

Abstract
The organometallic AuI bis‐N‐heterocyclic carbene complex [Au(9‐methylcaffeine‐8‐ylidene)2]+ (AuTMX2) was previously shown to selectively and potently stabilise telomeric DNA G‐quadruplex (G4) structures. This study sheds light on the molecular reactivity and mode of action of AuTMX2 in the cellular context using mass spectrometry‐based methods, including shotgun proteomics in A2780 ovarian cancer cells. In contrast to other metal‐based anticancer agents, this organogold compound is less prone to form coordinative bonds with biological nucleophiles and is expected to exert its drug effects mainly by non‐covalent interactions. Global protein expression changes of treated cancer cells revealed a multimodal mode of action of AuTMX2 by alterations in the nucleolus, telomeres, actin stress‐fibres and stress‐responses, which were further supported by pharmacological assays, fluorescence microscopy and cellular accumulation experiments. Proteomic data are available via ProteomeXchange with identifier PXD020560.
Keywords: cancer, gold complexes, G-quadruplexes, N-heterocyclic carbenes, proteomics, telomeres
Golden touch: A cationic organometallic gold(I) complex featuring N‐heterocyclic carbene ligands and endowed with selective anticancer activity towards human ovarian A2780 cancer cells was studied using an integrated investigational approach, including shotgun proteomics, mass spectrometry and pharmacological methods. The obtained results suggest a multimodal activity related to actin stress fibres, Nrf2‐mediated redox response and telomere disturbance.
Introduction
During the last decades, metal complexes have occupied a pivotal role in medicinal chemistry, either as imaging or as therapeutic agents. [1] Within the large pool of metal‐based compounds, gold complexes have been extensively investigated and are now recognized as promising experimental drugs for cancer treatment. [2] Among the various families of cytotoxic AuI compounds, those featuring N‐heterocyclic carbene (NHC) ligands can be tuned for their steric and electronic properties, which enable modulation of the compounds’ reactivity, solubility, lipophilicity and targeting properties. [3] Concerning possible mechanisms of anticancer action, AuI NHC complexes can affect numerous biological targets and metabolic pathways, also depending on their propensity to exchange some of their ligands to bind to intracellular nucleophiles, including sulfur and selenium‐containing amino acids in proteins. [4] For example, several homo‐ and heteroleptic AuI NHCs were developed as potent inhibitors of seleno‐enzymes. [5]
In addition, we have recently observed that cationic AuI bis‐NHC compounds, endowed with high stability in aqueous environment, can target and stabilize DNA secondary structures of pharmacological relevance, named G‐quadruplexes (G4s). [6] In detail, the AuI complex [Au(9‐methylcaffeine‐8‐ylidene)2]+ (AuTMX2, Figure 1) was shown to selectively stabilize telomeric G4s using fluorescence resonance energy transfer (FRET) DNA melting assays. [7] G4 structures may also form in promoter regions of oncogenes where they seem to control their transcription and expression. [8] Moreover, stabilizing the folding of G4s in telomeres indirectly inhibits telomerase activity, thus, affecting cancer cell mortality. [9] An interesting feature of AuTMX2 is its selective antiproliferative effect against ovarian cancer cell lines with respect to other human cancerous cells, as well as its scarce toxicity in non‐tumorigenic cells and tissues. [7] X‐ray diffraction analysis of the adduct formed by AuTMX2 and a model of telomeric G4 (Tel23) showed that the compound binds non‐covalently between neighbouring quadruplexes. [10] Further characterization of the binding modes of AuTMX2 with different G4 structures was achieved by metadynamics simulations, [11] highlighting the importance of π–π stacking and possibly electrostatic interactions in stabilizing the gold complex/G4 adducts. Of note, the presence of two NHC ligands is essential to achieve the highest possible stabilization of the G4 structure. [12]
Figure 1.
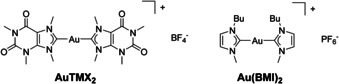
Chemical structures of AuTMX2 and Au(BMI)2, BMI=1‐butyl‐3‐methyl‐imidazole‐2‐ylidene. The latter was previously investigated using gel‐based proteomic methods in ref. [19a].
In parallel to their role in cancer biology, G4s are emerging and promising targets for anticancer drug discovery. Numerous studies described the efficient G4 stabilization by small molecules with associated anticancer potential, [13] including metal complexes. [6a] Since, to the best of our knowledge, AuTMX2 is the first organometallic complex reported so far for its selective G4 stabilization properties, we investigate here its cellular mode of action and pharmacological effects by an integrated approach, including mass spectrometry‐based shotgun proteomics.
Proteomic approaches are being successfully employed to study drug‐activity profiles and modes of action, including in vitro investigations of a variety of metal‐based pharmaceuticals, [14] as well as the effects of metal restriction on microbial physiology. [15] Using these techniques, different classes of metal‐based anticancer [16] or antimicrobial agents [17] were shown to trigger specific effects in target cells and evidenced that small changes in metallodrug's structure would affect dramatically its target spectrum. [18] A few AuI and AuIII anticancer compounds were investigated by gel‐based [19] and gel‐free [20] proteomic analyses to characterize their molecular mechanisms of action, and in a few cases to identify their intracellular targets.
For example, the AuI bis‐NHC complex Au(BMI)2 (BMI=1‐butyl‐3‐methyl‐imidazole‐2‐ylidene, Figure 1) is structurally similar to AuTMX2, and was investigated using gel‐based proteomics in the ovarian cancer cell line A2780. [19a] The compound was shown to regulate a larger number of proteins compared to its mono‐carbene analogue, and resulted in an impairment of metabolic activity, while being devoid of nuclear protein alterations. Here, we show that AuTMX2 features a multimodal mode of action by affecting nuclear and telomeric proteins in addition to inducing a stress response related to redox stress. The latter includes reactive oxygen species (ROS) and stress‐sensitive proteome alterations in addition to actin stress‐fibres.
Results and Discussion
The binding of the cationic AuI bis‐NHC complex AuTMX2 has stabilizing effects on G4 secondary DNA‐structures,[ 10 , 12 ] which should entail observable protein alterations due to the involvement of G4s in telomeres and promoter regions of many (onco‐)genes. Hence, mass spectrometry‐based shotgun proteomics was employed to comprehensively investigate the effects of AuTMX2 in cancer cells at the molecular level. The overall experimental workflow of this study is depicted in Figure S1 in the Supporting Information.
First, the reactivity of AuTMX2 was studied by electrospray ionization mass spectrometry (ESI‐MS) in order to evaluate potential coordinative interactions with biomolecules that may cause off‐target effects, in a similar setup as reported previously. [21] AuTMX2 was stable in water and in the presence of ascorbic acid over the entire incubation period (Figure S2 A). In the presence of the amino acid mixture containing l‐histidine (His), l‐cysteine (Cys), l‐methionine (Met), l‐glutamate (Glu) and l‐methylselenocysteine (MeSe‐Cys), AuTMX2 remained intact even after 24 h, forming minor adducts only with His and Cys via the release of one TMX ligand (Figure S2 B). Adducts corresponding to AuI coordinated to MeSe‐Cys, 9‐ethylguanine or nucleotides were not observed, and neither were adducts with model proteins ubiquitin or cytochrome C (Figure S3). [22] Thus, AuTMX2 appeared stable and largely unreactive towards biological nucleophiles, supporting the idea that its mode of action may reply primarily on non‐covalent interactions. This stands in contrast to benzimidazole‐based bis‐NHC AuI complexes, which appear to be less stable and tend to exchange their ligands.[ 7 , 23 ]
In cells, AuTMX2 features selectivity for cancerous over non‐cancerous cells and against the ovarian cancer cell line A2780 with respect to other human cancer cell lines. [7] We determined the antiproliferative effect of AuTMX2 against A2780 cancer cells and found a half maximal effective concentration (EC50) of 13.3±1.1 μm after 24 h treatment. This was similar to the published EC50 in A2780 cancer cells after a 72 h treatment. [7] Further experiments in this study were conducted by incubating A2780 cancer cells either at 8 μm (2/3 EC50), 13 μm (EC50) or 26 μm (2×EC50) concentrations.
The accumulation of AuTMX2 in A2780 cancer cells was studied by analysing 197Au using inductively‐coupled plasma mass spectrometry (ICP‐MS). The total cellular accumulation was evaluated after treating the cells for 3, 6 and 24 h and at concentrations corresponding to 8 μm and 26 μm (Figure 2 A). A significant accumulation of gold was detected only after 24 h when treating with 8 μm AuTMX2 and the amount of gold corresponded to 25.0±0.6 fg cell−1. A steady increase in gold levels was observed over time, when treating the cells with 26 μm, culminating in 47±2 fg cell−1 after 24 h. Subsequently, A2780 cells were treated with 8 μm of AuTMX2 to obtain information about its distribution between cytoplasmic and nuclear fractions. It turned out that the gold distributed to approximately one third to the nucleus and two thirds to the cytoplasmic fraction irrespective of the incubation time (Figure 2 B).
Figure 2.
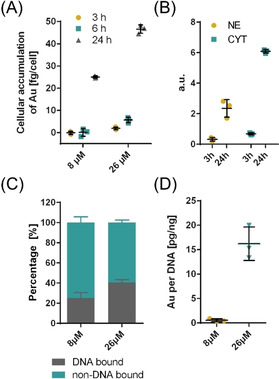
Cellular accumulation and distribution of AuTMX2 in A2780 cancer cells by analysing 197Au using ICP‐MS. (A) AuTMX2 accumulates in cells more efficiently with increasing dose or longer incubation time. (B) AuTMX2 distributes to one third in the nucleus (NE) and two thirds in the cytoplasmic (CYT) fraction irrespective of time when applied at 8 μm for 24 h. (C) Relative and (D) absolute amount of Au‐bound to DNA increases with applied dose after 24 h.
Since AuTMX2 was shown to be a potent and selective G4‐inhibitor, [10] it was of interest to determine the percentage of Au bound to DNA. DNA was isolated from A2780 cells treated with 8 μm and 26 μm of AuTMX2 using silica‐based spin columns. DNA‐bound Au was assumed to co‐elute with DNA, while free Au should be in the flow‐through and wash fractions. The ratio of Au to DNA was found to be constant in eluate 1 and 2 (Figure S4), indicating that Au detected in these samples was actually bound to DNA. The percentage of Au bound to DNA was 25±6 % and 41±3 % for 8 μm and 26 μm concentrations, respectively Figure 2 C). A total of 0.6±0.3 pg Au and 16±3 pg Au were found per ng DNA, respectively (Figure 2 D). Thus, the cellular accumulation experiments confirmed an efficient uptake of AuTMX2 into the nucleus of A2780 cancer cells.
Mass spectrometry‐based shotgun proteomics was applied to study the response of A2780 cancer cells to the AuTMX2 treatment. Global changes in protein expression upon treatment were obtained using a label‐free quantification (LFQ) approach.[ 16 , 18 ] The compound was administered at sub‐cytotoxic concentrations corresponding to 8 μm for 24 h. Six biological replicates were analysed in each condition and after treatment, the cells were fractionated into cytoplasmic (CYT) and nuclear (NE) fractions, similar to the uptake experiments (see Supporting Information). A total of 4202 proteins were detected with a false‐discovery rate (FDR) of 0.01 at the peptide and protein levels. In the cytoplasmic fraction, 36 proteins were significantly regulated, while 115 proteins were significantly regulated in the nuclear fraction (FDR=0.05, S0=0.1). The workflow spanning cell culture, nucleocytoplasmic fractionation, proteolytic digestion and LC‐MS analysis was robust with correlations between replicates typically around R 2=0.9 (Figure 3 A–B).
Figure 3.
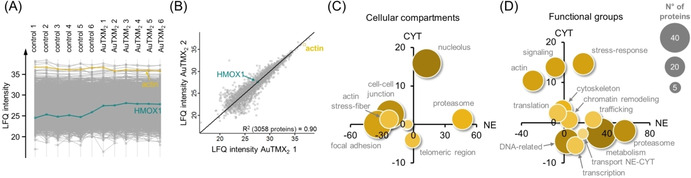
Response profiling by shotgun proteomics. The profile (A) and scatter (B) plots display the stability of the proteomic workflow, which contained six biological replicates per condition of AuTMX2‐treated A2780 cancer cells. The scatter plot highlights the global LFQ‐correlation between replicate 1 and 2. Global protein expression changes were visualized by grouping significantly regulated proteins according to cellular compartment (C) and functional groups (D). The position of the protein group reflects the summed regulation in the cytoplasmic (CYT) and nuclear (NE) fractions.
Global protein expression changes of the AuTMX2 treatment were visualized by grouping significantly regulated proteins according to their gene ontology terms of cellular compartments and molecular functions (Figure 3 C–D). The treatment with AuTMX2 induced pronounced changes in the nuclear compartment, especially regulating proteins belonging to the nucleolus and the telomeric region. In addition, structural elements like the actin cytoskeleton or the stress‐response were regulated (e.g., the latter including heat shock proteins, Nrf2 target genes and the proteasome). Although metabolism‐related proteins were regulated, it turned out that this was a very heterogeneous group largely lacking proteins of the same pathways. Interestingly, another gold(I) bis‐NHC compound Au(BMI)2 was previously reported to induce regulations in protein synthesis, metabolism, cytoskeleton and stress‐response as the most striking protein expression changes. [19a]
Upon treatment with AuTMX2, the most affected protein cluster in the nucleolus referred to stress‐induced transcriptional activation and was significantly up‐regulated (Table S1). This cluster is probably initiated by the up‐regulated protein heme oxygenase 1 (HMOX1), which led to the regulation of the transcriptional activator BRG1 (SMARCA4), lysine‐specific demethylase 2A (KDM2A) and signal transducer and activator of transcription 1‐alpha/beta (STAT1). A total of 14 out of 20 known telomere‐associated proteins were significantly regulated (Table S2). [24] Interestingly, this protein group was generally up‐regulated in the nuclear fraction, while down‐regulated in the cytoplasmic fraction. For example, protein PML acts as a negative regulator of human telomerase (hTERT) and was found up‐regulated upon treatment. [25] Three components of telomere maintenance were up‐regulated, including T‐complex protein 1 subunits alpha (TCP1), zeta (CCT6A) and theta (CCT8). This mirrors what seems to be a common behaviour for gold‐based chemotherapeutics since the AuI complex auranofin and the dinuclear AuIII compound Auoxo6 were previously observed to up‐regulate TCP1 and CCT8, respectively,[ 19b , 19d ] while the structurally similar Au(BMI)2 was found to up‐regulate CCT6A. [19a]
All significantly up‐regulated proteins were searched for common transcription factor activity using oPOSSUM (Figure S5). [26] This indicated the involvement of transcription factors responsible for mediating growth (Klf4, RREB1, ESR1), selenium metabolism (ZNF143) and oxidative stress (SP1). ZNF143 can activate the gene for selenocysteine tRNA to up‐regulate selenoproteins in redox defence. [27] Interestingly, the general transcription factor SP1 seems also to be responsible for regulating telomerase activity in cancer cells. [28]
The observation of regulated proteins within the telomeric region (Figure 3 C) together with the over‐expressed transcription factor SP1 regulating telomerase activity indicates that the mode of action of AuTMX2 may indeed involve targeting telomeric regions in A2780 cancer cells. As AuTMX2 stabilizes telomeric G4 structures, we hypothesized that this interaction would reduce the efficiency of hTERT during replication. The latter is the enzyme responsible for maintaining appropriate telomere lengths required for replicative immortality [9] and is often overexpressed in cancer cells. This stabilising effect may lead to a reduced rate of proliferation and possibly a reduced telomere length in treated A2780 cancer cells, which is expected to be observable after several rounds of duplication. Thus, a 14 d exposure study was planned and suitable concentrations of AuTMX2 had to be selected to avoid unspecific effects like cell cycle arrest or apoptosis induction. A single dose of AuTMX2 at 13 μm for 24 h had only a marginal effect on the cell cycle with a slight increase of cells in the G2/M fraction from 13±1 % to 22.0±0.3 % at the expense of a reduction in G0/G1 cell population (Figure 4 A). AuTMX2 also showed a minor capacity to induce apoptosis at 8 μm and 13 μm concentrations, with a small dose‐dependent increase of late apoptotic/necrotic cells after 72 h treatment: from 10±1 % in control cells to 22±3 % at 8 μm and 29±6 % at 13 μm concentrations (Figure 4 B). This contrasts Au(BMI)2, which was found to be a significant apoptosis inducer (>60 %) and also induced the G0/G1 phase of the cell cycle. [19a] Consequently, A2780 cells were treated with AuTMX2 at sub‐cytotoxic concentrations of 0.2, 1 and 5 μm for the long‐term exposure experiments.
Figure 4.
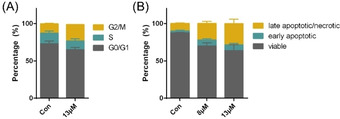
Effect of AuTMX2 on the cell cycle (A) of A2780 cancer cells after 24 h treatment and on their apoptosis induction (B) after 72 h treatment at the indicated concentrations measured by flow cytometry.
A biologically relevant stabilizing effect of AuTMX2 on telomeric G4 in A2780 cancer cells is expected to lead to shortened telomeres after a number of duplication steps due to perturbed hTERT function. Additionally, this interaction is also expected to reduce the proliferation rate of these cancer cells. Therefore, the effect of AuTMX2 on the telomere lengths in A2780 cancer cells was analysed after 14 d. In parallel, the effect on proliferation was evaluated by determining duplication times, number of duplications and cell cycle distributions after 7 d and 14 d. The cellular accumulation of Au was also assessed. The duplication time of the control cells was found to be 23 h throughout the experiment, resulting in 14.4±0.2 duplications in total (Figure 5 A). A gradual dose‐dependent decrease in the number of duplications to 8.6±0.2 at 5 μm AuTMX2 was observed. While 5 μm AuTMX2, which roughly corresponds to 1/3 EC50 (24 h), reduced the duplication time already during the first week, this effect was only observable in the second week for the lower concentrations, indicating gradual accumulation of AuTMX2.
Figure 5.
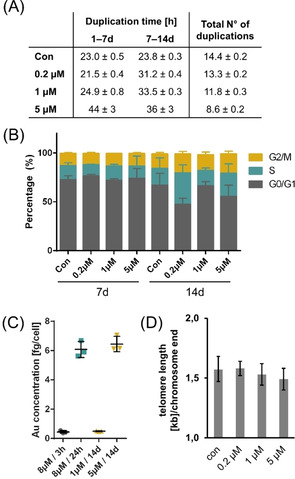
Effect of low dose treatments of AuTMX2 on A2780 cancer cells after 7 d and 14 d treatment: (A) Duplication times and number of duplications, (B) cell cycle distribution assessed by flow cytometry after the end of week 1 (7 d) and week 2 (14 d), (C) cellular accumulation of 197Au in trypsinized cells analysed by ICP‐MS and (D) absolute telomere lengths after 14 d. All experiments were performed in triplicate.
At 7 and 14 days treatment, we additionally determined the cell cycle distribution of A2780 cells by flow cytometry reasoning that the potential effects on telomeres would manifest in an altered cell cycle profile. The effect of AuTMX2 at low doses was also exclusively observable during the second week, while controls remained similarly distributed (Figure 5 B).
This result parallels the cellular accumulation of AuTMX2 assessed by quantifying Au in trypsinized A2780 cells by ICP‐MS (Figure 5 C). AuTMX2 seems to accumulate to a similar extent after 14 d treatment using 5 μm corresponding to 6.5±0.5 fg cell−1 Au, compared to the single dose of 8 μm over 24 h corresponding to 6.1±0.5 fg cell−1 Au. The 14 d treatment using 1 μm corresponded to 0.50±0.03 fg cell−1 Au, which was on the same order as an 8 μm treatment over 3 h corresponding to 0.45±0.08 fg cell−1 Au.
Finally, the absolute telomere length was assessed after the 14 d treatment period. A2780 cancer cells feature tightly controlled telomeres, [29] which was evidenced in our experiments by a low variation of the telomeric length even upon drug exposure (Figure 5 D). Although we were able to observe a slight reduction in telomere lengths in a dose‐dependent manner, this effect was not significant. This suggests that telomere stabilizing‐effects in A2780 cancer cells may contribute to the mode of action of AuTMX2, but may not be the sole determinants of their anticancer activity. For example, higher concentrations of AuTMX2 may achieve a significant reduction in telomere length, but may additionally increase stress‐responses that exert a stronger influence on the proliferative capacity of cancer cells. Thus, other mechanisms seem to contribute to the mode of action of AuTMX2.
The response profiling indicated a proteomic signature related to actin stress‐fibres and stress‐response upon treatment with AuTMX2 (Figure 3). The actin stress‐fibres were characterized by down‐regulation of their protein components (Table S3), suggesting a reduced pool of free building blocks and an increase in polymeric (insoluble) actin stress‐fibres. For example, actinin‐1 (ACTN1) and actinin‐4 (ACTN4) were downregulated, as well as cofilin‐2 (CFL2), septin components (SEP02, 06, 07, 09 and 11) and filamin A (FLNA). The interpretation that AuTMX2 could induce the formation of actin stress‐fibres was supported by the morphological appearance of actin filaments in A2780 cancer cells after treatment with either 13 μm or 26 μm drug concentrations for 24 h (Figure 6). Evidently, AuTMX2 induced a re‐organization of actin toward the cell periphery, resembling the formation of stress‐anchoring points.
Figure 6.
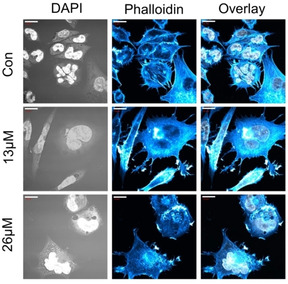
Fluorescence microscopy of nuclei (DAPI staining) and actin cytoskeleton (Alexa Fluor 488 phalloidin staining) of AuTMX2‐treated A2780 cancer cells at different concentrations for 24 h vs. untreated cells (Con). Scalebars=20 μm.
Importantly, beside the regulation of several heat shock proteins (Table S4), the stress response profile revealed a number of Nrf2‐induced genes, including glutathione related glutamate‐cysteine ligase catalytic subunit (GCLC) and regulatory subunit (GCLM), glutathione reductase (GSR), glutathione S‐transferase Mu 5 (GSTM5), but also thioredoxin reductase 1 (TXNRD1), peroxiredoxin 2 (PRDX2) and thioredoxin‐like protein 1 (TXNL1), besides the mentioned HMOX1. Nrf2 forms a cytoplasmic complex with its inhibitor Keap1. The Nrf2‐Keap1 complex is anchored to the actin cytoskeleton and mechanical stimulation may activate the Nrf2 transcription factor. [30] Thus, rearrangement of the actin network as observed by AuTMX2 treatment can per se activate Nrf2 and induce transcription of Nrf2‐induced genes. [31] We did not directly observe Nrf2 to be regulated in the proteome profiling experiment, but it is the known starting point of the upregulated glutathione‐related proteins. The induction of these redox‐active proteins is accompanied by a significant nearly 2‐fold increase in reactive oxygen species (ROS) after treating A2780 cancer cells with AuTMX2 for 24 h with 8 μm and 13 μm concentrations, respectively (one‐way ANOVA, p‐value=0.0004) Figure S6 A). This result contrasts again Au(BMI)2, which did not induce cellular ROS in A2780 cancer cells. [19a] Furthermore, the induction of proteins involved in the glutathione pathway was paralleled by an increased abundance of glutathione (GSH) in AuTMX2 treated cancer cells (Figure S6 B). This treatment also led to a significant 2‐fold reduction of the ratio of reduced vs. oxidized glutathione indicating that GSH is actively used in the cellular defence against ROS formation (Figure S6 C). It may be noted that a heterodimetallic titanium‐gold(I) drug was also found to induce HMOX1 and other Nrf2 target genes in a clear cell renal carcinoma cell line. [32]
Finally, it is known that the proteasome is affected by redox alterations. [33] Indeed, an up‐regulation of many proteasomal components, especially in the nuclear fraction, was observed during the proteomic response profiling experiment (Table S5).
Conclusions
Here, we report on the in‐depth investigation of the mechanisms of reactivity and anticancer action of the cationic organometallic AuI complex AuTMX2 via an integrated approach, including a combination of mass spectrometry‐based methods, such as shotgun proteomics, and pharmacological assays. The obtained results suggest a multimodal mode of action involving metallodrug binding to nuclear and cytoplasmic components in human ovarian cancer cells A2780. Proteomic studies revealed that treatment with AuTMX2 at sub‐cytotoxic concentrations induced the regulation of proteins involved in stress‐induced transcriptional activation, as well as in telomere function. In parallel, in the nuclear fraction, down‐regulation of proteins related to actin stress‐fibres was observed.
Further experiments have been conducted to gain insight into some of these mechanisms, including effects on telomere length, actin stress‐fibres and stress response signatures. Overall, while our study is not yet conclusive in validating the effects of the compound on telomeric DNA via stabilization of G4 structures, indications of stress response signatures via heat shock proteins and Nrf2‐target genes were obtained and suggest the concurrence of several mechanisms that contribute to the anticancer activity of AuTMX2. Future studies will be directed towards the elucidation of the proteasomal changes in the nuclear fraction of treated A2780 cancer cells.
Experimental Section
Materials: The gold‐complex [Au(9‐methylcaffeine‐8‐ylidene)2]BF4 (AuTMX2) was synthesized as previously described.[ 7 , 12 ] N‐ethylmaleimide, NH4OAc, tris(2‐carboxyethyl)phosphine hydrochloride, N‐ethyl‐D5‐maleimide and l‐methionine were purchased from Sigma–Aldrich. Dimethylsulfoxide (DMSO), cytochrome C (horse heart) and ubiquitin (from bovine erythrocytes) were obtained from Sigma. Formic acid (99 %), l‐glutamic acid and l‐cysteine were obtained from Fluka and l‐histidine from Merck. Ascorbic acid was purchased from Acros. MTT reagent powder was purchased from Cayman and dissolved in a 5 mg mL−1 stock solution in PBS and stored in the dark at −20 °C. All reagents were used as received.
Interaction studies: Stock solutions (10 mm) of AuTMX2 were prepared in DMSO and stored at −20 °C in the dark. Aliquots were diluted to 400 μm with water for the preparation of the reaction mixtures prior to incubation. Additional stock solutions in water were prepared of ascorbic acid (400 μm), ubiquitin and cytochrome C (each 200 μm), as well as a mixture of the amino acids His, Met, Cys, Glu and MeSe‐Cys (each 100 μm). The compounds were reacted with the proteins in a 2:1 metal‐to‐protein ratio and with the mixture of the amino acids in an equimolar ratio. Furthermore, reaction mixtures were prepared at a 1:4 molar ratio with ascorbic acid. Reaction mixtures contained a final metal concentration of 50 μm. They were incubated at 37 °C at constant shaking (400 rpm) in the dark and aliquots were taken after 10 min, 3, 6 and 24 h.
All samples were diluted to a final concentration of 1–5 μm. Small molecule samples were diluted with H2O and protein samples with H2O : MeOH : formic acid (50:50:0.2). Mass spectra were recorded over 0.5 min in the positive and negative ion mode and averaged. They were processed using Compass 1.3 and DataAnalysis 4.0 software (both Bruker Daltonics, Bremen, Germany). Mass spectra of the proteins were deconvoluted by the maximum entropy deconvolution algorithm using automatic data‐point spacing and a peak width of 0.2 (ion trap MS) or 30 000 instrument resolving power (time‐of‐flight MS).
The mass spectra were recorded on an AmaZon SL electrospray ionization ion trap mass spectrometer (ESI‐IT MS, Bruker Daltonics GmbH, Bremen, Germany) by direct infusion at a flow rate of 4 μL min−1. Positive and negative ionization modes were recorded. Typical instrument parameters were as follows: ±4.5 kV capillary voltage, −0.5 kV end plate offset, 6 L min−1 dry gas, 180–250 °C dry temperature, 8 psi nebulizer, 89 % RF level, 74.3 trap drive. Experimental mass signals include a standard deviation of m/z ±0.05.
High resolution mass spectra were recorded on a Maxis qTOF mass spectrometer (ESI‐TOF MS, Bruker Daltonics GmbH, Bremen, Germany) equipped with a Triversa nanomate (Advion Biosystems Inc., Ithaca, New York, USA). ChipSoft 8.3 (Advion Biosystems Inc.) was used to control the nanomate. Typical instrument parameters were as follows: −1.8 kV capillary voltage, 0.1 psi gas flow, 4 L min−1 dry gas, 250 °C dry heater, 3 eV quadrupole energy, 8 eV collision energy, 150 μs ion cooler transfer time, 10 μL sample volume and 15 °C nanomate sample plate temperature. Only the 24 h incubation samples were measured on the high resolution nESI‐TOF MS. Additional mass spectra were recorded by direct infusion (3 μL min−1) without the Triversa nanomate using the following parameters: −4.5 kV capillary voltage, 0.4 bar nebulizer, 4 L min−1 dry gas, 180 °C dry heater, 4 eV quadrupole energy, 8 eV collision energy, 80 μs ion cooler transfer time.
Cell culture: The ovarian cancer cell line (A2780) was kindly provided by Thomas Grunt (Medicinal University of Vienna, Austria). A2780 cells were cultured in RPMI‐1640 medium including l‐glutamine (Gibco, Life Technologies, UK). All media contained 10 % foetal calf serum (FCS, ATCC, USA) and 1 % penicillin/streptomycin (ATCC, USA). Cells were grown in a humidified atmosphere containing 5 % CO2 and 95 % air at 37 °C (HeraCell 150i, CO2 incubator, ThermoFisher Scientific). Before reaching confluency, the cells were detached using Trypsin/EDTA solution (0.25 %, Sigma) and split. Cells were counted using an automated cell counter (Moxi Z, Orflo, USA).
Viability assay: The MTT assay was used to investigate the cytotoxicity of AuTMX2 against A2780 cancer cells. Cells were seeded in densities of 3000–50 000 cells/well in a flat‐bottom 96‐well plate (Corning) in 100–200 μL of the respective medium. The medium was replaced after 24 h with control medium or medium containing the appropriate concentration of AuTMX2 and was incubated for 24 h or 72 h. AuTMX2 was first dissolved as a 10 mm stock solution in DMSO and was further diluted with medium directly prior to treatment. The DMSO concentration did not exceed 1 %. After the incubation time, MTT reagent (5 mg mL−1 in PBS) was added to each well in 10 % v/v and was further incubated for 4 h. Then, the medium was removed and the formazan crystals were dissolved in 50–100 μL of DMSO. The metabolic activity was measured photometrically using a UV/Vis photometer (Multiscan GO, ThermoFisher Scientific). Each assay was blank‐corrected performed in triplicates of triplicates and the half maximal effective concentration (EC50) necessary to achieve 50 % inhibition of cell growth was calculated using a sigmoidal fitting.
Cellular accumulation: The A2780 ovarian cancer cells were maintained in T25 or T75 culture flasks (Sarstedt, Germany) in RPMI‐1640 containing 10 % FCS and penicillin/streptomycin as indicated above. For accumulation studies upon AuTMX2 treatment, the cells were seeded at 3×105 cells per well in 1.5‐2 mL complete culture medium in flat‐bottom 6‐well plates (Becton Dickinson Co.). The cells were allowed to adhere for 24 h. Then, the medium was exchanged with medium containing freshly dissolved AuTMX2 (at 8 or 13 μm from a 10 mm stock solution) in 1.5 mL. The cells were treated for 3, 6 and 24 h, respectively, at the respective concentrations. Appropriate absorption blanks were included, and additional wells were used to determine the cell number.
Total cellular accumulation: To determine the total cellular accumulation, the medium was removed after the treatment time and the wells were washed three times with PBS (1×, 1 mL). PBS was removed and the remaining volume was completely removed with a Gilson pipette. The cells were lysed in ultrapure HNO3 (conc., 0.5 mL ) for 3 h after which, aliquots of 400 μL were transferred into a Falcon tube (15 mL) and diluted to a total volume of 8 mL using H2O (Millipore Advantage A10, 18.2 Ω, Merck). Alternatively, the cells were washed once with PBS (1×) and detached using Trypsin/EDTA. After transferring into Falcon tubes (15 mL), the cells were washed three times with PBS (1×). PBS was removed and the remaining PBS completely removed with a Gilson pipette. The cells were lysed in ultrapure HNO3 (conc., 0.5 mL ) for 3 h after which, aliquots of 400 μL were transferred into a Falcon tube (15 mL) and diluted to a total volume of 8 mL using H2O (Millipore Advantage A10, 18.2 Ω, Merck).
Subcellular fractionation: The treated cells were washed three times with PBS (1×) after the incubation time. PBS was completely removed. Then, isotonic lysis buffer (1 mL, s. fractionation protocol in proteomics) was added and the cells were scraped and collected into a Falcon tube (15 mL). The cells were lysed by shear stress by pressing through a syringe (15×). The nuclei were pelleted by centrifugation at 3000 g for 5 mins. The cytoplasm was precipitated in ice‐cold ethanol (abs., 99.9 %, AustrAlco, Austria) over night, while the nuclear pellets were lysed by adding ultrapure HNO3 (conc., 0.5 mL) for 3 h. The cytoplasm was pelleted by centrifugation at 3’000 g for 5 min. The supernatant was completely removed and the lysed with ultrapure HNO3 (conc., 0.5 mL) for 3 h. An aliquot of each sample (400 μL) was transferred into a Falcon tube (15 mL) and diluted to a total volume of 8 mL.
DNA‐based experiments
Treatment for Au‐DNA binding study: A total of 2×106 cells were seeded in T25 flasks and left to adhere for 24 h. Then, the medium was replaced with fresh complete medium or fresh complete medium containing AuTMX2 at doses of 8 μm (2/3 EC50) and 26 μm (2×EC50), respectively, and was incubated for another 24 h. After trypsinization and washing with PBS, the cells were counted. The cells were pelleted, isotonic fractionation buffer was added, and the cellular membrane was lysed by shear stress (8× through a 1 mL syringe with canula). After centrifugation (3500 rpm, 5 min, 4 °C), the supernatant was discarded and the pellet containing the nuclei was washed with PBS. After removal of PBS, the nuclear pellets were stored at −80 °C until DNA extraction. Each condition was performed in triplicates.
Treatment for telomere length quantification: A2780 cells were seeded at 3×105 cells per well in 6‐wells, which were coated with poly‐l‐lysine (0.01 %, Sigma). The cells were continuously treated with AuTMX2 at concentrations of 0.2, 1 and 5 μm (from a 10 mm DMSO stock solution) for 14 d. Cells treated with an equivalent DMSO served as controls and each condition was performed in triplicates. The cell number was followed over time. After 14 d, each sample was analysed for cell cycle distribution by flow cytometry and DNA was extracted for determination of telomere length. Additionally, the cellular accumulation of gold was determined in the samples of 1 and 5 μm AuTMX2 treatment after 14 d using ICP‐MS.
DNA extraction: Genomic DNA was isolated with silica‐based spin columns from nuclear pellets using the QIAamp DNA Mini Kit (Qiagen, Germany) according to manufacturer's instructions for cell lines with the following modifications. After incubation at 56 °C for 10 min, the lysate was additionally homogenized by passing it 5× through a 1 mL syringe with 27G canula. The filtrates after loading the lysate to the spin column (flow‐through) and after the first washing step (wash 1) were collected in 2 mL Eppendorf tubes (Eppendorf, Austria). DNA was eluted using 100 μl EB buffer containing 10 mm Tris‐Cl, pH 8.5 (Qiagen, Germany) and elution was repeated once. DNA concentration of eluates was determined fluorometrically with the Qubit dsDNA BR/HS Assay Kits and the Qubit 4 instrument (Thermo Scientific, Austria). All filtrates and eluates were stored at −20 °C until qPCR or digestion as described below.
Telomere length quantification: Real‐time quantitative polymerase chain reaction (qPCR) was performed in 20 μl reactions containing 2.5 ng genomic DNA template, 2 μl telomere or single copy reference primer and FastStart Essential DNA Green Master Mix (Roche, Austria) using the absolute human telomere length quantification qPCR assay Kit (ScienCell, USA) according to the manufacturer's protocol. Nuclease‐free water served as no template control‐ The reference sample contained in the kit with a telomere length of 695±16 kb per diploid cell was used for absolute quantification The Rotor‐Gene Q instrument (Qiagen, Germany) was used with the following temperature program: initial activation at 95 °C for 10 min followed by 32 cycles of denaturation at 95 °C for 20 s, annealing at 52 °C for 20 s and elongation at 72 °C for 45 s. Specificity of PCR was checked by subsequent melting curve analysis. Each sample was analysed in technical triplicates and in two independent runs. Absolute telomere length was calculated by the comparative ΔΔCT method following the manufacturer's instructions.
Inductively coupled plasma mass spectrometry: An ICP‐MS 7800 (Agilent) was used to quantify the cellular and subcellular accumulation of AuTMX2 and was tuned on a daily basis. The instrument was controlled with MassHunter (C.01.04, Agilent) together with the SPS 4 autosampler (Agilent). The following parameters were used: Cone material was nickel, RF power was 1550, Carrier gas 1.08 L min−1, Plasma gas 15 L min−1, Integration time 0.1 s, number of replicates 10, number of sweeps 100. 185Re was employed as internal standard and gold was assessed using 197Au. The tubing was washed with HCl (aq.) to remove carry‐over of gold for 3 mins after every sample. The instrumental limit of quantification was 1.6 pg mL−1.
Proteomics
Treatment: A2780 ovarian cancer cells were seeded in T25 flasks at 5×106 cells and left to adhere for 24 h in complete medium. Then, the medium was exchanged for fresh medium (controls) or fresh medium containing freshly dissolved AuTMX2 (treatments, 8 μm from a 10 mm stock solution in DMSO) and incubated for another 24 h. Six biological replicates were analysed per condition.
Fractionation: After the 24 h treatment, cells were fractionated into cytoplasmic and nuclear extracts as previously described.[ 16 , 18 , 34 ] All steps were performed on ice. Briefly, the cells were extensively washed with PBS (1×). Isotonic lysis buffer (10 mm HEPES, 10 mm NaCl, 3.5 mm MgCl2, 1 mm EGTA, 0.25 m Sucrose, 0.5 % Triton X‐100) containing protease inhibitors (1 % PMSF and 1 % protease and phosphatase inhibitor cocktail from Roche) was added, the cells were scraped off and transferred into labelled 15 mL Falcon tubes (17×120 mm, Corning). The cellular membrane was ruptured using shear stress by pressing the cell suspension through a syringe multiple times. After centrifugation (3500 rpm, 5 min), the supernatant was transferred into ice‐cold ethanol (1:5) and precipitated over night at −20 °C. The pellet containing the nuclei was incubated with a hypertonic solution (10 mm Tris‐HCl, 1 mm EDTA and 0.5 m NaCl) and subsequently with NP‐40 buffer (10 mm Tris‐HCl, 1 mm EDTA and 0.5 % NP‐40) containing protease inhibitors (1 % PMSF from Sigma and 1 % protease and phosphatase inhibitor cocktail from Roche). After centrifugation (3500 rpm, 5 min), the soluble nuclear proteins were also transferred into ice‐cold ethanol (1:5) and precipitated over night at −20 °C. The precipitated proteins were pelleted, dried under vacuum and dissolved in sample buffer (100 mm dithiothreitol (DTT), 7.5 m urea, 1.5 m thiourea, 4 % CHAPS, 0.05 % sodium dodecyl sulphate) and the protein concentration was determined using a Bradford assay.
Digestion protocol: The samples were digested in‐solution as previously described according to the FASP protocol.[ 16 , 18 , 34 ] Equal amounts of 20 μg protein per sample were reduced with DTT at 37 °C and then loaded on 10 kDa centrifugal filters (Microcon‐10, Merck, Millipore) and pre‐concentrated. The samples were carbamidomethylated with iodacetamide. The samples were then digested over night with trypsin/lys‐C (Promega, Germany) at 37 °C. Filters were washed with 0.5 % TFA and the eluates were dried with a miVac duo concentrator (GeneVac Ltd, UK) and stored at −20 °C until analysis.
LC‐MS/MS analysis: The data was acquired on a QExactive orbitrap mass spectrometer (Thermo Scientific, Germany), which was coupled with a nanoLC‐system (Dionex Ultimate 3000, Thermo Scientific, Germany), equipped with a C‐18 separation column (Dionex, Acclaim PepMap RSCL, 75 μm×50 cm) and C‐18 trapping column (2 cm×100 μm). Dried samples were dissolved in formic acid (30 %, 5 μL) containing four synthetic peptides (Glu1‐fribrinopeptide B, EGVNDNEEGFFSAR; M28, TTPAVLDSDGSYFLYSK; HK0, VLETKSLYVR and HK1, VLETK(ϵ‐AC)SLYVR) and diluted with mobile phase A (40 μL), which consisted of 98 % water, 2 % acetonitrile, 0.1 % formic acid. Mobile phase B consisted of 20 % water, 80 % acetonitrile and 0.1 % formic acid. Each biological sample was recorded once by injecting 2 μL per analysis. Samples were analysed over a 135 min chromatographic run containing a 90 min gradient from 8–40 % mobile phase B. MS1 resolution was 70k with 50 ms injection time and MS2 resolution was 17.5k with 75 ms injection time. A top 8 method was used in the mass range of m/z 400–1400. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD020560 and 10.6019/PXD020560.
Data analysis: MaxQuant (Version 1.6.3.4), including the in‐built Andromeda search engine, was used for label‐free quantification. For identification, we used only non‐redundant Swissprot entries with at least two peptides (of which one needed to be unique). The first and main search peptide tolerance was 50 and 25 ppm, respectively. The false discovery rate (FDR) was fixed to 0.01 on the peptide and protein level. Proteins were considered if they showed at least one unique peptide. The statistical evaluation was performed with Perseus software (Version 1.6.6.0) using LFQ intensities of the MaxQuant result file. After filtering potential contaminants the LFQ values were Log(2)‐transformed. Only proteins, which were found at least five times in either control and/or treatment groups were considered for data evaluation. Permutation‐based FDR set to 0.05 for truncation was used for t‐tests and gave multi‐parameter significant protein expression changes. The final data set was further analysed using the web‐based applications DAVID bioinformatics Resources 6.8, STRING 10.0 and oPOSSUM.
Flow cytometry
Three biological replicates were analysed per condition in A2780 cells (around 105 cells) on a FACS Canto II cytometer (BD Bioscience).
Assessment of reactive oxygen species (ROS): The DCFDA cellular ROS detection assay kit (Abcam) was used according to the manufacturer protocol. 2’,7’‐dichlorofluorescin diacetate (DCFDA) is deacetylated intracellularly by esterases and then oxidized by ROS to the fluorescent 2’,7’‐dichlorofluorescein (DCF), which can be detected by flow cytometry. In addition to controls, A2780 cancer cells were treated for 24 h with AuTMX2 at concentrations of 8 and 13 μm (from 10 mm stock solution in DMSO) using 2×105 cells per well in 6‐wells. They were detached and washed three times with PBS. Cells were resuspended in PBS + DCFDA (20 μm) and incubated for 30 min at 37 °C and were then immediately transferred on ice. Without further washing steps, ROS levels were quantified by flow cytometry in the FITC channel. Three biological replicates were analysed per condition.
Assessment of apoptosis induction: A2780 cancer cells were treated for 72 h with AuTMX2 at concentrations of 8 and 13 μm (from 10 mm stock solution in DMSO) using 2×105 cells per well in 6‐wells. The supernatant and the detached cells were combined and washed three times with PBS and once in Annexin binding buffer (eBioscience). Samples were resuspended in Annexin binding buffer (100 μL) and incubated with 5 μL Annexin V‐APC (eBioscience) for 10 min at room temperature, followed by washing once in Annexin binding buffer. Cells were then resuspended in Annexin binding buffer (200 μL) and propidium iodide was added (PI; Sigma Aldrich; final concentration 100 ng mL−1). Without further washing steps, cells were analysed by flow cytometry in the PE‐channel (PI) and APC‐channel (Annexin‐V). Annexin‐V−/PI− cells were identified as viable cells, Annexin‐V+/PI− as early apoptotic cells and Annexin‐V+/PI+ and Annexin‐V−/PI+ cells as late apoptotic/necrotic cells. Three biological replicates were analysed per condition.
Cell cycle analysis: For a short pulse analysis, A2780 cancer cells were treated for 24 h with AuTMX2 at 13 μm (from 10 mm stock solution in DMSO) using 3×105 cells per well in 6‐wells. They were detached and washed three times with PBS, resuspended and put on ice. Three biological replicates were analysed per condition. The cell cycle distributions of control and treatment groups were analysed with the BD cycletest Plus DNA kit according to the manufacturer's protocol. In brief, cells were washed three times and permeabilized. The amount of interfering RNA was reduced using RNase after which propidium iodide was added as a DNA stain. Propidium iodide was measured by flow cytometry in the PE‐channel and cells in G0/G1, S and G2/M‐phase were identified according to the manufacturer's protocol.
Confocal microscopy
A2780 cancer cells were seeded in chamber slides (μ‐Slide 8 well, Ibidi GmbH, Germany), pre‐coated with poly‐l‐lysine (0.01 %, Sigma) at 4×104 cells well−1 in complete RPMI‐1640 medium and were left to adhere for 24 h. For evaluation of actin morphology, the medium was exchanged, and the cells were treated at EC50 (13 μm) and 2×EC50 (26 μm) doses (from 10 mm stock solution in DMSO), respectively, for 24 h. Thereafter, the cells were fixed (3.7 % formaldehyde, 15 min), washed with PBS and permeabilized (0.2 % triton‐X, 10 min). Blocking was performed with Donkey serum (10 % in PBS‐A, 1 h). Primary antibodies were incubated overnight at 4 °C. Actin was stained alexa fluor 488 phalloidin (Molecular Probes, Life Technologies, ThermoFisher Scientific). The slides were washed with PBS and post‐fixed with 3.7 % formaldehyde (10 min). Finally, glycine (100 mm) was used to mask reactive sites and slides were mounted and sealed with Roti‐Mount FluoCare (Roth, Graz, Austria) with DAPI. All images were acquired on a confocal LSM Zeiss 710 equipped with ELYRA PS.1 system and a plan apochromat 63x/1.4 oil objective.
Glutathione determination
Sample preparation: The ratio of reduced versus oxidized glutathione (GSH) was measured according to a modified protocol from Tomin et al. [35] Therefore, 5×105 A2780 cancer cells were seeded in 6‐wells in triplicates per condition. The cells were left to adhere for 24 h. Then, the medium was replaced, and the treated cells were incubated with AuTMX2 (13 μm) for 24 h. After removing the supernatant and washing twice with PBS the cells were incubated with 500 μL N‐ethylmaleimide (NEM, 2.5 mm) for 20 min at RT. The NEM solution was removed and the cells were again washed twice with PBS. Then, 200 μL of 80 % methanol in water was added an incubated for 20 min at 4 °C. The cells were scraped off, transferred into an Eppendorf tube and centrifuged (10 min, 13’000 g, 4 °C). The supernatant was collected and dried under a nitrogen stream before resuspending in 100 μL NH4OAc (50 mm, pH 7). After adding 300 μL DCM and thoroughly vortexing, the samples were centrifuged (5 min, 13’000 g, 4 °C). An aliquot of the upper phase (45 μL) containing NEM‐GSH and GSSG was taken and reduced with 2.5 μL Tris(2‐carboxyethyl)phosphine hydrochloride (TCEP, 5 mm, pH 7) for 30 min at 37 °C. Afterwards, 2.5 μL of N‐ethyl‐D5‐Maleimide (D5‐NEM, 100 mm) was added and again incubated for 20 min at RT in the dark. The samples were finally diluted with 200 μL of NH4OAc (50 mm) for processing by HPLC‐MS.
LC‐MS analysis: Chromatographic separation was performed on a Kinetex XB‐C18 column (2.6 μm, 100×2.1 mm, 100 Å, Agilent) using a Vanquish UHPLC System (Thermo Fisher Scientific). Mobile Phase A consisted of water with 0.2 % formic acid and mobile phase B of methanol with 0.2 % formic acid. The following gradient profile was run: 0–30 % B in 5 min, 30–70 % B in 2 min, wash at 95 % B for 3 min and at 10.1 min the system was re‐equilibrated at 0 % B to give a total runtime of 12 min. The flow rate was 300 μL min−1, the column chamber and the preheater temperature were set to 40 °C and the injection volume was 5 μL. MS analysis was performed in the positive ion mode on a QExactive HF (Thermo Fisher Scientific). All samples were analysed in technical replicates. MS scans were acquired in the mass range of m/z 100–1000 and the resolution was set to 60’000 (at m/z 200). The four most abundant ions in the full scan were selected for further fragmentation in the HCD collision cell applying 30 eV dynamic collision energy. MS2 fragments were analysed at a resolution of 15’000 (at m/z 200). Dynamic exclusion was applied for 6 s. NEM‐GSH was found at m/z 433 and D5‐NEM‐GSH was found at m/z 438. The former corresponds to reduced GSH and the latter to oxidized GSH. The instrument was controlled using Xcalibur software (Thermo Fisher Scientific). Raw files generated by the Q Exactive HF were further analysed with Tracefinder Software 4.1 (Thermo Fisher Scientific).
Statistical analysis
Data was analysed using GraphPad Prism Version 6.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
S.M. and C.G. receive support from the Austrian Science Fund (FWF): P33238‐N. The authors are grateful for the support of the Core Facility of Mass Spectrometry, the Core Facility Multimodal Imaging (Faculty of Chemistry) and the Joint Metabolome Facility, members of the Vienna Life‐Science Instruments (VLSI). Authors acknowledge the help of Julia Brunmair with the measurements on the Q Exactive HF. Open access funding enabled and organized by Projekt DEAL.
S. M. Meier-Menches, B. Neuditschko, K. Zappe, M. Schaier, M. C. Gerner, K. G. Schmetterer, G. Del Favero, R. Bonsignore, M. Cichna-Markl, G. Koellensperger, A. Casini, C. Gerner, Chem. Eur. J. 2020, 26, 15528.
Contributor Information
Dr. Samuel M. Meier‐Menches, Email: samuel.meier@univie.ac.at.
Prof. Angela Casini, Email: angela.casini@tum.de.
Prof. Christopher Gerner, Email: christopher.gerner@univie.ac.at.
References
- 1.
- 1a. Casini A., Vessières A., Meier-Menches S. M., Metal-based Anticancer Agents, Royal Society of Chemistry, 2019, 355; [Google Scholar]
- 1b. Franz K. J., Metzler-Nolte N., Chem. Rev. 2019, 119, 727–729; [DOI] [PubMed] [Google Scholar]
- 1c. Kostelnik T. I., Orvig C., Chem. Rev. 2019, 119, 902–956; [DOI] [PubMed] [Google Scholar]
- 1d. Meier-Menches S. M., Gerner C., Berger W., Hartinger C. G., Keppler B. K., Chem. Soc. Rev. 2018, 47, 909–928. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Zou T., Lum C. T., Lok C.-N., Zhang J.-J., Che C.-M., Chem. Soc. Rev. 2015, 44, 8786–8801; [DOI] [PubMed] [Google Scholar]
- 2b. Bertrand B., Casini A., Dalton Trans. 2014, 43, 4209–4219. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Mora M., Gimeno M. C., Visbal R., Chem. Soc. Rev. 2019, 48, 447–462; [DOI] [PubMed] [Google Scholar]
- 3b. Porchia M., Pellei M., Marinelli M., Tisato F., Del Bello F., Santini C., Eur. J. Med. Chem. 2018, 146, 709–746; [DOI] [PubMed] [Google Scholar]
- 3c. Zhang C., Maddelein M.-L., Wai-Yin Sun R., Gornitzka H., Cuvillier O., Hemmert C., Eur. J. Med. Chem. 2018, 157, 320–332; [DOI] [PubMed] [Google Scholar]
- 3d. Zou T., Lok C.-N., Wan P.-K., Zhang Z.-F., Fung S.-K., Che C.-M., Curr. Opin. Chem. Biol. 2018, 43, 30–36; [DOI] [PubMed] [Google Scholar]
- 3e. Jürgens S., Casini A., Chimia 2017, 71, 92–101; [DOI] [PubMed] [Google Scholar]
- 3f. Fernández-Gallardo J., Elie B. T., Sanaú M., Contel M., Chem. Commun. 2016, 52, 3155–3158; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3g. Muenzner J. K., Biersack B., Albrecht A., Rehm T., Lacher U., Milius W., Casini A., Zhang J.-J., Ott I., Brabec V., Stuchlikova O., Andronache I. C., Kaps L., Schuppan D., Schober R., Chem. Eur. J. 2016, 22, 18953–18962; [DOI] [PubMed] [Google Scholar]
- 3h. Mui Y. F., Fernandez-Gallardo J., Elie B. T., Gubran A., Maluenda I., Sanau M., Navarro O., Contel M., Organometallics 2016, 35, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Tolbatov I., Coletti C., Marrone A., Re N., Int. J. Mol. Sci. 2019, 20, 820; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Casini A., Sun R. W. Y., Ott I., Metal Ions in Life Sciences, Vol. 18, Eds.: Sigel A., Sigel H., Freisinger E., Sigel R. K. O., 2018, pp. 351–386. [Google Scholar]
- 5.
- 5a. Karaca Ö., Scalcon V., Meier-Menches S. M., Bonsignore R., Brouwer J. M. J. L., Tonolo F., Folda A., Rigobello M. P., Kühn F. E., Casini A., Inorg. Chem. 2017, 56, 14237–14250; [DOI] [PubMed] [Google Scholar]
- 5b. Schmidt C., Karge B., Misgeld R., Prokop A., Franke R., Brönstrup M., Ott I., Chem. Eur. J. 2017, 23, 1869–1880; [DOI] [PubMed] [Google Scholar]
- 5c. Rubbiani R., Salassa L., de Almeida A., Casini A., Ott I., ChemMedChem 2014, 9, 1205–1210; [DOI] [PubMed] [Google Scholar]
- 5d. Schuh E., Pflüger C., Citta A., Folda A., Rigobello M. P., Bindoli A., Casini A., Mohr F., J. Med. Chem. 2012, 55, 5518–5528; [DOI] [PubMed] [Google Scholar]
- 5e. Bertrand B., Citta A., Franken I. L., Picquet M., Folda A., Scalcon V., Rigobello M. P., Le Gendre P., Casini A., Bodio E., J. Biol. Inorg. Chem. 2015, 20, 1005–1020; [DOI] [PubMed] [Google Scholar]
- 5f. Bindoli A., Rigobello M. P., Scutari G., Gabbiani C., Casini A., Messori L., Coord. Chem. Rev. 2009, 253, 1692–1707. [Google Scholar]
- 6.
- 6a. Karaca O., Meier-Menches S. M., Casini A., Kuhn F. E., Chem. Commun. 2017, 53, 8249–8260; [DOI] [PubMed] [Google Scholar]
- 6b. Monchaud D., Teulade-Fichou M.-P., Org. Biomol. Chem. 2008, 6, 627–636; [DOI] [PubMed] [Google Scholar]
- 6c. Vilar R. in Metallo-Drugs: Development and Action of Anticancer Agents (Eds.: Sigel A., Sigel H., Freisinger E., Sigel R. K. O.), Metal Ions in Life Sciences Vol. 18, Walter de Gruyter GmbH, Berlin/Boston, 2018, pp. 325–350. [Google Scholar]
- 7. Bertrand B., Stefan L., Pirrotta M., Monchaud D., Bodio E., Richard P., Le Gendre P., Warmerdam E., de Jager M. H., Groothuis G. M. M., Picquet M., Casini A., Inorg. Chem. 2014, 53, 2296–2303. [DOI] [PubMed] [Google Scholar]
- 8. Rhodes D., Lipps H. J., Nucleic Acids Res. 2015, 43, 8627–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bernal A., Tusell L., Int. J. Mol. Sci. 2018, 19, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bazzicalupi C., Ferraroni M., Papi F., Massai L., Bertrand B., Messori L., Gratteri P., Casini A., Angew. Chem. Int. Ed. 2016, 55, 4256–4259; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4328–4331. [Google Scholar]
- 11. Wragg D., de Almeida A., Bonsignore R., Kühn F. E., Leoni S., Casini A., Angew. Chem. Int. Ed. 2018, 57, 14524–14528; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14732–14736. [Google Scholar]
- 12. Meier-Menches S. M., Aikman B., Döllerer D., Klooster W. T., Coles S. J., Santi N., Luk L., Casini A., Bonsignore R., J. Inorg. Biochem. 2020, 202, 110844. [DOI] [PubMed] [Google Scholar]
- 13. Neidle S., Nat. Rev. Chem. 2017, 1, 0041. [Google Scholar]
- 14.
- 14a. Kreutz D., Gerner C., Meier-Menches S. M., in Metal-based Anticancer Agents, Vol. 14 (Eds.: Casini A., Vessières A., Meier-Menches S. M.), Royal Society of Chemistry, 2019, pp. 246–270; [Google Scholar]
- 14b. Wang Y., Li H., Sun H., Inorg. Chem. 2019, 58, 13673–13685; [DOI] [PubMed] [Google Scholar]
- 14c. Wang H., Zhou Y., Xu X., Li H., Sun H., Curr. Opin. Chem. Biol. 2020, 55, 171–179. [DOI] [PubMed] [Google Scholar]
- 15. Wang J., Lonergan Z. R., Gonzalez-Gutierrez G., Nairn B. L., Maxwell C. N., Zhang Y., Andreini C., Karty J. A., Chazin W. J., Trinidad J. C., Skaar E. P., Giedroc D. P., Cell Chem. Biol. 2019, 26, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kreutz D., Bileck A., Plessl K., Wolrab D., Groessl M., Keppler B. K., Meier S. M., Gerner C., Chem. Eur. J. 2017, 23, 1881–1890. [DOI] [PubMed] [Google Scholar]
- 17. Wang R., Lai T.-P., Gao P., Zhang H., Ho P.-L., Woo P. C.-Y., Ma G., Kao R. Y.-T., Li H., Sun H., Nat. Commun. 2018, 9, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meier-Menches S. M., Zappe K., Bileck A., Kreutz D., Tahir A., Cichna-Markl M., Gerner C., Metallomics 2019, 11, 118–127. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Magherini F., Fiaschi T., Valocchia E., Becatti M., Pratesi A., Marzo T., Massai L., Gabbiani C., Landini I., Nobili S., Mini E., Messori L., Modesti A., Gamberi T., Oncotarget 2018, 9, 28042–28068; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Guidi F., Landini I., Puglia M., Magherini F., Gabbiani C., Cinellu M. A., Nobili S., Fiaschi T., Bini L., Mini E., et al., Metallomics 2012, 4, 307–314; [DOI] [PubMed] [Google Scholar]
- 19c. Guidi F., Puglia M., Gabbiani C., Landini I., Gamberi T., Fregona D., Cinellu M. A., Nobili S., Mini E., Bini L., Modesti P. A., Modesti A., Messori L., Mol. BioSyst. 2012, 8, 985–993; [DOI] [PubMed] [Google Scholar]
- 19d. Magherini F., Modesti A., Bini L., Puglia M., Landini I., Nobili S., Mini E., Cinellu M. A., Gabbiani C., Messori L., J. Biol. Inorg. Chem. 2010, 15, 573–582. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Fung S. K., Zou T., Cao B., Lee P.-Y., Fung Y. M. E., Hu D., Lok C.-N., Che C.-M., Angew. Chem. Int. Ed. 2017, 56, 3892–3896; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3950–3954; [Google Scholar]
- 20b. Hu D., Liu Y., Lai Y.-T., Tong K.-C., Fung Y.-M., Lok C.-N., Che C.-M., Angew. Chem. Int. Ed. 2016, 55, 1387–1391; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1409–1413; [Google Scholar]
- 20c. Wang Y., He Q.-Y., Che C.-M., Chiu J.-F., Proteomics 2006, 6, 131–142. [DOI] [PubMed] [Google Scholar]
- 21. Meier S. M., Gerner C., Keppler B. K., Cinellu M. A., Casini A., Inorg. Chem. 2016, 55, 4248–4259. [DOI] [PubMed] [Google Scholar]
- 22. Wenzel M., Casini A., Coord. Chem. Rev. 2017, 352, 432–460. [Google Scholar]
- 23. Rubbiani R., Can S., Kitanovic I., Alborzinia H., Stefanopoulou M., Kokoschka M., Monchgesang S., Sheldrick W. S., Wolfl S., Ott I., J. Med. Chem. 2011, 54, 8646–8657. [DOI] [PubMed] [Google Scholar]
- 24. Ganesan K., Xu B., Int. J. Mol. Sci. 2017, 19, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oh W., Ghim J., Lee E. W., Yang M. R., Kim E. T., Ahn J. H., Song J., J. Cell Sci. 2009, 122, 2613–2622. [DOI] [PubMed] [Google Scholar]
- 26. Ho Sui S. J., Fulton D. L., Arenillas D. J., Kwon A. T., Wasserman W. W., Nucleic Acids Res. 2007, 35, W245–W252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu W., Chen Z., Zhang H., Wang Y., Luo Y., Huang P., Cell Death Dis. 2012, 3, e422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao J., Bilsland A., Hoare S. F., Keith W. N., FEBS Lett. 2003, 536, 111–119. [DOI] [PubMed] [Google Scholar]
- 29. Dahlgren P. N., Bishop K., Dey S., Herbert B.-S., Tanaka H., Neoplasia 2018, 20, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.
- 30a. Del Favero G., Zaharescu R., Marko D., Arch. Tox. 2018, 92, 3535–3547; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30b. Hosoya T., Maruyama A., Kang M.-I., Kawatani Y., Shibata T., Uchida K., Itoh K., Yamamoto M., J. Biol. Chem. 2005, 280, 27244–27250. [DOI] [PubMed] [Google Scholar]
- 31. Kang M.-I., Kobayashi A., Wakabayashi N., Kim S.-G., Yamamoto M., Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elie B. T., Hubbard K., Layek B., Yang W. S., Prabha S., Ramos J. W., Contel M., ACS Pharmacol. Transl. Sci. 2020, 3, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lefaki M., Papaevgeniou N., Chondrogianni N., Redox Biol. 2017, 13, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meier S. M., Kreutz D., Winter L., Klose M. H. M., Cseh K., Weiss T., Bileck A., Alte B., Mader J. C., Jana S., Chatterjee A., Bhattacharyya A., Hejl M., Jakupec M. A, Heffeter P., Berger W., Hartinger C. G., Keppler B. K., Wiche G., Gerner C., Angew. Chem. Int. Ed. 2017, 56, 8267–8271; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8379–8383. [Google Scholar]
- 35. Tomin T., Schittmayer M., Birner-Gruenberger R., Metabolites 2020, 10, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary