

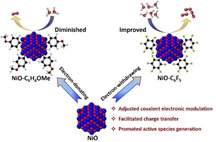
Abstract
Tuning the local environment of nanomaterial‐based catalysts has emerged as an effective approach to optimize their oxygen evolution reaction (OER) performance, yet the controlled electronic modulation around surface active sites remains a great challenge. Herein, directed electronic modulation of NiO nanoparticles was achieved by simple surface molecular modification with small organic molecules. By adjusting the electronic properties of modifying molecules, the local electronic structure was rationally tailored and a close electronic structure‐activity relationship was discovered: the increasing electron‐withdrawing modification readily decreased the electron density around surface Ni sites, accelerating the reaction kinetics and improving OER activity, and vice versa. Detailed investigation by operando Raman spectroelectrochemistry revealed that the electron‐withdrawing modification facilitates the charge‐transfer kinetics, stimulates the catalyst reconstruction, and promotes abundant high‐valent γ‐NiOOH reactive species generation. The NiO−C6F5 catalyst, with the optimized electronic environment, exhibited superior performance towards water oxidation. This work provides a well‐designed and effective approach for heterogeneous catalyst fabrication under the molecular level.
Keywords: catalyst self-reconstruction, electrocatalysis, molecular modification, nanomaterials, water oxidation
Electronic modulation: A simple molecular modification approach is explored to rationally modulate the electronic structure of NiO nanoparticles, by which the catalyst self‐reconstruction is directly regulated, and the water oxidation activity is systematically tailored. An efficient NiO−C6F5 catalyst is fabricated by the adjusted molecular functionalization and shows outstanding water oxidation activity.
Introduction
The oxygen evolution reaction (OER) plays a pivotal role as the “anodic half‐reaction” in sustainable energy conversion strategies, such as electrochemical water splitting, CO2 reduction, and nitrogen reduction. [1] Limited by the multi‐proton coupled electron transfer process, the OER suffers from sluggish kinetics and is the bottleneck for most renewable energy systems. To lower the OER overpotential, first‐row transition‐metal oxides have been investigated as cost‐effective heterogeneous catalysts over the past years. Plenty of studies suggested that the local structure around surface active sites is essential to heterogeneous catalysis.[ 1a , 2 ] Adjustment of the local environment is thus considered an effective approach to improve the OER activity.
To date, two approaches have been developed for local electronic modulation on OER catalysts, including heteroatom doping and defect engineering. Several efficient OER catalysts have been fabricated by these methods, such as NiFe, CoFe, NiFeCr, NiV catalysts. [3] However, since these methodologies are mainly based on bulk materials modification, it remains challenging to rationally tailor the local electronic structure at catalyst surface, which can directly impact the OER catalysis at the electrocatalyst/electrolyte interface. Moreover, the structural complexity of multi‐metal OER catalysts renders it difficult to gain deeper mechanistic understanding, for example, the identification of the real active center, the investigation of electronic structure‐activity relationships. [4] In addition, recent reports revealed that a self‐reconstruction of catalyst commonly occurs at catalyst surfaces during the OER process, deriving the oxy(hydroxide) layer as the true OER active phase. [5] It is thus highly favorable to rationally adjust the surface electronic structure of the catalyst, and thereby directly influence the catalyst reconstruction, tuning the OER activity. Hence, exploration of simple and effective strategies for directed electronic modulation at catalyst surface are greatly desired.
In this work, we turn to molecular functionalization approach to rationally modulate the surface electronic structure of NiO nanocatalyst, and thereby directly tailor the catalyst self‐reconstruction process, improving the OER activity. Through the covalent grafting reaction between organic diazonium salts and metal oxides, ultrasmall (1–3 nm) NiO nanoparticles are functionalized with a series of aryl groups bearing different substituents (Figure 1). By adjusting the electronic properties of the substituents, the electronic structure around surface Ni sites has been explicitly modulated and the OER activity has been systematically regulated, that is, the introduction of electron‐withdrawing groups decreases the electron density around Ni and improves the OER activity, and vice versa. Detailed investigations indicated that the strong electron‐withdrawing molecular modification affects the charge transfer kinetics and facilitates the catalyst surface reconstruction, generating abundant γ‐NiOOH active phase. The NiO−C6F5 catalyst, possessing the strongest electron‐withdrawing functionalization, exhibits superior performance towards OER.
Figure 1.
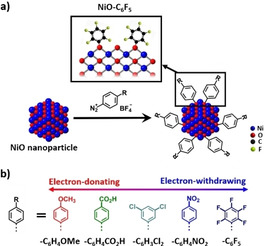
a) Schematic presentation of the molecular modification process on NiO nanoparticles, inset: the local surface structure of NiO−C6F5. b) Schematic presentation of the aryl groups with different electronic properties used as molecular modification.
Results and Discussion
Ultrasmall NiO nanoparticles were synthesized as a model heterogeneous OER catalyst, as the small particle size is expected to enlarge the catalyst surface area, promote coordinatively unsaturated site exposure, and facilitate the surface molecular modification via covalent bonds. [6] The HRTEM image of the obtained NiO sample shows non‐aggregated ultrasmall nanoparticles with particle size of 1–3 nm (Figures 2a and S3), and the XRD pattern presents typical nickel oxide diffraction peaks at 37.2, 43.2, and 62.8° (Figure S1). Based on the XRD peak width, the averaged particle size was calculated to be 2.4 nm, illustrating the formation of ultrasmall NiO nanocrystals. [7]
Figure 2.
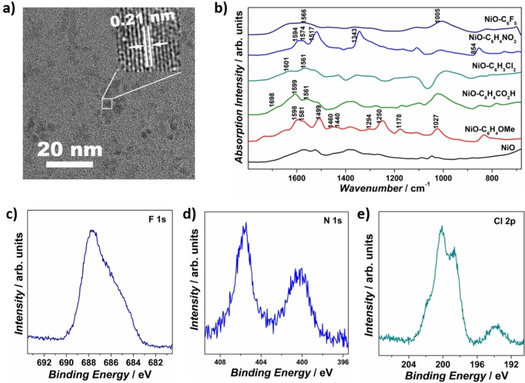
a) TEM image of as‐prepared ultrasmall NiO nanoparticles. b) DRIFTS spectra of bare NiO and all modified NiO samples. High‐resolution core XPS spectra of c) F 1s of NiO−C6F5, d) N 1s of NiO−C6H4NO2, and e) Cl 2p of NiO−C6H3Cl2.
A series of aryl‐groups with different substitution were introduced to the NiO nanoparticle surface through the grafting reaction between diazonium salts and nickel oxide (Figure 1a). [8] The substitution groups were adjusted to be 4‐MeO‐, 4‐CO2H‐, 3,5‐Cl2‐, 4‐NO2‐, and 2,3,4,5,6‐F5‐groups, giving molecularly modified samples of NiO−C6H4OMe, NiO−C6H4CO2H, NiO−C6H3Cl2, NiO−C6H4NO2, and NiO−C6F5 (Figure 1b). Dictated by the varied Hammett parameters of substitutions, the electronic property of modifying aryl‐groups can be systematically regulated: the highest Hammett parameter (0.98) of 2,3,4,5,6‐F5 demonstrates the strongest electron‐withdrawing ability of −C6F5 group in this series, while the lowest Hammett parameter (−0.27) of 4‐MeO− expresses the highest electron‐donating property of the −C6H4OMe group (Figure S5). [9] The different electronic features of the molecular modification is expected to distinctively influence the local electronic environment at NiO nanoparticles surface.
XRD, TEM, and high‐resolution (HR)TEM were firstly performed to characterize the crystalline structure of modified NiO samples. The XRD pattern of all modified NiO samples show typical NiO diffraction patterns, without apparent peak change compared with the parent NiO nanoparticles (Figure S6). The TEM images display ultrasmall nanoparticles with particle sizes of 1∼4 nm, and the HRTEM images show distinct lattice fringes with an interplanar spacing of 0.21 nm (Figures S7–S11). No obvious particle aggregation or surface amorphousness was observed after molecular modification. The TEM energy‐dispersive X‐ray spectroscopy (EDS) spectra of NiO−C6F5 and NiO−C6H3Cl2 samples exhibit distinct F and Cl fluorescence signals, illustrating the successful molecular modification.
Diffuse reflectance infrared Fourier‐transform spectroscopy (DRIFTS) was employed to study the molecular modification at NiO nanoparticle surface (Figure 2b). All the modified NiO samples show additional characteristic peaks of the organic modification compared to bare NiO nanoparticles: the NiO−C6F5 spectrum shows C−F stretch at 1005 cm−1; [10] the NiO−C6H4NO2 spectrum shows NO2 stretching and scissoring absorption modes at 1517, 1343, and 854 cm−1;[ 8b , 10c ] the NiO−C6H4CO2H spectrum exhibits C=O stretching at 1698 cm−1; [10c] and the NiO−C6H4OMe spectrum presents OMe stretches at 1460, 1440 cm−1, Ar−O stretches at 1294, 1250 cm−1, Ar−O−CH3 stretch at 1027 cm−1, and O−CH3 rocking mode at 1178 cm−1.[ 8b , 10c ] Aryl‐group‐related characteristic vibrations are also present at 1550–1600 cm−1 and 1450–1500 cm−1 for all modified NiO samples, yet distinctly affected by the different electronic properties of substituents.[ 8b , 10c ] The NiO−C6H4OMe sample presents a strong absorption band at 1499 cm−1, which is absent for other samples, consistent with the principle that this aryl‐group IR band is only IR active when the aryl‐group has an electron‐donating substituent.[ 8b , 10c ] In 1550–1600 cm−1 region, the NiO−C6H3Cl2 and NiO−C6F5 samples show much lower peak intensity at 1601 cm−1 compared to the peak at 1564 cm−1, in sharp contrast to NiO−C6H4NO2 (comparable peak intensity at 1594 and 1574 cm−1) and NiO−C6H4OMe, NiO−C6H4CO2H (higher peak intensity at 1598 cm−1 compared to the peak at 1581 cm−1).[ 8b , 10c ] The different electron‐withdrawing strengths influence the symmetry of the phenyl stretch mode by altering the intrinsic dipole, inducing different IR absorption behavior.[ 8b , 10c , 11 ]
UV/Vis absorption spectroscopy was conducted to approximate the loading of the organic modifications at the NiO nanoparticle surface (Figures S12–S15). By comparing the absorption intensity of the diazonium salt stock solutions before and after the grafting process, the functionalization amount of −C6F5, −C6H4NO2, −C6H4CO2H, and −C6H4OMe were estimated to be 2.11, 2.55, 3.31, and 2.37 nm−2, indicating their comparable coverage at the NiO surface. [12] The molecular modification can be further confirmed by the X‐ray photoelectron spectroscopy (XPS) spectra of NiO−C6F5, NiO−C6H4NO2, and NiO−C6H3Cl2, which present distinct F, N, and Cl signals, respectively (Figure 2c–e). Together with EDS and DRIFTS, these results demonstrate the successful molecular modification of the NiO by aryl‐groups with comparable coverage.
Then, to investigate the electronic modulation by molecular modification, XPS was performed on all the modified samples. As shown in Figure 3, the bare NiO sample shows Ni peaks at 856.1, 853.9 eV, and O peaks at 529.1, 531.2 eV, which can be attributed to the Ni2+−O and Ni2+−OH species, [13] indicating the generation of Ni(OH)2 at NiO surface when exposed to air. [6a] Interestingly, the nickel oxide‐related Ni and O peaks were markedly shifted upon functionalization of the NiO particles. The NiO−C6H4OMe sample shows Ni peaks at 856.0 and 853.8 eV and an O peak at 531.1 eV, which are 0.1 eV lower in energy compared with bare NiO, indicating the higher electron density on the surface Ni species.[ 3a , 14 ] In contrast, the NiO−C6H4CO2H, NiO−C6H3Cl2, and NiO−C6H4NO2 samples show Ni peaks at 856.4 eV and O peaks at 531.6 eV, which show 0.3 and 0.4 eV shifts towards higher energy compared with the bare NiO, illustrating their decreased electron density around surface Ni species.[ 3a , 14 ] The strongest electron‐withdrawing pentafluorophenyl‐modified NiO sample shows a Ni peak at 856.7 eV and an O peak at 531.7 eV, which are 0.6 and 0.5 eV positively shifted compared with bare NiO, presenting the highest value among all modified NiO samples. To sum up, the Ni and O binding energies increase with an order of NiO−C6H4OMe<NiO<NiO−C6H4CO2H≈NiO−C6H3Cl2≈NiO−C6H4NO2<NiO−C6F5, demonstrating the successive electronic structure modulation by the varied electron‐withdrawing abilities of −C6H4OMe<−C6H4CO2H≈−C6H3Cl2≈−C6H4NO2<−C6F5.[ 3a , 14a ] All the molecular modified NiO samples show an additional peak in the O spectra in the range of 532.8–533.2 eV, which can be assigned to the O−C bond, indicating the formation of O−C bond linkage between NiO and the organic molecules. [15]
Figure 3.
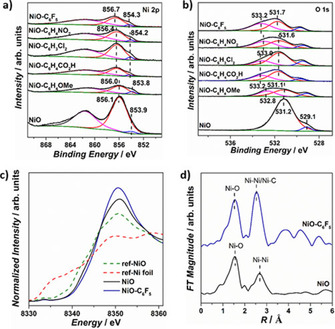
High‐resolution core XPS spectra of a) Ni 2p and b) O 1s of bare NiO and all modified NiO samples. c) Normalized Ni K‐edge XANES spectra of NiO, NiO−C6F5, reference Ni foil, and reference NiO samples. d) Fourier transformations of k 3‐weighted EXAFS spectra of bare NiO and NiO−C6F5 samples.
X‐ray absorption of near‐edge structure (XANES) and extended X‐ray absorption fine structure (EXAFS) spectroscopy were performed on the strongest electron‐withdrawing modified NiO−C6F5 samples to investigate its local structure. In the XANES region, a characteristic pre‐edge peak is presented at 8333 eV, which corresponds to the 1 s to 3d transition of Ni in a disordered octahedral geometry (Figure 3c). The absorption edge energy value (E 0) is comparable to that of reference NiO, indicating the Ni2+ oxidation state in the samples. The Fourier transformed k 3‐weighted EXAFS spectra presents two main peak maximums in the range of 1–3 Å (Figure 3d). The first peak maximum can be attributed to the Ni−O backscattering path, which exhibits comparable radial distance (R) and coordination number (CN) for bare NiO and NiO−C6F5, in agreement with the disordered octahedral geometry of NiO6 (Table S1). [16] For the second peak maximum, two backscattering paths are required for the perfect peak fitting of NiO−C6F5, which can be attributed to the Ni−Ni and Ni−C backscattering. The R of Ni−Ni backscattering was fitted to be 2.97 Å for NiO−C6F5, lower than 3.10 Å of NiO, illustrating the structural disorder induced by the molecular modification. The CN of Ni−Ni backscattering path is fitted to be 7.96, much lower than the value of 12 in standard bulk NiO, indicating the exposure of coordinatively unsaturated sites at the nanoparticle surface. [17] The CN and R of Ni−C backscattering was fitted to be 0.48 and 2.72 Å, which can be attributed to the Ni−O−C linkage between NiO and grafting molecules, confirming the molecular modification. Overall, these XPS and XAS results illustrate the systematical local electronic structure modulation of NiO nanoparticles by the simple molecular modification. The controlled electronic modulation is expected to directly tailor the Ni active species behavior and water oxidation activity.
Next, the electrocatalytic OER activity of bare NiO and modified NiO samples was evaluated under alkaline conditions (1 m KOH) with glassy carbon electrode (GCE) as the working electrode (Figure 4a). The bare NiO nanoparticles show an overpotential of 315 mV for reaching 1 mA cm−2 current density and achieve 6.85 mA cm−2 current density under 450 mV overpotential, consistent with its moderate activity towards OER.[ 3b , 18 ] All the modified NiO samples exhibit distinctly altered OER activity, which is systematically affected by the molecular modification. The NiO−C6H4OMe sample requires 340 mV overpotential for 1 mA cm−2 current density and achieves 5.62 mA cm−2 current density under 450 mV overpotential, which presents a repressed catalytic activity compared to the bare NiO. In sharp contrast, the NiO−C6H3Cl2, NiO−C6H4NO2, and NiO−C6F5 samples show lower overpotential requirements of 293, 287, and 257 mV for 1 mA cm−2 current density, and higher catalytic current densities of 13.17, 18.98, and 23.17 mA cm−2 under 450 mV overpotential, demonstrating their sequentially improved OER activity. The NiO−C6H4CO2H exhibits 310 mV overpotential for 1 mA cm−2 and 8.93 mA cm−2 current density under 450 mV overpotential, which is approximate to the bare NiO nanoparticles and can be related to the electron‐neutral property of −C6H4CO2 − under alkaline conditions.
Figure 4.
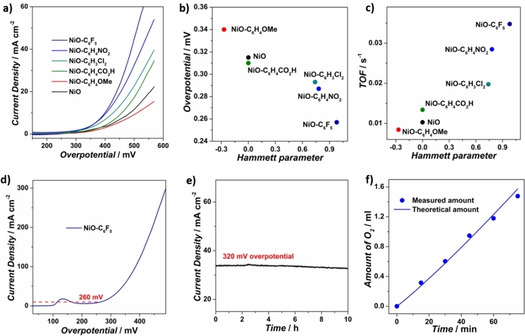
a) Polarization curves of bare NiO and modified NiO samples on glassy carbon electrode in 1 m KOH. b) Overpotentials at 1 mA cm−2 current density and c) calculated TOFs at 450 overpotential of bare NiO and all modified NiO samples. d) Polarization curves of bare NiO and NiO−C6F5 on carbon fiber paper electrode in 1 m KOH. e) Chronopotentiometric curve of NiO−C6F5 on carbon fiber paper electrode at 320 mV overpotential. f) Quantification and comparison of the experimental and theoretical O2 evolution amount by electrolysis of NiO−C6F5 for OER under 250 mV overpotential.
Turnover frequencies (TOFs) were calculated by assuming that all the Ni ions are active sites (Figure 4c and Table 1). The TOF values of NiO−C6H4CO2H, NiO−C6H3Cl2, NiO−C6H4NO2, and NiO−C6F5 are calculated to be 0.013, 0.020, 0.028, and 0.035 s−1, which are sequentially increased compared with 0.010 s−1 for bare NiO and indicate their monotonically improved intrinsic activity. Conversely, NiO−C6H4OMe shows a decreased TOF value of 0.008 s−1, showing a suppressed intrinsic activity compared to bare NiO. Tafel slopes were calculated by plotting overpotential against log (J) to obtain insight on the electrocatalytic OER kinetics (Figure S17). The NiO−C6H4CO2H, NiO−C6H3Cl2, NiO−C6H4NO2, and NiO−C6F5 samples show Tafel slopes of 126, 129, 89, and 87 mV dec−1, which are beneficially decreased compared with 133 mV dec−1 of bare NiO, illustrating the accelerated OER kinetics. In contrast, the NiO−C6H4OMe sample shows a Tafel slope of 156 mV dec−1, much higher than the native NiO nanoparticles and signifies the retardation of OER kinetics with electron‐donating group. Altogether, the overpotential, current density, TOF, and Tafel slope performances follow a similar OER activity trend with NiO−C6H4OMe<bare NiO≤NiO−C6H3CO2H<NiO−C6H3Cl2<NiO−C6H4NO2<NiO−C6F5. By plotting the OER performances versus the Hammett sigma constants, a reasonable correlation can be presented between catalytic activity and the electronic properties of the modifying molecules: increasing electron‐withdrawing strength readily decreases the electron density around surface Ni sites, accelerating catalytic kinetics and improving OER performance (Figures 4b, c and S18).
Table 1.
Summary of Hammett sigma constants and calculated TOF values under 450 mV overpotential of bare NiO and all molecule modified NiO samples on GCE.
|
Sample |
Hammett parameter |
TOF [s−1] |
|---|---|---|
|
NiO−C6H4OMe |
−0.27 |
0.008 |
|
NiO |
0 |
0.010 |
|
NiO−C6H4CO2H[a] |
0 |
0.013 |
|
NiO−C6H3Cl2 |
0.74 |
0.020 |
|
NiO−C6H4NO2 |
0.78 |
0.028 |
|
NiO−C6F5 |
0.98 |
0.035 |
[a] The −C6H4CO2H group is deprotonated under alkaline conditions (−C6H4CO2 −), behaving as electron‐neutral property.
The OER performance of the best catalyst, NiO−C6F5, was further evaluated on carbon fiber paper (CFP) as a 3D electrode substrate. The CFP/NiO−C6F5 electrode shows an overpotential of 260 mV for 10 mA cm−2 current density (Figure 4d). High current densities of 50 and 100 mA cm−2 can be achieved for CFP/NiO−C6F5 by increasing the overpotential to 350 and 380 mV, confirming its prominent activity. In chronoamperometry measurements under 320 mV overpotential (Figure 4e), no obvious deterioration in current density was observed during 10 h electrolysis, which indicates the excellent stability of the catalyst. This high stability is further confirmed by the TEM image of the NiO−C6F5 catalyst after long‐term electrolysis, which indicated that nanoparticle size remained between 1–4 nm, displaying no apparent aggregation (Figure S19). Typical F, Ni, and O signals can be observed on the EDS spectrum, verifying the durability of the modified NiO−C6F5 catalyst. By comparison of experimental and theoretical O2 evolution during electrolysis under 250 mV overpotential, the OER faradaic efficiency was estimated to be 93.6 % for the CFP/NiO−C6F5 electrode, indicating that the vast majority of consumed charges was contributed to catalytic water oxidation (Figure 4f).
The above OER performance results illustrate that, through our simple covalent functionalization approach, the water oxidation activity is rationally tailored by the directed electronic modulation and an efficient NiO−C6F5 water oxidation catalyst is achieved with the adjusted molecular modification. Then, we performed a series of experiments to gain insightful understanding on the influence of −C6F5 modification on the activity.
Electrochemical surface area (ECSA) is one of the key factors that may impacted by the surface molecular modification. The ECSA values of bare NiO and NiO−C6F5 were estimated from their double‐layer capacitance (C dl) (Figures S20 and S21). The linear slope of NiO−C6F5 was calculated to be 0.16 mF cm−2, close to 0.19 mF cm−2 for bare NiO. This negligible difference in ECSA indicates that the molecular modification does not impact the active surface area of the catalyst.
The wettability of bare NiO and molecular modified NiO samples were evaluated by water contact angle measurement (Figure S22). The NiO−C6F5 sample presents contact angle of 103.9°, which is higher than 77.7° of bare NiO. The decreased hydrophilicity of NiO−C6F5 can be attributed to the hydrophobicity of grafted molecules. No clear correlation was observed between the hydrophilicity and OER performance, which indicate that the change of wettability cannot account for the significantly influenced activity.
Charge transfer also plays an essential role in water oxidation reaction at catalyst surface. To investigate the influence of molecular modification on charge‐transfer kinetics, electrochemical impedance spectroscopy (EIS) was conducted on NiO and NiO−C6F5 (Figure 5a). The NiO−C6F5 sample shows a charge transfer resistance (R ct) of 56.3 Ω, which is much lower compared with 152.4 Ω of bare NiO. The diminished resistance demonstrates that the −C6F5 molecular modification greatly facilitates the charge‐transfer kinetics.
Figure 5.
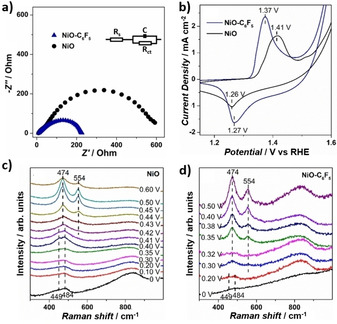
a) Nyquist diagram of NiO−C6F5 and NiO nanoparticles with a bias set at 350 mV overpotential. b) CV of bare NiO nanoparticles and the NiO−C6F5 sample at a scan rate of 100 mV s−1 in 1 m KOH electrolyte. Operando Raman spectra of c) bare NiO and d) NiO−C6F5 samples under OER conditions.
A catalyst self‐reconstruction of Ni2+→NiOOH is reported to commonly occur at Ni‐based water oxidation catalyst, generating γ‐NiOOH as the surface active phase.[ 5a , 19 ] To study the influence of molecular modification on the surface self‐reconstruction, cyclic voltammetry (CV) was performed on bare NiO and NiO−C6F5 samples (Figure 5b). The bare NiO sample shows a pair of redox peaks with an anodic peak (E a,p) at 1.41 V, cathodic peak (E c,p) at 1.26 V, and mid‐peak potential [E mid=(E a,p+E c,p)/2] of 1.34 V, which can be attributed to the transformation of surface Ni2+ moieties to NiOOH active species.[ 5a , 19 ] Notably, for NiO−C6F5, the anodic peak shows peak potential at 1.37 V, which is 40 mV lower compared with bare NiO, and the Emid of the Ni redox peaks was calculated to be 1.32 V, 20 mV lower compared with bare NiO. The decrease peak potential indicate the promoted Ni2+→NiOOH transformation in the NiO−C6F5 sample.[ 3a , 5a , 18 ] Besides, along with the shift of the redox peak potential, the peak difference (ΔE p) between the anodic and cathodic peaks was estimated to be 100 mV for NiO−C6F5, much lower compared with 150 mV of bare NiO, illustrating the accelerated transformation kinetics of Ni2+→NiOOH on NiO−C6F5. [20]
Then, operando Raman spectroscopy was further performed to investigate the relationship between molecular modification, active species behavior, and catalytic activity (Figure 5c, d). Under 0 V potential (vs. Ag/AgCl), both NiO and NiO−C6F5 samples show two vibration bands at 449 and 484 cm−1, which can be attributed to the A1g stretching modes of Ni−OH and Ni−O respectively. [21] Upon increasing the applied potential, both samples present γ‐NiOOH related bands at 474 and 554 cm−1, yet the potential requirement is greatly altered.[ 21a , 21d , 21e ] For NiO−C6F5, the vibration bands at 474 and 554 cm−1 started to appear at a potential of 0.30 V, which is 120 mV lower compared with 0.42 V of bare NiO. The maximum‐steady state of these two bands was achieved at 0.40 V for NiO−C6F5, which is 40 mV less compared with 0.44 V of bare NiO. These decreased potential requirements clearly demonstrate the lower formation energy barrier of γ‐NiOOH, illustrating the facilitated generation of this active species at the NiO−C6F5 catalyst surface. Combined with the EIS and CV, we can conclude that the −C6F5 group modification effectively accelerates the charge transfer kinetics, promotes the catalyst reconstruction, and enhances the OER activity.[ 5a , 19 ]
Although the catalytic mechanism of Ni‐based OER catalysts remains a long‐term scientific discussion topic, the formation of NiOOH species is accepted to play a pivotal role as the key intermediate in water oxidation. The nucleophilic addition of water molecules on the catalysts would be the first key step in the catalytic cycle. The electron‐withdrawing modification can promote the local electron‐delocalization at NiO surface, decreasing the electron density around the active Ni species and modulating the catalyst local structure. On one hand, the more electrophilic Ni site can favor the nucleophilic addition of water as well as the deprotonation in the Ni2+→NiOOH reconstruction, achieving improved OER activity. On the other hand, with specific proton affinity, the −C6F5 group is proposed to promote proton shuttling at the catalyst surface, which can accelerate the deprotonation process, stimulate the generation of key intermediate, and improve the catalytic performance. [22] Benefiting from the synergistic promotion effect, the NiO−C6F5 catalyst exhibits outstanding performance towards water oxidation. Further OER activity improvements and deeper mechanistic understanding can be achieved by directed molecular functionalization with optimized electronic properties.
Conclusions
We achieve directed electronic modulation of NiO through surface molecular modification, and thereby directly tailor the oxygen evolution reaction (OER) activity. By adjusting the electronic properties of the modifying molecules, the electronic environment around active sites is rationally modulated and the OER activity is systematically regulated: the electron‐withdrawing modification decreases the electron density at the surface Ni species and improves the OER activity, and vice versa. The strong electron‐withdrawing −C6F5 molecular modification can effectively facilitate the charge‐transfer kinetics, promoting the reconstruction of Ni2+ moieties to the active γ‐NiOOH phase, and thus greatly enhancing the OER performance. This work opens a new avenue for heterogeneous OER catalyst design at the molecular level, demonstrating the significant impact of local electronic structure on the OER active species generation and catalytic activity.
Experimental Section
Materials
Nickel(II) acetylacetonate [Ni(C5H7O2)2], anhydrous tert‐butanol [(CH3)3COH], potassium hydroxide (KOH), 4‐nitrobenzenediazonium tetrafluoroborate (C6H4N3O2BF4), 3,5‐dichlorophenyldiazonium tetrafluoroborate (C6H3Cl2N2BF4), 4‐methoxybenzenediazonium tetrafluoroborate (CH3OC6H4N2BF4), 2,3,4,5,6‐pentafluoroaniline (C6F5NH2), nitrosyl tetrafluoroborate (NOBF4), anhydrous acetone (CH3COCH3), anhydrous dichloromethane (CH2Cl2), and acetonitrile‐d3(CD3CN) were purchased from Sigma‐Aldrich. 4‐carboxylbenzenediazonium tetrafluoroborate (CO2HC6H4N2BF4) was purchased from Chemtronica AB (Sweden). The water used for electrolyte solutions was deionized using a Millipore Milli‐Q UF Plus system (15–18 MΩ‐cm resistivity).
Synthesis of pentafluorophenyldiazonium tetrafluoroborate (C6F5N2BF4)
The pentafluorophenyldiazonium tetrafluoroborate was synthesized according to an available literature procedure. [23] 5 mmol pentafluoroaniline was firstly dissolved in 1 mL acetonitrile, and then added dropwise over the course of 30 min to a mixture of pulverized nitrosyl tetrafluoroborate (5 mmol, 915 mg) in dry acetonitrile (1 mL) at −30 °C in 30 min. After stirring at −30 °C for 1 h, 7.5 mL dry dichloromethane was added to the mixture. The desired pentafluorophenyldiazonium tetrafluoroborate could be obtained by filtration and drying in vacuum. 19F NMR spectroscopy confirmed formation of the desired product: 19F NMR (377 MHz, CD3CN): δ=−119.54 (tt, J=21.4, 16.6 Hz), −123.43 (m), −151.31 (broad singlet, BF4 −), −152.47 ppm (m).
Synthesis of ultrasmall NiO nanoparticles
The ultrasmall NiO nanoparticles were synthesized following a reported solvothermal method. [6a] 0.4 g Ni(acac)2 was firstly mixed with 35 mL tert‐butanol under vigorous stirring at 50 °C to form a suspension. The suspension was transferred into an autoclave reactor and hermetically sealed. The reaction was performed by heating at 205 °C for 20 h. After the reaction, the ultrasmall nanoparticles were obtained by washing with ethanol for 3 times and drying at 80 °C.
Functionalization of NiO nanoparticles with organic molecule
The NiO nanoparticles were functionalized by suspending approximately 100 mg of NiO nanoparticles in Milli‐Q water with 0.1 m NaOH under N2 atmosphere, and then adding dropwise approximately 5 mL of a 10 mg mL−1 solution of the corresponding diazonium tetrafluoroborate salt: pentafluorophenyldiazonium tetrafluoroborate, 4‐nitrobenzenediazonium tetrafluoroborate, 3,5‐dichlorophenyldiazonium tetrafluoroborate, 4‐carboxylbenzenediazonium tetrafluoroborate, or 4‐methoxybenzenediazonium tetrafluoroborate. The mixed solutions were stirred overnight, after which the products were collected by centrifugation and washing with water. The resulting functionalized NiO samples were obtained by drying under vacuum.
Physical characterization
Powder XRD was performed on Bruker D5000 X‐ray diffraction diffractometer with CuKα radiation (λ=1.5406 Å). SEM images and EDS spectra were recorded with JEOL JSM 7401 equipped with an EDS system. TEM images and HRTEM images were taken on a JEOL JEM2100F transition electron microscope. DRIFTS spectra were measured on a Thermo Scientific Nicolet iS5 FT‐IR spectrometer. UV/Vis spectra were carried on PerkinElmer Lambda 750 UV/Vis spectrophotometer. XPS spectra were acquired by a Thermo VG ESCALAB250 surface analysis system with monochromatized AlKα small‐spot source and 500 μm concentric hemispherical energy analyzer. All the spectra energy was calibrated by setting the adventitious carbon peak to 284.6 eV.
Determination of the molecule loading amount
The degree of molecular modification was determined by the UV/Vis absorption method. A standard curve was obtained by plotting the UV/Vis absorption of stock solutions of the respective diazonium slats at different concentrations. The remaining concentration of diazonium salt of the solution after NiO functionalization was determined based on their UV/Vis absorption intensity. The molecule loading amount was calculated by the difference between the initial molecular amount and the molecular concentration after NiO functionalization.
Electrode preparation
4 mg of the respective samples were dispersed in a mixed solution of 32 μL 5 % Nafion (ethanol solution), 200 μL ethanol, and 768 μL H2O and sonicated for over 1 h to make a finely dispersed suspension. For GCEs, 10 μL of the obtained suspension was drop‐casted on pre‐polished GCE and the resulting electrodes were dried at 50 °C for 30 min. For the CFP electrode, 100 μL of the catalyst suspension was drop‐casted on pre‐cleaned GFP electrode and the resulting electrodes were dried at 50 °C for 30 min.
Electrochemical characterization
All electrochemical experiments were performed on a CH Instrument 660E potentiostat. The electrochemical cell was built by a three‐electrode system with the as‐prepared samples electrode as the working electrode, a standard Ag/AgCl electrode as the reference electrode and a platinum foil (4 cm2) as the counter electrode. All the applied potentials were converted to reversible hydrogen electrode (RHE) using the equation E (vs. RHE)=E (vs. Ag/AgCl)+0.059pH+0.197 V. The overpotential was calculated with equation η=E (vs. RHE) −1.23 V. For evaluation of the OER activity, linear sweep voltammetry (LSV) was performed from low initial potential to high final potential with a scan rate of 5 mV s−1. 95 % iR compensation was applied with the current interrupt method by the software supplied with the potentiostat. The Tafel slopes were calculated by plotting the overpotential η against log (j) from the LSV curves. Chronoamperometry measurement was performed to assess the durability of the catalyst. CV was operated under scan rate of 100 mV s−1 to investigate the catalyst redox behavior. The catalytic TOF values are determined according to TOF=jA/(4Fm), where j is the catalytic current density, A is the surface area of the electrode, F is the Faraday constant, and m is the number of moles of the active sites. In this work, we assume that all the Ni ions are active sites for water oxidation. By catalyst loading amount calculation, the m is calculated to be 5.36×10−7 mole. The faradaic efficiency was measured in a N2‐purged sealed cell. The quantity of O2 gas evolution was determined every 15 min by GC (Shimadzu GC‐2014) every 15 min. The faradaic efficiency was calculated by the equation: faradaic efficiency=4F×n O2/Q, where F is the Faraday constant, n O2 is the measured O2 amount, and Q is the consuming charge amount. ECSA was measured by the non‐faradaic Cdl method. CV was performed at the potential window of 0–0.1 V vs. Ag/AgCl, with different scan rates of 30, 50, 70, and 90 mV s−1. By plotting ΔJ=(J a−J c) at 0.05 V vs. Ag/AgCl against the scan rate, the linear slope, which is twice C dl, is used to represent ECSA. EIS was carried out in the catalytic OER region with the frequency ranged from 0.1–105 Hz under an applied potential of 1.58 V.
XAS data analysis
The XAS spectra were measured at P‐64 beamline, PETRA III, DESY, Hamburg. The samples and reference Ni foil were measured simultaneously in transmission mode. The data processing (pre‐edge, post‐edge background removal and normalization) and structural modelling (Fourier transformation and data fitting) were performed using EXAFS PAK. [24] The k 3‐weighted EXAFS oscillations were analyzed by nonlinear least‐squares fit of the data to the EXAFS equation. The various model parameters: average coordination number (CN), mean interatomic distances (R), Debye‐Waller factor coefficients (σ 2), and threshold energy (E 0) were refined. The theoretical phases and amplitudes used in the refinements were calculated using the FEFF7.
In order to directly compare all of the NiO modified samples, a two‐shell model based on bulk NiO rock salt structure was used to fit the EXAFS data. The Fourier transform fitting of the EXAFS spectra was performed until approximately 3 Å which contains the main single scattering contributions. Hence, a single Ni−O scattering path was used to describe the 6 nearest neighboring oxygen atoms (R eff=2.08 Å) in the first coordination shell. For fitting the second shell, a single Ni−Ni scattering path (R eff=2.954 Å) was used for both the samples. In addition, a third Ni−C scattering path (R eff=2.72 Å) was necessary to obtain a better fit in NiO−C6F5 (best fit in Figure S16). The S0 2 was refined separately for each sample, while the number of single scattering paths for Ni−O, Ni−Ni and Ni−C as well as their lengths, and Debye‐Waller factors were allowed to refine together. In the present study, the single scattering contributions up to approximately 3 Å have been used for structural modelling and have been found sufficient to describe the local structure as reported in literature. [25]
Operando Raman spectra
Operando Raman spectra of bare NiO and NiO−C6F5 samples were collected by a confocal Raman microscope (RM 1000, Renishaw) under OER in an ambient air condition. The reference electrode and counter electrode were Ag/AgCl (3 m KCl) and Pt wire, respectively. The excitation source was emitted by a frequency doubled Nd: YAG laser with the wavelength of 532 nm. The power of laser through the Olympus 10× objective was about 12.5 mW measured by PM160T power meter. A grating of 1800 lines mm−1 was used for operando Raman tests. All operando Raman measurements were performed by a custom‐made spectroelectrochemical cell, measuring the surface of the electrode from the side to avoid the bubble scattering. Each operando Raman spectra was recorded with an acquisition time of 10 s with 5–10 sweeps under current‐time (i–t) measurement at a constant potential using a confocal Raman microscope coupled with a 90‐degree angled Olympus 10× objective. The electrochemical cell was made of quartz and stood in front of the objective. All spectra were calibrated against the value of 520.7 cm−1 of a silicon wafer. The resolution of each spectra is 1 cm−1.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was financially supported by the Swedish Research Council (2017‐00935), the Swedish Energy Agency, the Knut and Alice Wallenberg Foundation (KAW 2016.0072), the National Natural Science Foundation of China (21120102036). The X‐ray absorption experiments were performed at the Deutsches Elektronen‐Synchrotron (DESY), PETRA III, P64. We sincerely thank Beamline scientists Dr. Wolfgang A. Caliebe and Dr. Akhil Tayal at P64, PETRA III, DESY, and Dr. Ning Yuan at Stockholm University for assistance with the XAS measurements. L. Fan, T. Liu and Q. Meng thank the China Scholarship Council (CSC) for Ph.D. student scholarship.
L. Fan, B. Zhang, Z. Qiu, N. V. R. A. Dharanipragada, B. J. J. Timmer, F. Zhang, X. Sheng, T. Liu, Q. Meng, A. K. Inge, T. Edvinsson, L. Sun, ChemSusChem 2020, 13, 5901.
References
- 1.
- 1a. Song J., Wei C., Huang Z.-F., Liu C., Zeng L., Wang X., Xu Z. J., Chem. Soc. Rev. 2020, 49, 2196–2214; [DOI] [PubMed] [Google Scholar]
- 1b. Zhang B., Sun L., Chem. Soc. Rev. 2019, 48, 2216–2264. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Suen N. T., Hung S. F., Quan Q., Zhang N., Xu Y. J., Chen H. M., Chem. Soc. Rev. 2017, 46, 337–365; [DOI] [PubMed] [Google Scholar]
- 2b. Gong J., Li C., Wasielewski M. R., Chem. Soc. Rev. 2019, 48, 1862–1864. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Fan L., Zhang P., Zhang B., Daniel Q., Timmer B. J. J., Zhang F., Sun L., ACS Energy Lett. 2018, 3, 2865–2874; [Google Scholar]
- 3b. Gong M., Li Y., Wang H., Liang Y., Wu J. Z., Zhou J., Wang J., Regier T., Wei F., Dai H., J. Am. Chem. Soc. 2013, 135, 8452–8455; [DOI] [PubMed] [Google Scholar]
- 3c. Xu X., Song F., Hu X., Nat. Commun. 2016, 7, 12324; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Liu R., Wang Y., Liu D., Zou Y., Wang S., Adv. Mater. 2017, 29, 1701546; [DOI] [PubMed] [Google Scholar]
- 3e. Fan K., Chen H., Ji Y., Huang H., Claesson P. M., Daniel Q., Philippe B., Rensmo H., Li F., Luo Y., Sun L., Nat. Commun. 2016, 7, 11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Lee S., Banjac K., Lingenfelder M., Hu X., Angew. Chem. Int. Ed. 2019, 58, 10295–10299; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 10401–10405; [Google Scholar]
- 4b. Gorlin M., Chernev P., Ferreira de Araujo J., Reier T., Dresp S., Paul B., Krahnert R., Dau H., Strasser P., J. Am. Chem. Soc. 2016, 138, 5603–5614; [DOI] [PubMed] [Google Scholar]
- 4c. Hunter B. M., Thompson N. B., Müller A. M., Rossman G. R., Hill M. G., Winkler J. R., Gray H. B., Joule 2018, 2, 747–763. [Google Scholar]
- 5.
- 5a. Wu T., Sun S., Song J., Xi S., Du Y., Chen B., Sasangka W. A., Liao H., Gan C. L., Scherer G. G., Zeng L., Wang H., Li H., Grimaud A., Xu Z. J., Nat. Can. 2019, 2, 763–772; [Google Scholar]
- 5b. Duan Y., Sun S., Sun Y., Xi S., Chi X., Zhang Q., Ren X., Wang J., Ong S. J. H., Du Y., Gu L., Grimaud A., Xu Z. J., Adv. Mater. 2019, 31, 1807898. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Fominykh K., Feckl J. M., Sicklinger J., Döblinger M., Böcklein S., Ziegler J., Peter L., Rathousky J., Scheidt E.-W., Bein T., Fattakhova-Rohlfing D., Adv. Funct. Mater. 2014, 24, 3123–3129; [Google Scholar]
- 6b. Fominykh K., Chernev P., Zaharieva I., Sicklinger J., Stefanic G., Döblinger M., Müller A., Pokharel T., Böcklein S., Scheu C., Bein T., Fattakhova-Rohlfing D., ACS Nano 2015, 9, 5180–5188. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Thamaphat K., Limsuwan P., Ngotawornchai B., Nat. Sci. 2008, 42, 357–361; [Google Scholar]
- 7b. Banerjee R., Jayakrishnan R., Ayyub P., J. Phys. Condens. Matter 2000, 12, 10647–10654. [Google Scholar]
- 8.
- 8a. Biju V., Chem. Soc. Rev. 2014, 43, 744–764; [DOI] [PubMed] [Google Scholar]
- 8b. Benson E. E., Zhang H., Schuman S. A., Nanayakkara S. U., Bronstein N. D., Ferrere S., Blackburn J. L., Miller E. M., J. Am. Chem. Soc. 2018, 140, 441–450; [DOI] [PubMed] [Google Scholar]
- 8c. Chaussé A., Chehimi M. M., Karsi N., Pinson J., Podvorica F., Vautrin-Ul C., Chem. Mater. 2002, 14, 392–400; [Google Scholar]
- 8d. Brymora K., Fouineau J., Eddarir A., Chau F., Yaacoub N., Grenèche J.-M., Pinson J., Ammar S., Calvayrac F., J. Nanopart. Res. 2015, 17, 438; [Google Scholar]
- 8e. Bangle R., Sampaio R. N., Troian-Gautier L., Meyer G. J., ACS Appl. Mater. Interfaces 2018, 10, 3121–3132; [DOI] [PubMed] [Google Scholar]
- 8f. Griffete N., Herbst F., Pinson J., Ammar S., Mangeney C., J. Am. Chem. Soc. 2011, 133, 1646–1649; [DOI] [PubMed] [Google Scholar]
- 8g. Xie L., Li X., Wang B., Meng J., Lei H., Zhang W., Cao R., Angew. Chem. Int. Ed. 2019, 58, 18883–18887; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 19059–19063. [Google Scholar]
- 9.
- 9a. Hansch C., Leo A., Taft R. W., Chem. Rev. 1991, 91, 165–195; [Google Scholar]
- 9b. Mille J., Yeung W. H., Aust. J. Chem. 1967, 2, 379–381; [Google Scholar]
- 9c. Sheppard A. W., J. Am. Chem. Soc. 1970, 92, 5419–5422. [Google Scholar]
- 10.
- 10a. Sellevåg S. R., Kelly T., Sidebottom H., Nielsen C. J., Phys. Chem. Chem. Phys. 2004, 6, 1243–1252; [Google Scholar]
- 10b. Liang Z., Chen W., Liu J., Wang S., Zhou Z., Li W., Sun G., Xin Q., J. Membr. Sci. 2004, 233, 39–44; [Google Scholar]
- 10c. Lin-Vien D., Colthup N. B., Fateley W. G., Grasselli J. G., The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press: San Diego, 1991. [Google Scholar]
- 11. Jain S. M., Tripathi S., Tripathi S., Spoto G., Edvinsson T., RSC Adv. 2016, 6, 104782–104792. [Google Scholar]
- 12.
- 12a. Doub L., Vandenbelt J. M., J. Am. Chem. Soc. 1947, 69, 2714–2723; [Google Scholar]
- 12b. Doub L., Vandenbelt J. M., J. Am. Chem. Soc. 1949, 71, 2414–2420. [Google Scholar]
- 13. Ke Q., Guan C., Zheng M., Hu Y., Ho K.-H., Wang J., J. Mater. Chem. A 2015, 3, 9538–9542. [Google Scholar]
- 14.
- 14a. Fan L., Long J., Gu Q., Huang H., Lin H., Wang X., J. Catal. 2014, 320, 147–159; [Google Scholar]
- 14b. Scheffler M., Stampfl C., Horn E. K., Handbook of Surface Science 2000, 2, 749—862, Elsevier, Amsterdam; [Google Scholar]
- 14c. Greczynski G., Hultman L., Prog. Mater. Sci. 2020, 107, 100591; [Google Scholar]
- 14d.“Electron Spectroscopy for Chemical Analysis”: K. Siegbahn in Atomic Physics 3 (Eds.: S. J. Smith, G. K. Walters), Springer, Boston, MA, 1973;
- 14e. Lindgren I., J. Electron Spectrosc. Relat. Phenom. 2004, 137–140, 59–71. [Google Scholar]
- 15.
- 15a. Bae D. R., Han W. S., Lim J. M., Kang S., Lee J. Y., Kang D., Jung J. H., Langmuir 2010, 26, 2181–2185; [DOI] [PubMed] [Google Scholar]
- 15b. Soderlind F., Pedersen H., R. M. Petoral, Jr. , Kall P. O., Uvdal K., J. Colloid Interface Sci. 2005, 288, 140–148. [DOI] [PubMed] [Google Scholar]
- 16. Mansour A .N., Melendres C. A., J. Phys. Chem. A 1998, 102, 65–81. [Google Scholar]
- 17.
- 17a. Peck M. A., Langell M. A., Chem. Mater. 2012, 24, 4483–4490; [Google Scholar]
- 17b. Zhu Y., Guo H., Wu Y., Cao C., Tao S., Wu Z., J. Mater. Chem. A 2014, 2, 7904–7911. [Google Scholar]
- 18. Stevens M. B., Trang C. D. M., Enman L. J., Deng J., Boettcher S. W., J. Am. Chem. Soc. 2017, 139, 11361–11364. [DOI] [PubMed] [Google Scholar]
- 19. Diaz-Morales O., Ferrus-Suspedra D., Koper M. T. M., Chem. Sci. 2016, 7, 2639–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Yang Q., Nie Y., Zhu X., Liu X., Li G., Electrochim. Acta 2009, 55, 276–280; [Google Scholar]
- 20b. Elgrishi N., Rountree K. J., McCarthy B. D., Rountree E. S., Eisenhart T. T., Dempsey J. L., J. Chem. Educ. 2017, 95, 197–206. [Google Scholar]
- 21.
- 21a. Louie M. W., Bell A. T., J. Am. Chem. Soc. 2013, 135, 12329–12337; [DOI] [PubMed] [Google Scholar]
- 21b. Trzesniewski B. J., Diaz-Morales O., Vermaas D. A., Longo A., Bras W., Koper M. T., Smith W. A., J. Am. Chem. Soc. 2015, 137, 15112–15121; [DOI] [PubMed] [Google Scholar]
- 21c. Lee S., Banjac K., Lingenfelder M., Hu X., Angew. Chem. Int. Ed. 2019, 58, 10295–10299; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 10401–10405; [Google Scholar]
- 21d. Qiu Z., Tai C.-W., Niklasson G. A., Edvinsson T., Energy Environ. Sci. 2019, 12, 572–581; [Google Scholar]
- 21e. Zhu K., Zhu X., Yang W., Angew. Chem. Int. Ed. 2019, 58, 1252–1265; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1264–1277. [Google Scholar]
- 22.
- 22a. Yamaguchi A., Inuzuka R., Takashima T., Hayashi T., Hashimoto K., Nakamura R., Nat. Commun. 2014, 5, 4256; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Li W., Li F., Yang H., Wu X., Zhang P., Shan Y., Sun L., Nat. Commun. 2019, 10, 5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fehler S. K., Pratsch G., Östreicher C., Fürst M. C. D., Pischetsrieder M., Heinrich M. R., Tetrahedron 2016, 72, 7888–7893. [Google Scholar]
- 24.G. N. George, I. J. Pickering, EXAFSPAK – A Suite of Computer Programs for Analysis of X-ray Absorption spectra, SSRL, 1993.
- 25.
- 25a. Zhuang F., Tang J. C., He J. P., Wang L., Phys. Chem. Chem. Phys. 2000, 2, 3571–3575; [Google Scholar]
- 25b. Agegnehu A. K., Pan C.-J., Rick J., Lee J.-F., Su W.-N., Hwang B.-J., J. Mater. Chem. 2012, 22, 13849–13854; [Google Scholar]
- 25c. Anspoks A., Kalinko A., Kalendarev R., Kuzmin A., Phys. Rev. B 2012, 86, 174114. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary