

Abstract
Background
In patients with non‐severe hemophilia A, we lack detailed knowledge on the timing of treatment with factor VIII (FVIII) concentrates. This knowledge could provide information about the expected treatment timing in patients with severe hemophilia A treated with non‐replacement therapies.
Objective
To assess the FVIII treatment history in patients with non‐severe hemophilia A.
Methods
Patients with non‐severe hemophilia (baseline FVIII activity [FVIII:C] 2‐40 IU/dL) were included from the INSIGHT study. The primary outcome was median age at first FVIII exposure (ED1). In a subgroup of patients for whom more detailed information was available, we analyzed the secondary outcomes: median age at first 20 EDs, annualized bleeding rate for all bleeds (ABR), joint bleeds (AJBR), and major spontaneous bleeds (ASmBR).
Results
In the total cohort (n = 1013), median baseline FVIII activity was 8 IU/dL (interquartile range [IQR] 4‐15) and the median age at ED1 was 3.7 years (IQR 1.4‐7.7). Median age at ED1 rose from 2.5 years (IQR 1.2‐5.7) in patients with FVIII:C 2‐5 IU/dL to 9.7 years (IQR 4.8‐16.0) in patients with FVIII:C 25‐40 IU/dL. In the subgroup (n = 104), median age at ED1, ED5, ED10, and ED20 was 4.0 years (IQR 1.4‐7.6), 5.6 years (IQR 2.9‐9.3), 7.5 years (IQR 4.4‐11.3), and 10.2 years (IQR 6.5‐14.2), respectively. Median ABR, AJBR, and ASmBR were 1.1 (IQR 0.5‐2.6), 0.3 (IQR 0.1‐0.7), and 0 (IQR 0‐0), respectively.
Conclusion
This study demonstrates that in non‐severe hemophilia A, the age at first FVIII exposure increases with baseline FVIII:C and that major spontaneous bleeds rarely occur.
Keywords: factor VIII, hemophilia A, hemorrhage, joint bleed, treatment
Essentials.
Patients with non‐severe hemophilia A (HA) receive factor VIII (FVIII) treatment on demand.
Timing of FVIII treatment was assessed in a large international non‐severe HA cohort.
In non‐severe HA, the median age at first FVIII exposure was 3.7 years.
Patients with non‐severe HA reached their 20th FVIII exposure at a median age of 10.2 years.
1. INTRODUCTION
Hemophilia A is an X‐linked inherited bleeding disorder, caused by a mutation in the F8 gene, leading to a deficiency of functional clotting factor VIII (FVIII). The severity of disease is classified according to the amount of residual FVIII activity. 1 , 2
In patients with non‐severe hemophilia A (FVIII activity 2‐40 IU/dL), bleeding is elicited by trauma or surgery. Therefore, treatment is given on demand in order to treat or to prevent these provoked bleeds. 3 A review of treatment burden in non‐severe hemophilia A identified 44% of subjects requiring hemostatic treatment at least once in a 2‐year observation period. In 69% of the patients, treatment consisted of FVIII concentrate. 4
In contrast to non‐severe hemophilia A, patients with severe hemophilia A (FVIII activity < 1.0 IU/dL) experience spontaneous joint and muscle bleeds. To prevent chronic joint damage, children with severe hemophilia A are treated prophylactically with regular intravenous FVIII infusions. 5 , 6
To reduce treatment burden on patients with severe hemophilia A, non‐replacement therapies, such as emicizumab, are becoming available, offering less frequent subcutaneous infusions for prophylaxis. 7 Non‐replacement therapies have the potential to convert a severe bleeding phenotype into a non‐severe bleeding phenotype with a more continuous pharmacodynamics profile and infrequent, on demand requirement for FVIII replacement. When used in previously untreated patients (PUPs) with severe hemophilia A, it is anticipated that non‐replacement therapies will reduce the bleeding frequency and subsequent FVIII administration, thus postponing initial FVIII exposure.
Data on the treatment history of patients with non‐severe hemophilia A could provide important information about the expected bleeding phenotype and time course to treatment in PUPs with severe hemophilia A when treated with such novel therapies from a very early age onward. Unfortunately, the bleeding phenotype of patients with non‐severe hemophilia A is not well known, as previous studies on this topic are limited by their cross‐sectional and single‐center designs and/or by the inclusion of a selected patient cohort. 8 , 9 , 10 , 11
Therefore, the aim of this study is to investigate the treatment history of patients with non‐severe hemophilia A in a large, international, and multi‐center non‐severe hemophilia A cohort (the INSIGHT cohort). This study will focus on the age at the first FVIII exposure, the time interval until the first 20 FVIII exposures, and the annual bleeding rate, stratified for subgroups of patients based on their residual FVIII level.
2. METHODS
2.1. Patients and setting
Patients were included from the INSIGHT study—a cohort of patients with non‐severe hemophilia A (FVIII activity 2‐40 IU/dL) who received at least one exposure to FVIII concentrate between 1980 and 2011 in 1 of the 34 participating hemophilia treatment centers. For the present study we analyzed all patients born after the 1 January 1980. As FVIII concentrates were not widely available before 1980, patients born before this time point had delayed access to treatment with FVIII concentrates, which would bias the age at the first exposure to FVIII concentrates to a later time point. The follow‐up for all patients was from birth until either the end of the study, emigration, loss to follow‐up, or death. We refer to previously published papers for further information on the INSIGHT cohort. 12 , 13 , 14 , 15
2.2. Data collection
In the total INSIGHT cohort, the following clinical baseline characteristics were available: baseline FVIII activity, date of the first FVIII exposure, the reason for the first FVIII treatment, total number of FVIII exposure days (EDs) during follow‐up, F8 genotype, ethnicity, and family history of hemophilia A. Baseline FVIII activity was defined as the lowest FVIII activity during follow‐up. The definitions of a clinically relevant inhibitor development and an ED to FVIII concentrate are described in the supplemental list of definitions in supporting information.
In the subgroup of the INSIGHT cohort, detailed information per ED was available. The subgroup consists of patients who developed inhibitors (24%) and their matched controls (76%; matched for age, exposure days, and hemophilia treatment center or country). In the subgroup, detailed clinical data for all FVIII exposures during follow‐up until inhibitor development were available, including: the calendar date of the ED and the reason for treatment (including type, location, and cause of the bleed or type of surgical procedure).
All bleeds that were treated with FVIII concentrate were included in the analysis. Bleeds were classified as spontaneous if there were no provoking factors (sport activity or trauma) in the preceding 72 hours. Major bleeding events were defined as bleeds that occurred intracranially, in muscles, or in major joints (elbow, hip, knee, ankle) and required more than 1 ED to FVIII concentrate to resolve. The nature of spontaneous major bleeding episodes was verified by sending all investigators a verification list containing the spontaneous bleeding episodes on file of patients that were treated in their center. Unverified bleeds remained classified as spontaneous.
The definitions for new bleeds were taken from the standard criteria defined by the Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. 16
2.3. Study analysis
2.3.1. Primary outcome
The primary outcome of the study was the age at first exposure to FVIII treatment. We classified the reason for first FVIII treatment as: bleed, surgery, prophylaxis, or unknown. The age at the first FVIII treatment is presented for seven baseline FVIII activity categories: patients with moderate hemophilia A (FVIII:C 2 and 3‐5 IU/dL) and patients with mild hemophilia A (FVIII:C 5‐10, 10‐15, 15‐20, 20‐25, and 25‐40 IU/dL).
2.3.2. Secondary outcomes
The following secondary outcomes were investigated in the subgroup: (a) the age at the first 20 FVIII EDs, (b) the annualized bleeding rate (ABR) for all treated bleeds, (c) ABR for spontaneous major treated bleeds (ASmBR), and (d) the annualized joint bleeding rate (AJBR) for all treated joint bleeds. The ABR, ASmBR, and AJBR were based on bleeds treated with FVIII concentrates. The age at the first 20 EDs was investigated both for EDs with all treatment indications (bleed, surgery, prophylaxis, or unknown) and exclusively for EDs due to bleeding episodes.
The ABR was calculated as: (total number of treated bleeding episodes during follow‐up/the follow‐up period in days) x 365.25. In this analysis we excluded patients (N = 22) with a follow‐up period of less than 180 days. The results of the secondary outcomes are presented for two baseline FVIII activity categories: patients with moderate hemophilia A (FVIII:C 2‐5 IU/dL) and patients with mild hemophilia A (FVIII:C 5‐40 IU/dL). It was not possible to divide patients into six categories as in the cohort study, due to the smaller population size of the subgroup.
2.3.3. Statistical analysis
All statistical analyses were performed using IBM SPSS Statistics, version 25 (IBM SPSS Statistics, Chicago, IL, USA). Continuous data were reported as median with interquartile range (IQR) and categorical data as frequencies with percentage. To evaluate the age at the first FVIII exposure, Kaplan‐Meier cumulative incidence distributions were calculated for each of the six baseline FVIII activity categories. To compare the median age at first FVIII treatment among the six baseline FVIII activity categories, the Mann Whitney U test was performed. Cases with missing data in the primary and secondary outcomes were removed from analysis. A P value less than or equal to 0.05 was considered statistically significant.
3. RESULTS
3.1. Patient characteristics
In total, 1013 patients were included in the total cohort and 104 of them were included in the subgroup (see flowchart of patients in Figure 1). The median baseline FVIII activity was 8 IU/dL (IQR 4‐15) and 6 IU/dL (IQR 4‐11) for the total cohort and the subgroup, respectively. The median age at the end of follow‐up was 16 years (IQR 10‐23) in the total cohort and 10 years (IQR 6‐15) in the subgroup. Additional baseline characteristics are described in Table S1.
Figure 1.
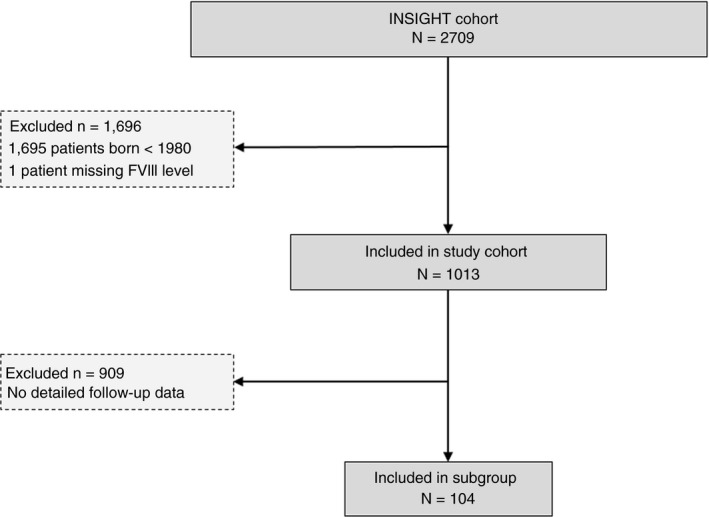
Flowchart of patients included in the study
3.2. Age at the first exposure to FVIII treatment
The age at the first ED to FVIII concentrates was known in 873 patients. The median age at the first ED was 2.5 years (IQR 1.2‐5.7) for patients with a baseline FVIII activity between 2 and 5 IU/dL, increasing up to 9.7 years (IQR 4.8‐16.0) for patients with a baseline FVIII activity between 25 and 40 IU/dL (Table 1 and Figure 2). In nearly all categories of baseline FVIII activity, the age at the first exposure to FVIII treatment occurred earlier in patients with a positive family history of hemophilia (Table 1). The reason for the first exposure to FVIII concentrates was known in 81% of patients (817/1013), with the majority of patients (74%) receiving FVIII concentrates for treatment of a bleed. (Table S2). At the age of 4 years, 48% of patients in the cohort had not yet been exposed to FVIII concentrates. The proportion of untreated patients increased with higher baseline levels of FVIII (Table S3).
Table 1.
Age at first exposure to FVIII concentrates stratified for FVIII activity and for family history of hemophilia
| Baseline FVIII:C, IU/dL | Age at ED 1, years | |||
|---|---|---|---|---|
|
All patients N = 873 |
Positive FH N = 534 |
Negative FH N = 258 |
||
| Moderate, 2‐5 | N = 292 | 2.5 (1.2‐5.7) | 2.0 (1.0‐5.0) | 3.4 (1.8‐6.6) |
| Moderate, in categories | ||||
| 2 | N = 65 | 1.7 (1.1‐5.0) | 1.4 (0.9‐2.5) | 3.3 (1.4‐6.9) |
| 3‐5 | N = 227 | 2.8 (1.2‐6.0) | 2.4 (1.0‐5.0) | 3.4 (2.0‐6.5) |
| Mild, 5‐40 | N = 581 | 4.4 (1.7‐9.4) | 3.9 (1.4‐7.9) | 6.5 (2.4‐13.0) |
| Mild, in categories | ||||
| 5‐10 | N = 221 | 3.8 (1.4‐7.5) | 3.0 (1.2‐5.7) | 6.4 (2.7‐10.8) |
| 10‐15 | N = 149 | 4.2 (1.7‐9.2) | 3.3 (1.5‐6.6) | 7.2 (1.8‐15.9) |
| 15‐20 | N = 93 | 4.2 (1.7‐8.9) | 4.5 (2.3‐11.0) | 4.2 (1.4‐8.9) |
| 20‐25 | N = 57 | 4.3 (1.9‐11.2) | 3.3 (1.7‐9.2) | 6.1 (2.0‐13.3) |
| 25‐40 | N = 61 | 9.7 (4.8‐16.0) | 8.0 (3.9‐15.2) | 12.1 (6.5‐19.0) |
| Total, 2‐40 | N = 873 | 3.7 (1.4‐7.7) | 3.1 (1.3‐6.5) | 5.2 (2.1‐9.9) |
Values are given in medians and interquartile ranges (IQR).
Abbreviations: ED, exposure day; FH, family history of hemophilia; FVIII, factor VIII.
Figure 2.
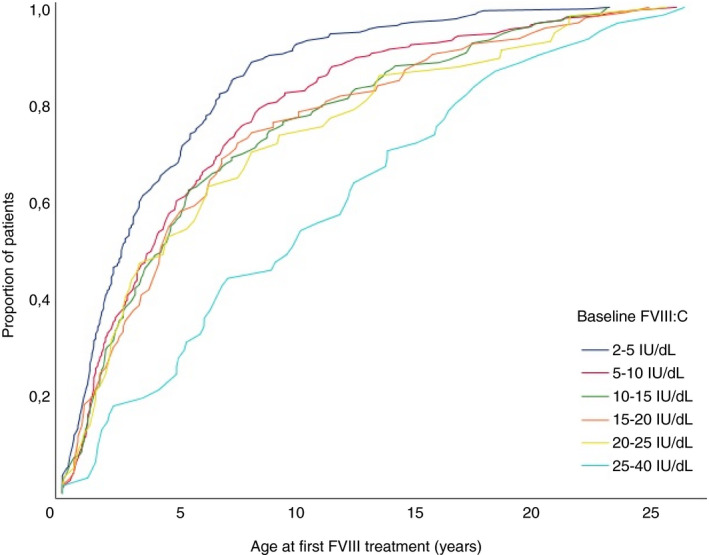
Age at first exposure to factor VIII (FVIII) treatment stratified for FVIII baseline activity in categories
3.3. Age at the first 20 exposures to FVIII treatment
In the subgroup, a total of 2824 EDs were recorded in 104 patients. The median number of EDs per patient was 20 (IQR 12‐38). The median age was 4.0 years (IQR 1.4‐7.6) at ED1, 5.6 years (IQR 2.9‐9.3) at ED5, 7.5 years (IQR 4.4‐11.3) at ED10, 8.9 years (IQR 4.9‐12.1) at ED15, and 10.2 (IQR 6.5‐14.2) at ED20 for all treatment indications. The timing of the first 20 FVIII EDs was similar when bleeding was selected as treatment indication.
In Table 2 the timing of the first 20 EDs is presented for the two FVIII activity categories.
Table 2.
Median age at first 20 exposures to FVIII concentrates stratified for FVIII activity and for treatment indication
| Baseline FVIII:C, IU/dL | Treatment indication |
Age at ED1 N = 104 |
Age at ED5 N = 95 |
Age at ED10 N = 82 |
Age at ED15 N = 68 |
Age at ED20 N = 52 |
|
|---|---|---|---|---|---|---|---|
| Moderate, 2‐5 | N = 48 | All | 4.5 (1.4‐7.5) | 5.2 (3.2‐9.4) | 5.9 (3.8‐10.7) | 7.6 (3.8‐11.2) | 10.9 (4.9‐14.2) |
| N = 41 | Bleeds | 3.2 (1.4‐7.4) | 4.8 (2.7‐8.7) | 5.1 (2.5‐9.5) | 5.2 (3.3‐10.9) | 9.1 (4.7‐12.3) | |
| Mild, 5‐40 | N = 56 | All | 3.8 (1.3‐8.4) | 5.7 (2.8‐9.1) | 8.6 (4.4‐11.7) | 9.0 (5.8‐12.8) | 9.6 (6.7‐14.3) |
| N = 48 | Bleeds | 4.0 (2.2‐8.8) | 7.7 (3.9‐12.5) | 9.6 (5.8‐14.6) | 10.3 (7.3‐14.2) | 10.8 (8.5‐15.5) | |
| Total, 2‐40 | N = 104 | All | 4.0 (1.4‐7.6) | 5.6 (2.9‐9.3) | 7.5 (4.4‐11.3) | 8.9 (4.9‐12.1) | 10.2 (6.5‐14.2) |
| N = 89 | Bleeds | 3.9 (1.5‐7.6) | 5.8 (3.5‐11.0) | 7.6 (4.3‐12.0) | 9.0 (4.9‐13.0) | 10.2 (7.1‐13.0) | |
Values are given in medians (interquartile ranges).
Abbreviation: ED, exposure day; FVIII, factor VIII.
3.4. Annualized bleeding rate
During follow‐up, a total of 710 bleeding episodes were treated in the subgroup. The median ABR for all treated bleeds was 1.1 (IQR 0.5‐2.6) and the AJBR was 0.3 (IQR 0.1‐0.7). Patients with moderate hemophilia A (N = 36) experienced bleeds more frequently when compared to patients with mild hemophilia A (N = 46). For moderate and mild hemophilia A, the median ABR was 1.6 (IQR 0.6‐3.5) and 0.8 (IQR 0.3‐2.5), respectively. Of the 33 major spontaneous bleeds that occurred in 23 patients, 11 bleeds could be verified to be indeed spontaneous (33%). The median ASmBR was 0 (IQR 0‐0).
4. DISCUSSION
4.1. Age at the first exposure to FVIII treatment
In this multicenter cohort study among 1013 patients with non‐severe hemophilia A, we demonstrated that the median age at the first exposure to FVIII treatment increases with baseline FVIII activity. At first exposure, the median age was 2.5 years (IQR 1.2‐5.7) and 4.4 years (IQR 1.7‐9.4) in moderate and mild patients, respectively.
These results are in line with a previous Dutch single‐center study that included 195 patients with non‐severe hemophilia A (baseline FVIII:C 1‐40 IU/dL), demonstrating a median age of 2.9 years (IQR 1.1‐12.2) in moderates and a median age of 5.5 years (IQR 1.8‐15.0) in mild patients. 9
We were able to further stratify the timing of initial FVIII treatment for baseline FVIII activity in patients with mild hemophilia A.
We demonstrated that patients with baseline FVIII activity categories from 10 to 15 IU/dL to 20 to 25 IU/dL received their first FVIII treatment at comparable median ages (4.2‐4.3 years). Interestingly, patients with baseline FVIII activity between 25 and 40 IU/dl reached this milestone at a significantly older age (9.7 years, IQR 4.8‐16.0). Acknowledging the relatively small number of patients with baseline FVIII activity between 25 and 40 IU/dL, these results may be influenced by the use of desmopressin as hemostatic agent of choice with a greater likelihood of sufficient response and efficacy to cover bleeds or procedures without recourse to FVIII concentrate. Patients with a positive family history received their first exposure to FVIII at an earlier age than patients with a negative family history. This is in line with previous results that demonstrated an earlier age of diagnosis in non‐severe hemophilia when a positive family history is present. 9 , 17
4.2. Longitudinal observations
Our descriptive data from the subgroup demonstrated that the age at each ED given for a bleed was higher in patients with mild hemophilia A when compared to patients with moderate hemophilia A.
Although we found that patients with mild hemophilia A were slightly younger at the first and the twentieth ED when compared to patients with moderate hemophilia A (median age of, respectively, 3.8 and 4.5 years at ED1 and, respectively, 9.6 and 10.9 years at ED20), this difference was not statistically significant. To our knowledge this is the first study to investigate the age at the first 20 exposures to FVIII treatment in patients with non‐severe hemophilia A.
The median ABR for all bleed types varied from 1.6 (IQR 0.6‐3.5) in patients with moderate hemophilia A to 0.8 (IQR 0.3‐2.5) in patients with mild hemophilia A. These results are in line with the previous studies which reported ABRs of 0.6 for all bleed types in mild hemophilia A, acknowledging the wide ranges (0‐31) in the study by Aznar et al. 4 , 8 , 10
When evaluating the incidence of joint bleeds, we found a median AJBR of 0.3 (IQR 0.1‐0.7) for all patients included in the subgroup. Stratified for severity, the median AJBR was 0.5 (IQR 0.15‐1.1) and 0.1 (IQR 0.0‐0.6) in patients with moderate and mild hemophilia A, respectively. Three recent studies evaluated the AJBR in non‐severe hemophilia A. 8 , 10 , 11 Although our results are comparable with two of these studies, they are considerably lower than in the study by Soucie et al, 11 which reported a mean AJBR of 2.8 in patients with moderate hemophilia A (FVIII activity 2‐5 IU/dL) and a mean AJBR of 0.97 in patients with baseline FVIII activity between 5 and 40 IU/dL. This difference may be explained by the fact that the latter study collected data on joint bleeds through self‐reporting at annual visits, which could have led to an overestimation of the AJBR. 11
In line with previous observations in patients with non‐severe hemophilia A, patients in our study rarely experienced spontaneous major bleeding episodes (median ABR 0, IQR 0.0‐0.0). 18
4.3. Strengths and limitations of the study
The INSIGHT cohort is an international, multi‐center study containing a large population of patients with moderate and mild hemophilia A. This allows further classification of FVIII activity, providing detailed information in the broad range of FVIII activity levels among patients with mild hemophilia A.
However, when interpreting the results of our study, there are certain limitations that should be kept in mind. Although the proportion of patients with moderate hemophilia A in our cohort is only slightly higher than in most cohorts, it is important to note that our patients may have a relatively more severe bleeding phenotype 10 , 19 , 20 due to the inclusion criteria of the INSIGHT study, which required patients to have received at least one exposure to FVIII concentrate. 13 Therefore, the patients with very mild phenotypes or a good response to desmopressin treatment were excluded. Thus, the results of the present study are not applicable to this group of patients.
In this regard, it is important to mention that in the present study no information was collected on the use of desmopressin in our population. As desmopressin is the first‐choice treatment in non‐severe hemophilia A when possible, it may be expected that a significant proportion of patients in our study were treated with desmopressin prior to FVIII concentrate. Therefore, the median age at first treatment may be lower than the median age of 3.7 years reported in this study, although many countries would not expose children less than 2 years of age to desmopressin.
We acknowledge the limitations of retrospective data collection, especially with respect to the differentiation between spontaneous and non‐spontaneous bleeding episodes. Despite our efforts, we could not verify the nature of all major spontaneous bleeds. However, of the bleeds that we could verify, the majority was recoded as a traumatic bleed. Therefore, we expect that the ABR for major spontaneous bleeds may be overestimated in this study.
Another potential limitation is that the baseline FVIII activity value was determined in participating centers local laboratories rather than in a central laboratory. Significant variability may be present among laboratories in performing FVIII:C assays, possibly due to differences in reagents and assays as well as due to patient‐related variables. 21 , 22
4.4. Clinical implications
More insight into the natural course of non‐severe hemophilia A may provide us with valuable information to predict the treatment course of severe hemophilia A patients who are treated with prophylactic non‐replacement prophylaxis therapies. Compared with severe hemophilia A patients, who receive their first FVIII treatment at a median age of 10 to 12 months, our results suggest a considerable postponement of the age at first FVIII treatment when severe patients are converted to a non‐severe bleeding phenotype. 9 , 23 , 24
If the bleed and treatment profile in this non‐severe hemophilia A cohort would be equivalent to severe hemophilia A receiving prophylaxis with non‐replacement therapy, the postponement of the first 20 exposures could have implications for inhibitor surveillance in patients with severe hemophilia A.
Currently, pediatric centers are very methodical in inhibitor surveillance in the first 20 EDs through to 50 EDs, which happens within a relatively short time frame of months or only a couple of years after the first exposure. 25 Our data presented here suggest this time period for inhibitor surveillance will be spread out over years rather than months for the majority in the context of a non‐replacement prophylaxis agent. This would require important attention to detail and change in practice for continuity of care and inhibitor screening after each exposure.
In conclusion, this study on the treatment history of patients with non‐severe hemophilia A demonstrates that the age at first exposure to FVIII treatment increases with baseline FVIII activity and suggests that these patients rarely experience major spontaneous bleeds. Our data may provide insight into the expected effect of non‐replacement therapies in patients with severe hemophilia A, suggesting a considerable postponement of the age at initial FVIII treatment.
CONFLICTS OF INTEREST
FK, AA, CE, EB, KP, CH, HP, JB, SG have nothing to disclose. CM has received research grant awards from Biotest and CSL Behring. He and/or his institution has received advisory of lecturing honoraria from Bayer, Biotest, CSL Behring, Novo Nordisk, Roche. GC received fees to act as a speaker at or to participate in Advisory Board meetings from Ablynx, Bayer, CSL Behring, Kedrion, Novo Nordisk, Shire/Takeda, Sobi, Roche, Uniqure, and Werfen. GC received unrestricted research grants from CSL Bhering, Pfizer, and Sobi. K. Fischer has received speaker's fees from Bayer, Baxter/Shire, Sobi/Biogen, CSL Behring, Octapharma, Pfizer, NovoNordisk, and has performed consultancy for Bayer, Baxter, Biogen, CSL‐Behring,Freeline, Novo Nordisk, Pfizer, Roche, and Sobi. She and/or her institution has received research support from Bayer, Pfizer, Baxter/Shire, and Novo Nordisk. MK received unrestricted research grants from ZonMW, Innovatiefonds, Bayer, Boehringer‐Ingelheim, Pfizer, Daiichi‐Sankyo for research outside of this work. MM received fees to act as an advisor/consultant/speaker for Bayer, CSL Behring, Novo Nordisk, Pfizer, Roche, Sobi, Bioverativ, Shire/Takeda, Octapharma, Kedrion, Grifols, Biotest, and Catalyst. CS has received consultancy or speaker fees by Bayer, CSL Behring, Shire/Takeda, Novo Nordisk, Sobi, Novartis, Pfizer, Amgen, and Roche. The institution of K. Fijnvandraat has received unrestricted research grants from CSL Behring, Novo Nordisk, and received consultancy fees from Grifols, Takeda, and Novo Nordisk. DPH has received research grant awards from Bayer, Octapharma, and Takeda. He and/or his institution have received advisory or lecturing honoraria from Bayer, Biomarin, Biotest, Grifols, Octapharma, Pfizer, Roche, Sanofi, Sobi, Takeda, UniQure.
AUTHOR CONTRIBUTIONS
AA and FK contributed equally to this work; AA and FK interpreted the data, analyzed the data, and wrote the manuscript; FK edited the final version of the manuscript; DPH and KF conceived and designed the study; CLE, CM, GC, K. Fischer, EB, MK, KP, MM, CS, CH, HP, JGB, SG, K. Fijnvandraat, and DPH collected data or supervised data collection, and reviewed and approved the final version of the manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by an unrestricted research grant from CSL Behring (K. Fijnvandraat). The sponsors had no role in the choice of members of the steering committee and the participating centers nor the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, and approval of the manuscript.
Abdi A, Kloosterman FR, Eckhardt CL, et al; INSIGHT Study Group . The factor VIII treatment history of non‐severe hemophilia A. J Thromb Haemost 2020;18:3203–3210. 10.1111/jth.15076
Amal Abdi and Fabienne Kloosterman contributed equally to this work as first authors.
Karin Fijnvandraat and Dan Hart contributed equally to this work as senior authors.
A complete list of the members of the INSIGHT Study Group appears in the supporting information.
Manuscript handled by: David Lillicrap
Final decision: David Lillicrap, 18 August 2020
REFERENCES
- 1. Fijnvandraat K, Cnossen MH, Leebeek FW, Peters M. Diagnosis and management of haemophilia. BMJ (Clinical research ed). 2012;344:e2707. [DOI] [PubMed] [Google Scholar]
- 2. White GC 2nd, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001;85(03):560. [PubMed] [Google Scholar]
- 3. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1‐e47. [DOI] [PubMed] [Google Scholar]
- 4. Batty P, Austin SK, Khair K, et al. Treatment burden, haemostatic strategies and real world inhibitor screening practice in non‐severe haemophilia A. Br J Haematol. 2017;176(5):796‐804. [DOI] [PubMed] [Google Scholar]
- 5. Donadel‐Claeyssens S. Current co‐ordinated activities of the PEDNET (European Paediatric Network for Haemophilia Management). Haemophilia. 2006;12(2):124‐127. [DOI] [PubMed] [Google Scholar]
- 6. Manco‐Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535‐544. [DOI] [PubMed] [Google Scholar]
- 7. Castaman G, Linari S. Current and emerging biologics for the treatment of hemophilia. Exp Opin Biol Ther. 2019;19(8):801‐810. [DOI] [PubMed] [Google Scholar]
- 8. Tagliaferri A, Di Perna C, Riccardi F, Pattacini C, Rivolta GF, Franchini M. The natural history of mild haemophilia: a 30‐year single centre experience. Haemophilia. 2012;18(2):166‐174. [DOI] [PubMed] [Google Scholar]
- 9. Den Uijl IE, Mauser Bunschoten EP, Roosendaal G, et al. Clinical severity of haemophilia A: does the classification of the 1950s still stand? Haemophilia. 1950;17(6):849‐853. [DOI] [PubMed] [Google Scholar]
- 10. Aznar JA, Lucia F, Abad‐Franch L, et al. Haemophilia in Spain. Haemophilia. 2009;15(3):665‐675. [DOI] [PubMed] [Google Scholar]
- 11. Soucie JM, Monahan PE, Kulkarni R, Konkle BA, Mazepa MA. The frequency of joint hemorrhages and procedures in nonsevere hemophilia A vs B. Blood Adv. 2018;2(16):2136‐2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eckhardt CL, Loomans JI, van Velzen AS, et al. Inhibitor development and mortality in non‐severe hemophilia A. J Thromb Haemost. 2015;13(7):1217‐1225. [DOI] [PubMed] [Google Scholar]
- 13. Eckhardt CL, van Velzen AS, Peters M, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122(11):1954‐1962. [DOI] [PubMed] [Google Scholar]
- 14. van Velzen AS, Eckhardt CL, Hart DP, et al. Inhibitors in nonsevere haemophilia A: outcome and eradication strategies. Thromb Haemost. 2015;114(1):46‐55. [DOI] [PubMed] [Google Scholar]
- 15. van Velzen AS, Eckhardt CL, Streefkerk N, et al. The incidence and treatment of bleeding episodes in non‐severe haemophilia A patients with inhibitors. Thromb Haemost. 2016;115(3):543‐550. [DOI] [PubMed] [Google Scholar]
- 16. Blanchette VS, Key NS, Ljung LR, Manco‐Johnson MJ, van den Berg HM, Srivastava A. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935‐1939. [DOI] [PubMed] [Google Scholar]
- 17. MacLean PE, Fijnvandraat K, Beijlevelt M, Peters M. The impact of unaware carriership on the clinical presentation of haemophilia. Haemophilia. 2004;10(5):560‐564. [DOI] [PubMed] [Google Scholar]
- 18. Venkateswaran L, Wilimas JA, Jones DJ, Nuss R. Mild hemophilia in children: prevalence, complications, and treatment. J Pediatr Hematol Oncol. 1998;20(1):32‐35. [DOI] [PubMed] [Google Scholar]
- 19. Walker IR, Julian JA. Causes of death in Canadians with haemophilia 1980–1995. Association of Hemophilia Clinic Directors of Canada. Haemophilia. 1998;4(5):714‐720. [DOI] [PubMed] [Google Scholar]
- 20. Plug I, van der Bom JG, Peters M, et al. Thirty years of hemophilia treatment in the Netherlands, 1972–2001. Blood. 2004;104(12):3494‐3500. [DOI] [PubMed] [Google Scholar]
- 21. Loomans JI, van Velzen AS, Eckhardt CL, et al. Variation in baseline factor VIII concentration in a retrospective cohort of mild/moderate hemophilia A patients carrying identical F8 mutations. J Thromb Haemost. 2017;15(2):246‐254. [DOI] [PubMed] [Google Scholar]
- 22. Nagler M, Bachmann LM, Alberio L, et al. Variability between laboratories performing coagulation tests with identical platforms: a nationwide evaluation study. Thromb J. 2013;11(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gouw SC, van den Berg HM, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121(20):4046‐4055. [DOI] [PubMed] [Google Scholar]
- 24. Gouw SC, van der Bom JG. Marijke van den Berg H. Treatment‐related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. 2007;109(11):4648‐4654. [DOI] [PubMed] [Google Scholar]
- 25. Collins PW, Chalmers E, Hart DP, et al. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: UK Haemophilia Centre Doctors Organization. Br J Haematol. 2013;160(2):153‐170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material