

SUMMARY
Trifoliate orange (Poncirus trifoliata), a deciduous close relative of evergreen Citrus, has important traits for citrus production, including tolerance/resistance to citrus greening disease (Huanglongbing, HLB) and other major diseases, and cold tolerance. It has been one of the most important rootstocks, and one of the most valuable sources of resistance and tolerance genes for citrus. Here we present a high‐quality, chromosome‐scale genome assembly of P. trifoliata. The 264.9‐Mb assembly contains nine chromosomal pseudomolecules with 25 538 protein‐coding genes, covering 97.2% of the estimated gene space. Comparative analyses of P. trifoliata and nine Citrus genomes revealed 605 species‐specific genes and six rapidly evolving gene families in the P. trifoliata genome. Poncirus trifoliata has evolved specific adaptation in the C‐repeat/DREB binding factor (CBF)‐dependent and CBF‐independent cold signaling pathways to tolerate cold. We identified candidate genes within quantitative trait loci for HLB tolerance, and at the loci for resistance to citrus tristeza virus and citrus nematode. Genetic diversity analysis of Poncirus accessions and Poncirus/Citrus hybrids shows a narrow genetic base in the US germplasm collection, and points to the importance of collecting and preserving more natural genetic variation. Two phenotypically divergent Poncirus accessions are found to be clonally related, supporting a previous conjecture that dwarf Flying Dragon originated as a mutant of a non‐dwarfing type. The high‐quality genome reveals features and evolutionary insights of Poncirus, and it will serve as a valuable resource for genetic, genomic and molecular research and manipulation in citrus.
Keywords: citrus, cold tolerance, disease resistance, evolution, genome, Huanglongbing, Poncirus
Significance Statement
A high‐quality, chromosome‐scale reference genome of trifoliate orange has enabled a comprehensive comparative genomics analysis of Citrus‐related genomes, discovery of gene families under expansion or positive selection, identification of gene families associated with important disease resistance and cold tolerance, and localization of candidate genes for tolerance to Huanglongbing, the most destructive disease in citrus. This genome will be a valuable resource for genomic research and manipulation in citrus and related plants.
INTRODUCTION
Citrus is one of the most economically important fruit crops grown in more than 114 countries (Talon and Gmitter, 2008). In 2017, the world citrus acreage was 9.3 million hectares, and citrus production was 146.6 million tons (FAO statistics, http://www.fao.org/faostat/en/). The most commonly cultivated citrus species include sweet orange (Citrus × sinensis), mandarin (C. reticulata), grapefruit (C. × paradisi), pummelo (C. maxima) and lemon (C. × limon). Citrus is mainly consumed as fresh fruit or juice, which provide abundant vitamin C and other phytochemicals for human nutrition. However, commercial production of citrus is confronted with many challenges from biotic and abiotic stresses. Currently, the most devastating disease in citrus is Huanglongbing (HLB) or citrus greening disease, which is presumably caused by the bacterium Candidatus Liberibacter asiaticus (CLas) transmitted by the Asian citrus psyllid (Diaphorina citri, ACP; Bové, 2006). HLB has caused tremendous yield loss in citrus throughout the world (Bové, 2006). In Florida alone, the estimated economic loss due to HLB was at least $4 billion during the period from 2012−2013 to 2015−2016 (Trejo‐pech et al., 2018). Other diseases that limit citrus productivity include several species of Phytophthora and nematodes, and citrus tristeza virus (CTV; Radhika et al., 2008; Kamiri et al., 2018). Additionally, catastrophic economic losses caused by cold (freezes) have been recorded in several regions of the world (Sahin‐Cevik and Moore, 2006). Therefore, improving disease resistance and cold tolerance of cultivated citrus has been a major objective of major citrus breeding programs in the world.
Trifoliate orange (Poncirus trifoliata, 2n = 2x = 18) is a close relative of Citrus species, both belonging to the Rutaceae family. It was considered the only species in Poncirus until the discovery of Poncirus polyandra in the 1980s (Ding et al., 1984; Pang et al., 2003). Native to China, P. trifoliata has become naturalized in other regions of Asia, USA, Australia and Europe (Nesom, 2014). Poncirus trifoliata is sexually compatible with Citrus species (Spiegel‐Roy and Goldschmidt, 1996), but distinct from Citrus in many characteristics. It is deciduous (versus evergreen in Citrus), has three leaflets on each leaf and large thorns on the shoots, and produces bitter, inedible fruit containing many seeds. Poncirus trifoliata and its hybrids have been widely utilized as rootstocks in citrus production due to several advantageous features, including cold hardiness and resistance/tolerance to several major diseases and pests (Boava et al., 2011; Gong and Liu, 2013). Poncirus trifoliata is resistant to CTV, the most important viral pathogen of citrus (Kitajima et al., 1964; Tanaka et al., 1971; Garnsey and Barrett, 1987; Gmitter et al., 1996), and citrus nematodes (Tylenchulus semipenetrans; Ducharme, 1948; Cameron et al., 1954; Ling et al., 2000), and highly tolerant to Phytophthora species causing citrus seedling damping‐off, root and foot rot, and gummosis (Graham, 1990; Yang et al., 2001; Kamiri et al., 2018; Tian et al., 2018). A single dominant locus (Ctv) has been identified for the CTV resistance in P. trifoliata (Gmitter et al., 1996), and genetically and physically mapped (Deng et al., 2001; Yang et al., 2003). Poncirus trifoliata can provide high levels of tolerance to HLB and confers resistance to ACP (George and Lapointe, 2019). Poncirus trifoliata has been a major source of valuable genes for use in citrus sexual hybridization and somatic hybridization. It is expected to be a very important source of valuable genes for using cisgenic approaches to improving Citrus toward enhanced resistance to important biotic and abiotic stresses. Poncirus trifoliata also has considerable medicinal values, with anti‐inflammatory, anti‐tumor and anti‐anaphylactic activities (Yi et al., 2004; Rahman et al., 2015).
Another important feature of P. trifoliata is its cold tolerance. Poncirus trifoliata is extremely cold‐hardy and can survive −26°C (Spiegel‐Roy and Goldschmidt, 1996). This capability was probably acquired during its evolution in a cold habitat in northeastern Asia (Oustric et al., 2017). Other than P. trifoliata, Ichang papeda (Citrus ichangensis) has been considered the most cold‐hardy among evergreen Citrus species (Gmitter and Hu, 1990). Kumquats (Fortunella spp.) are considered cold tolerant within the Aurantioideae sub‐family of the citrus family (Krueger and Navarro, 2007). On the other hand, C. maxima and C. medica are the most cold‐sensitive species in Citrus (Nordby and Yelenosky, 1982; Crifò et al., 2011). Therefore, a comparative analysis of these cold‐tolerant and cold‐sensitive genomes would yield insights into the evolutionary signatures leading to their differences in cold tolerance. Studies in Arabidopsis have shown that the C‐repeat/DREB binding factor (CBF) pathway plays an important role in plant tolerance to cold stresses (Shi et al., 2018). When plants are under cold environmental conditions, CBF genes are induced rapidly to express and then activate the downstream target cold‐regulated (COR) genes or CBF regulons (Gilmour et al., 1998; Liu et al., 1998; Shi et al., 2018). However, CBFs only regulate less than 20% of the COR genes (Park et al., 2015; Shi et al., 2015). Therefore, CBF‐independent pathways are also critical for cold signaling regulation (Park et al., 2015). In P. trifoliata, a few genes have been well characterized and involved in cold tolerance, including bHLH (Huang et al., 2013), PRP (Peng et al., 2015), ICE1 (Huang et al., 2015) and ERF109 (Wang et al., 2018b). A previous transcriptome study revealed differentially expressed P. trifoliata genes (DEGs) in response to cold stresses at three time points (6, 24 and 72 h) after cold treatment (Wang et al., 2015). Overall, the molecular mechanisms of cold tolerance in P. trifoliata remain poorly understood. Thus, more studies are needed to fully exploit this unusual and striking feature of P. trifoliata, which makes it a perfect model and valuable source of candidate genes for improving cold tolerance in citrus. With the potential to confer to citrus these many favorable traits, including resistance to several major diseases and cold tolerance, a reference genome of P. trifoliata would greatly expedite the genetic and genomic research and improvement of citrus.
In order to explore the unique features of P. trifoliata, we performed genome sequencing and de novo assembly, and obtained a high‐quality, chromosome‐scale reference genome for this important species. We investigated the gene family evolution and positive selection in P. trifoliata by performing a comparative genomic analysis of the P. trifoliata genome and publicly available genomes of Citrus. To further understand Poncirus, we obtained and analyzed the genome sequences of P. polyandra, the only other species in Poncirus, and a number of important accessions of P. trifoliata and its hybrid cultivars. Moreover, we identified candidate genes in P. trifoliata by using previously identified quantitative trait loci (QTLs) for HLB tolerance, and loci for resistance to CTV and citrus nematodes. The high‐quality genome assembly has uncovered features and evolutionary insights of Poncirus, and it will serve as a valuable resource for genetic, genomic and molecular research and manipulation of citrus.
RESULTS
Genome sequencing and assembly
The HLB‐tolerant P. trifoliata accession DPI 50‐7 was selected as the reference genotype for whole‐genome sequencing and assembly. We sequenced the genome of DPI 50‐7 de novo combining ~140 × PacBio single‐molecule long‐read sequence reads with ~80 × Illumina shotgun sequence (Table S1). To achieve chromosome‐scale scaffolding, we used Hi‐C chromosome conformation capture data (Figure S1). The nine chromosomal pseudomolecules were numbered and oriented according to the convention of C. × clementina (Wu et al., 2014; Figure S2). These sequences represent a non‐redundant mosaic of the two haplotypes of diploid P. trifoliata, representing more than 99% of the assembled contigs (Figure 1). The largest scaffold was 42.7 Mb (Chr3), and the overall N50 length was 27.7 Mb, corresponding to the median chromosome length (Table 1). The GC content was estimated to be 33.9%.
Figure 1.
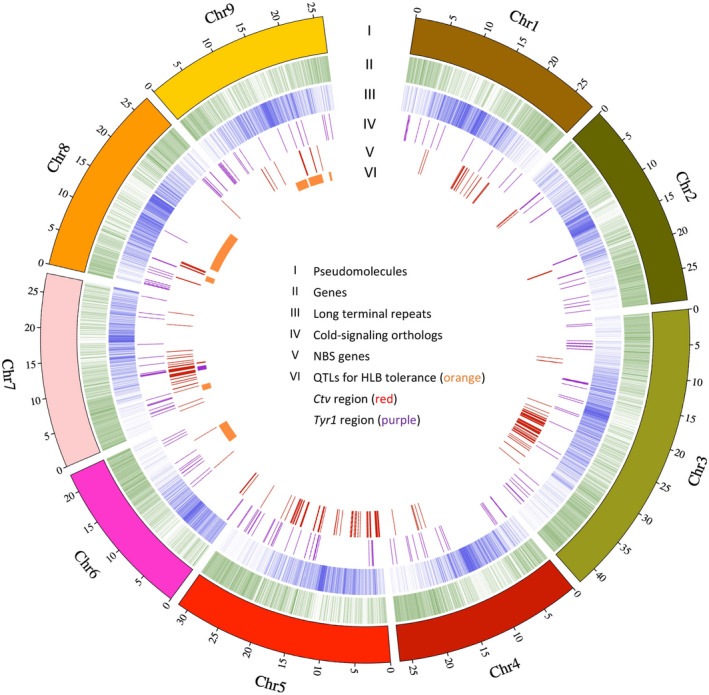
Characterization of the Poncirus trifoliata genome.
I. The nine assembled pseudomolecules (in Mb) corresponding to the nine chromosomes (Chr1−Chr9) of Citrus × clementina.
II. Locations of predicted gene models.
III. Locations of predicted long terminal repeat (LTR) transposable elements (TEs).
IV. Locations of predicted homologous cold signaling genes.
V. Locations of predicted nucleotide‐binding site (NBS)‐containing genes.
VI. Orange: locations of quantitative trait loci (QTLs) associated with Huanglongbing (HLB) tolerance and loci for resistance to citrus tristeza virus (Ctv) and citrus nematode (Tyr1). Red: Ctv region. Purple: Tyr1 region.
Table 1.
Summary statistics of the Poncirus trifoliata genome assembly and annotation
| Total size of genome assembly | 264.9 Mb |
| Chromosome number (2n) | 18 |
| Number of chromosomal pseudomolecules | 9 |
| Number of scaffolds | 152 |
| Longest scaffold | 42.7 Mb |
| Scaffold N50 length | 27.7 Mb |
| Number of contigs | 809 |
| Contig N50 length | 842.8 kb |
| GC content | 33.9% |
| Number of gene models | 25 538 |
| Number of genes with isoforms | 4398 |
| Mean transcript length | 1667 bp |
| Mean coding sequence length | 1268 bp |
| Mean exon length | 296 bp |
| Percentage of TEs | 42.6% |
TE, transposable element.
Genome annotation
A large proportion (42.6%) of the P. trifoliata genome was comprised of transposable elements (TEs). Long terminal repeats (LTRs) were the most predominant repeats (24.62%), including LTR/Gypsy (9.42%) and LTR/Copia (6.37%; Table S2). Other types of repeats accounted for smaller proportions of the genome, including DNA transposons (10.40%), LINE (2.57%) and SINE (0.25%). An integrated strategy combining ab initio‐ and homology‐based methods was applied to predict gene models in the P. trifoliata genome. In total, we identified 25 538 protein‐coding genes with 33 230 transcripts. The transcripts for primary gene models had a mean length of 1667 bp, a mean coding sequence length of 1268 bp, and a mean exon length of 296 bp (Table 1). In addition, 4398 genes had spliced isoforms with an average of 2.7 isoforms per gene. To evaluate the completeness of our genome, the predicted protein sequences were compared with the 1440 eukaryotic genes in the BUSCO OrthoDB9 embryophyta dataset and obtained a completeness of 97.2%. The completeness of this genome is currently the highest compared with 10 other publicly available Citrus genomes (Table S3). The high completeness and N50 length support the quality of this genome assembly.
Identification of cold signaling‐related genes, nucleotide‐binding site (NBS)‐leucine‐rich repeat (LRR) genes and transcription factors (TFs)
To identify cold signaling‐related genes in Poncirus and Citrus, we focused on homologs of Arabidopsis genes that are involved in cold signaling (Guo et al., 2018; Liu et al., 2018a; Shi et al., 2018) and P. trifoliata genes known to respond to cold stress. Most of the Arabidopsis thaliana cold signaling pathway genes and P. trifoliata cold tolerance‐related genes had homologs in Citrus species. Based on analysis using existing annotations of the 10 Citrus‐related genomes, the homologs of A. thaliana cold signaling pathway genes were assigned to 45 gene families containing 69 P. trifoliata genes (Figure 1; Table S4). In addition, there were 49 gene families containing either ‘first‐wave’ TF genes or CBF regulon genes in A. thaliana, in which 119 P. trifoliata genes were included. Several gene families containing cold signaling‐related genes from A. thaliana found no members in Citrus‐related species. This may be a consequence of comparing a perennial woody plant with an herbaceous annual plant, and that different genetic players have been adopted to confer cold tolerance in Citrus species compared with A. thaliana. After re‐assigning gene families using re‐annotated gene models, an average of ~88% of genes or gene families containing cold signaling‐related genes were recovered for the 10 genomes (Tables S4 and S5). If only considering low‐copy genes (less than two genes for each species), a higher percentage (~92%) of genes or gene families were recovered. Therefore, the vast majority of genes were not influenced (when it comes to finding the gene) by the bias of different annotation pipelines of genomes. By matching P. trifoliata genes from the current study to previously identified DEGs upon cold treatment in P. trifoliata (Wang et al., 2015), a total of 118, 467 and 1292 genes were found to be differentially expressed at 6, 24 and 72 h after cold treatment, respectively. The evolutionary dynamics of these candidate cold signaling‐related genes were further investigated in the following sections.
A total of 348 NBS genes was identified in the genome of P. trifoliata (Figure 1; Table S6). These NBS genes were further classified into different classes based on the presence of Toll/Interleukin‐1 receptor (TIR), coiled coil (CC) and LRR domains. The two largest classes with at least one of these domains were the ‘CC‐NBS’ (84; 24.1%) and ‘CC‐NBS‐LRR’ (77; 22.1%) types. A small proportion (58; 16.7%) contained the TIR domain compared with other domains. Through phylogenetic analysis, these NBS genes could be classified into three major groups (Figure S3). The TF analysis revealed a total of 1493 putative TF genes in 58 families, representing 5.8% of the predicted gene models in the P. trifoliata genome (Table S7). The most predominant TF family in P. trifoliata is the bHLH family (124), followed by NAC (123), ERF (110) and MYB (109) families.
Gene family analysis and evolutionary dynamics among Citrus ‐related species
The gene family assignment using existing annotations was first explored to have a global view of gene families among the 10 genomes. A total of 253 788 (86.7%) genes from the 10 species, including P. trifoliata and nine other Citrus and Citrus‐related species, were assigned to 28 226 gene families, among which 15 763 (55.8%; 98 960 genes) represented one‐to‐one orthologous gene families (Figure 2a; Table S8). There were 11 597 (41.1%) gene families shared by all 10 species, which implied their conservation. In P. trifoliata, 9467 (37.1%) genes were single‐copy orthologs, 208 genes were assigned to 67 gene families specific to P. trifoliata, while 1529 genes were unclustered (Figure 2a; Table S9). Among these genes, analysis using re‐annotations supported that 605 genes are specific to P. trifoliata. These exonerate recoverable genes are not present in other genomes under the same annotation method. By calculating the percentage of shared gene families relative to the total (unique) gene families of each pair of species, P. trifoliata shared a higher proportion of its gene families with C. × clementina (Figure 2b). To explore the phylogenetic relationship of the 10 Citrus‐related species, a high‐confidence phylogenetic tree was constructed using the protein sequences of 5747 single‐copy orthologs (Figure 2c). The species tree topology obtained from the coalescence method was the same as that of the concatenation method. The phylogenetic tree revealed that P. trifoliata diverged from the common ancestor of Citrus clade about 9.8 million years ago (MYA). This result is close to previous estimations on Citrus−Poncirus divergence time (4.0–9.6 MYA in Pfeil and Crisp, 2008; 9.1–9.2 MYA in Wu et al., 2018). Within the Citrus clade, C. ichangensis is located at the basal position, while C. maxima (pummelo) and C. medica (citron) form a distinct cluster. Fortunella hindsii (kumquat) is located at a basal position relative to C. × clementina (Clementine), C. reticulata (mandarin), C. unshiu (Satsuma) and C. × sinensis (sweet orange). It is noteworthy that the cold‐tolerant species, P. trifoliata, C. ichangensis and F. hindsii all seem to be located at a more basal position than other cold‐sensitive species. The re‐annotation and analysis of single‐copy gene families showed a high agreement between existing annotations and re‐annotations (Table S10). The same species tree topology was obtained after re‐constructing the tree using exonerate models. Therefore, the phylogenetic tree obtained from existing annotations was used for further analysis.
Figure 2.
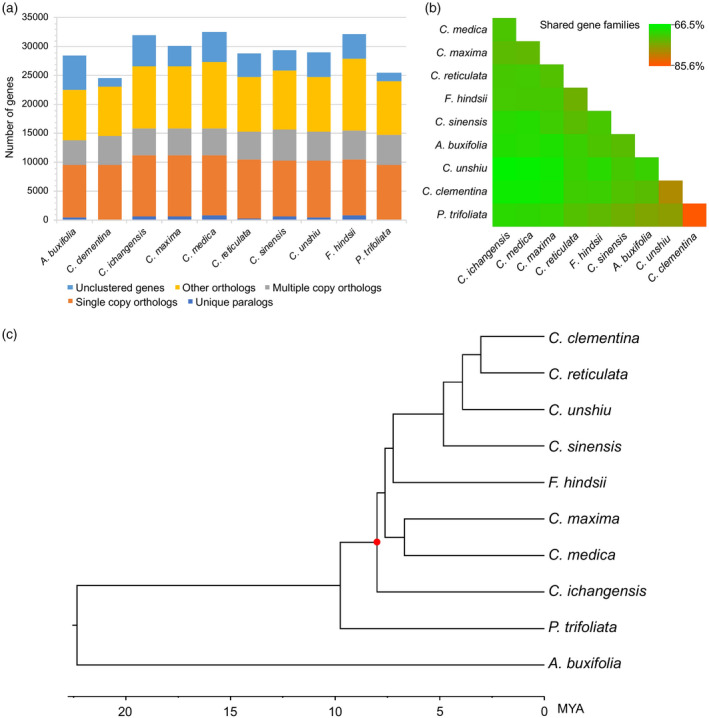
Comparative genomics of the 10 Citrus and Citrus‐related species analyzed in this study using existing annotations.
(a) Gene numbers for different types of orthologous/paralogous gene family relationships in 10 species. Detailed classification of orthologous/paralogous types is provided in Table S5 (column ‘Classification’).
(b) A heatmap for comparison of shared gene families for each pair of species.
(c) A phylogenetic tree containing 10 analyzed species. Note that some citrus, e.g. Citrus × sinensis, arose as the offspring of previously admixed individuals, and thus divergence times between admixtures are not meaningful for these so‐called species.
Through gene family expansion and contraction analysis using existing annotations, in P. trifoliata there were 45 rapid evolving gene families that were significantly expanded (41) or contracted (4) across the species tree with a family‐wide P ≤ 0.01 and branch‐specific P ≤ 0.01 (Table S11). We performed further filtering on these gene families through re‐analysis using re‐annotated gene models. An expanded family was considered supported by re‐annotation if it met the following thresholds: (i) at least half of the P. trifoliata genes from this family were recovered by re‐annotation and the exonerate models were assigned to the same family; (ii) the re‐analysis using exonerate models also showed a family‐wide P ≤ 0.01 and branch‐specific P ≤ 0.01. No significantly contracted families were supported by re‐annotation. We finally identified six significantly expanded gene families for P. trifoliata, which were supported by both existing annotations and re‐annotations (Table S11). To further investigate the gene functions, gene ontology (GO) enrichment analysis was performed on the 605 genes specific to P. trifoliata, and on the six rapid evolving gene families in P. trifoliata, respectively (Table S12). The structural constituents of cell wall (GO:0005199) and plant‐type cell wall organization (GO:0009664) were significantly enriched for genes specific to P. trifoliata. Three GO terms were significantly enriched for the rapid evolving genes, including protein binding (GO:0005515), DNA binding (GO:0003677) and protein dimerization activity (GO:0046983). Specifically, among the six rapid evolving gene families, one family (OG1.5_1006) encodes a group of LRR proteins, whose only homolog in Arabidopsis (AT2G34930) encodes a disease resistance family protein involved in defense response. Interestingly, two gene families were associated with TEs, including a gag protein family associated with retrotransposon (OG1.5_1264) and a Tam3‐transposase family associated with DNA transposon (OG1.5_1413). Another family was a group of zinc finger proteins with hAT dimerization domain. The remaining two families had unknown functions.
As shown in Table S4, most of the gene families containing cold signaling‐related genes had similar gene numbers across all Citrus‐related species, showing no significant differential gene gain/loss between cold‐hardy and other Citrus species, including cold‐sensitive C. maxima, C. medica and C. × sinensis. However, a larger number of genes was observed in cold‐tolerant P. trifoliata and F. hindsii for an amino acid transporter family (OG1.5_1258) containing a DEG responsive to cold stress in P. trifoliata. This gene family contains fewer gene members in the cold‐sensitive Citrus species. Interestingly, for an FAR1 TF family (OG1.5_1201) containing two DEGs responsive to cold stress, a much larger number of genes was observed in all three cold‐tolerant species than in the cold‐sensitive Citrus species. A similar trend was observed for gene assignment using re‐annotated gene models. These P. trifoliata genes may potentially play an important role in increasing its cold tolerance.
Whole‐genome duplication (WGD) and divergence between the two Poncirus species
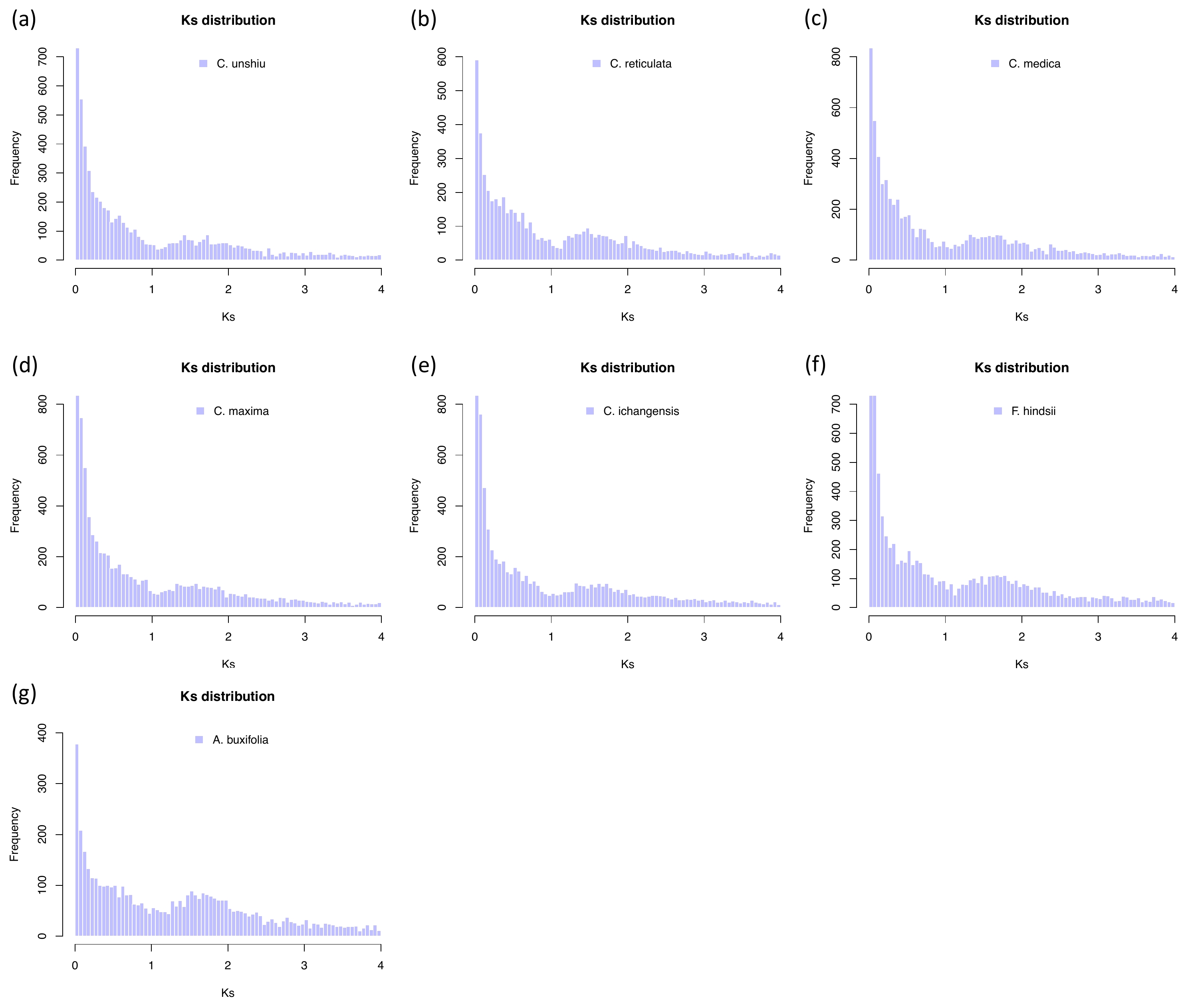
To investigate the evolutionary history of the P. trifoliata genome and other Citrus genomes, the Ks was estimated for paralogous gene groups in each species. A clear secondary peak (Ks = ~1.5) was identified for P. trifoliata, similar with C. × clementina, C. × sinensis, as well as with other Citrus‐related genomes (Figures S4a and S5a–g). Similar results have been observed previously in C. × clementina (Ks peak = 1.5; Wu et al., 2014) and C. × sinensis (Ks mean = 1.27; Xu et al., 2013). Thus, as expected, we confirm that the Citrus−Poncirus lineage shared an ancient WGD event, which is also shared with other eudicots. To study the divergence between Poncirus species, we predicted a total of 8326 gene models for P. polyandra based on the existing re‐sequencing data, and calculated the Ks values between these genes and their P. trifoliata orthologs. We observed a clear peak at Ks = 0.007 (Figure S4b). By assuming the neutral substitution rate r to be 1 ~ 2 × 10−9 (Wu et al., 2014), the divergence time between P. trifoliata and P. polyandra was estimated to be approximately 1.75 ~ 3.5 MYA. Among the 8326 genes identified from P. polyandra, 2170 match to the 5747 single‐copy orthologs among 10 Citrus‐related species. If we include P. polyandra gene models when constructing the Poncirus−Citrus phylogenetic tree (using 2170 single‐copy orthologs including P. polyandra), the estimated divergence time between P. trifoliata and P. polyandra is about 2.82 MYA, which falls into the range of the Ks‐based method (Figure 2c). These results indicate that from an evolutionary perspective the P. polyandra/P. trifoliata split is one of the most recent divergences among the 11 Citrus‐related species.
Positive selection (dN/dS analysis)
To gain more insight into adaptive evolution in the P. trifoliata genome, positive selection was first tested for the 5747 single‐copy orthologs from P. trifoliata and nine Citrus species using a branch‐site model. Our analysis revealed that 35 genes underwent positive selection specifically in the P. trifoliata lineage, which was supported by both existing annotations and re‐annotations (Table S13; ‘Category 4’). To further compare the positively selected genes among the three cold‐tolerant species, additional analyses were performed using the three cold‐sensitive species as the background branch (Table S13; ‘Category 1–3’). Similar with C. ichangensis, P. trifoliata has evolved particular adaptation in both the CBF‐dependent and CBF‐independent cold signaling pathways to tolerate the cold environment from a more northern and temperate natural habitat (Figure 3a). Four genes (shown in Figure 3a) from P. trifoliata and C. ichangensis were supported by both existing annotations and re‐annotations. Strikingly, the homolog of a CBF regulon, LOW‐TEMPERATURE‐INDUCED 65 (LTI65), was positively selected in two cold‐tolerant species P. trifoliata and C. ichangensis (Figure 3a). However, P. trifoliata has a larger number of positively selected sites (25) than C. ichangensis (1). We further noticed LTI65 remains a positively selected gene when treating the nine species as the background branch and P. trifoliata as the foreground branch. Therefore, P. trifoliata, the most cold‐hardy species among the 10, underwent extensive and lineage‐specific adaptations in LTI65, a player in the CBF‐dependent cold signaling pathway. The other two positively selected genes in P. trifoliata were COR413‐PM1, another CBF regulon, and hybrid proline‐rich protein (PRP). Importantly, the PRP gene has already been characterized in P. trifoliata previously and confirmed to confer cold tolerance (Peng et al., 2015). Additionally, three DEGs responsive to cold stress were also positively selected uniquely in the P. trifoliata genome, which was supported by both existing annotations and re‐annotations (Table S13; ‘Category 4’). These species‐unique adaptations together with the unique expansions of genes responsive to cold stress may explain why P. trifoliata is the most cold‐tolerant species among the 10 species analyzed.
Figure 3.
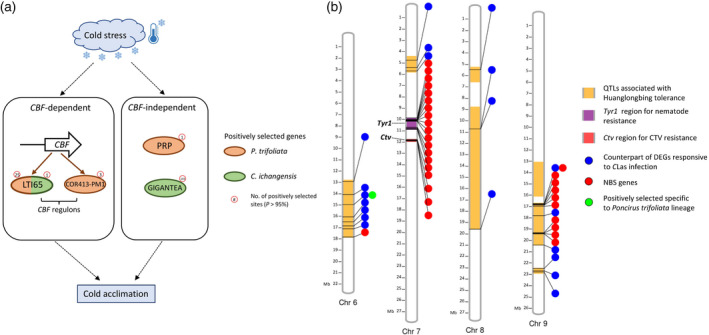
Candidate genes associated with cold tolerance and disease resistance.
(a) Positively selected genes associated with cold signaling pathways and specific to two cold‐tolerant species. Genes in brown ovals were specifically positively selected in the Poncirus trifoliata lineage. Genes in green ovals were specifically positively selected in the Citrus ichangensis lineage.
(b) A graphic map showing quantitative trait loci (QTL) and candidate genes in the P. trifoliata genome associated with tolerance to the Huanglongbing (HLB) disease, as well as candidate genes associated with citrus tristeza virus (CTV) and nematode resistance within Ctv and Tyr1 regions.
Candidate genes associated with HLB tolerance and resistance to CTV and citrus nematodes
To further exploit the HLB tolerance feature of P. trifoliata, we mapped the markers flanking 14 QTL intervals associated with HLB tolerance that were previously placed on linkage maps of P. trifoliata (Huang et al., 2018). Their genomic locations were found in the P. trifoliata genome with a physical size ranging from 0.24 Mb (QTL: FS‐2016‐t8) to 10.61 Mb (QTL: CD‐2015‐t8; Table S14). Some of the QTLs are overlapping; after overlapping QTLs were merged, seven unique genomic regions for the 14 QTLs were observed (highlighted in Figure 3b). By searching the P. trifoliata counterparts (reciprocal best hits) of DEGs responsive to CLas infection within these QTL regions, we identified 20 P. trifoliata genes that are counterparts of previously identified DEGs potentially associated with HLB (Figure 3b; Table S15; Fu et al., 2016; Hu et al., 2017; Yu et al., 2017). Interestingly, a TF WRKY70 (orange1.1g020291m/Ptrif.0006s1042) was upregulated upon CLas infection in sweet orange and positively selected uniquely in P. trifoliata (considering nine species as the background branch). The positive selection of this gene was also supported by re‐annotations. Considering the evidence, this TF gene may play an important role in P. trifoliata tolerance to HLB. Strikingly, 11 NBS genes (putative disease resistance genes) were found in these QTL regions, and 10 of them (as two clusters) are located in a single QTL FS‐2015‐t9a (phenotypic variation explained 24.5%; physical size 3.75 Mb) on Chromosome 9 (Table S16). Moreover, one of them (Ptrif.0009s1449) is a counterpart of a DEG (orange1.1t00706.1) responsive to CLas infection. Overall, these genes can serve as candidate genes for further investigation of the genetic factors conferring HLB tolerance in P. trifoliata.
By mapping the Ctv locus controlling CTV resistance and Tyr1 locus controlling nematode resistance to the P. trifoliata genome, candidate disease resistance genes (NBS genes) within these regions were identified (Figure 3b; Table S16). Strikingly, there was a total of 15 NBS genes (as two clusters) located within the Tyr1 region (1 Mb in size). Overall, these major loci controlling disease resistance in P. trifoliata have clusters of NBS genes. Chromosomes 7 and 9 of P. trifoliata may play a very important role in conferring resistance/tolerance to HLB, CTV and citrus nematodes.
Genetic diversity and relatedness of Poncirus and hybrids
All four P. trifoliata accessions (DPI 50‐7, Flying Dragon, Little Leaf and Rubidoux) have genome‐wide average heterozygosity at 0.5–0.6% (Figure 4a), similar to that observed in most pure Citrus species (Wu et al., 2018). By contrast, the heterozygosity of the P. polyandra accession is much lower (~0.2%). The Poncirus/Citrus hybrids are characterized by inter‐specific/generic sequence divergence with heterozygosity 2.3–2.4% (Figure 4a, where the three‐letter codes for the 17 accessions are also defined). Admixture analysis using 194 598 nuclear genic single nucleotide polymorphisms (SNPs) revealed the genetic composition of the 17 re‐sequenced accessions. At k = 3, the three source populations correspond to Poncirus, C. maxima (pummelos) and C. reticulata (mandarins). With k = 4, P. polyandra (PCP) separates from P. trifoliata, with the six Poncirus/Citrus hybrids showing no genetic contribution from P. polyandra (Figure 4b). These six hybrids exhibit three patterns of admixture, with four being mandarin/Poncirus hybrids, one as pummelo/Poncirus hybrid (PXP), and the Carrizo citrange (PXO) being consistent with its parentage C. × sinensis × P. trifoliata. The sweet orange (SWO, C. × sinensis) is an admixture of C. maxima and C. reticulata with a complex origin (Figure 4b; Wu et al., 2014). Note that the ADMIXTURE software did not reveal the small amount of pummelo admixture in C. reticulata cultivars Cleopatra (CLP) and Sunki (SNK) as identified with local ancestry analysis (Wu et al., 2018). Principle component analysis with the same set of nuclear genic SNPs revealed a population structure that is consistent with admixture analysis (Figure 4c). In particular, PC1 separates Poncirus from Citrus with Poncirus/Citrus hybrids at intermediate positions. PC2 separates pummelos from mandarins with sweet orange situated between the two parental species (Figure 4c). Poncirus polyandra splits from the other accessions with PC3.
Figure 4.
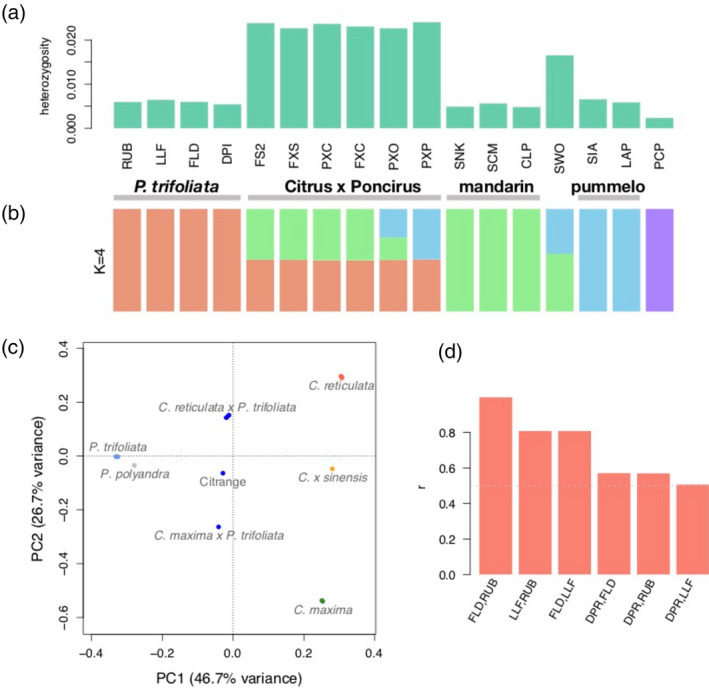
Genetic diversity and relatedness of Poncirus and hybrids.
(a) Heterozygosity of five Poncirus accessions and six Citrus accessions, including Poncirus trifoliata (RUB = Rubidoux, LLF = Little Leaf, FLD = Flying Dragon, and DPI = DPI 50‐7), Poncirus polyandra (PCP), Citrus reticulata (CLP = Cleopatra, SNK = Sunki, and SCM = Sun Chun Sha Kat), Citrus maxima (LAP = low acid and SIA = Siamese) and Citrus sinensis (SWO), and six Poncirus/Citrus hybrids [PXO = Carrizo citrange (C. × sinensis × P. trifoliata), PXP = Citrus maxima × P. trifoliata, and four C. reticulata/Poncirus hybrids FXC, PXC, FXS, FS2]. Accession IDs are provided in Table S1.
(b) Admixture analysis at k = 4 reveals the genetic makeup of the re‐sequenced accessions in terms of four species. Each bar represents the ancestry proportion of one accession, and the accessions are ordered as in panel. Note that Poncirus polyandra (PCP) represents a genetic isolate.
(c) Principle component analysis (PCA) shows a population structure that is consistent with admixture analysis.
(d) Genetic relatedness among the four P. trifoliata accessions. The coefficient of relatedness is shown for each pair of accessions. In particular, FLD and RUB are clonally related (r = 1), and all pairs show high degrees of relatedness (r ≥ 0.5).
Genetic relatedness among the P. trifoliata accessions and Poncirus/Citrus hybrids are computed following the method of Wu et al. (2018). The four P. trifoliata accessions are genetically related to each other with coefficient of relatedness > 0.5 (Figure 4d). Throughout the genome, each Poncirus pair shares either one or both haplotypes (Figure S6). In particular, Flying Dragon and Rubidoux are related by somatic mutations with r = 1, though their phenotypes are quite different, including the dwarfing trait of Flying Dragon (Cheng and Roose, 1995). This clonal relatedness is in line with an earlier proposal that Flying Dragon originated as a mutant of a non‐dwarfing genotype (Cheng and Roose, 1995). We also note that a previously published Poncirus accession (P. trifoliata Pomeroy; Wu et al., 2018) is clonally related to Little Leaf. The high degree of relatedness among the four P. trifoliata accessions indicates a very narrow genetic base for the Poncirus accessions in the US germplasm, and points to the importance of exploring additional genetic variation that exists in the native habitat of Poncirus (Zhu et al., 2015).
Among the six Poncirus/Citrus hybrids, genome‐wide haplotype sharing analysis verified the parentage of Carrizo citrange with C. × sinensis as the maternal parent and P. trifoliata Flying Dragon/Rubidoux (or its somatic mutant) being the paternal parent. The pummelo/Poncirus hybrid (PXP) has P. trifoliata Little Leaf as the paternal parent, with the maternal parent being an unknown C. maxima that is different from the Siamese pummelos LAP and SIA. Of the four mandarin/Poncirus hybrids, FXC = Cleopatra mandarin × P. trifoliata Flying Dragon, PXC = P. trifoliata Little Leaf × Cleopatra mandarin, FXS = unknown mandarin × P. trifoliata Little Leaf, and FS2 = unknown mandarin × P. trifoliata Flying Dragon, where the maternal parents are listed first, we did not distinguish between somatic mutants in listing the parentage (e.g. FLD and RUB). By ‘unknown mandarin’ in the parents of FXS and FS2, we meant that the parents cannot be found in the three mandarins (CLP, SNK, SCM) of our analysis.
DISCUSSION
The availability of a high‐quality reference genome is a prerequisite for effective and efficient mining of a crop genome. As a major rootstock in the citrus industry and a major source of resistance genes for citrus breeding, P. trifoliata has many desirable traits, including resistance to several major diseases, especially tolerance to HLB, and cold tolerance. Since more than a century ago, many of these traits have been targeted for introducing into citrus for crop improvement. However, a P. trifoliata genome assembly was not available until now. Our study filled this gap by providing a high‐quality and chromosome‐scale reference genome of P. trifoliata to the citrus research community. This genome should lay a solid foundation for future genetic, genomic and molecular studies in this important species.
De novo assembly and TE
Assembling heterozygous genomes de novo has been a challenge (Kajitani et al., 2014). High contents of repetitive sequences often present a major barrier to reliable assembly of such genomes from short‐read sequences. We applied an integrated assembly strategy combining Illumina short‐read sequencing, PacBio long‐read sequencing and Hi‐C scaffolding technology to facilitate the assembly process and improve the accuracy. Our assembly strategy has proven to be effective. The Hi‐C data and resulting scaffolding improved the assembly N50 length by approximately 32‐fold. To our knowledge, this genome is the fourth reference genome assembly at chromosome‐scale or pseudomolecule‐level in Citrus‐related genera or, more broadly speaking, in the family Rutaceae, following C. × sinensis (Xu et al., 2013), C. × clementina (Wu et al., 2014) and C. maxima (Wang et al., 2018a). The completeness of this genome is 97.2% based on the BUSCO score, which is higher than that of previous reports, such as C. reticulata (96%; Wang et al., 2018a) and F. hindsii (95.1%; Zhu et al., 2019). A considerable proportion of the P. trifoliata genome is comprised of TEs (42.63%), and the most abundant repeat types are LTR retrotransposons (Copia and Gypsy most frequent). This seems a shared feature among Citrus‐related genomes.
Re‐annotation of genes of interest
One limitation of using existing gene model annotations of other citrus genomes for comparative analysis is that some findings could be potentially caused by the different annotation pipelines. Therefore, we first used existing annotations to gain a global view of gene family assignment and identified a preliminary set of genes or gene families of interest, including homologs of cold signaling‐related genes, rapid evolving gene families, P. trifoliata‐specific genes and single‐copy genes across 10 genomes. To address and minimize potential biases from different annotation pipelines, we therefore applied a homology‐based gene annotation pipeline (Blast/Exonerate) to all the genomic regions potentially containing the preliminary set of genes of interest for all the 10 genomes, including P. trifoliata itself. Findings from existing annotations were further filtered according to the re‐analysis using the re‐annotated gene models (under the same pipeline).
Phylogenetic analysis
A phylogenetic tree is needed to understand the evolutionary relationships among species. The use of highly conserved and low‐copy nuclear genes in resolving phylogenetic relationships has long been recognized (Duarte et al., 2010; Zhang et al., 2012). Previously, as the genome of P. trifoliata was unavailable, a phylogenetic tree was constructed by using SNPs located within low‐copy genes for nine Citrus‐related species, including P. trifoliata (Zhu et al., 2019). With the availability of 10 Citrus‐related genomes including this P. trifoliata genome, we were able to use protein sequences of 5747 conserved and single‐copy nuclear genes (genome‐wide) across the 10 species to re‐construct a highly confident species tree. The species tree topology was supported by both the concatenation method and the coalescence method after using either existing annotations or re‐annotations. To test the effect of gene numbers on tree structure for the concatenation method, we also constructed additional trees by using a subset of these single‐copy genes, including using 100, 1000, 2000, 3000 and 4000 randomly selected genes. It was found that when using more than 2000 genes, the general tree structure became stable and remained unchanged even if more genes were included. Therefore, this species tree can be considered relatively confident and stable. In this analysis, P. trifoliata showed the closest relationship to C. ichangensis among the analyzed species, which is consistent with a previous report (Wu et al., 2018). The P. trifoliata genome will be useful for phylogenetic studies in citrus.
Poncirus polyandra
Poncirus polyandra is the only existing species within Poncirus other than P. trifoliata. Almost extinct in the wild, P. polyandra has been classified as a protected wild plant (national second‐class) in China (http://rep.iplant.cn/protlist). Recently, the chloroplast genome of P. polyandra was sequenced and its 105 predicted gene models were used for phylogenetic analysis (Li et al., 2019; Yang et al., 2019). In this study, the genome sequences of P. polyandra were reported, and a pipeline combining reference‐based assembly using RGAAT and gene prediction using Genewise was applied for gene model predictions. We obtained 8326 high‐quality predicted protein‐coding genes in P. polyandra. This pipeline can also be applied to other species with a reference genome available from a close relative. On the basis of these 8326 genes, we proposed a divergence time of 2.82 MYA between P. trifoliata and P. polyandra. Our study provided additional genomic resources to this rare and endangered species, which can be useful for future research of P. polyandra and may provide guidance to develop conservation strategies.
What’s unique about the genome of Poncirus trifoliata ?
Our analysis using existing annotations revealed that 11 597 gene families were shared by the 10 analyzed Poncirus and Citrus species, indicating that these gene families may have conserved functions and represent the essence of Citrus‐related species. However, there were 605 genes in P. trifoliata that are not shared with the remaining nine species. These P. trifoliata‐specific genes are likely not required for the basic growth and development of Citrus species, but they may represent the diversity or uniqueness of P. trifoliata. Notably, the GO enrichment analysis revealed that two cell‐wall‐related GO terms were significantly enriched in P. trifoliata‐specific genes, which may indicate a unique cell wall structure and organization in P. trifoliata. LRR proteins play an important role in plant innate immunity against pathogens, and most immune receptors contain the LRR domain for pathogen recognition (Padmanabhan et al., 2009). While the NBS‐LRR genes are the most common type of disease resistance genes in plants (McHale et al., 2006), interestingly, we identified a rapid evolving gene family in P. trifoliata comprised of non‐NBS type LRR genes. The expansion of this family in the P. trifoliata genome may have a potential role in its disease resistance as their single homolog in Arabidopsis belongs to a disease resistance gene family. TEs provide an abundant source of genetic variations, thereby contributing to the evolution and genetic diversity of plants (Kidwell and Lisch, 1997; Butelli et al., 2012; Borredá et al., 2019). In P. trifoliata, we identified two significantly expanded gene families encoding the retrotransposon gag protein and Tam3‐transposase. Interestingly, in contrast to most TEs whose regulations are epigenetic dependent, the activity of Tam3 is controlled by its own Tam3‐transposase as characterized in Antirrhinum majus (Zhou et al., 2017). Moreover, the transposition of Tam3 is closely controlled by temperature, with more transposition activities at lower temperatures and repressed at higher temperatures (Hashida et al., 2003, 2006). Therefore, it is likely that the cold habitat of P. trifoliata is associated with the expansion of Tam3‐transposase gene families. Overall, the enriched functions for lineage‐specific genes and rapid evolving genes in P. trifoliata may be essential for its adaptive evolution, and contribute to the characters specific to this species.
Adaptive evolution of Poncirus trifoliata and Citrus
As the most cold‐hardy species among all Citrus‐related species (Spiegel‐Roy and Goldschmidt, 1996), the P. trifoliata genome provides a valuable candidate gene pool to improve cold tolerance in citrus. Our analysis revealed specific adaptions potentially associated with cold tolerance not only in P. trifoliata, but also in the other well‐known cold‐tolerant species, C. ichangensis. Our analysis indicated that P. trifoliata and C. ichangensis had adaptations in both the CBF‐independent and CBF‐dependent pathways. One positively selected gene homologous to LTI65 was shared by P. trifoliata and C. ichangensis. Therefore, LTI65 could be important in regulating cold signaling as it was positively selected in two cold‐tolerant species. These positively selected genes can be good candidates for further studies and genetic engineering to improve cold tolerance in citrus. Apart from the above adaptations, the DEGs responsive to cold and positively selected specifically in P. trifoliata branch may reveal genetic factors and mechanisms of cold tolerance in P. trifoliata.
Tolerance to HLB, CTV and citrus nematode
A recent study identified QTLs for HLB tolerance in P. trifoliata (Huang et al., 2018), based on linkage maps of P. trifoliata and reference genomes of C. × clementina and C. × sinensis. Because the P. trifoliata genome was not available then, the study was not able to associate the QTLs with P. trifoliata genes or genomic sequences. With the availability of this P. trifoliata genome, candidate genes located within these QTLs could be mined. To be considered as a candidate gene controlling HLB tolerance, it would be reasonable to assume that its expression likely will change after infection with CLas. Therefore, we catalogued P. trifoliata genes that are counterparts of DEGs responsive to CLas infection based on three previous transcriptome studies. The WRKY70 TF gene under positive selection on Chromosome 6 and QTL FS‐2015‐t9a harboring two NBS gene clusters on Chromosome 9 may be promising candidates for further studies. Similarly, NBS gene clusters were also observed within Ctv and Tyr1 regions on Chromosome 7. The NBS genes identified in this study may facilitate understanding the disease resistance in Poncirus and expedite introgression of resistance genes into Citrus.
Need to preserve the genetic diversity in Poncirus
Poncirus is extremely important for current citrus breeding and future genetic improvement of citrus. Our study reveals a surprisingly narrow genetic base in the US germplasm collection, and indicates a strong need for further exploration and preservation of its natural genetic variation. A previous study (Zhu et al., 2015) indicates a wide genetic diversity in the native habitat of P. trifoliata in China. While still in existence, preserving and sharing this natural variation should be considered as an important item on the agenda of citrus germplasm researchers in the world to prevent the tragedy that had happened to P. polyandra, the only other species in Poncirus.
Conclusion
In summary, our study significantly enriched the genomic resources for P. trifoliata, an extremely important relative of Citrus with remarkable disease resistance and cold tolerance. Our reference genome and comprehensive evolutionary analysis are expected to contribute to a better understanding of disease resistance, cold tolerance and other unique biological features of P. trifoliata, and play an important role in breeding, genetic, genomic and molecular research of Citrus.
EXPERIMENTAL PROCEDURES
Plant materials
Poncirus trifoliata accessions DPI 50‐7, Flying Dragon, Little Leaf and Rubidoux, six P. trifoliata/Citrus hybrids, including Carrizo citrange, US‐802, US‐812, US‐897 and US‐942, and XCitroncirus X639, and one P. polyandra accession were sequenced (Table S1). Poncirus trifoliata DPI 50‐7 is an accession maintained by Florida Department of Plant Industry (DPI; https://www.fdacs.gov/Divisions‐Offices/Plant‐Industry). This accession has been used in various studies (Graham et al., 2003; Albiach‐Marti et al., 2004). Based on genotyping data (F.G. Gmitter Jr.), its heterozygosity was lower than some other Poncirus accessions and it has a high percentage of nucellar seedlings that develop from nucellar embryos, a type of apomixis that is important for P. trifoliata to be used as citrus rootstock. According to the most recent 2018–2019 report from Florida Department of Agriculture and Consumer Services Bureau of Citrus Budwood Registration (2019; https://www.fdacs.gov/Divisions‐Offices/Plant‐Industry/Bureaus‐and‐Services/Citrus‐Budwood‐Registration), Poncirus accessions and Poncirus hybrids were used as the rootstock for 3 224 857 citrus trees in Florida in 2018–2019. Poncirus and Poncirus hybrid rootstocks accounted for 82.2% of the top 20 rootstocks used in the 2018–2019 citrus propagation cycle (a total of 3 918 440 citrus plants budded on the top 20 rootstocks). Six Poncirus hybrids were included in this study, including Carrizo citrange, US‐802, US‐812, US‐897, US‐942 (Castle et al., 2009; Bowman et al., 2016) and XCitroncirus X639 (Table S1). Nucellar embryony is widespread in Citrus and Poncirus (Wang et al., 2017). It is very important and valuable for rootstock cultivars, because it allows for production of highly uniform plants (because of origin from the nucellar tissue) from seeds for use as rootstock for grafting (Castle et al., 2009). On the other hand, this may have led to high levels of relatedness among Poncirus accessions in germplasm collections. The selected and preserved accessions may have some differences in morphology and have different names, but might have originated from nucellar seedlings and have little to minor differences at the DNA level or in the genome. Young leaf tissues were collected from the above accessions and immediately put into liquid nitrogen for subsequent DNA extraction using the CTAB method (Cheng et al., 2003).
Genome sequencing and assembly
The genome of P. trifoliata accession DPI 50‐7 was sequenced using PacBio, Illumina HiSeq 2500 (125 bp paired‐end reads) and Hi‐C technology. High‐molecular‐weight DNA was extracted from approximately 30 g of young leaves of P. trifoliata DPI 50‐7 seedlings as described by Kim et al. (2014) and sent to the University of Florida Interdisciplinary Center for Biotechnology Research (UF/ICBR; Gainesville, FL, USA), where library construction and SMRT sequencing were performed. A total of 10 µg of DNA was sheared in a Covaris g‐tube and converted to a sequencing library using the PacBio SMRT‐bell template kit following the manufacturer’s instructions (20 kb template preparation using BluePippin size selection) with a low threshold of 15 kb. A total of two continuous long‐read (CLR) libraries were made and sequenced. A total of 41 SMRT cells were run on an RS II sequencer using the P6‐C4 chemistry and a 4−6‐h movie length. Illumina whole‐genome shotgun sequencing was performed by Novogene Corporation (Beijing, China) for P. trifoliata accessions DPI 50‐7, Flying Dragon, Little Leaf and Rubidoux, P. polyandra and P. trifoliata hybrids using the Illumina HiSeq 2500 platform (125‐bp paired‐end reads). The standard Illumina sequencing protocol was followed for short‐insert paired‐end libraries. PacBio sequencing yielded 4 953 819 polymerase reads with an N50 length of 10 874 bp and a total of 37.1 Gb polymerase read bases (approximately 140 × coverage) for DPI 50‐7. A total of 21.2 Gb of raw short‐read sequences (approximately 80 × coverage) was obtained for DPI 50‐7, and 8.6–12.4 Gb of raw short‐read sequences (approximately 32–46 × coverage) was obtained for each of the other P. trifoliata accessions and hybrids (Table S1).
The Poncirus trifoliata DPI 50‐7 genome was assembled using ~140 × depth of sequencing coverage in PacBio CLR data as input to Falcon (Chin et al., 2016; v2018.03.12‐04.00) using the following parameters: genome_size=300000000 input_type=raw length_cutoff=5000 length_cutoff_pr=5000 pa_HPCdaligner_option=[‐v ‐B20 ‐t16 ‐k14 ‐e.70 ‐l3000 ‐s100] ovlp_HPCdaligner_option=[‐v ‐B20 ‐t32 ‐k25 ‐w5 ‐h70 ‐e.94 ‐l3000 ‐s100] pa_DBsplit_option=[‐x500 ‐s350] ovlp_DBsplit_option=[‐x500 ‐s350] falcon_sense_option=[‐‐output_multi ‐‐min_idt 0.70 ‐‐min_cov 2 ‐‐min_cov_aln 4 ‐‐max_cov_aln 30 ‐‐n_core 4] overlap_filtering_setting=[‐‐max_diff 38 ‐‐min_cov 3 ‐‐max_cov 58 ‐‐bestn 10 ‐‐min_len 5000]. Short‐insert paired‐end Illumina sequencing reads from DPI 50‐7 were aligned to the primary contigs with BWA‐MEM (Li, 2013; v0.7.17‐r1188) and filtered for properly‐paired primary alignments using SAMtools (Li et al., 2009; v1.6), then the median depth of each contig was calculated using BEDtools (Quinlan and Hall, 2010; v2.27.0). Primary contigs with median depth approximately half the expected depth (or lower) were aligned to all other primary contigs with minimap2 (Li, 2018; v2.5‐284‐g1739a26). Contigs with alignments at 90% similarity and aligned over 90% of their lengths were deemed redundant and set aside as alternate haplotigs. The remaining contigs were then scaffolded using the Dovetail Genomics HiRise Hi‐C‐based scaffolder. The assembly was curated for correctness and residual redundancy in JuiceBox (Durand et al., 2016a; Dudchenko et al., 2018; v1.9.0) with the JuiceBox Assembly Tools using Hi‐C alignments performed with Juicer (Durand et al., 2016b; v1.5.6‐64‐g94ec691). PacBio long reads were aligned to the scaffolds and alternate haplotigs with BLASR (Chaisson and Tesler, 2012) via pbalign (smrtlink v2.3.0, patch 5; parameters: ‐‐nproc 8 ‐‐algorithm blasr ‐‐maxHits 1 ‐‐algorithmOptions “‐useQuality ‐minReadLength 5000 ‐minSubreadLength 5000”) and the scaffolds polished using the Arrow (Chin et al., 2013) polishing algorithm (parameters: ‐‐diploid ‐‐noEvidenceConsensusCall lowercasereference ‐‐refineDinucleotideRepeats). A total of two long‐read signal polishing iterations were performed. The DPI 50‐7 short‐insert paired reads were aligned to the scaffolds and filtered (as described above), variants were called with FreeBayes (Garrison and Marth, 2012; v1.3.1‐17‐gaa2ace8; parameters: ‐‐genotype‐qualities ‐‐use‐mapping‐quality ‐‐report‐genotype‐likelihood‐max ‐‐standard‐filters ‐‐haplotype‐length 2 ‐‐strict‐vcf), and the resulting variants selected for supporting depth within the range of 10–135 ×, which were then used to patch small‐scale errors using a custom script (ILEC v0.2.0‐28‐g190e036; https://bitbucket.org/rokhsar‐lab/map4cns). This short‐read‐based polishing process was performed twice to yield the intermediate v1.2 assembly, and a total of six times to produce the final v1.3 assembly. The final polished scaffolds (v1.3) were then ordered, oriented and named to correspond to that of the Citrus × clementina (v1.0) assembly (downloaded from Phytozome).
Genome annotation
A de novo TE library was constructed and annotated by RepeatModeler (Smit and Hubley, 2010). For LTR retrotransposons, we applied LTR_finder (Xu and Wang, 2007) and LTR_harvest (Ellinghaus et al., 2008) to identify candidates in the P. trifoliata genome. Outputs were processed using LTR_retriever (Ou and Jiang, 2018). The unknown predicted TEs were further classified using TEClass (Abrusán et al., 2009). The above predicted TEs were integrated into RepBase (Bao et al., 2015). Finally, RepeatMasker (Tarailo‐Graovac and Chen, 2009) was used for TE annotation in P. trifoliata genome assembly.
For gene model annotation, transcript assemblies for DPI 50‐7 were first generated from ~39 million pairs of 125‐bp (paired‐end) Illumina RNA‐seq reads using PERTRAN (Shu et al., 2013). A total of 46 423 transcript assemblies was generated using PASA (Haas et al., 2003) from above RNA‐seq transcript assemblies. Protein‐coding gene loci were determined based on transcript assembly alignments and/or EXONERATE alignments of protein sequences from Arabidopsis, poplar, cotton, Clementine mandarin, citrus, Kitaake rice, green foxtail, grape and Swiss‐Prot proteomes, aligned to the repeat‐soft‐masked P. trifoliata genome using RepeatMasker. Alignment footprints were extended up to 2 kb sequences in both directions unless extending into another locus on the same strand. Gene models were predicted by the homology‐based predictors, FGENESH+ (Salamov and Solovyev, 2000), FGENESH_EST, GenomeScan (Yeh et al., 2001), PASA assembly open reading frames (ORFs), and from AUGUSTUS via BRAKER1 (Hoff et al., 2015). For each locus, the best scored predictions were selected considering several positive factors (EST and protein support) and one negative factor (overlapping with repeats). PASA was used to further improve the selected gene predictions, including adding UTRs, splicing correction and adding alternative transcripts. A protein homology analysis was performed by comparing the PASA‐improved gene model proteins with the above‐mentioned proteomes to obtain the Cscore and protein coverage. The selected gene models were subject to Pfam analysis. The gene models whose protein sequences matched more than 30% of the Pfam TE domains were filtered out.
Comprehensive search for cold signaling pathway and cold signaling‐related genes
For the analysis of cold signaling genes in P. trifoliata, the CBF‐dependent and CBF‐independent cold signaling pathway genes characterized in A. thaliana were summarized from three most recent reports (Guo et al., 2018; Liu et al., 2018a; Shi et al., 2018). The protein sequences of a total of 66 non‐redundant genes from the three reports were downloaded from NCBI (https://www.ncbi.nlm.nih.gov) or UniProt (https://www.uniprot.org/). In addition, 44 CBF regulon genes contributing to freezing tolerance and 27 cold‐induced ‘first‐wave’ TF genes that are induced in parallel with CBF genes in A. thaliana were also included in the analysis (Park et al., 2015, 2018). The homologs of the above genes in Citrus‐related genomes were determined using the Blast/OrthoMCL method as described in gene family analysis below. A previous transcriptome study revealed P. trifoliata DEGs in response to cold stress at three time points (6, 24 and 72 h) after cold treatment (Wang et al., 2015). To find P. trifoliata genes corresponding to the expressed sequence tag (EST) assemblies in that report, the EST sequences were compared with the transcript sequences of P. trifoliata gene models using BLAST. To ensure finding the correct corresponding genes, a high cutoff of alignment was used: (i) best hit from BLAST; (ii) [(alignment length × percentage of identity)/(query or subject length)] ≥ 99%.
Analysis of NBS‐LRR genes
To identify NBS‐containing genes in the P. trifoliata genome, the protein sequences were first scanned using the NBS Hidden Markov Model (HMM) profile (PF00931) and ‘hmmsearch’ in Hmmer under E‐value 1e‐04 (Finn et al., 2011). In addition, a P. trifoliata‐specific NBS domain HMM profile was made by using the high‐quality hits from ‘hmmsearch’ (E‐value 1e‐60) and ‘hmmbuild’, which was also used to scan the protein sequences. The presence of NBS domain was further confirmed using PfamScan (Finn et al., 2016). The TIR and LRR domains were identified using the NCBI conserved domain tool. The CC domain was identified using Marcoil (probability > 90%; Delorenzi and Speed, 2002). To construct a phylogenetic tree for NBS genes, the NBS domain sequences for those with a complete NBS domain were included for tree construction using RAxML same as below. The phylogenetic tree was visualized using MEGA7.
TF gene analysis
To identify genes encoding TFs in P. trifoliata, the protein sequences of predicted gene models were searched against the Plant Transcription Factor Database v5.0 (http://planttfdb.cbi.pku.edu.cn).
Candidate gene identification within regions associated with disease resistance
Our previous report of QTLs associated with HLB tolerance (Huang et al., 2018) was based on linkage maps of P. trifoliata and either C. clementina or C. sinensis as the reference genomes. With the availability of this P. trifoliata reference genome, we mapped the markers flanking the QTL intervals directly to their own P. trifoliata reference genome. To search for candidate genes associated with CLas infection and HLB tolerance, three transcriptome studies reporting DEGs responsive to CLas infection in C. sinensis, C. jambhiri (rough lemon) and/or C. hystrix (Kaffir lime) were utilized (Fu et al., 2016; Hu et al., 2017; Yu et al., 2017). To find the counterpart genes of these DEGs in P. trifoliata, a combination of Reciprocal Best Hit and OrthoMCL (same as below) methods were applied. Thus, a P. trifoliata gene was considered as the counterpart if it is the reciprocal best hit of and orthologous to a DEG responsive to CLas infection. Other genes were also searched within the QTL regions, including NBS genes and positively selected genes specific to the P. trifoliata lineage among the 10 species described above. The two major loci associated with CTV (Ctv) and nematode resistance (Tyr1) were also mapped to the genome for candidate gene identification (Ling et al., 2000; Yang et al., 2003).
Gene family analysis
The protein sequences of collected cold signaling‐related genes from above and gene models from C. clementina (v1.0), A. thaliana TAIR10 (downloaded at Phytozome), Citrus × sinensis (v2), C. ichangensis (v1.0), C. reticulata (v1.0), C. maxima (v1.0), C. medica (v1.0), Atalantia buxifolia (v1.0), Fortunella hindsii (v1.0; downloaded from http://citrus.hzau.edu.cn/orange/download/index.php), C. unshiu (http://www.citrusgenome.jp) and P. trifoliata (from this study) were compared using all‐against‐all BLAST (E‐value 1e‐5). For gene models with multiple isoforms, protein sequences corresponding to the longest ORF were selected for further analysis. Homologous/orthologous gene families were assigned using OrthoMCL (inflation value 1.5; Fischer et al., 2011). The gene families were compared between the three cold‐hardy species (P. trifoliata, C. ichangensis and F. hindsii) and three cold‐sensitive Citrus species, including C. maxima, C. medica (Nordby and Yelenosky, 1982; Crifò et al., 2011) and C. × sinensis. The functions of genes were retrieved from the genome annotation files. GO enrichment analysis was performed using FatiGO software (http://babelomics.org).
Phylogenetic analysis
A total of 5747 single‐copy gene families (one‐to‐one) across all 10 Citrus‐related species were used to construct a phylogenetic tree. The species tree topology was constructed using two methods, including a concatenation method and a coalescence method. For the concatenation method, the protein sequences for each gene family were first aligned using MAFFT (Katoh et al., 2005). Then, alignments were trimmed using Gblocks (Talavera and Castresana, 2007). The resulting sequence alignments were concatenated into a supermatrix to construct a species tree using RAxML with 1000 bootstraps (Stamatakis, 2014). The best protein substitution model ‘PROTGAMMAJTTF’ was selected using a perl script ProteinModelSelection.pl (https://cme.h‐its.org/exelixis/web/software/raxml/). Atalantia buxifolia was treated as the outgroup. The evolution time was calibrated using r8s by fixing the Citrus root age at 8 MYA (Wu et al., 2018). The phylogenetic tree was visualized using MEGA7 (Kumar et al., 2016). In addition, a coalescence method was applied using the same set of single‐copy gene families to compare the tree topologies with that of the concatenation method. Firstly, individual gene trees were constructed for each of the single‐copy gene families using the same method described for the concatenation method (using RAxML under the ‘PROTGAMMAJTTF’ model with 1000 bootstraps). The best‐scoring maximum likelihood (ML) gene trees were further analyzed using ASTRAL (v5.7.3; Zhang et al., 2018) to estimate a species tree under a multi‐species coalescent model with default parameters.
Gene family evolution analysis
CAFÉ was used to investigate gene family expansion and contraction across all branches of the species tree (De Bie et al., 2006). Only gene families containing at least one gene in no less than two species were included for the analysis. We also excluded from this analysis gene families containing more than 100 genes for one or more species. CAFÉ was run with ‘‐s’ option to estimate the gene birth‐death parameter lambda and P‐value cutoff 0.01.
Positive selection analysis
We first performed a positive selection analysis using the single‐copy gene families across all 10 Citrus‐related species (Table S13; ‘Category 4’). The protein alignments were converted to their corresponding CDS alignments. The P. trifoliata branch was used as the foreground branch, while other branches were used as background branches under the branch‐site model in PAML (Yang, 2007; Jeffares et al., 2015). The likelihood ratio test (LRT) was conducted to compare the two models (model A and A1). Genes with an FDR < 0.05 and at least one positively selected site (Bayes probability > 95%) were considered positively selected. To further compare the positively selected genes among cold‐tolerant species, three additional phylogenetic trees were constructed using RAxML and single‐copy genes from three combinations of species, including the three cold‐sensitive species (background branch) and each of the cold‐tolerant species (foreground branch), for positive selection analysis (Table S13; ‘Category 1–3’).
Re‐annotation of the 10 genomes using the same gene prediction pipeline
To further filter the findings from existing annotations that were produced previously by different annotation pipelines, a homology‐based gene prediction pipeline (Blast/Exonerate) was applied on all the potential genomic regions containing genes of interest for all the genomes, including P. trifoliata itself. The protein sequences from P. trifoliata were used as the seeding peptides to search for potential genomic regions containing their homologs, including candidate cold signaling‐related genes, significantly expanded gene families, P. trifoliata‐specific genes and single‐copy genes used for positive selection analysis. For significantly contracted gene families, the proteins from the species with the largest gene number were used as seeding peptides. The seeding peptides were compared with the 10 genomes using tBlastn/Blastx under E‐value 1e‐05. The hits in each genome were extended for 1 kb on each side (if available), and sequences were extracted for gene prediction using Exonerate (v2.2.0) under the ‘protein2genome’ model. Exonerate was run twice under either a stringent cutoff (‐‐percent 80 ‐n 1) or a less stringent cutoff (‐‐percent 50 ‐n 5). The best scoring predicted proteins were used for further analysis. The exonerate models derived from the less stringent cutoff were used to re‐run gene family assignment and expansion/contraction analysis (mainly involving gene numbers) following the same methods described above. The exonerate models derived from the stringent cutoff were used to re‐run gene family assignment, phylogenetic analysis and positive selection analysis (involving gene CDS). For a few homologs of the key cold signaling pathway genes whose exonerate models were not available for all 10 species, they were manually re‐annotated using AUGUSTUS for all species.
Prediction of Poncirus polyandra gene models
To obtain P. polyandra gene models for phylogenetic analysis, a genome assembly of P. polyandra short reads was performed using the reference‐based genome assembly and annotation tool (RGAAT; Liu et al., 2018b). The reads from whole‐genome sequencing of P. polyandra were aligned to the P. trifoliata genome using BWA‐MEM. Only uniquely mapped reads were retained for further analysis. Sequence variants were identified using Samtools and Bcftools (Li et al., 2009) according to the instructions of RGAAT. RGAAT was run using the sequence variant result with default parameters, except ‘‐m 50’ for comparison with different species. Subsequently, the resulting assembled sequences were used for gene prediction if their matching P. trifoliata genomic regions contain a gene with a depth coverage ≥ 3 for all positions. Poncirus polyandra genes were predicted by comparing between P. polyandra assembled sequences and their corresponding P. trifoliata proteins using Genewise in Wise2 (Birney et al., 2004). Only predicted genes from Genewise without breaks or frameshift errors were included for downstream analysis.
WGD analysis and estimation of divergence within Poncirus
We estimated the WGDs of P. trifoliata genome and other Citrus‐related genomes based on the Ks (number of synonymous substitutions per synonymous site) method using a python pipeline GenoDup (Mao, 2019). For each species, the paralogous group information obtained from results of OrthoMCL, proteins and CDS of genes were used as input with ‘‐n 15’ and other parameters as default. The model plant A. thaliana was also included for comparison of results. To estimate the divergence time between P. trifoliata and P. polyandra, the gene pair information from the previous step, proteins and CDS from the two species were also input into the GenoDup pipeline. The divergence time was estimated using the formula T = Ks/2r, where the neutral substitution rate r was assumed to be 1 ~ 2 × 10−9 (Wu et al., 2014).
Re‐sequencing data analysis
Illumina paired‐end reads from five Poncirus accessions, six Poncirus/Citrus hybrids and six citrus cultivars (including sweet orange, two pummelos and three mandarins) were mapped to the haploid Poncirus reference sequence V1.3 using BWA‐MEM. Sequences of sweet orange (SWO), Sunki mandarin (SNK) and low acid pummelo (LAP) were from previous publications (Wu et al., 2014, 2018). Duplicate reads are removed with sambamba before calling variants with GATK HaplotypeCaller (version 3.7‐0‐gcfedb67; McKenna et al., 2010). To reduce variant call errors, the following filters are applied as done previously (Wu et al., 2014): variants are bi‐allelic, read mapping quality ≥ 20, base quality ≥ 20, genotype quality ≥ 20, read depth between 1/2 × and 2 × the genome‐wide median, allele balance on heterozygous sites. To determine the chloroplast type of each accession, the Illumina paired‐end reads are mapped to the chloroplast genome sequence of sweet orange (Bausher et al., 2006), and variants are called using GATK as above.
For principal component analysis (PCA), Eigensoft (Patterson et al., 2006) was used on SNPs in the genic regions with missing rate < 0.05. Admixture analysis was performed with ADMIXTURE (Alexander et al., 2009), using the same set of SNPs as in PCA. For relatedness analysis, genome‐wide haplotype sharing between accession pairs were calculated in sliding windows of 100 kb, and the coefficient of relatedness was estimated following Wu et al. (2014).
Author contributions
ZD, FG and DR designed and supervised this project. ZP, JB, GW and SS analyzed the data. JB performed genome assembly. SS and ZP performed genome annotation. ZP performed comparative genomics and evolutionary analyses. GW analyzed re‐sequencing data of Poncirus accessions. NR, DD and QY (Qibin Yu) prepared and collected plant materials, isolated DNA and RNA, and/or coordinated sequencing. SP confirmed SNP data of Poncirus accessions. QY (Qian You) participated in genome analysis. ZP, GW and JB wrote the original manuscript; ZD, DR, FG, ZP, GW and Qibin Yu critically revised the manuscript. All authors approved the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Genome‐wide Hi‐C contact map of the Poncirus trifoliata V1.3 assembly (X and Y axes) at 250 kb matrix resolution. The nine chromosomes and 143 unplaced scaffolds are numbered and oriented according to Clementine V1.0, and are presented as blue boxes in descending order (from upper left to lower right). Contacts (red pixels) within chromosomes are denser than between chromosomes, with contacts between adjacent genomic loci (pixels nearer to the diagonal) being denser than those at greater inter‐locus distances (pixels farther off of the diagonal). This feature of Hi‐C was exploited to perform the scaffolding shown.
{kind=link}
Figure S2. Dot‐plot showing segmental correspondence between P. trifoliata V1.3 reference sequence (y‐axis) and haploid Clementine reference sequence V1.0 (x‐axis). Aligned segments between assemblies shown with lines; purple lines indicate alignments between sequences in the same orientation, while light‐blue lines indicate alignments in the reversed orientation.
{kind=link}
Figure S3. A phylogenetic tree of identified NBS genes in Poncirus trifoliata. Three major clusters were identified (blue, orange and purple dashed lines).
{kind=link}
Figure S4. WGD and divergence between Poncirus trifoliata and Poncirus polyandra. (a) The Ks distribution for paralogous gene groups in P. trifoliata, C. × clementina, C. × sinensis and Arabidopsis (as a control). (b) The Ks distribution between P. trifoliata and P. polyandra obtained from the 8326 gene pairs.
{kind=link}
Figure S5. WGD in other Citrus‐related species.
{kind=link}
Figure S6. Genetic relatedness among four P. trifoliata accessions. Each row represents one pair of accessions with codes explained in Figure S5(a). Dark and light green colors denote sharing two and one haplotypes, respectively, whereas gray stands for the absence of haplotype sharing.
{kind=link}
Table S1. Summary of sequenced Poncirus accessions using PacBio and Illumina Hiseq 2500 platforms
Table S2. TEs in P. trifoliata genome
Table S3. Comparison of completeness and statistics of assemblies among all published Citrus‐related genomes
Table S4. Summary of gene families containing cold signaling‐related genes
Table S5. Summary of cold signaling‐related genes recovered by re‐annotation
Table S6. Classification of identified NBS genes in P. trifoliata
Table S7. Summary of identified TF genes in P. trifoliata genome
Table S8. Gene family assignment of 10 Citrus‐related species based on existing annotations
Table S9. Genes specific to P. trifoliata
Table 10. Summary of re‐annotation of single‐copy gene families across 10 genomes
Table S11. Summary of rapid evolving gene families in P. trifoliata lineage from CAFÉ analysis
Table S12. GO enrichment analysis of P. trifoliata‐specific genes and fast evolving genes in P. trifoliata
Table S13. Summary of positively selected genes
Table S14. QTLs associated with HLB tolerance mapped to P. trifoliata genome and locations of Ctv and Tyr1 regions
Table S15. Candidate P. trifoliata genes responsive to CLas infection and located within QTLs associated with HLB tolerance
Table S16. NBS genes located within QTL associated with HLB tolerance and Ctv and Tyr1 regions
ACKNOWLEDGEMENTS
The sequencing and assembly of Poncirus genomes were supported by the Citrus Research and Development Foundation (CRDF, Project Number 724) and the USDA Specialty Crop Research Initiative/Citrus Disease Research and Extension (SCRI/CDRE; Award Number 2015‐70016‐23027) grants to F.G.G. and Z.D, and the mining of HLB tolerance candidate genes was supported by CRDF, Project Number 18‐010, grant to F.G.G., Z.D., G.A.W. and D.S.R. The authors thank Mr Benjamin Rosson at the Florida Department of Agriculture and Consumer Services Division of Plant Industry, Bureau of Citrus Budwood Registration for help collecting some leaf tissue samples for DNA extraction, Dr Kim Bowman and Dr Ed Stover for access to citrus materials in their collection, Drs Savita Shanker and David Moraga at the University of Florida Interdisciplinary Center for Biotechnology Research for performing PacBio sequencing, Novogene Corporation Inc. for performing Illumina‐based DNA and RNA sequencing, and Sierra McWilson and Dovetail Genomics for performing Hi‐C sequencing and completing the HiRise assembly of the Poncirus genome.
Contributor Information
Daniel S. Rokhsar, Email: dsrokhsar@gmail.com.
Frederick G. Gmitter, Jr, Email: fgmitter@ufl.edu.
Zhanao Deng, Email: zdeng@ufl.edu.
Data Availability Statement
The whole‐genome sequencing and re‐sequencing data from this study have been deposited at GenBank (BioProject: PRJNA648176, SRA accession number: SRR12323696‐SRR12323706). The Poncirus trifoliata genome v1.3 has been deposited in Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html).
REFERENCES
- Abrusán, G. , Grundmann, N. , DeMester, L. and Makalowski, W. (2009) TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics, 25, 1329–1330. [DOI] [PubMed] [Google Scholar]
- Albiach‐Marti, M.R. , Grosser, J.W. , Gowda, S. , Mawassi, M. , Satyanarayana, T. , Garnsey, S.M. and Dawson, W.O. (2004) Citrus tristeza virus replicates and forms infectious virions in protoplasts of resistant citrus relatives. Mol. Breed. 14, 117–128. [Google Scholar]
- Alexander, D.H. , Novembre, J. and Lange, K. (2009) Fast model‐based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, W. , Kojima, K.K. and Kohany, O. (2015) Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausher, M.G. , Singh, N.D. , Lee, S.B. , Jansen, R.K. and Daniell, H. (2006) The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple’: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol. 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney, E. , Clamp, M. and Durbin, R. (2004) GeneWise and genomewise. Genome Res. 14, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boava, L.P. , Cristofani‐Yaly, M. , Mafra, V.S. , Kubo, K. , Kishi, L.T. , Takita, M.A. , Ribeiro‐Alves, M. and Machado, M.A. (2011) Global gene expression of Poncirus trifoliata, Citrus sunki and their hybrids under infection of Phytophthora parasitica . BMC Genom., 12, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borredá, C. , Pérez‐Román, E. , Ibanez, V. , Terol, J. and Talon, M. (2019) Reprogramming of retrotransposon activity during speciation of the genus Citrus . Genome Biol. Evol. 11, 3478–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman, K.D. , Faulkner, L. and Kesinger, M. (2016) New citrus rootstocks released by USDA 2001–2010: field performance and nursery characteristics. HortScience, 51, 1208–1214. [Google Scholar]
- Bové, J.M. (2006) Huanglongbing: a destructive, newly emerging, century‐old disease of citrus. J. Plant Pathol. 88, 7–37. [Google Scholar]
- Butelli, E. , Licciardello, C. , Zhang, Y. , Liu, J. , Mackay, S. , Bailey, P. , Reforgiato‐Recupero, G. and Martin, C. (2012) Retrotransposons control fruit‐specific, cold‐dependent accumulation of anthocyanins in blood oranges. Plant Cell, 24, 1242–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle, W.S. , Nunnallee, J. and Manthey, J.A. (2009) Screening citrus rootstocks and related selections in soil and solution culture for tolerance to low‐iron stress. HortScience, 44, 638–645. [Google Scholar]
- Cameron, J.W. , Baines, R.C. and Oscar, F.C. (1954) Resistance of hybrid seedlings of the trifoliate orange to infestation by the citrus nematode. Phytopathology, 44, 456–458. [Google Scholar]
- Chaisson, M.J. and Tesler, G. (2012) Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics, 13, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F.S. and Roose, M. (1995) Origin and inheritance of dwarfing by the citrus rootstock Poncirus trifoliata ‘Flying Dragon’. J. Am. Soc. Hort. Sci. 120, 286–291. [Google Scholar]
- Cheng, Y. , Guo, W. , Yi, H. , Pang, X. and Deng, X. (2003) An efficient protocol for genomic DNA extraction from Citrus species. Plant Mol. Biol. Rep. 21, 177–178. [Google Scholar]
- Chin, C.S. , Alexander, D.H. , Marks, P. et al (2013) Nonhybrid, finished microbial genome assemblies from long‐read SMRT sequencing data. Nat. Methods, 10, 563. [DOI] [PubMed] [Google Scholar]
- Chin, C.S. , Peluso, P. , Sedlazeck, F.J. et al (2016) Phased diploid genome assembly with single‐molecule real‐time sequencing. Nat. Meth. 13, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crifò, T. , Puglisi, I. , Petrone, G. , Recupero, G.R. and Lo Piero, A.R. (2011) Expression analysis in response to low temperature stress in blood oranges: implication of the flavonoid biosynthetic pathway. Gene, 476, 1–9. [DOI] [PubMed] [Google Scholar]
- De Bie, T. , Cristianini, N. , Demuth, J.P. and Hahn, M.W. (2006) CAFE: a computational tool for the study of gene family evolution. Bioinformatics, 22, 1269–1271. [DOI] [PubMed] [Google Scholar]
- Delorenzi, M. and Speed, T. (2002) An HMM model for coiled‐coil domains and a comparison with PSSM‐based predictions. Bioinformatics, 18, 617–625. [DOI] [PubMed] [Google Scholar]
- Deng, Z. , Huang, S. , Ling, P. , Yu, C. , Tao, Q. , Chen, C. , Wendell, M.K. , Zhang, H.B. and Gmitter, F.G. Jr (2001) Fine genetic mapping and BAC contig development for the citrus tristeza virus resistance gene locus in Poncirus trifoliata (Raf.). Mol. Genet. Genomics, 265, 739–747. [DOI] [PubMed] [Google Scholar]
- Ding, S. , Zhang, X. , Bao, Z. and Ling, M. (1984) A new species of Poncirus from China. Acta Bot. Yunnan. 6, 292–293. [Google Scholar]
- Duarte, J.M. , Wall, P.K. , Landherr, L.L. , Ma, H. , Depamphilis, C.W. , Edger, P.P. , Pires, J.C. and Leebens‐Mack, J. (2010) Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 10, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducharme, E.P. (1948) Resistance of Poncirus trifoliata rootstock to nematode infestation in Argentina. Citrus Ind. 29, 9–15. [Google Scholar]
- Dudchenko, O. , Shamim, M.S. , Batra, S. et al (2018) The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome‐length scaffolds for under $1000. BioRxiv, 254797 10.1101/254797 [DOI] [Google Scholar]
- Durand, N.C. , Robinson, J.T. , Shamim, M.S. , Machol, I. , Mesirov, J.P. , Lander, E.S. and Aiden, E.L. (2016a) Juicebox provides a visualization system for Hi‐C contact maps with unlimited zoom. Cell Syst. 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, N.C. , Shamim, M.S. , Machol, I. , Rao, S.S.P. , Huntley, M.H. , Lander, E.S. and Aiden, E.L. (2016b) Juicer provides a one‐click system for analyzing loop‐resolution Hi‐C experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus, D. , Kurtz, S. and Willhoeft, U. (2008) LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics, 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Clements, J. and Eddy, S.R. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R.D. , Coggill, P. , Eberhardt, R.Y. , Eddy, S.R. , Mistry, J. , Mitchell, A.L. , Potter, S.C. , Punta, M. , Qureshi, M. and Sangrador‐Vegas, A. (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, S. , Brunk, B.P. , Chen, F. , Gao, X. , Harb, O.S. , Iodice, J.B. , Shanmugam, D. , Roos, D.S. and Stoeckert, C.J. Jr (2011) Using OrthoMCL to assign proteins to OrthoMCL‐DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinformatics, 35, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florida Department of Agriculture and Consumer Services Bureau of Citrus Budwood Registration (2019) Citrus budwood registration annual report 2018–2019. Chiefland, FL: Available at: https://www.fdacs.gov/Divisions‐Offices/Plant‐Industry/Agriculture‐Industry/Citrus‐Health‐Response‐Program/Citrus‐Budwood‐Program/Annual‐Reports. [Google Scholar]
- Fu, S. , Shao, J. , Zhou, C. and Hartung, J.S. (2016) Transcriptome analysis of sweet orange trees infected with ‘Candidatus Liberibacter asiaticus’ and two strains of Citrus Tristeza Virus . BMC Genom., 17, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnsey, D.J. and Barrett, S.M. (1987) Identification of citrus tristeza virus resistance in citrus relatives and its potential applications. Phytophylactica, 19, 187–192. [Google Scholar]
- Garrison, E. and Marth, G. (2012) Haplotype‐based variant detection from short‐read sequencing. arXiv preprint, arXiv, 1207–3907. [Google Scholar]
- George, J. and Lapointe, S.L. (2019) Host‐plant resistance associated with Poncirus trifoliata influence oviposition, development and adult emergence of Diaphorina citri (Hemiptera: Liviidae). Pest Manag. Sci. 75, 279–285. [DOI] [PubMed] [Google Scholar]
- Gilmour, S.J. , Zarka, D.G. , Stockinger, E.J. , Salazar, M.P. , Houghton, J.M. and Thomashow, M.F. (1998) Low temperature regulation of the Arabidopsis CBF family of AP2 transcriptional activators as an early step in cold‐induced COR gene expression. Plant J. 16, 433–442. [DOI] [PubMed] [Google Scholar]
- Gmitter, F.G. Jr and Hu, X. (1990) The possible role of Yunnan, China, in the origin of contemporary Citrus species (Rutaceae). Econ. Bot. 44, 267–277. [Google Scholar]
- Gmitter, F.G. Jr , Xiao, S. , Huang, S. , Hu, X.L. , Garnsey, S.M. and Deng, Z. (1996) A localized linkage map of the citrus tristeza virus resistance gene region. Theor. Appl. Genet. 92, 688–695. [DOI] [PubMed] [Google Scholar]
- Gong, X.Q. and Liu, J. (2013) Genetic transformation and genes for resistance to abiotic and biotic stresses in Citrus and its related genera. Plant Cell Tissue Organ Cult. 113, 137–147. [Google Scholar]
- Graham, J.H. (1990) Evaluation of tolerance of citrus rootstocks to Phytophthora root rot in chlamydospore‐infested soil. Plant Dis. 74, 743–746. [Google Scholar]
- Graham, J.H. , Bright, D.B. and McCoy, C.W. (2003) Phytophthora‐Diaprepes weevil complex: Phytophthora spp. relationship with citrus rootstocks. Plant Dis. 87, 85–90. [DOI] [PubMed] [Google Scholar]
- Guo, X. , Liu, D. and Chong, K. (2018) Cold signaling in plants: insights into mechanisms and regulation. J. Integr. Plant Biol. 60, 745–756. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. , Delcher, A.L. , Mount, S.M. , Wortman, J.R. , Smith, R.K. Jr , Hannick, L.I. , Maiti, R. , Ronning, C.M. , Rusch, D.B. and Town, C.D. (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashida, S.N. , Uchiyama, T. , Martin, C. , Kishima, Y. , Sano, Y. and Mikami, T. (2006) The temperature‐dependent change in methylation of the Antirrhinum transposon Tam3 is controlled by the activity of its transposase. Plant Cell, 18, 104–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashida, S.N. , Kitamura, K. , Mikami, T. and Kishima, Y. (2003) Temperature shift coordinately changes the activity and the methylation state of transposon Tam3 in Antirrhinum majus. Plant Physiol. 132, 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff, K.J. , Lange, S. , Lomsadze, A. , Borodovsky, M. and Stanke, M. (2015) BRAKER1: unsupervised RNA‐Seq‐based genome annotation with GeneMark‐ET and AUGUSTUS. Bioinformatics, 32, 767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y. , Zhong, X. , Liu, X. , Lou, B. , Zhou, C. and Wang, X. (2017) Comparative transcriptome analysis unveils the tolerance mechanisms of Citrus hystrix in response to 'Candidatus Liberibacter asiaticus' infection. PLoS One, 12, e0189229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, M. , Roose, M.L. , Yu, Q. , Du, D. , Yu, Y. , Zhang, Y. , Deng, Z. , Stover, E. and Gmitter, F.G. Jr (2018) Construction of high‐density genetic maps and detection of QTLs associated with Huanglongbing tolerance in citrus. Front. Plant Sci. 9, 1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Li, K. , Jin, C. and Zhang, S. (2015) ICE1 of Pyrus ussuriensis functions in cold tolerance by enhancing PuDREBa transcriptional levels through interacting with PuHHP1. Sci. Rep. 5, 17620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. , Wang, W. , Zhang, Q. and Liu, J. (2013) A basic helix‐loop‐helix transcription factor, PtrbHLH, of Poncirus trifoliata confers cold tolerance and modulates peroxidase‐mediated scavenging of hydrogen peroxide. Plant Physiol. 162, 1178–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffares, D.C. , Tomiczek, B. , Sojo, V. and dos Reis, M. (2015) A beginners guide to estimating the non‐synonymous to synonymous rate ratio of all protein‐coding genes in a genome. Methods Mol. Biol. 1201, 65–90. [DOI] [PubMed] [Google Scholar]
- Kajitani, R. , Toshimoto, K. , Noguchi, H. et al (2014) Efficient de novo assembly of highly heterozygous genomes from whole‐genome shotgun short reads. Genome Res. 24, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiri, M. , Stift, M. , Costantino, G. , Dambier, D. , Kabbage, T. , Ollitrault, P. and Froelicher, Y. (2018) Preferential homologous chromosome pairing in a tetraploid intergeneric somatic hybrid (Citrus reticulata + Poncirus trifoliata) revealed by molecular marker inheritance. Front. Plant Sci. 9, 1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell, M.G. and Lisch, D. (1997) Transposable elements as sources of variation in animals and plants. Proc. Natl Acad. Sci. USA, 94, 7704–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.E. , Peluso, P. , Babayan, P. et al (2014) Long‐read, whole‐genome shotgun sequence data for five model organisms. Sci. Data, 1, 14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima, E.W. , Silva, D.M. , Oliveira, A.R. , Müller, G.W. and Costa, A.S. (1964) Thread‐like particles associated with tristeza disease of citrus. Nature, 201, 1011–1012. [DOI] [PubMed] [Google Scholar]
- Katoh, K. , Kuma, K. , Toh, H. and Miyata, T. (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger, R.R. and Navarro, L. (2007) Citrus germplasm resources In Citrus Genetics, Breeding and Biotechnology (Khan I.A., ed). Wallingford, UK: CAB International, pp. 45–140. [Google Scholar]
- Kumar, S. , Stecher, G. and Tamura, K. (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Zong, D. , Zhou, A. and He, C. (2019) The complete chloroplast genome sequence of Poncirus polyandra (Rutaceae), an endangered species endemic to Yunnan Province, China. Mitochondrial DNA B, 4, 766–768. [Google Scholar]
- Li, H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv preprint, arXiv, 1303–3997. [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. and Durbin, R. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, P. , Duncan, L.W. , Deng, Z. , Dunn, D. , Hu, X. , Huang, S. and Gmitter, F.G. Jr (2000) Inheritance of citrus nematode resistance and its linkage with molecular markers. Theor. Appl. Genet. 100, 1010–1017. [Google Scholar]
- Liu, J. , Shi, Y. and Yang, S. (2018a) Insights into the regulation of C‐repeat binding factors in plant cold signaling. J. Integr. Plant Biol. 60, 780–795. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Wu, S. , Lin, Q. , Gao, S. , Ding, F. , Zhang, X. , Aljohi, H.A. , Yu, J. and Hu, S. (2018b) RGAAT: a reference‐based genome assembly and annotation tool for new genomes and upgrade of known genomes. Genomics Proteomics Bioinformatics, 16, 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Q. , Kasuga, M. , Sakuma, Y. , Abe, H. , Miura, S. , Yamaguchi‐Shinozaki, K. and Shinozaki, K. (1998) Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought‐ and low‐ temperature‐responsive gene expression, respectively, in Arabidopsis . Plant Cell, 10, 1391–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, Y. (2019) GenoDup Pipeline: a tool to detect genome duplication using the dS‐based method. PeerJ, 7, e6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale, L. , Tan, X. , Koehl, P. and Michelmore, R.W. (2006) Plant NBS‐LRR proteins: adaptable guards. Genome Biol. 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesom, G. (2014) Citrus trifoliata (Rutaceae): Review of biology and distribution in the USA. Phytoneuron, 46, 1–14. [Google Scholar]
- Nordby, H.E. and Yelenosky, G. (1982) Relationships of leaf fatty acids to cold hardening of citrus seedlings. Plant Physiol. 70, 132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, S. and Jiang, N. (2018) LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oustric, J. , Morillon, R. , Luro, F. , Herbette, S. , Lourkisti, R. , Giannettini, J. , Berti, L. and Santini, J. (2017) Tetraploid Carrizo citrange rootstock (Citrus sinensis Osb. × Poncirus trifoliata L. Raf.) enhances natural chilling stress tolerance of common clementine (Citrus clementina Hort. ex Tan). J. Plant Physiol. 214, 108–115. [DOI] [PubMed] [Google Scholar]
- Padmanabhan, M. , Cournoyer, P. and Dinesh‐Kumar, S.P. (2009) The leucine‐rich repeat domain in plant innate immunity: a wealth of possibilities. Cell. Microbiol. 11, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, X. , Hu, C. and Deng, X. (2003) Phylogenetic relationships among citrus and its relatives as revealed by SSR markers. Acta Genet. Sin. 30, 81–87. [PubMed] [Google Scholar]
- Park, S. , Gilmour, S.J. , Grumet, R. and Thomashow, M.F. (2018) CBF‐dependent and CBF‐independent regulatory pathways contribute to the differences in freezing tolerance and cold‐regulated gene expression of two Arabidopsis ecotypes locally adapted to sites in Sweden and Italy. PLoS One, 13, e0207723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. , Lee, C. , Doherty, C.J. , Gilmour, S.J. , Kim, Y. and Thomashow, M.F. (2015) Regulation of the Arabidopsis CBF regulon by a complex low‐temperature regulatory network. Plant J. 82, 193–207. [DOI] [PubMed] [Google Scholar]
- Patterson, N. , Price, A.L. and Reich, D. (2006) Population structure and eigenanalysis. PLOS Genet. 2, 2074–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, T. , Jia, M. and Liu, J. (2015) RNAi‐based functional elucidation of PtrPRP, a gene encoding a hybrid proline rich protein, in cold tolerance of Poncirus trifoliata . Front. Plant Sci. 6, 808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeil, B.E. and Crisp, M.D. (2008) The age and biogeography of Citrus and the orange subfamily (Rutaceae: Aurantioideae) in Australasia and New Caledonia. Am. J. Bot. 95, 1621–1631. [DOI] [PubMed] [Google Scholar]
- Quinlan, A.R. and Hall, I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhika, G. , Sudha, V. , Mohan Sathya, R. , Ganesan, A. and Mohan, V. (2008) Association of fruit and vegetable intake with cardiovascular risk factors in urban south Indians. Br. J. Nutr. 99, 398–405. [DOI] [PubMed] [Google Scholar]
- Rahman, A. , Siddiqui, S.A. , Jakhar, R. and Kang, S.C. (2015) Growth inhibition of various human cancer cell lines by imperatorin and limonin from Poncirus trifoliata Raf. Seeds. Anticancer Agents Med. Chem. 15, 236–241. [DOI] [PubMed] [Google Scholar]
- Sahin‐Cevik, M. and Moore, G.A. (2006) Identification and expression analysis of cold‐regulated genes from the cold‐hardy Citrus relative Poncirus trifoliata (L.) Raf. Plant Mol. Biol. 62, 83–97. [DOI] [PubMed] [Google Scholar]
- Salamov, A.A. and Solovyev, V.V. (2000) Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10, 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Y. , Ding, Y. and Yang, S. (2018) Molecular regulation of CBF signaling in cold acclimation. Trends Plant Sci. 23, 623–637. [DOI] [PubMed] [Google Scholar]
- Shi, Y. , Ding, Y. and Yang, S. (2015) Cold signal transduction and its interplay with phytohormones during cold acclimation. Plant Cell Physiol. 56, 7–15. [DOI] [PubMed] [Google Scholar]
- Shu, S. , Goodstein, D.M. and Rokhsar, D. (2013) PERTRAN: genome‐guided RNA‐seq Read Assembler. Methods, 40, 50. [Google Scholar]
- Smit, A.F. and Hubley, R. (2010) RepeatModeler Open‐1.0. Available at: www.repeatmasker.org.
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel‐Roy, P. and Goldschmidt, E.E. (1996) The Biology of Citrus. Cambridge, UK: Cambridge University Press. [Google Scholar]
- Talavera, G. and Castresana, J. (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. [DOI] [PubMed] [Google Scholar]
- Talon, M. and Gmitter, F.G. (2008) Citrus genomics. Int. J. Plant Genomics, 2008, 528361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, H. , Yamada, S. and Nakanishi, J. (1971) Approach to eliminating tristeza virus from citrus trees by using trifoliate orange seedlings. Bull. Hort. Res. Sta. Jpn. 11, 157–165. [Google Scholar]
- Tarailo‐Graovac, M. and Chen, N. (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics, 25, 1–14. [DOI] [PubMed] [Google Scholar]
- Tian, L. , Wu, Q.S. , Kuča, K. and Rahman, M.M. (2018) Responses of four citrus plants to phytophthora‐induced root rot. Sains. Malays. 47, 1693–1700. [Google Scholar]
- Trejo‐Pech, C.J.O. , Spreen, T.H. and Zansler, M.L. (2018) Is growing oranges in Florida a good investment? Am. J. Agric. Econ. 100, 625–639. [Google Scholar]
- Wang, L. , He, F. , Huang, Y. et al (2018a) Genome of wild mandarin and domestication history of mandarin. Mol. Plant, 11, 1024–1037. [DOI] [PubMed] [Google Scholar]
- Wang, M. , Dai, W. , Du, J. , Ming, R. , Dahro, B. and Liu, J. (2018b) ERF 109 of trifoliate orange (Poncirus trifoliata (L.) Raf.) contributes to cold tolerance by directly regulating expression of Prx1 involved in antioxidative process. Plant Biotechnol. J. 17, 1316–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Zhang, X. and Liu, J. (2015) Deep sequencing‐based characterization of transcriptome of trifoliate orange (Poncirus trifoliata (L.) Raf.) in response to cold stress. BMC Genom., 16, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Xu, Y. , Zhang, S. et al (2017) Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nat. Genet. 49, 765–772. [DOI] [PubMed] [Google Scholar]
- Wu, G.A. , Prochnik, S. , Jenkins, J. et al (2014) Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat. Biotechnol. 32, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G.A. , Terol, J. , Ibanez, V. et al (2018) Genomics of the origin and evolution of Citrus . Nature, 554, 311–316. [DOI] [PubMed] [Google Scholar]
- Xu, Q. , Chen, L. , Ruan, X. et al (2013) The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 45, 59–66. [DOI] [PubMed] [Google Scholar]
- Xu, Z. and Wang, H. (2007) LTR_FINDER: an efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Wu, H. , Zhao, L. and He, S. (2019) Analysis of Poncirus polyandra (Rutaceae) chloroplast genome and its phylogenetic implications. Mitochondrial DNA B, 4, 2287–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang, Z. , Ye, X. , Choi, S. , Molina, J. , Moonan, F. , Wing, R.A. , Roose, M.L. and Mirkov, T. (2001) Construction of a 1.2‐Mb contig including the citrus tristeza virus resistance gene locus using a bacterial artificial chromosome library of Poncirus trifoliata (L.) Raf. Genome, 44, 382–393. [PubMed] [Google Scholar]
- Yang, Z. , Ye, X. , Molina, J. , Roose, M.L. and Mirkov, T.E. (2003) Sequence analysis of a 282‐kilobase region surrounding the citrus tristeza virus resistance gene (Ctv) locus in Poncirus trifoliata L. Raf. Plant Physiol. 131, 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh, R.F. , Lim, L.P. and Burge, C.B. (2001) Computational inference of homologous gene structures in the human genome. Genome Res. 11, 803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, J.M. , Kim, M. , Koo, H.N. , Song, B.K. , Yoo, Y.H. and Kim, H.M. (2004) Poncirus trifoliata fruit induces apoptosis in human promyelocytic leukemia cells. Clin. Chim. Acta. 340, 179–185. [DOI] [PubMed] [Google Scholar]
- Yu, Q. , Chen, C. , Du, D. , Huang, M. , Yao, J. , Yu, F. , Brlansky, R.H. and Gmitter, F.G. (2017) Reprogramming of a defense signaling pathway in rough lemon and sweet orange is a critical element of the early response to ‘Candidatus Liberibacter asiaticus’. Hortic. Res. 4, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Rabiee, M. , Sayyari, E. and Mirarab, S. (2018) ASTRAL‐III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics, 19, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, N. , Zeng, L. , Shan, H. and Ma, H. (2012) Highly conserved low‐copy nuclear genes as effective markers for phylogenetic analyses in angiosperms. New Phytol. 195, 923–937. [DOI] [PubMed] [Google Scholar]
- Zhou, H. , Hirata, M. , Osawa, R. , Fujino, K. and Kishima, Y. (2017) Detainment of Tam3 transposase at plasma membrane by its BED‐zinc finger domain. Plant Physiol. 173, 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, C. , Zheng, X. , Huang, Y. et al (2019) Genome sequencing and CRISPR /Cas9 gene editing of an early flowering mini‐Citrus (Fortunella hindsii). Plant Biotechnol. J. 17, 2199–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, S. , Wang, F. , Shen, W. , Jiang, D. , Hong, Q. and Zhao, X. (2015) Genetic diversity of Poncirus and phylogenetic relationships with its relatives revealed by SSR and SNP/InDel markers. Acta Physiol. Plant. 37, 141. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Genome‐wide Hi‐C contact map of the Poncirus trifoliata V1.3 assembly (X and Y axes) at 250 kb matrix resolution. The nine chromosomes and 143 unplaced scaffolds are numbered and oriented according to Clementine V1.0, and are presented as blue boxes in descending order (from upper left to lower right). Contacts (red pixels) within chromosomes are denser than between chromosomes, with contacts between adjacent genomic loci (pixels nearer to the diagonal) being denser than those at greater inter‐locus distances (pixels farther off of the diagonal). This feature of Hi‐C was exploited to perform the scaffolding shown.
Figure S2. Dot‐plot showing segmental correspondence between P. trifoliata V1.3 reference sequence (y‐axis) and haploid Clementine reference sequence V1.0 (x‐axis). Aligned segments between assemblies shown with lines; purple lines indicate alignments between sequences in the same orientation, while light‐blue lines indicate alignments in the reversed orientation.
Figure S3. A phylogenetic tree of identified NBS genes in Poncirus trifoliata. Three major clusters were identified (blue, orange and purple dashed lines).
Figure S4. WGD and divergence between Poncirus trifoliata and Poncirus polyandra. (a) The Ks distribution for paralogous gene groups in P. trifoliata, C. × clementina, C. × sinensis and Arabidopsis (as a control). (b) The Ks distribution between P. trifoliata and P. polyandra obtained from the 8326 gene pairs.
Figure S5. WGD in other Citrus‐related species.
Figure S6. Genetic relatedness among four P. trifoliata accessions. Each row represents one pair of accessions with codes explained in Figure S5(a). Dark and light green colors denote sharing two and one haplotypes, respectively, whereas gray stands for the absence of haplotype sharing.
Table S1. Summary of sequenced Poncirus accessions using PacBio and Illumina Hiseq 2500 platforms
Table S2. TEs in P. trifoliata genome
Table S3. Comparison of completeness and statistics of assemblies among all published Citrus‐related genomes
Table S4. Summary of gene families containing cold signaling‐related genes
Table S5. Summary of cold signaling‐related genes recovered by re‐annotation
Table S6. Classification of identified NBS genes in P. trifoliata
Table S7. Summary of identified TF genes in P. trifoliata genome
Table S8. Gene family assignment of 10 Citrus‐related species based on existing annotations
Table S9. Genes specific to P. trifoliata
Table 10. Summary of re‐annotation of single‐copy gene families across 10 genomes
Table S11. Summary of rapid evolving gene families in P. trifoliata lineage from CAFÉ analysis
Table S12. GO enrichment analysis of P. trifoliata‐specific genes and fast evolving genes in P. trifoliata
Table S13. Summary of positively selected genes
Table S14. QTLs associated with HLB tolerance mapped to P. trifoliata genome and locations of Ctv and Tyr1 regions
Table S15. Candidate P. trifoliata genes responsive to CLas infection and located within QTLs associated with HLB tolerance
Table S16. NBS genes located within QTL associated with HLB tolerance and Ctv and Tyr1 regions
Data Availability Statement
The whole‐genome sequencing and re‐sequencing data from this study have been deposited at GenBank (BioProject: PRJNA648176, SRA accession number: SRR12323696‐SRR12323706). The Poncirus trifoliata genome v1.3 has been deposited in Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html).