidentate auxiliary were reported. [11] Although these reactions allow the formation of the target compounds, they are still far from ideal with respect to the atom‐ and step‐economy.
Scheme 1.

Previous studies on the indirect C−H alkynylation of aliphatic acids (Path B) and direct C(sp3)−H alkynylation of free carboxylic acids developed in this study (Path A).
Recently, ourselves and others have demonstrated that the challenging direct C(sp3)−H activation/functionalization of free carboxylic acids can be enabled through ligand design and, within a short period of time, protocols have been developed that enable valuable transformations, such as direct arylations, [12] acetoxylations, [13] lactonizations, [14] and olefinations [15] of such substrates. Based on our experience in the β‐ and γ‐C(sp3)−H functionalization of free carboxylic acids, we thus expected that the identification of a suitable ligand would be crucial for the development of the desired alkynylation process.
We initiated our studies using pivalic acid 1 a as model substrate. Different ligands, that have been used for the direct C(sp3)−H activation of free carboxylic acids for other transformations were tested. The amino acid derivatives L1‐L3,[ 12c , 12d ] thioether ligand L4, [15a] monoprotected aminoethyl amine ligand L5,[ 12f , 12g ] anthranilic acid ligand L6, [15b] and oxazoline ligand L7 all delivered discouraging results (Scheme 2).[ 8 , 16 ]
Scheme 2.

Identification of suitable ligands for the β‐ and γ‐C(sp3)−H alkynylation of free carboxylic acids. Reactions were conducted on a 0.2 mmol scale. Yields were determined by 1H NMR analysis of the crude reaction mixture using 1,3,5‐trimethoxybenzene as an internal standard. The mass balance of the reactions is fully accounted for by the product formed and unreacted starting material. No di‐ or tri‐substituted product was observed in any case during the optimization studies.
Finally, we identified a novel class of diamine‐derived ligands which proved highly efficient in this transformation. Starting from our initial discovery (L8, 10 %), the catalyst efficiency improved dramatically when an aryl substituent was introduced on the sulfonamide (L9, 65 %). Further optimization on the ligand structure showed that various substitution on the aromatic ring do not have a significant influence on the yield (L10–L13). Substitution on the ethylenediamine backbone combined with varied arenes on the sulfonamide likewise delivered no substantial further improvement in yield for the standard substrate (L14–L18), such that the comparably simple and easy to access ligand L9 was chosen for further studies.
Having identified a suitable ligand for the β‐alkynylation of free carboxylic acids, we started to investigate the substrate scope (Scheme 3). As expected from the optimization studies, the product 2 a derived from pivalic acid 1 a was obtained in 65 % yield. Variation of the aliphatic chain in 2 b (70 %), 2 c (63 %), and 2 d (65 %) is not detrimental for the reaction outcome. A fluorine substituent was well tolerated (2 e, 57 %) as well as a TBS‐protected (2 f, 55 %) and free hydroxy group (2 g, 45 %). We furthermore evaluated various aryl‐containing carboxylic acids. Our protocol showed a broad functional group tolerance irrespective of the substitution pattern on the arene. Both electron‐donating (2 i, 2 j) and electron‐withdrawing substituents (2 k–2 r) were well tolerated under our reaction conditions. Interestingly, we did not observe competing C(sp2)−H alkynylation under our reaction conditions and unsubstituted phenyl (2 h, 58 %) and naphthyl rings (2 s, 59 %) provided good yields as well. Importantly, a series of functional groups that are of relevance in biologically active substances was well tolerated, such as ethers (2 j, 63 %), nitriles (2 k, 57 %), trifluoromethyl‐ and trifluoromethylthio‐groups (2 l, 58 % and 2 m, 55 %), esters (2 n, 57 %), sulfonates (2 o, 68 %), nitro groups (2 p, 75 %) and ketones (2 q, 67 %). Additionally, we achieved the β‐methylene‐C(sp3)−H alkynylation of cyclopropane carboxylic acid (2 t, 45 %, 98:2 dr). [17]
Scheme 3.
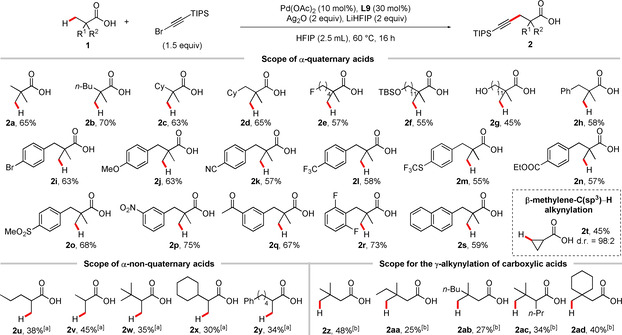
Reaction scope. Reactions were conducted on a 0.2 mmol scale. [a] L14 was used instead of L9, LiHFIP (1 equiv), H2O (1.5 equiv), and HFIP (2.0 mL) were used at 60 °C for 24 h; [b] Pd(OAc)2 (20 mol %), L14 (60 mol %), LiHFIP (2 equiv), Ag2O (2 equiv), 1‐bromo‐2‐(triisopropylsilyl)acetylene (3 equiv), and HFIP (4 mL) were used at 60 °C for 24 h. The structures of the respective starting materials are shown for simplicity with the position that is C−H alkynylated highlighted.
To further broaden the applicability of our method, we proceeded to study the use of notoriously challenging α‐non‐quaternary acid substrates. Notably, due to the absence of an accelerating Thrope–Ingold effect and additional possible side reactions, such substrates are either not included or have proved particularly challenging in the majority of studies on the direct C−H activation/functionalization of aliphatic carboxylic acids.[ 4a , 4b ]
Interestingly, a re‐optimization of the reaction conditions, using L14 instead of L9 alongside further adaptions, enabled us to alkynylate α‐non‐quaternary acid substrates.
Various substrates with varying chain lengths and steric demands could be alkynylated (2 u–2 y). It should be noted that, although the yields of these reactions remained moderate, the fact that the functionalization can be achieved on the free acid substrate rather than using an exogenous directing group nevertheless provides a substantial increase in efficiency.
Building upon the advances in the ligand‐enabled C(sp3)−H bond activation of free carboxylic acids in the proximal β‐position, ourselves and others have recently strived to expand this reactivity to the more distal γ‐position.[ 12e , 12h , 15b , 15c ] Such reactions face additional challenges with respect to activation entropies and enthalpies, as well as potential side reactions.
We were thus highly pleased to find that our alkynylation protocol could be adapted to the γ‐C(sp3)−H alkynylation of carboxylic acids. As for the β‐alkylnylation of α‐non quaternary acids, the key to enable this transformation was the use of L14 as ligand, under partially re‐optimised conditions. The alkyl substituted products 2 z–2 ad were obtained in moderate to good yield.
It should be noted that these yields, despite remaining moderate, compare well with those that have previously been achieved by using exogenous directing groups.[ 6a , 7b , 10b ] Additionally, the yields in these indirect routes would in practice be reduced further through losses of material in the steps required for the introduction and removal of the DG, which adds further value to the direct protocol described herein.
After completing the investigation on the reaction scope, we became interested to explore the possibility of achieving enantioinduction in the reaction of substrates that become chiral upon alkynylation by using ligands with a chiral, enantiopure ethylenediamine backbone (Scheme 4).
Scheme 4.

Initial findings on the enantioselective β‐C(sp3)−H alkynylation of carboxylic acids 1 c and 1 t.
We observed that the introduction of a benzyl group (L19) enabled the desymmetrization of 1 c, giving 2 c in 66 % yield and with 19 % ee (Scheme 4 a). Similarly, the introduction of an isopropyl unit (L20) induced a moderate enantioselectivity in the desymmetrization of 1 t, giving 2 t in 35 % yield and with a promising 39 % ee (Scheme 4 b).
To further demonstrate the synthetic utility of our protocol, we proceeded to probe the scalability of our reaction. The product 2 a was obtained with a virtually identical yield of 60 % on a 5 mmol scale (Scheme 5 a). Next, we investigated possible derivatizations of product 2 a. [18] Compound 3 could be synthesized via a deprotection/Sonogashira coupling sequence. Interestingly, both steps could be performed in the presence of the free carboxylic acid moiety. In parallel, 2 a was esterified and subsequently submitted to a deprotection/coupling sequence to yield compound 4 in a high overall yield of 67 % over 3 steps (Scheme 5 b).
Scheme 5.

Synthetic utility of the β‐C(sp3)‐H alkynylation of aliphatic carboxylic acids.
Additionally, we were able to synthesize the bioactive valproic acid (VPA) derivative 5, starting from the easily accessible starting material 1 ae (Scheme 5 c). [19]
It should be noted that this sequence could be conducted with synthetically useful yields despite requiring the alkynylation of a challenging α‐non‐quaternary substrate.
Finally, we focused on obtaining a preliminary mechanistic understanding of this transformation. We began by determining the kinetic isotope effect (KIE) both in a competition experiment and parallel experiments. The clear primary KIE observed in both experiments indicates that the C−H activation is the rate determining step (Scheme 6 A). [20] Next, we evaluated the reversibility of the C−H activation step. We conducted two reversibility experiments (Scheme 6 B), one in presence and one in absence of the TIPS‐alkynyl bromide reagent. In both cases the deuteration of the remaining starting material was analyzed. When no 1‐bromo‐2‐(triisopropylsilyl)acetylene was added, a strong de‐deuteration was observed, showing that the C−H activation step is in principle reversible under the reaction conditions. In contrast, when the reagent was present, no loss of deuteration was observed in the remaining starting material.
Scheme 6.

Preliminary mechanistic studies.
Based on these observations we conclude that the reaction occurs through a rate‐limiting C−H activation that, although in principle reversible, is typically not reversed under conditions suitable for product formation. The subsequent steps of the catalytic cycle can, based on literature precedent, be assumed to follow one of two pathways.[ 2 , 21 ] Either a PdIV/PdII‐pathway through an oxidative addition into the C−Br bond followed by a C−C bond‐forming reductive elimination, or through a PdII pathway consisting of an alkyne insertion followed by Pd‐Br elimination. In both cases the silver salt would act to activate the alkyne and subsequently bind the bromide ions that could otherwise poison the catalyst. Finally, the role of the LiHFIP base can be rationalized in analogy to our recent studies on the acyloxylation of carboxylic acids: By producing only a solvent molecule as byproduct, this base avoids possible detrimental effects exerted by the conjugate acids of many common bases. Additionally, the presence of an alkali metal ion is likely required to induce a κ1‐coordination mode between the carboxylate and palladium, which is in turn a prerequisite for the C−H activation to occur. [13a]
In summary, we have developed a protocol for the palladium‐catalyzed β‐ and γ‐C(sp3)−H alkynylation of free carboxylic acids. The reaction is enabled by a newly discovered ligand class that is derived from an ethylenediamine backbone. Our protocol gives access to a broad range of alkynylated products, starting from the respective acids in one step, without the need to introduce an exogenous directing group. Importantly, the scope of this reaction could be extended to α‐non‐quaternary acid substrates as well as distal γ‐C(sp3)−H bonds. Finally, our study encompasses initial results on the feasibility of an enantioselective variant of this transformation and synthetic applications of the products obtained. We expect that the synthetic method described herein will prove helpful for example in the synthesis of bioactive molecules or components for functional materials. Additionally, the novel ligand class disclosed is expected to prove useful in further studies towards the development of challenging C−H functionalization protocols.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors gratefully acknowledge financial support from the DFG (Funding through the Emmy Noether Programme, M.v.G.), the Studienstiftung des deutschen Volkes (fellowship to F.G.) and the WWU Münster. We thank the members of our NMR and MS departments for their excellent service. Furthermore we are indebted to Prof. F. Glorius for his generous support. Open access funding enabled and organized by Projekt DEAL.
F. Ghiringhelli, A. Uttry, K. K. Ghosh, M. van Gemmeren, Angew. Chem. Int. Ed. 2020, 59, 23127.
Dedicated to Prof. Antonio M. Echavarren on the occasion of his 65th birthday
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.12770468.v1).
References
- 1.
- 1a. Modern Acetylene Chemistry, Wiley-VCH, Weinheim, 1995; [Google Scholar]
- 1b. Larock R. C., Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, Weinheim, 1999; [Google Scholar]
- 1c. Gorzynski Smith J., Org. Chem. McGraw-Hill Education, 2008; [Google Scholar]
- 1d. Lutz J. F., Zarafshani Z., Adv. Drug Delivery Rev. 2008, 60, 958. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Brand J. P., Waser J., Chem. Soc. Rev. 2012, 41, 4165; [DOI] [PubMed] [Google Scholar]
- 2b. Caspers L. D., Nachtsheim B. J., Chem. Asian J. 2018, 13, 1231. [DOI] [PubMed] [Google Scholar]
- 3. Falbe J., Bauer W., Büchel K. H., Houben J., Weyl T., Carboxylic Acids and Carboxylic Acid Derivatives, Thieme, Stuttgart, 1985. [Google Scholar]
- 4.
- 4a. Uttry A., van Gemmeren M., Synlett 2018, 29, 1937; [Google Scholar]
- 4b. Uttry A., van Gemmeren M., Synthesis 2020, 52, 479; [Google Scholar]
- 4c. Pichette Drapeau M., Gooßen L. J., Chem. Eur. J. 2016, 22, 18654. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Engle K. M., Mei T. S., Wasa M., Yu J.-Q., Acc. Chem. Res. 2012, 45, 788; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Rouquet G., Chatani N., Angew. Chem. Int. Ed. 2013, 52, 11726; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11942; [Google Scholar]
- 5c. Chen Z., Wang B., Zhang J., Yu W., Liu Z., Zhang Y., Org. Chem. Front. 2015, 2, 1107; [Google Scholar]
- 5d. Zhu R.-Y., Farmer M. E., Chen Y.-Q., Yu J.-Q., Angew. Chem. Int. Ed. 2016, 55, 10578; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10734; [Google Scholar]
- 5e. Sambiagio C., Schönbauer D., Blieck R., Dao-Huy T., Pototschnig G., Schaaf P., Wiesinger T., Zia M. F., Wencel-Delord J., Besset T., Maes B. U. W., Schnürch M., Chem. Soc. Rev. 2018, 47, 6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Ano Y., Tobisu M., Chatani N., J. Am. Chem. Soc. 2011, 133, 12984; [DOI] [PubMed] [Google Scholar]
- 6b. Al-Amin M., Arisawa M., Shuto S., Ano Y., Tobisu M., Chatani N., Adv. Synth. Catal. 2014, 356, 1631. [Google Scholar]
- 7.
- 7a. He J., Wasa M., Chan K. S. L., Yu J.-Q., J. Am. Chem. Soc. 2013, 135, 3387; [DOI] [PubMed] [Google Scholar]
- 7b. Fu H., Shen P.-X., He J., Zhang F., Li S., Wang P., Liu T., Yu J.-Q., Angew. Chem. Int. Ed. 2017, 56, 1873; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1899. [Google Scholar]
- 8. Wu Q. F., Shen P. X., He J., Wang X. B., Zhang F., Shao Q., Zhu R. Y., Mapelli C., Qiao J. X., Poss M. A., Yu J.-Q., Science 2017, 355, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu T., Qiao J. X., Poss M. A., Yu J.-Q., Angew. Chem. Int. Ed. 2017, 56, 10924; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11064. [Google Scholar]
- 10.
- 10a. Wang B., Lu C., Zhang S. Y., He G., Nack W. A., Chen G., Org. Lett. 2014, 16, 6260; [DOI] [PubMed] [Google Scholar]
- 10b. Wang B., He G., Chen G., Sci. China Chem. 2015, 58, 1345; [Google Scholar]
- 10c. Ye X., Xu C., Wojtas L., Akhmedov N. G., Chen H., Shi X., Org. Lett. 2016, 18, 2970; [DOI] [PubMed] [Google Scholar]
- 10d. Lou J., Wang Q., He Y., Yu Z., Adv. Synth. Catal. 2018, 360, 4571; [Google Scholar]
- 10e. Han Y. Q., Ding Y., Zhou T., Yan S. Y., Song H., Shi B. F., J. Am. Chem. Soc. 2019, 141, 4558; [DOI] [PubMed] [Google Scholar]
- 10f. Jerhaoui S., Djukic J. P., Wencel-Delord J., Colobert F., ACS Catal. 2019, 9, 2532; [Google Scholar]
- 10g. Balliu A., Strijker A. R. F., Oschmann M., Pourghasemi Lati M., Verho O., CHEMRXIV 2020, 10.26434/CHEMRXIV.12034743.V1. [DOI] [Google Scholar]
- 11.
- 11a. Zhang J., Chen H., Lin C., Liu Z., Wang C., Zhang Y., J. Am. Chem. Soc. 2015, 137, 12990; [DOI] [PubMed] [Google Scholar]
- 11b. Luo F. X., Cao Z. C., Zhao H. W., Wang D., Zhang Y. F., Xu X., Shi Z. J., Organometallics 2017, 36, 18. [Google Scholar]
- 12.
- 12a. Giri R., Maugel N., Li J. J., Wang D. H., Breazzano S. P., Saunders L. B., Yu J.-Q., J. Am. Chem. Soc. 2007, 129, 3510; [DOI] [PubMed] [Google Scholar]
- 12b. Chen G., Zhuang Z., Li G., Saint-Denis T. G., Hsiao Y., Joe C. L., Yu J.-Q., Angew. Chem. Int. Ed. 2017, 56, 1506; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1528; [Google Scholar]
- 12c. Zhu Y., Chen X., Yuan C., Li G., Zhang J., Zhao Y., Nat. Commun. 2017, 8, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Ghosh K. K., van Gemmeren M., Chem. Eur. J. 2017, 23, 17697; [DOI] [PubMed] [Google Scholar]
- 12e. Dolui P., Das J., Chandrashekar H. B., Anjana S. S., Maiti D., Angew. Chem. Int. Ed. 2019, 58, 13773; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13911; [Google Scholar]
- 12f. Shen P. X., Hu L., Shao Q., Hong K., Yu J.-Q., J. Am. Chem. Soc. 2018, 140, 6545; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12g. Hu L., Shen P., Shao Q., Hong K., Qiao J. X., Yu J.-Q., Angew. Chem. Int. Ed. 2019, 58, 2134; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 2156; [Google Scholar]
- 12h. Liu L., Liu Y. H., Shi B. F., Chem. Sci. 2020, 11, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Ghosh K. K., Uttry A., Koldemir A., Ong M., van Gemmeren M., Org. Lett. 2019, 21, 7154; [DOI] [PubMed] [Google Scholar]
- 13b. Zhuang Z., Herron A. N., Fan Z., Yu J.-Q., J. Am. Chem. Soc. 2020, 142, 6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Kao L. C., Sen A., J. Chem. Soc. Chem. Commun. 1991, 1242; [Google Scholar]
- 14b. Dangel B. D., Johnson J. A., Sames D., J. Am. Chem. Soc. 2001, 123, 8149; [DOI] [PubMed] [Google Scholar]
- 14c. Janssen M., De Vos D. E., Chem. Eur. J. 2019, 25, 10724; [DOI] [PubMed] [Google Scholar]
- 14d. Lee J. M., Chang S., Tetrahedron Lett. 2006, 47, 1375; [Google Scholar]
- 14e. Novák P., Correa A., Gallardo-Donaire J., Martin R., Angew. Chem. Int. Ed. 2011, 50, 12236; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12444; [Google Scholar]
- 14f. Zhuang Z., Yu J.-Q., Nature 2020, 577, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Zhuang Z., Bin Yu C., Chen G., Wu Q. F., Hsiao Y., Joe C. L., Qiao J. X., Poss M. A., Yu J.-Q., J. Am. Chem. Soc. 2018, 140, 10363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Ghosh K. K., Uttry A., Mondal A., Ghiringhelli F., Wedi P., van Gemmeren M., Angew. Chem. Int. Ed. 2020, 59, 12848–12852; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12948–12952; [Google Scholar]
- 15c. Park H. S., Fan Z., Zhu R., Yu J.-Q., Angew. Chem. Int. Ed. 2020, 59, 12853–12859; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12953–12959. [Google Scholar]
- 16. He J., Shao Q., Wu Q., Yu J.-Q., J. Am. Chem. Soc. 2017, 139, 3344. [DOI] [PubMed] [Google Scholar]
- 17.Unfortunately, other β-methylene-C(sp3)−H bonds were found to be unreactive under our reaction conditions presumably due to the lower C−H acidity and increased steric hindrance that both contribute to the well-known lower reactivity of such groups in C−H activation processes.
- 18.During the course of our studies we found that TIPS and TBS substituted alkynyl bromides gave satisfactory results, while other substituents were unsuccessful For further details, see the Supporting Information.
- 19.
- 19a. Gotfryd K., Hansen M., Kawa A., Ellerbeck U., Nau H., Berezin V., Bock E., Walmod P. S., Basic Clin. Pharmacol. 2011, 109, 164; [DOI] [PubMed] [Google Scholar]
- 19b. Leng Y., Marinova Z., Reis-Fernandes M. A., Nau H., Chuanga D.-M., Neurosci. Lett. 2010, 476, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simmons E. M., Hartwig J. F., Angew. Chem. Int. Ed. 2012, 51, 3066; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3120. [Google Scholar]
- 21.
- 21a. Tobisu M., Ano Y., Chatani N., Org. Lett. 2009, 11, 3250; [DOI] [PubMed] [Google Scholar]
- 21b. Dudnik A. S., Gevorgyan V., Angew. Chem. Int. Ed. 2010, 49, 2096; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2140; [Google Scholar]
- 21c. Mondal A., Chen H., Flämig L., Wedi P., van Gemmeren M., J. Am. Chem. Soc. 2019, 141, 18662. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary