

Abstract
A gene cluster encoding a cryptic trans‐acyl transferase polyketide synthase (PKS) was identified in the genomes of Burkholderia gladioli BCC0238 and BCC1622, both isolated from the lungs of cystic fibrosis patients. Bioinfomatics analyses indicated the PKS assembles a novel member of the glutarimide class of antibiotics, hitherto only isolated from Streptomyces species. Screening of a range of growth parameters led to the identification of gladiostatin, the metabolic product of the PKS. NMR spectroscopic analysis revealed that gladiostatin, which has promising activity against several human cancer cell lines and inhibits tumor cell migration, contains an unusual 2‐acyl‐4‐hydroxy‐3‐methylbutenolide in addition to the glutarimide pharmacophore. An AfsA‐like domain at the C‐terminus of the PKS was shown to catalyze condensation of 3‐ketothioesters with dihydroxyacetone phosphate, thus indicating it plays a key role in polyketide chain release and butenolide formation.
Keywords: anticancer agents, biosynthesis, enzymology, genome mining, natural products
Genome mining enabled the discovery of gladiostatin, a novel glutarimide antibiotic from the cystic fibrosis associated bacterium Burkholderia gladioli. Gladiostatin contains an unusual 2‐acyl‐3‐hydroxy‐4‐methylbutenolide and exhibits promising activity against human cancer cells. The polyketide synthase responsible for its biosynthesis employs a novel mechanism for chain release, resulting in formation of a phosphorylated butenolide intermediate.
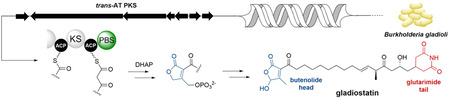
Introduction
The constant competition between microbes and their environment has driven the evolution of specialised metabolite production in bacteria, enabling rapid ecological adaptation. [1] Such metabolites frequently find important applications in medicine, as antibiotics, anticancer agents and immune modulators, and agriculture, as insecticides, herbicides and fungicides. Gram‐negative bacteria belonging to the Burkholderia genus produce a wide array of bioactive specialised metabolites, including the respiratory toxin bongkrekic acid, anti‐proliferative agents such as thailanstatin and spliceostatin, and the antibiotics enacyloxin IIa and gladiolin.[ 2 , 3 , 4 , 5 , 6 ] Despite their structural complexity and diversity, these molecules are biosynthesised from simple building blocks by modular polyketide synthase (PKS) and non‐ribosomal peptide synthetase (NRPS) assembly lines, often harbouring non‐canonical characteristics.[ 7 , 8 ] Recently, we have shown that the opportunistic pathogen Burkholderia gladioli BCC0238, isolated from the lung of a cystic fibrosis (CF) patient, produces a range of specialised metabolites, dependent on the carbon source. When glycerol is used as the carbon source, this strain produces gladiolin, a novel macrolide with promising activity against Mycobacterium tuberculosis, and the swarming inhibitor icosalide A1, which was originally isolated from a filamentous fungus, but has subsequently been shown to originate from a Burkholderia symbiont.[ 6 , 9 , 10 ] Switching to a mixture of glycerol and ribose as carbon sources, induces the production of bolagladins A and B, novel lipodepsipeptides containing a unique citrate‐primed fatty acid and an unusual dehydro‐β‐alanine residue. [11]
Although several specialised metabolites and their associated biosynthetic gene clusters (BGCs) have already been identified in B. gladioli BCC0238, analysis of the complete genome sequence of this strain indicated that it contains numerous cryptic BGCs, the metabolic products of which are currently unknown. [6] Here we report the discovery of gladiostatin, a novel member of the glutarimide class of polyketide antibiotics, as the metabolic product of a cryptic trans‐acyl transferase (trans‐AT) PKS‐encoding BGC in B. gladioli BCC0238. This unusual metabolite, which contains a rare 2‐acyl‐4‐hydroxy‐3‐methylbutenolide in addition to the 2, 6‐piperidinedione common to all glutarimides, is active against yeast and has promising activity against several human cancer cell lines. Gene disruption experiments confirmed that the BGC directs the biosynthesis of gladiostatin, and chain release from the PKS was reconstituted in vitro, providing insights into the mechanism for formation of the unusual butenolide moiety. These experiments enabled us to propose a biosynthetic pathway for gladiostatin, the first glutarimide to be isolated from Gram‐negative bacteria, illuminating the role played by horizontal gene transfer in trans‐AT PKS evolution.
Results and Discussion
Genome Mining Identifies a Cryptic trans‐AT PKS Predicted to Assemble a Novel Glutarimide
Building on our previous work in B. gladioli BCC0238, in silico analysis of the genome sequence revealed a ≈50 kb cryptic BGC encoding a trans‐AT PKS on the second chromosomal replicon (Figure S1). Bioinformatics analyses indicated that several of the proteins encoded by this BGC are very similar to enzymes known to be involved in the biosynthesis of the glutarimide class of polyketide antibiotics in Streptomyces species (Figure 1 a and b), including cycloheximide (1),[ 12 , 13 ] 9‐methylstreptimidone (2),[ 14 , 15 ] migrastatin/iso‐migrastatin (3)[ 16 , 17 , 18 ] and lactimidomycin (4; Figure 1 c, Figure S2).[ 19 , 20 ] Glutarimides have potent antifungal activity and inhibit eukaryotic translation by blocking the binding of tRNA to the E‐site of the 60S ribosomal subunit.[ 21 , 22 ] They also possess promising anticancer activity and several members of the family have been reported to inhibit tumour cell‐migration.[ 23 , 24 ]
Figure 1.
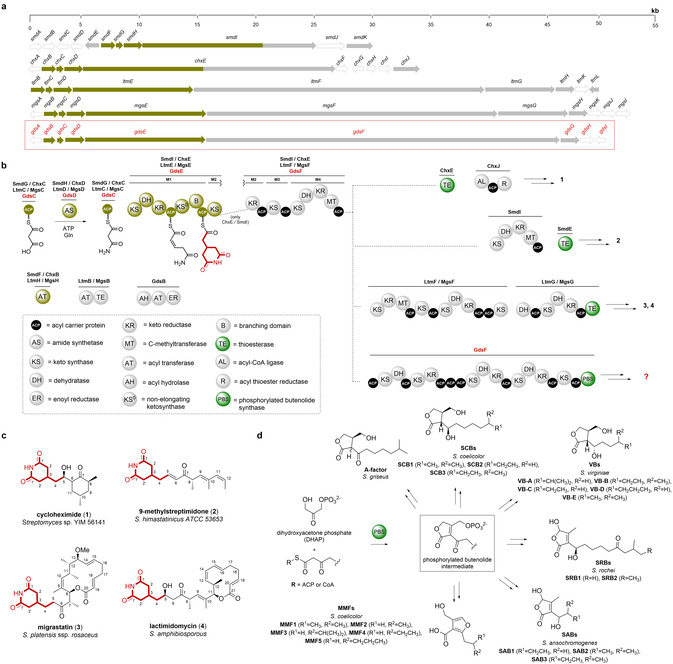
Comparison of Streptomyces glutarimide biosynthetic gene clusters and polyketide synthases they encode with that encoded by the cryptic B. gladioli gene cluster and role played by AfsA‐like phosphorylated butenolide synthases (PBSs) in Streptomyces signalling molecule biosynthesis. a) Comparison of biosynthetic gene clusters (BGCs) that direct glutarimide biosynthesis in Streptomyces species (9‐methylstreptimidone (smd), cycloheximide (chx), lactimidomycin (ltm) and migrastatin (mgs)) with the cryptic B. gladioli BCC0238/BCC1622 gene cluster (highlighted by the red box). The genes (or regions thereof) encoding the machinery responsible for assembling the common 2‐(2,6‐dioxopiperidin‐4‐yl)acetyl thioester biosynthetic intermediate are highlighted in gold. b) Comparison of the 9‐methylstreptimidone, cycloheximide, lactimidomycin and migrastatin PKS architectures with the PKS encoded by the cryptic B. gladioli BGC. Domains highlighted in dark yellow are involved in constructing the common 2‐(2,6‐dioxopiperidin‐4‐yl)acetyl thioester intermediate. Domains responsible for chain release (TE and PBS) are highlighted in green. Abbreviations for protein domains are displayed in the dashed box. c) Structures of glutarimide antibiotics produced by Streptomyces species. d) Structures of Streptomyces signalling molecules known or proposed to originate from a phosphorylated butenolide intermediate resulting from PBS‐catalysed condensation of DHAP with a 3‐ketoacyl‐ACP/CoA thioester.
Three conserved genes (smdFGH, chxBCD, ltmHCD and mgsHCD in the 9‐methylstreptimidone, cycloheximide, lactimidomycin and migrastatin BGCs, respectively) are proposed to be involved in the creation of a common malonamyl thioester starter unit for the PKSs that assembles these metabolites (Figure 1 a).[ 12 , 15 , 16 , 17 , 18 , 19 , 20 ] These genes encode: an acyltransferase (AT; SmdF/ChxB/LtmH/MgsH) that is proposed to malonylate an acyl carrier protein (ACP; SmdG/ChxC/LtmC/MgsC); and an asparagine synthetase homologue (SmdH/ChxD/LtmD/MgsD), which is hypothesised to convert the resulting malonyl thioester to a malonamyl thioester. The first module in each PKS (located in the SmdI, ChxE, LtmE and MgsE subunits) contains seven conserved domains that are proposed to elaborate the malonamyl starter unit into a common 2‐(2,6‐dioxopiperidin‐4‐yl)acetyl thioester intermediate (Figure 1 b). [25]
The gdsB, gdsC and gdsD genes in the cryptic B. gladioli BGC encode homologues of the three proteins hypothesised to create the malonamyl thioester starter unit (Figure 1 b). Moreover, the first module of the PKS encoded by this cluster (situated in the GdsE subunit) has an identical seven‐domain architecture to the corresponding modules of the PKSs that assemble the Streptomyces glutarimides. However, the domain architecture of subsequent modules in the B. gladioli PKS differs significantly from the Streptomyces glutarimide assembly lines and a unique AfsA‐like domain (pfam03756—identified by a conserved domain search) is appended to the C‐terminus of the final PKS module (Figure 1 b). AfsA catalyses the condensation of dihydroxyacetone phosphate (DHAP) with a β‐keto thioester to form a phosphorylated butenolide intermediate in the biosynthesis of A‐factor, a signalling molecule that controls morphological differentiation and antibiotic production in Streptomyces griseus (Figure 1 d). [26] AfsA homologues are proposed to catalyse analogous reactions in the biosynthesis of other γ‐butyrolactones (GBLs), such as the Streptomyces coelicolor butyrolactones and the Streptomyces virginiae butanolides (Figure 1 d).[ 27 , 28 ] Members of this enzyme family are also involved in the biosynthesis of other classes of Streptomyces signalling molecules, such as 2‐alkyl‐4‐hydroxymethylfuran‐3‐carboxylic acids (AHFCAs), typified by the methylenomycin furans, and 2‐alkyl‐4‐hydroxy‐3‐methylbutenolides (AHMBs), exemplified by the Streptomyces rochei butenolides and the Streptomyces ansachromogenes butenolides (Figure 1 d).[ 29 , 30 , 31 ] Incorporation experiments with stereospecifically 13C‐labelled glycerols indicate that an analogous phosphorylated butenolide to that formed by AfsA in A‐factor biosynthesis is an intermediate in the biosynthesis of the methylenomycin furans. [32] This suggests that the AfsA family of enzymes involved in GBL, AHFCA and AHMB assembly all produce analogous phosphorylated butenolides that are diversified by subsequent biosynthetic enzymes (Figure 1 d).
PKSs typically have a thioesterase (TE) domain appended to the C‐terminus of the last module, which catalyses release of the fully assembled polyketide chain via hydrolysis or macrocyclisation, but several other chain release mechanisms are known.[ 33 , 34 , 35 , 36 ] Based on the role played by AfsA family enzymes in Streptomyces signalling molecule biosynthesis, we hypothesised that the AfsA‐like domain appended to the C‐terminus of the last module in the cryptic B. gladioli PKS releases the fully assembled polyketide chain by condensing it with DHAP to form a phosphorylated butenolide (Figure 1 d). Accordingly, we designated this new type of chain release enzyme a phosphorylated butenolide synthase (PBS) domain. Overall, our in silico analyses indicated that the cryptic B. gladioli PKS assembles a novel glutarimide‐containing polyketide with significant structural differences to the glutarimide antibiotics assembled by Streptomyces species.
Isolation and Structure Elucidation of Metabolic Products of the Cryptic PKS
We originally identified the BGC encoding the cryptic trans‐AT PKS in B. gladioli BCC0238. However, due to difficulties with creating in‐frame deletions in this strain, we searched the genomes of other B. gladioli strains to see if they contain this BGC. Another CF isolate, B. gladioli BCC1622, which is more amenable to genetic manipulation, [11] was also found to contain the cluster. UHPLC‐ESI‐Q‐TOF‐MS analysis of an ethyl acetate extract from a culture of B. gladioli BCC1622, grown for 3 days on a minimal agar medium containing glycerol as the sole carbon source, identified a metabolite with the molecular formula C23H37NO5 (Figure S3).
The planar structure of this metabolite (5) was elucidated using 1H, 13C, COSY, HSQC and HMBC NMR experiments (Figure 2 a, Figure S4–S8, Table S1). HMBC correlations between H‐3 and C‐1/C‐1′ and the exchangeable NH proton and C‐2/C‐2′ confirmed the presence of a 2, 6‐piperidinedione, and the chemical shift values for this moiety were in good agreement with the literature.[ 18 , 19 , 37 , 38 ] Two networks of COSY correlations established the structures of the C‐2/C‐2′ to C‐6 and C‐8 to C‐18 regions of the molecule, and HMBC correlations further confirmed the locations of the C‐8 Me group and the C‐9/C‐10 double bond. HMBC correlations also showed that C‐6 and C‐8 are connected via a keto group and that a methyl ketone is attached to C‐18. Based on a 3 J HH coupling constant of 15 Hz, the C‐9/C‐10 double bond was assigned the E configuration.
Figure 2.
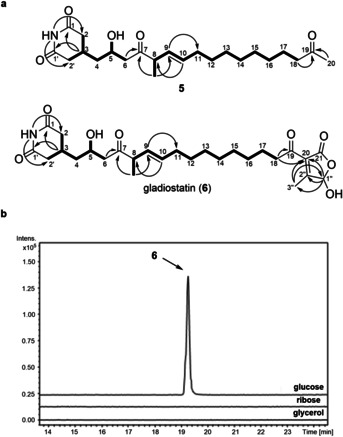
Identification and structure elucidation of metabolic products of the cryptic trans‐AT PKS in B. gladioli BCC1622. a) Structure elucidation of degradation product (5) (top) and gladiostatin (6) (bottom). COSY and key HMBC correlations observed for each compound are indicated by bold lines and arrows, respectively. b) Extracted ion chromatograms at m/z=506.27±0.02, (corresponding to the [M+H]+ ion of gladiostatin) from UHPLC‐ESI‐Q‐TOF‐MS analysis of ethyl acetate extracts from B. gladioli BCC1622 grown on a minimal medium containing different carbon sources.
Although compound 5 contains the anticipated 2, 6‐piperidinedione, it lacks the butenolide predicted to be installed by the PBS domain (Figure 1 d). We thus postulated that this compound results from degradation of the true metabolic product of the BGC. A range of carbon sources (glycerol, glucose, ribose) and growth periods were explored to investigate whether other metabolic products of the BGC could be identified. UHPLC‐ESI‐Q‐TOF‐MS analysis of an ethyl acetate extract from a 24 hour culture on a minimal medium containing glucose as the sole carbon source identified a new metabolite with the molecular formula C27H39NO8 (Figure 2 b and Figure S9). A time course showed that production of this metabolite peaked at 20 hours and fell off rapidly over more protracted growth periods (Figure S10). After 55 hours, small amounts of compound 5 could be detected, consistent with this being a degradation product of the new metabolite.
Comparison of the molecular formulae for compound 5 and the newly identified metabolite showed the latter contains four additional carbon atoms, and three additional hydrogen and oxygen atoms. The planar structure of the new metabolite (6) was elucidated using 1H, 13C, COSY, HSQC and HMBC NMR experiments (Figure 2 a, Figure S11–S15, Table S2). The 13C NMR spectrum of 6 lacked the signal due to methyl ketone in 5 and contained additional signals assigned to a carbonyl group (C‐21), two fully substituted alkene carbons (C‐20 and C‐2′′), a methyl group (C‐3′′) and a hemiacetal (C‐1′′). HMBC correlations between the C‐3′′ protons and C‐1′′/C‐20 and the C‐1′′ proton and C‐2′′/C‐21 led us to propose that this molecule contains a 2‐subsituted 4‐hydroxy‐3‐methylbutenolide. The NMR data for this moiety are similar to those reported for the AHMBs isolated from S. rochei and S. ansachromogenes (Figure 1 d).[ 30 , 31 ] An HMBC correlation between the C‐18 protons and C‐20 established the connectivity between the butenolide and the rest of the structure. The juxtaposition of a 2‐acyl‐4‐hydroxy‐3‐methylbutenolide and 2, 6‐piperidinedione in this molecule, coupled with the time course data for its production, led us to conclude that this compound, which we named gladiostatin, is the true metabolic product of the cryptic glutarimide‐like BGC in B. gladioli BCC1622 and BCC0238. Gladiostatin (6) is likely degraded to 5 via conjugate addition of water to the butenolide, followed by ring opening, decarboxylation and retro‐Aldol cleavage (Figure S16).
Biological Activity of Gladiostatin
Initially, we tested the activity of gladiostatin (6) against representative members of the ESKAPE panel of bacterial pathogens, Candida albicans and Saccharomyces cerevisiae. While no activity was detected against the bacterial pathogens or C. albicans at concentrations up to 64 μg mL−1, gladiostatin was found to be active against S. cerevisiae, with an MIC of 4 μg mL−1 (Table 1).
Table 1.
Antimicrobial activity of gladiostatin (MIC=minimum inhibitory concentration).
|
Strain |
MIC [μg mL−1] |
|---|---|
|
Gram‐negative Bacteria |
|
|
Klebsiella pneumonia DSM 26371 |
>64 |
|
Acinetobacter baumannii DSM25645 |
>64 |
|
Pseudomonas aeruginosa DSM29239 |
>64 |
|
Enterobacter cloacae DSM 16690 |
>64 |
|
Gram‐positive Bacteria |
|
|
Enterococcus faecium DSM25390 |
>64 |
|
Staphylococcus aureus DSM21979 |
>64 |
|
Fungi |
|
|
Candida albicans SC 5314 |
>64 |
|
Yeast |
|
|
S. cerevisiae W303‐1 a |
4 |
Prompted by reports that several glutarimides have anti‐tumour activity,[ 23 , 24 ] we investigated the activity of gladiostatin (6) against a range of cancer cell lines (Table 2). It was found to be active against ovarian, pancreatic and colon cancer cell lines (Table 2). These values are in the same range as those reported for cycloheximide (1), migrastatin (3) and lactimidomycin (4) against various other cell lines (Table S6).[ 17 , 19 , 38 ] Interestingly, gladiostatin (6) was found to be inactive against the A549 lung cancer cell line, indicating it may exhibit some selectivity.
Table 2.
Anticancer activity of gladiostatin.
|
|
Gladiostatin (6) |
|---|---|
|
Cancer cell line |
IC50 [μΜ] |
|
A2780 (ovarian) |
0.24±0.03 |
|
A549 (lung) |
no activity |
|
MiaPaca2 (pancreatic) |
0.57±0.06 |
|
HCTT116 P53 ‐/‐ (colon) |
0.82±0.04 |
|
SKOV3 (ovarian) |
0.6±0.1 |
|
PEA1 (ovarian) |
1.4±0.1 |
Some glutarimides have also been reported to inhibit tumour cell migration.[ 23 , 24 ] Thus, we used a wound‐healing assay to test whether gladiostatin (6) can inhibit the migration of A2780 ovarian cancer cells. Strong suppression of cell migration was observed after 24 h exposure to 240 nM gladiostatin (Figure 3, Figure S17).
Figure 3.
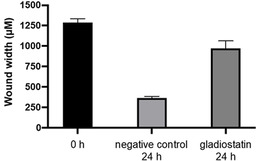
Inhibition of A2780 ovarian tumour cell migration by gladiostatin. The bar chart compares wound‐widths after 0 and 24 h in the absence of gladiostatin with the wound width after 24 h in the presence of 240 nM gladiostatin.
Proposed Pathway for Gladiostatin Biosynthesis.
To establish that the gds cluster directs the biosynthesis of gladiostatin (6), gdsE, which encodes the first polyketide synthase subunit, was disrupted in B. gladioli BCC1622 by insertional mutagenesis. LC‐MS comparison of extracts from the wild type and mutant strains confirmed that gladiostatin (6) production is abolished in the mutant (Figure 4).
Figure 4.
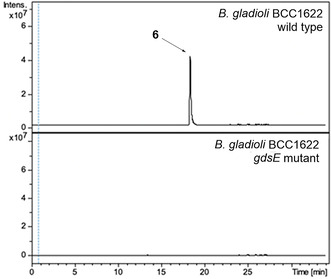
Insertional inactivation of gdsE abolishes gladiostatin production in B. gladioli BCC1622. Extracted ion chromatograms at m/z 506.27±0.02 (corresponding to [M+H]+ for gladiostatin) from UHPLC‐ESI‐Q‐TOF‐MS analysis of ethyl acetate extracts of B. gladioli BCC1622 (top) and the gdsE mutant (bottom).
The early stages of gladiostatin (6) biosynthesis appear to parallel the assembly of glutarimides in Streptomyces species. GdsB and GdsD are proposed to create a GdsC‐bound malonamyl thioester starter unit, which is elaborated to a 2‐(2,6‐dioxopiperidin‐4‐yl)acetyl thioester intermediate by the first PKS module (Figure 5). Modules 2, 3 and 4 have identical domain architectures in all glutarimide PKSs (Figure 1 b). In the Streptomyces assembly lines, a single MT domain in module 4 seems to be responsible for methylating the α‐carbon of the β‐keto‐ACP thioester intermediate generated by the ketosynthase (KS) domain in both modules 3 and 4 of the PKSs, installing methyl branches at C‐8 and C‐10 of the resulting glutarimides. In contrast, the MT domain in module 4 of the gladiostatin PKS appears to methylate only the β‐keto thioester intermediate attached to the module 3 ACP domain (Figure 5). Consequently, gladiostatin (6) has a methyl branch at C‐8, but not C‐10. Additional studies are needed to understand how PKSs with seemingly identical architectures are able to produce the distinct C‐methylation patterns observed in gladiostatin (6) and the Streptomyces glutarimides.
Figure 5.
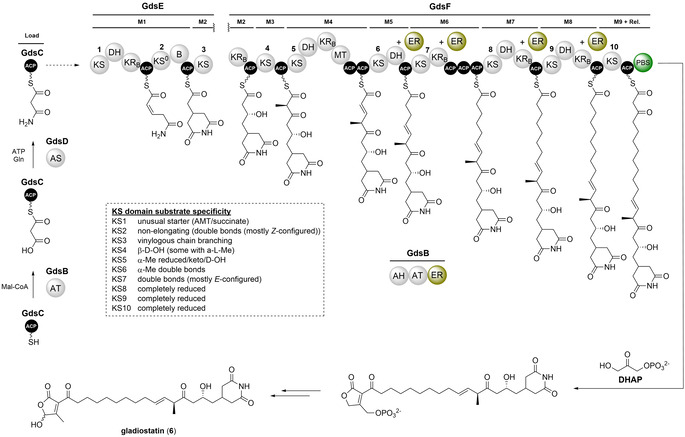
Proposed pathway for gladiostatin biosynthesis. Domain and module organization of the gladiostatin trans‐AT PKS showing the proposed structure of each ACP‐bound thioester intermediate. KS domains have been numbered sequentially, and the TransATor [42] predictions for their acyl‐ACP substrates are shown in the dashed box. Modules are labelled M1, M2 etc. The A/B subscripts denote the predicted stereospecificity of KR domains. The trans‐acting ER domain (gold) of GdsB is predicted to reduce enoyl thioesters attached to the ACP domains in modules 5, 6, 7 and 8. Domain abbreviations are as defined in Figure 1. The putative functions of the proteins encoded by the gladiostatin biosynthetic gene cluster are listed in Table S3. The stereochemistry of C‐5 and C‐8 is hypothesised to be the same as the corresponding stereocentres in lactimidomycin. Comparative sequence analysis of the module 2 KR domain predicts that it produces an R‐configured alcohol (Table S5), consistent with this hypothesis.
The domain architecture of modules 5–9 of the gladiostatin PKS diverges significantly from the corresponding modules in the Streptomyces glutarimide assembly lines. Indeed the C‐terminus of module 4 appears to be a branch point in all glutarimide‐producing PKSs (Figure 1 b), and could be a recombination hotspot. Based on comparisons between the domain architecture and the predicted biosynthetic intermediates, module 5 of the gladiostatin PKS appears to lack both a ketoreductase (KR) domain (which is required for installation of the β‐hydroxy group) and an enoyl reductase (ER) domain (required to saturate the double bond introduced by the dehydratase (DH) domain). Similarly, module 6 lacks a DH domain and modules 6, 7 and 8 lack ER domains, suggesting a high degree of non‐linear programming in the gladiostatin PKS, which is a common feature of glutarimide assembly lines (Figure 1). [39]
While it is unclear which domains are responsible for the keto reduction and dehydration reactions in modules 5 and 6, respectively, in silico analysis of the gladiostatin BGC identified two genes encoding putative reductases that could function as trans‐acting ERs in modules 5–8. The gdsB gene encodes a tri‐domain protein with acyl hydrolase (AH) and AT domains fused to a flavin‐dependent ER domain and gdsH encodes an NAD(P)H‐dependent oxidoreductase. We therefore propose that one, or both, of these reductases catalyse enoyl reduction in modules 5, 6, 7 and 8 of the PKS. A recently reported co‐evolutionary categorisation of trans‐AT PKS ACP domains supports this hypothesis, indicating that the module 6, 7 and 8 ACP domains in the gladiostatin PKS clade with ACP domains in modules with a similar architecture from other assembly lines (i.e. KS‐DH‐KR‐ACP and a trans‐acting ER). [40] Moreover, the TransATor software predicts that the KS domains in modules 7, 8 and 9 are selective for an α,β‐saturated thioester intermediate. [41] However, this software also predicts that the module 6 KS domain prefers a α,β‐unsaturated thioester, highlighting potential limitations of such predictive bioinformatics analyses (Figure 5, Figure S18 and S19; Table S4 and S7).
The most striking difference between the gladiostatin PKS and the other glutarimide assembly lines is the mechanism for polyketide chain release. All of the Streptomyces glutarimide PKSs are proposed to use TE domains that catalyse hydrolysis or macrolactonisation. In contrast, the gladiostatin PKS appears to employ a PBS domain to catalyse condensation of the β‐ketothioester attached to the last module of the PKS with DHAP. The resulting phosphorylated butenolide is proposed to undergo rearrangement and dephosphorylation to afford a 2‐acyl‐4‐hydroxy‐3‐methylbutenolide (Figure 5). This hypothesis is consistent with the observed increase in gladiostation production levels when glucose is used as the carbon source, because DHAP is an intermediate in glycolysis. To our knowledge, there is no precedent for PKS chain release by a PBS domain. However, PBSs are known to catalyse the condensation of DHAP with a range of β‐ketothioesters in the biosynthesis of several distinct classes of Streptomyces signalling molecule (Figure 1 d). These include the SRBs and the SABs, which contain a similar butenolide moiety to gladiostatin (6). In addition to the PBS (SabA), a phosphatase (SabP) and a ketoreductase (SabD) are known to be required for SAB biosynthesis, but the mechanism for elaboration of the putative phosphorylated butenolide produced by SabA to the 2‐akyl‐4‐hydroxy‐3‐methylbutenolide remains to be elucidated. The gdsA and gdsG genes in the gladiostatin BGC encode putative phosphatases that could play a similar role to SabP in the biosynthesis of the SABs (Table S3).
In vitro Reconstitution of Chain Release from the Gladiostatin PKS
To validate the proposed role of the PBS domain in gladiostatin (6) biosynthesis, we investigated its catalytic activity using simplified mimics of the fully assembled polyketide chain. A synthetic gene encoding the C‐terminal ACP and PBS domains of GdsF was used to overproduce the ACP‐PBS di‐domain in E. coli as an N‐terminal His8‐fusion, which was purified to homogeneity using immobilised metal‐ion affinity chromatography. The identity of the purified protein was confirmed by ESI‐Q‐TOF‐MS analysis (Figure S20).
The ability of the PBS domain to offload a 3‐ketothioester from the PKS was examined by loading a 3‐ketobutyryl mimic of the fully assembled polyketide chain onto the apo‐ACP domain using the promiscuous phosphopantetheinyl transferase Sfp (Figure S21). The mass of the protein decreased by 86 Da when DHAP was added (Figure S21), consistent with cleavage of the 3‐ketobutyryl group from the ACP domain.
Incubation of the apo‐ACP‐PBS di‐domain with the N‐acetylcystetamine (NAC) thioester of 3‐ketooctanoate and DHAP, followed by treatment with shrimp alkaline phosphatase yielded a product, absent from the negative control, that gave rise to ions with m/z=213.11 and m/z=235.09 (corresponding to the [M+H]+ and [M+Na]+, respectively, of the 2‐acyl‐3‐hydroxymethyl butenolide (8)) in UHPLC‐ESI‐Q‐TOF‐MS analyses (Figure 6). A synthetic standard of 8 had the same retention time and MS/MS fragmentation pattern as the product of the enzymatic reaction (Figure 6).
Figure 6.
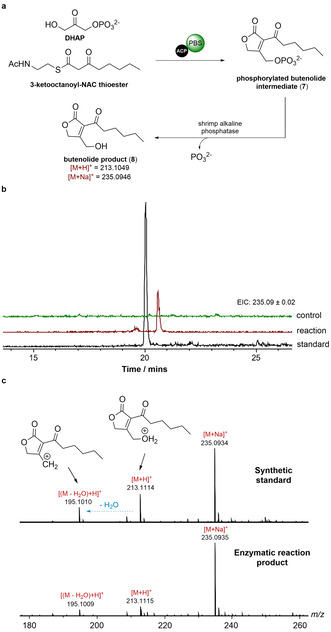
In vitro reconstitution of chain release by the PBS domain. a) Condensation of DHAP and the NAC thioester of 3‐ketooctanoate by the purified recombinant GdsF ACP‐PBS di‐domain and dephosphorylation of the resulting phosphorylated butenolide (7) using shrimp alkaline phosphatase. b) Extracted‐ion chromatograms at m/z=235.0925 (corresponding to [M+Na]+ for butenolide 8) from UHPLC‐ESI‐Q‐TOF‐MS comparisons of the enzymatic reaction cascade and a synthetic standard of 8. The ACP‐PBS di‐domain was omitted from the control reaction. c) Comparison of MS/MS spectra for the synthetic standard of butenolide 8 ([M+H]+=213.11) and the product of the enzymatic reaction cascade.
Conclusion
The discovery of gladiostatin (6) as the product of a cryptic trans‐AT PKS assembly line in B. gladioli BCC0238 and BCC1622 further expands the already rich specialised metabolic repertoire of these and related CF isolates.[ 6 , 9 , 11 ] As the first glutarimide antibiotic to be isolated from a Gram‐negative bacterium, gladiostatin differs significantly from other members of this family, all of which are produced by Streptomyces species. While some of these structural differences (such as the fully saturated C‐11 to C‐18 chain and the 2‐acyl‐4‐hydroxy‐3‐methylbutenolide) are reflected by alterations in PKS architecture, others (e.g. the lack of a C‐10 methyl group) are not. The discovery of gladiostatin thus offers a golden opportunity to develop a better understanding of the role played by horizontal gene transfer in trans‐AT PKS evolution, which could facilitate biosynthetic engineering approaches to polyketide structural diversification.
In vitro characterisation of the AfsA‐like PBS domain appended to the C‐terminus of the gladiostatin PKS shows that it releases 3‐keto thioesters from the upstream ACP domain by condensing them with DHAP. This constitutes a new mechanism for polyketide chain release, which could prove to be a valuable addition to the synthetic biology toolbox. The phosphorylated butenolide product of the PBS domain is analogous to the intermediate in A‐factor biosynthesis produced by AfsA. The same kind of intermediate has been shown to be formed by MmfL (another AfsA homologue) in the biosynthesis of the methylenomycin furans. [42] Thus, it appears that phosphorylated 2‐acyl‐3‐hydroxymethylbutenolide intermediates are involved in the biosynthesis of structurally diverse natural products, including GBLs, AHFCAs, AHMBs and gladiostatin. However, with the exception of GBLs, further work is needed to understand the mechanisms by which these intermediates get elaborated into the final metabolic products.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by grants from the BBSRC (Grant References BB/L021692/1 and BB/L023342/1) to E.M. and G.L.C. The Dionex 3000RS/Bruker MaXis Impact and Dionex 3000RS/Bruker MaXis II instruments used in this work were purchased with grants to G.L.C. from the BBSRC (BB/K002341/1 and BB/M017982/1). I.T.N. was supported by the BBSRC through the Midlands Integrative Bioscience Doctoral Training Partnership (BB/M01116X/1). M.J. is supported by a BBSRC Future Leader Fellowship (BB/R012121/1). Y.D. was supported by a grant from the MRC (MR/N501839/1 to G.L.C.). J.M. was supported by a Marie Sklodowska‐Curie Fellowship (Contract No. 656067). G.L.C. is the recipient of a Wolfson Research Merit Award from the Royal Society (WM130033). The authors thank Dr. Shanshan Zhou for providing the N‐acetylcysteamine thioester of 3‐ketooctanoate and the synthetic standard of the 2‐hexanoyl‐3‐hydroxymethylbutenolide used in the enzymatic assays.
I. T. Nakou, M. Jenner, Y. Dashti, I. Romero-Canelón, J. Masschelein, E. Mahenthiralingam, G. L. Challis, Angew. Chem. Int. Ed. 2020, 59, 23145.
References
- 1. Raffatellu M., Nat. Med. 2018, 24, 1097–1103. [DOI] [PubMed] [Google Scholar]
- 2. Moebius N., Ross C., Scherlach K., Rohm B., Roth M., Hertweck C., Chem. Biol. 2012, 19, 1164–1174. [DOI] [PubMed] [Google Scholar]
- 3. Liu X., Biswas S., Berg M. G., Antapli C. M., Xie F., Wang Q., Tang M.-C., Tang G.-L., Zhang L., Dreyfuss G., et al., J. Nat. Prod. 2013, 76, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. He H., Ratnayake A. S., Janso J. E., He M., Yang H. Y., Loganzo F., Shor B., O'Donnell C. J., Koehn F. E., J. Nat. Prod. 2014, 77, 1864–1870. [DOI] [PubMed] [Google Scholar]
- 5. Mahenthiralingam E., Song L., Sass A., White J., Wilmot C., Marchbank A., Boaisha O., Paine J., Knight D., Challis G. L., Chem. Biol. 2011, 18, 665–677. [DOI] [PubMed] [Google Scholar]
- 6. Song L., Jenner M., Masschelein J., Jones C., Bull M. J., Harris S. R., Hartkoorn R. C., Vocat A., Romero-Canelon I., Coupland P., et al., J. Am. Chem. Soc. 2017, 139, 7974–7981. [DOI] [PubMed] [Google Scholar]
- 7. Esmaeel Q., Pupin M., Jacques P., Leclère V., Environ. Sci. Pollut. Res. 2018, 25, 29794–2980. [DOI] [PubMed] [Google Scholar]
- 8. Esmaeel Q., Pupin M., Kieu N. P., Chataigné G., Béchet M., Deravel J., Krier F., Höfte M., Jacques P., Leclère V., Microbiologyopen 2016, 5, 512–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jenner M., Jian X., Dashti Y., Masschelein J., Hobson C., Roberts D. M., Jones C., Harris S., Parkhill J., Raja H. A., et al., Chem. Sci. 2019, 10, 5489–5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dose B., Niehs S. P., Scherlach K., Flórez L. V., Kaltenpoth M., Hertweck C., ACS Chem. Biol. 2018, 13, 2414–2420. [DOI] [PubMed] [Google Scholar]
- 11. Dashti Y., Nakou I. T., Mullins A. J., Webster G., Jian X., Mahenthiralingam E., Challis G. L., Angew. Chem. Int. Ed. 2020, 10.1002/anie.202009110; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 10.1002/ange.202009110. [DOI] [Google Scholar]
- 12. Yin M., Yan Y., Lohman J. R., Huang S. X., Ma M., Zhao G. R., Xu L. H., Xiang W., Shen B., Org. Lett. 2014, 16, 3072–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang S. X., Yu Z., Robert F., Zhao L. X., Jiang Y., Duan Y., Pelletier J., Shen B., J. Antibiot. 2011, 64, 163–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Allen M. S., Becker A. M., Rickards R. W., Aust. J. Chem. 1976, 29, 673–679. [Google Scholar]
- 15. Wang B., Song Y., Luo M., Chen Q., Ma J., Huang H., Ju J., Org. Lett. 2013, 15, 1278–1281. [DOI] [PubMed] [Google Scholar]
- 16. Ju J., Lim S. K., Jiang H., Shen B., J. Am. Chem. Soc. 2005, 127, 1622–1623. [DOI] [PubMed] [Google Scholar]
- 17. Takemoto Y., Nake K., Kawatani M., Takahashi Y., Naganawa H., Imoto M., J. Antibiot. 2001, 54, 1104–1107. [DOI] [PubMed] [Google Scholar]
- 18. Lim S. K., Ju J., Zazopoulos E., Jiang H., Seo J. W., Chen Y., Feng Z., Rajski S. R., Farnet C. M., Shen B., J. Biol. Chem. 2009, 284, 29746–29756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sugawara K., Nlshiyama Y., Toda S., Komiyama N., Hatori M., Moriyama T., Sawada Y., Kamei H., Konishi M., Oki T., J. Antibiot. 1992, 45, 1433–1441. [DOI] [PubMed] [Google Scholar]
- 20. Seo J. W., Ma M., Kwong T., Ju J., Lim S. K., Jiang H., Lohman J. R., Yang C., Cleveland J., Zazopoulos E., et al., Biochemistry 2014, 53, 7854–7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gaul C., Njardarson J. T., Shan D., Dorn D. C., Da Wu K., Tong W. P., Huang X. Y., Moore M. A. S., Danishefsky S. J., J. Am. Chem. Soc. 2004, 126, 11326–11337. [DOI] [PubMed] [Google Scholar]
- 22. Garreau De Loubresse N., Prokhorova I., Holtkamp W., Rodnina M. V., Yusupova G., Yusupov M., Nature 2014, 513, 517–522. [DOI] [PubMed] [Google Scholar]
- 23. Schneider-Poetsch T., Ju J., Eyler D. E., Dang Y., Bhat S., Merrick W. C., Green R., Shen B., Liu J. O., Nat. Chem. Biol. 2010, 6, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ju J., Rajski S. R., Lim S. K., Seo J. W., Peters N. R., Hoffmann F. M., Shen B., J. Am. Chem. Soc. 2009, 131, 1370–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bretschneider T., Heim J. B., Heine D., Winkler R., Busch B., Kusebauch B., Stehle T., Zocher G., Hertweck C., Nature 2013, 502, 124–128. [DOI] [PubMed] [Google Scholar]
- 26. Kato J. Y., Funa N., Watanabe H., Ohnishi Y., Horinouchi S., Proc. Natl. Acad. Sci. USA 2007, 104, 2378–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hsiao N.-H., Söding J., Linke D., Lange C., Hertweck C., Wohlleben W., Takano E., Microbiology 2007, 153, 1394–1404. [DOI] [PubMed] [Google Scholar]
- 28. Lee Y. J., Kitani S., Nihira T., Microbiology 2010, 156, 206–210. [DOI] [PubMed] [Google Scholar]
- 29. Corre C., Song L. J., O'Rourke S., Chater K. F., Challis G. L., Proc. Natl. Acad. Sci. USA 2008, 105, 17510–17515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arakawa K., Tsuda N., Taniguchi A., Kinashi H., ChemBioChem 2012, 13, 1447–1457. [DOI] [PubMed] [Google Scholar]
- 31. Wang W., Zhang J., Liu X., Li D., Li Y., Tian Y., Tan H., J. Biol. Chem. 2018, 293, 20029–20040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Corre C., Haynes S. W., Malet N., Song L., Challis G. L., Chem. Commun. 2010, 46, 4079–4081. [DOI] [PubMed] [Google Scholar]
- 33. Fischbach M. A., Walsh C. T., Chem. Rev. 2006, 106, 3468–3496. [DOI] [PubMed] [Google Scholar]
- 34. Du L., Lou L., Nat. Prod. Rep. 2010, 27, 255–278. [DOI] [PubMed] [Google Scholar]
- 35. Mo S., Sydor P. K., Corre C., Alhamadsheh M. M., Stanley A. E., Haynes S. W., Song L., Reynolds K. A., Challis G. L., Chem. Biol. 2008, 15, 137–148. [DOI] [PubMed] [Google Scholar]
- 36. Lin S., Van Lanen S. G., Shen B., Proc. Natl. Acad. Sci. USA 2009, 106, 4183–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Larsen B. J., Sun Z., Lachacz E., Khomutnyk Y., Soellner M. B., Nagorny P., Chem. Eur. J. 2015, 21, 19159–19167. [DOI] [PubMed] [Google Scholar]
- 38. Micoine K., Persich P., Llaveria J., Lam M. H., Maderna A., Loganzo F., Fürstner A., Chem. Eur. J. 2013, 19, 7370–7383. [DOI] [PubMed] [Google Scholar]
- 39. Helfrich E. J. N., Piel J., Nat. Prod. Rep. 2016, 33, 231–316. [DOI] [PubMed] [Google Scholar]
- 40. Vander Wood D. A., Keatinge-Clay A. T., Proteins Struct. Funct. Bioinf. 2018, 86, 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Helfrich E. J. N., Ueoka R., Dolev A., Rust M., Meoded R. A., Bhushan A., Califano G., Costa R., Gugger M., Steinbeck C., et al., Nat. Chem. Biol. 2019, 15, 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou S., Malet N. R., Song L., Corre C., Challis G. L., Chem. Commun. 2020, https://www.doi.org/10.1039/D0CC05658H. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary