

Abstract
Graphitic carbon nitride quantum dots (g‐CNQDs) are highly promising photoresponsive materials. However, synthesis of monodispersed g‐CNQDs remains challenging. Here we report the dual function of MOF [Cu3BTC2] (HKUST‐1) as a catalyst and template simultaneously to prepare g‐CNQDs under mild conditions. Cyanamide (CA), a graphitic carbon nitride precursor, catalytically dimerized inside the larger MOF cavities at 90 °C and condensed into g‐CNQDs at 120 °C in a controlled fashion. The HKUST‐1 template was stable under the reaction conditions, leading to uniform g‐CNQDs with a particle size of 2.22±0.68 nm. The as prepared g‐CNQDs showed photoluminescence emission with a quantum yield of 3.1 %. This concept (MOF dual functionality) for catalyzing CA polycondensation (open metal sites (OMSs) effect) and controlling the produced particle size (pore‐templating effect), together with the tunable MOF porosity, is expected to produce unique g‐CNQDs with controllable size, morphology, and surface functionality.
Keywords: cyanamide polycondensation, g-CNQDs, HKUST-1, MOF templating, photoluminescence
The first example of a MOF as a dual‐function material for templating and catalyzing the formation of monodispersed graphitic carbon nitride quantum dots (g‐CNQDs) under very mild conditions is reported. g‐CNQDs with tailorable sizes and properties can be prepared using this synthetic protocol, taking the advantages of the huge MOF family with tunable pore structures.
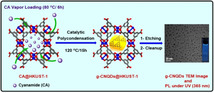
Graphite phase carbon nitride (g‐C3N4) has shown pronounced performance in numerous fields such as photocatalysis, fluorescence probes, filtration membranes, etc. [1] When the dimensions are downsized to few nanometers, namely g‐C3N4 quantum dots (g‐CNQDs), they exhibit enhanced photoabsorption and photoresponse comparing with the bulk material due to the quantum confinement effect.[ 1b , 2 ] Because of these properties, g‐CNQDs had wide range of applications that include photocatalysis, fluorescence probes, drug delivery, bioimaging, security Ink, etc.[ 3 , 4 ] They have been prepared utilizing two main strategies: top‐to‐down or bottom‐to‐up. [5] Top‐to‐down strategy involves fragmentation of the bulk g‐C3N4 either by chemical exfoliation, thermal treatment or ultrasonication into QDs.[ 3b , 3c , 6 ] The bottom‐to‐up strategy is usually based on thermal treatment of low molecular weight nitrogen‐rich organic precursors to polymerize into well dispersed g‐CNQDs.[ 3a , 7 ] Despite these efforts, it is still very challenging to produce monodispersed g‐CNQDs. Uniform size distribution ensures narrow absorption and emission optical range as the band gap between the valance and conduction bands correlates to the QDs size. [8] Synthesis of monodispersed QDs using a templating strategy has superior advantages. Following this strategy, the particle size control could be easily achieved by selecting appropriate template with well‐defined and uniform cavities. [9]
Metal‐organic frameworks (MOFs) are particular interesting due to their tunable pore size and reticular structure. [10] In the first report of MOF as a template, its structure collapsed as self‐scarified template at the carbonization temperature of carbon precursor of furfuryl alcohol. [11] After that, MOFs were used as chemically inert non‐scarified template for syntheses of nanoparticles, nanoclusters and quantum dots of noble metals/ metal oxides/ semiconductors in the pores following the same thermodynamic requirements with or without MOFs.[ 9b , 12 ] In 2017, the synthesis of carbon nanodots using MOF templating methods was reported with glucose as carbon precursor, where glucose was converted into carbon at low temperature of 200 °C and MOF template remained intact; whereas glucose can be carbonized at the same temperature without MOF. [13] Generally, the reported MOF template strategies could be classified into two categories: non‐scarified template and self‐scarified template, depending on preservation of MOF skeletons or not. Note that in both cases, MOF as template will not change the reaction thermodynamics. Transfer to g‐CNQDs templating by the MOF pores is not straight forward. Most MOFs will collapse at the temperature of g‐C3N4 production via thermal condensation of its precursors (>500 °C), and unexpected side reactions could take place at such high temperature. [14]
The applications of MOFs in heterogeneous catalysis, for example by taking advantage of Lewis acid centers pointing inside the cavities of the (activated) framework inspired us to think about a catalytic synthesis of g‐CNQDs at lower temperature in the cavities. Thus, we envisaged a combination of catalytic function and templating effect on the g‐CNQDs synthesis (MOF dual functionality). Lewis acidity originating from the open metal sites (OMSs) have been observed in specific kinds of MOFs such as [Cu3BTC2] (BTC=1,3,5‐benzenetricarboxylate, HKUST‐1). [15] Due to its accessible coordinatively unsaturated, open and hard Cu2+ Lewis acid centers, HKUST‐1 represents an outstanding catalyst for many reactions.[ 15 , 16 ] HKUST‐1 as a template contains three kinds of pores depending on size, the largest at ca. 1.35 nm. [13] Pores are interconnected by three dimensional channels and are accessible via pore windows of ca. 6 Å in diameter nicely suited to allow diffusion of small g‐C3N4 precursor molecules. [15]
In searching for appropriate precursor for g‐CNQDs synthesis, we found that cyanamide (CA) could be selected as a promising candidate. CA derivatives catalytically trimerized to yield cyclic 1,3,5‐triazene derivatives in presence of Lewis acids such as tris(dimethylamido) aluminum, triflic anhydride, etc.[ 17 , 18 , 19 ] Taking advantage of these properties, we proposed that HKUST‐1 could be used as a catalyst to efficiently activate CA under mild conditions and a non‐scarified template to control its condensation and assembly in the confined pore space simultaneously to yield uniform g‐CNQDs at low temperature (Scheme 1). The proposed strategy is rather different from the traditional MOF templating procedures, where MOF changes the morphologies of the products but does not affect thermodynamics of the reaction. Therefore, our report represents a new type of MOF templating with high novelty and significance, which introduce a new trend of MOF dual functionality. To the best of our knowledge, this is the first example to catalytically prepare g‐CNQDs under such mild conditions using MOFs.
Scheme 1.
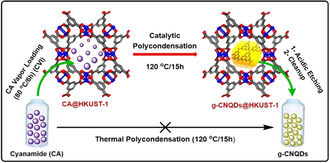
Cyanamide (NC‐NH2, CA) loading by chemical vapour infiltration (CVI) inside [Cu3BTC2] (HKUST‐1) cavities followed by Cu2+ Lewis acid sites catalysed polycondensation (liberation of ammonia) under mild conditions (90–120 °C) to produce uniform graphitic carbon nitride quantum dots (2.22±0.68 nm).
The procedure for producing uniform g‐CNQDs inside the cavities of HKUST‐1 is divided into two main steps: Firstly, loading the precursor in the pores of the activated framework; secondly, catalytic polycondensation by increasing the temperature to achieve high degree of condensation. CA with its advantageous low melting point (47 °C) and high vapour pressure facilitated the direct loading inside the cavities of the activated MOF at 80 °C/6 h via the chemical vapour infiltration technique (CVI).[ 11 , 12b ] At this temperature, a stoichiometric amount of CA will attach to the Cu2+ OMSs at the paddle wheel nodes and excess amount will occupy the cavity space. The polycondensation step had been initiated via increasing the temperature to 90, 120, and 150 °C. Because a continuous supply of the monomer during building up of the polymer (g‐CNQDs) is required, we kept the loaded HKUST‐1 in CA vapour during the condensation step (CA adsorption equilibrium). Successful loading of CA was evidenced by the Fourier transform infrared (FT‐IR) analysis and carbon/nitrogen ratio (C/N) in addition to visual observations (Figure 1 a, S1, and S2 and Table S1). FT‐IR pattern of bulk CA showed strong peak at 2260 cm−1 due to nitrile group (C≡N) stretching vibration. [20] Similar vibration was observed with weak red shift at 2245 cm−1 after loading CA inside the MOF cavities. The observed red shift of nitrile vibration may be assigned to the coordination with the Lewis acid centers of HKUST‐1.[ 18 , 21 ] Also, new peak at 2202 cm−1 had appeared which refers to dimerization of a small fraction of CA. The nitrogen content (N %) tested with elemental analyzer was found to be 3.69 % and the calculated C/N ratio was 0.50, which is similar to C/N ratio of CA. MOF color has changed from dark blue to greenish, confirming the coordination interaction of CA with the active sites. [22] Shifting from loading to polycondensation at 90 °C led to changes in the FT‐IR pattern where the peak at 2245 cm−1 strongly weakened and the vibration at 2202 cm−1 became stronger with red shift to 2198 cm−1. In addition, new vibration signal observed at 2152 cm−1 confirming the formation of dicyandiamide, accompanied by increase in the N % to 10.10 %. This increase might be attributed to loading during catalytic dimerization at this temperature.
Figure 1.

a) FT‐IR spectra of (i) pristine HKUST‐1 and composites after loading at (ii) 80 °C for 6 h, then polycondensation at (iii) 90 °C, (iv) 120 °C, and (v) 150 °C for 15 h in comparison with (vi) CA; b) PXRD pattern, c) TGA plot and d) SEM images of (1) pristine HKUST‐1 and the composites prepared at (2) 90 °C, (3) 120 °C and (4) 150 °C.
Dimerization of bulk CA could happen at 150 °C while cyclotrimerization occurs approximately at 240 °C. [23] Consequently, dimerization at such low temperature (90 °C) in HKUST‐1 cavities point to the role of the OMSs in activating CA. The coordination bonding between CA nitrile group and the active centers increases the electrophilicity of the carbon atom, hence initiating the polycondensation reaction at lower temperature. [18] When the temperature was increased to 120 °C, the relative intensity of the vibration peaks decreased dramatically, indicating conversion of the dimer to g‐CNQDs inside the cavities. As a result, the N % decreased to 6.62 % due to release of ammonia during condensation while the C/N ratio increased to 0.59. Further increase in the polycondensation temperature to 150 °C led to more decline in the relative intensity of the vibration peaks, confirming complete monomer consumption. The C/N ratio and produced g‐CNQDs size were used to find out the optimum reaction temperature. Because higher C/N ratio refers to more condensation, we selected 120 °C as the optimum polymerization temperature. The lower temperature degree (90 °C) led to incomplete condensation of the monomer (CA) with C/N ratio of 0.48, while higher degree (150 °C) resulted in larger particle size (Figure S3). Note that, the C/N ratio calculated after catalytic polycondensation at 120 and 150 °C was found to be lower than the theoretical value (0.75 for g‐C3N4) that could be ascribed to presence of less condensed fragments and dimerized CA in the framework channels in addition to well‐condensed g‐CNQDs inside the cavities. [24] the optimum reaction time was found to be 15 h depending on the FT‐IR data and N % in the composite (see Supporting Information for details). The N2 sorption isotherm analysis at 77 K was used to confirm the successful formation of g‐CNQDs inside HKUST‐1 pores (Figure S6) where the volume of adsorbed N2 and Brunauer‐Emmett‐Teller (BET) surface area had decreased from 403 cm3 g−1 and 1571 m2 g−1 in case of pristine HKUST‐1 to 231 cm3 g−1 and 762 m2 g−1 after CA condensation at 120 °C, respectively. These findings indicate that a fraction of HKUST‐1 pores had been filled with g‐CNQDs.
To further confirm the catalytic role of the copper ions (Cu2+), we tested the ability of two copper forms (acetate and nitrate) to catalytically initiate CA condensation under the established conditions (120 °C/15 h). The FT‐IR and powder X‐ray diffraction (PXRD) patterns of the bulk product confirmed the formation of g‐C3N4 under these mild conditions as shown in Figure S8 and S9.
MOF stability was tested after the catalytic condensation of CA using PXRD as depicted in Figure 1 b where freshly prepared HKUST‐1 pattern had completely matched with the simulated one. The diffraction patterns of the composites have shown some changes in the relative intensities of the low diffraction angle peaks by increasing the condensation temperature from 90 to 150 °C. The intensity of (002) diffraction peak at 6.8° had decreased relative to the (004) peak at 13.6° comparing with the guest‐free HKUST‐1. These changes confirm that, increasing the temperature led to more condensation and size growth.[ 12b , 25 ] Also, no new peaks were observed confirming the stability of the framework under the reaction conditions. Thermogravimetric analysis (TGA) (Figure 1 c) showed that HKUST‐1 framework was stable up to around 300 °C in N2 atmosphere after condensation at 150 °C indicating high thermal stability. [13] The sharp decrease in the weight loss percentage in the temperature range below 200 °C has declined with increasing the polymerization temperature referring to less moisture adsorption by the composite. This observation indicates that, the cavities occupied with g‐CNQDs and adsorb less moisture comparing with the pristine HKUST‐1 which confirm the N2 sorption results. For more focus on the framework stability, the octahedral crystals of HKUST‐1 had been tested after condensation using SEM to check the probability of surface etching or framework collapse. Figure 1 d shows the persistency of the framework shape and surface smoothing.
To further check the validity of our hypothesis to catalytically produce g‐CNQDs in HKUST‐1 cavities at low temperature, we isolated the produced material by etching the framework in acidic medium followed by cleanup using liquid‐liquid extraction and dialysis techniques. According to the PXRD analysis (Figure 2 a), the as prepared g‐CNQDs have one diffraction peak at 27.02° originates from the inter‐planer stacking of carbon nitride layers. [1a] The small size and few layers of the g‐CNQDs led to disappearance of the (100) peak that appear in case of bulk g‐C3N4 in addition to decrease in the intensity at 27.02°. [4a]
Figure 2.
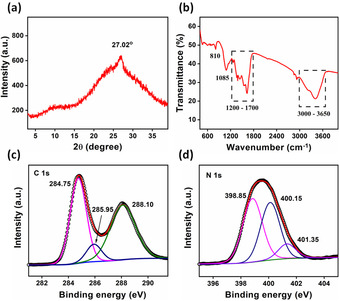
a) PXRD pattern, b) FT‐IR spectrum and c) deconvoluted C 1s and d) N 1s spectra of the produced g‐CNQDs.
FT‐IR spectrum (Figure 2 b) included sharp absorption peak at 810 cm−1 that may be originates from the tri‐s‐triazine ring‐sextant out‐of‐plane bending vibration confirming the presence of the heptazine rings in the g‐CNQDs structure. [1c] The stretching band at 1085 cm−1 is related to C‐O vibration while the range between 1200 to 1700 cm−1 correspond to the vibrations of the C‐NH‐C or C‐N‐(C)‐C in case of partial or full condensation, respectively.[ 4c , 26 ] The absorption band from 3000 to 3650 cm−1 is primarily attribute to the vibrations of NH2, NH, and OH groups. [3b]
The composition of the QDs was analyzed using X‐ray photoelectron spectroscopy (XPS) (Figure 2 c and d and Figure S11). The XPS survey included three main peaks related to carbon, nitrogen, and oxygen at binding energies of 284.80, 399.47 and 532.47 eV, respectively. The convoluted carbon 1 s spectrum was divided into three peaks at 284.75, 285.95, and 288.10 eV that could be assigned to the signals of standard reference carbon, C‐O and sp2 hybridized carbon (N−C=N) inside the aromatic structure, respectively.[ 1b , 27 ] N 1s spectrum included three peaks at: 398.85 eV related to the sp2 N atoms involved in the tri‐s‐triazine rings (C−N=C), 400.15 eV that refers to the tertiary nitrogen bonded to carbon (N−(C)3) or carbon and hydrogen (H−N−(C)2) and 401.35 eV attributed to the amino functional group (C−N−H).[ 1e , 28 ]
Particle size and dimensions of the g‐CNQDs were examined using transmission electron microscope (TEM) and atomic force microscope (AFM). TEM image (Figure 3 a) showed highly monodispersed QDs with lateral dimensions averaged around 2.22±0.68 nm as presented in the size distribution histogram (Figure 3 b) calculated depending on randomly selected 100 particles. However, HKUST‐1 contains three kinds of pores (0.5, 1.1 and 1.35) interconnect with each other, no g‐CNQDs with particle size smaller than 1 nm had been detected. [15] Absence of the smaller sized particles might be caused by the less favorable formation of g‐CNQDs inside the smallest pore. CA polycondensation under the established mild conditions (120 °C) depends mainly on the catalytic role of the OMSs which are directed toward the interior of one of the two larger pores.[ 15 , 22 ] Consequently, catalytic polycondensation inside that pore only is strongly preferred leading to templating a QDs with size larger than 1 nm. On the other hand, the obtained g‐CNQDs having typical particle sizes of 2.22±0.68 nm are somewhat larger than the HKUST‐1 cavity (1.35 nm). We assign this observation to some pore expansion (defect formation) caused by the ammonia gas evolved as a by‐product of the polycondensation reaction. [1d] Pore expansion was confirmed by the N2 sorption isotherm measurements (Figure S6), where narrow hysteresis ring was observed in the sorption pattern of the composite prepared at 120 °C/15 h. Note, in related MOF‐templated (autocatalytic) metal nanoparticle growth (e.g. Pd NPs), the obtained particle sizes are also slightly larger than the original MOF cavity. [12c]
Figure 3.
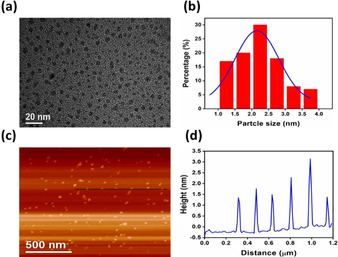
a) TEM image and b) particle size distribution histogram of g‐CNQDs; c) AFM picture and d) height profile of the produced g‐CNQDs along the black line in the AFM picture.
The high resolution TEM (HRTEM) showed highly crystalline g‐CNQDs with lattice spacing of 0.21 nm (Figure S12) referring to another advantage for the stablished catalytic condensation protocol. The topographic heights obtained from the AFM analysis (Figure 3 c and d) showed that g‐CNQDs height ranged from 1.4 to 3 nm suggesting that the QDs consists of few stacked C‐N layers, in perfect correlation with the TEM data. [1b] Combining the particle size measurements from the TEM and AFM, we could predict spherical QDs morphology completely matching with the HKUST‐1 cavities shape. As a trial for imaging the spatial distribution of g‐CNQDs inside the framework before cleanup, the scanning transmission electron microscope (STEM) could not identify clearly the QDs particulates because of its low contrast inside the framework. Also, elemental mapping depending on nitrogen element did not help due to presence of less condensed fragments in addition to dimerized CA in the MOF channels which could interfere with the signals from the g‐CNQDs inside the large cavities. This explanation was confirmed by the FT‐IR analysis done for the composite prepared at 120 °C. We still could observe the signals related to the nitrile group vibrations in the dimerized CA at 2152 and 2202 cm−1. Also, the C/N ratio calculated for the condensation product inside the MOF and before cleanup (0.59) found to be less than the theoretical value of carbon nitride (0.75) due to presence of less condensed matter in addition to g‐CNQDs. [24]
The UV/Visible absorption and photoluminescence (PL) spectra of g‐CNQDs were recorded to understand their spectroscopic properties and electronic structure. UV/Visible absorption pattern (Figure 4 a) showed obvious absorption in the range from 200 to 360 nm due to the π‐π* and n‐π* transitions in the conjugated C‐N system of the g‐CNQDs. [29] They showed symmetrical PL emission at 420 nm with excitation wavelength of 310 nm. By increasing the excitation wavelength up to 400 nm, the emission pattern red shifted from 420 to 465 nm accompanied by decrease in the PL intensity (Figures 4 b and S13) referring to the excitation wavelength dependent PL phenomenon reported earlier. [7a] It is expected that such phenomenon might happen due to the optical selection for the deferentially sized nanoparticles in addition to the distribution of the emissive sites on the surface of the QDs. [30] In addition to these characterizations, the pH effect on the PL intensity, zeta potential and quantum yield of the g‐CNQDs were investigated (see Supporting Information for more details).
Figure 4.
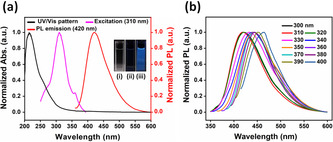
a) UV/Visible, excitation, and emission spectra of g‐CNQDs in aqueous solution (Insets i, ii, and iii are photographs for g‐CNQDs solution in ambient light, 255, and 365 nm UV light); b) g‐CNQDs PL response at different excitation wavelengths.
In summary, we demonstrated a proof‐of‐concept study using MOF as dual function material for catalytic synthesis and size control by templating to yield highly monodispersed g‐CNQDs. In comparison with the bench‐mark methods, our g‐CNQDs could be produced at temperature as low as 90 °C (optimized at 120 °C) owing to the catalytic function of the coordinatively unsaturated Cu2+ sites in HKUST‐1. Under such mild conditions, HKUST‐1 is kept intact so that g‐CNQDs are size confined by the pore structure and by the matrix properties of HKUST‐1 which yields an unusual narrow size range of 2.22±0.68 nm. On the other hand, the low temperature led to the production of less defective and crystalline g‐CNQDs. As properties of g‐CNQDs are closely related with their size and surface functionality, we are currently exploiting even more tailored g‐CNQDs synthesis in terms of size, shape, and chemical functionality. By our concept, MOFs are introduced as a platform for g‐CNQDs production, pointing out the huge parameter space of MOF design in terms of catalytic function and pore structure for size confined templating purposes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge support from Hefei National Laboratory for Physical Sciences at the Microscale, Hefei Science Center of Chinese Academy of Sciences, Fujian Institute of Innovation of Chinese Academy of Sciences, the National Natural Science Foundation of China (NSFC, 21571167, 51502282), the Fundamental Research Funds for the Central Universities (WK2060190053) and Anhui Province Natural Science Foundation (1608085MB28). Also, M.K.A. acknowledges the CAS‐TWAS President's PhD fellowship for the financial support and W.L. is grateful for the Alexander von Humboldt post‐doctoral research fellowship. This work was additionally supported by the DFG Priority Program 1928 COORNETs (http://www.coornets.tum.de). Open access funding enabled and organized by Projekt DEAL.
M. K. Albolkany, Y. Wang, W. Li, S. Arooj, C.-H. Chen, N. Wu, Y. Wang, R. Zbořil, R. A. Fischer, B. Liu, Angew. Chem. Int. Ed. 2020, 59, 21499.
Contributor Information
Prof. Dr. Roland A. Fischer, Email: roland.fischer@tum.de.
Prof. Dr. Bo Liu, Email: liuchem@ustc.edu.cn.
References
- 1.
- 1a. Niu P., Zhang L., Liu G., Cheng H.-M., Adv. Funct. Mater. 2012, 22, 4763–4770; [Google Scholar]
- 1b. Zhang X., Xie X., Wang H., Zhang J., Pan B., Xie Y., J. Am. Chem. Soc. 2013, 135, 18–21; [DOI] [PubMed] [Google Scholar]
- 1c. Wang Y., Li Y., Ju W., Wang J., Yao H., Zhang L., Wang J., Li Z., Carbon 2016, 102, 477–486; [Google Scholar]
- 1d. Wang Y., Hou X., Zhang J., Xu T., Liu S., Liu B., ChemPhotoChem 2018, 2, 490–497; [Google Scholar]
- 1e. Wang Y., Wu N., Wang Y., Ma H., Zhang J., Xu L., Albolkany M. K., Liu B., Nat. Commun. 2019, 10, 2500–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kagan C. R., Lifshitz E., Sargent E. H., Talapin D. V., Science 2016, 353, aac5523. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Li H., Shao F.-Q., Huang H., Feng J.-J., Wang A.-J., Sens. Actuators B 2016, 226, 506–511; [Google Scholar]
- 3b. Yin Y., Zhang Y., Gao T., Yao T., Han J., Han Z., Zhang Z., Wu Q., Song B., Mater. Chem. Phys. 2017, 194, 293–301; [Google Scholar]
- 3c. Zhan Y., Liu Z., Liu Q., Huang D., Wei Y., Hu Y., Liana X., Hu C., New J. Chem. 2017, 41, 3930–3938; [Google Scholar]
- 3d. Dong J., Zhao Y., Wang K., Chen H., Liu L., Sun B., Yang M., Sun L., Wang Y., Yu X., Dong L., ChemistrySelect 2018, 3, 12696–12703. [Google Scholar]
- 4.
- 4a. Zhang S., Li J., Zeng M., Xu J., Wang X., Hu W., Nanoscale 2014, 6, 4157–4162; [DOI] [PubMed] [Google Scholar]
- 4b. Zhang X., Wang H., Wang H., Zhang Q., Xie J., Tian Y., Wang J., Xie Y., Adv. Mater. 2014, 26, 4438–4443; [DOI] [PubMed] [Google Scholar]
- 4c. Wang Y., Wang J., Ma P., Yao H., Zhang L., Li Z., New J. Chem. 2017, 41, 14918–14923; [Google Scholar]
- 4d. Patir K., Gogoi S. K., ACS Sustainable Chem. Eng. 2018, 6, 1732–1743. [Google Scholar]
- 5. Wang A.-J., Li H., Huang H., Qian Z.-S., Feng J.-J., J. Mater. Chem. C 2016, 4, 8146–8160. [Google Scholar]
- 6.
- 6a. Li Y., Cai J., Liu F., Yu H., Lin F., Yang H., Lin Y., Li S., Microchim. Acta 2018, 185, 134–140; [DOI] [PubMed] [Google Scholar]
- 6b. Song Z., Lin T., Lin L., Lin S., Fu F., Wang X., Guo L., Angew. Chem. Int. Ed. 2016, 55, 2773–2777; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2823–2827. [Google Scholar]
- 7.
- 7a. Barman S., Sadhukhan M., J. Mater. Chem. 2012, 22, 21832–21837; [Google Scholar]
- 7b. Tang Y., Su Y., Yang N., Zhang L., Lv Y., Anal. Chem. 2014, 86, 4528–4535. [DOI] [PubMed] [Google Scholar]
- 8. Brus L. E., J. Chem. Phys. 1984, 80, 4403. [Google Scholar]
- 9.
- 9a. Kruk M., Jaroniec M., Ryoo R., Joo S. H., J. Phys. Chem. B 2000, 104, 7960–7968; [Google Scholar]
- 9b. Li X., Goh T. W., Xiao C., Stanton A. L. D., Pei Y., Jain P. K., Huang W., ChemNanoMat 2016, 2, 810–815. [Google Scholar]
- 10. Jacobs B. W., Houk R. J. T., Anstey M. R., House S. D., Robertson I. M., Talinc A. A., Allendorf M. D., Chem. Sci. 2011, 2, 411–416. [Google Scholar]
- 11. Liu B., Shioyama H., Akita T., Xu Q., J. Am. Chem. Soc. 2008, 130, 5390–5391. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Esken D., Turner S., Wiktor C., Kalidindi S. B., Tendeloo G. V., Fischer R. A., J. Am. Chem. Soc. 2011, 133, 16370–16373; [DOI] [PubMed] [Google Scholar]
- 12b. Esken D., Noei H., Wang Y., Wiktor C., Turner S., Tendeloo G. V., Fischer R. A., J. Mater. Chem. 2011, 21, 5907–5915; [Google Scholar]
- 12c. Kalidindi S. B., Oh H., Hirscher M., Esken D., Wiktor C., Turner S., Van Tendeloo G., Fischer R. A., Chem. Eur. J. 2012, 18, 10848–10856; [DOI] [PubMed] [Google Scholar]
- 12d. Esken D., Turner S., Lebedev O. I., Van Tendeloo G., Fischer R. A., Chem. Mater. 2010, 22, 6393–6401; [Google Scholar]
- 12e. Kratzl K., Kratky T., Günther S., Tomanec O., Zbořil R., Michalička J., Macak J. M., Cokoja M., Fischer R. A., J. Am. Chem. Soc. 2019, 141, 13962–13969. [DOI] [PubMed] [Google Scholar]
- 13. Gu Z.-G., Li D.-J., Zheng C., Kang Y., Wöll C., Zhang J., Angew. Chem. Int. Ed. 2017, 56, 6853–6858; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 6957–6962. [Google Scholar]
- 14. Wang X., Maeda K., Thomas A., Takanabe K., Xin G., Carlsson J. M., Domen K., Antonietti M., Nat. Mater. 2009, 8, 76. [DOI] [PubMed] [Google Scholar]
- 15. Alaerts L., Séguin E., Poelman H., Thibault-Starzyk F., Jacobs P. A., Vos D. E. D., Chem. Eur. J. 2006, 12, 7353–7363. [DOI] [PubMed] [Google Scholar]
- 16. Schlichte K., Kratzke T., Kaskel S., Microporous Mesoporous Mater. 2004, 73, 81–88. [Google Scholar]
- 17. Larraufie M.-H., Maestri G., Malacria M., Ollivier C., Fensterbank L., Lacôte E., Synthesis 2012, 1279–1292. [DOI] [PubMed] [Google Scholar]
- 18. Dornan P., Rowley C. N., Priem J., Barry S. T., Burchell T. J., Woo T. K., Richeson D. S., Chem. Commun. 2008, 3645–3647. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Chen X., Bai S.-D., Wang L., Liu D.-S., Heterocycles 2005, 65, 1425–1430; [Google Scholar]
- 19b. Herrera A., Martínez-Alvarez R., Ramiro P., Chioua M., Chioua R., Synthesis 2004, 503–505. [Google Scholar]
- 20. Calandra P., Longo A., Ruggirello A., Liveri V. T., J. Phys. Chem. B 2004, 108, 8260–8268. [Google Scholar]
- 21.
- 21a. Lee G., Kossowska D., Lim J., Kim S., Han H., Kwak K., Cho M., J. Phys. Chem. B 2018, 122, 4035–4044; [DOI] [PubMed] [Google Scholar]
- 21b. Koroleva E. V., Ignatovich Z. V., Ignatovich S. V., Gusak K. N., Russ. J. Org. Chem. 2011, 47, 1222–1226. [Google Scholar]
- 22.
- 22a. Chui S. S.-Y., Lo S. M.-F., Charmant J. P. H., Orpen A. G., Williams I. D., Science 1999, 283, 1148–1150; [DOI] [PubMed] [Google Scholar]
- 22b. Vishnyakov A., Ravikovitch P. I., Neimark A. V., Bülow M., Wang Q. M., Nano Lett. 2003, 3, 713–718. [Google Scholar]
- 23. Groenewolt M., Antonietti M., Adv. Mater. 2005, 17, 1789–1792. [Google Scholar]
- 24. Lau V. W.-h., Mesch M. B., Duppel V., Blum V., Senker J., Lotsch B. V., J. Am. Chem. Soc. 2015, 137, 1064–1072. [DOI] [PubMed] [Google Scholar]
- 25. Guo W., Liu J., Weidler P. G., Li J., Neumann T., Danilov D., Wenzel W., Feldmann C., Wöll C., Phys. Chem. Chem. Phys. 2014, 16, 17918–17923. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Wang H., Jiang S., Chen S., Li D., Zhang X., Shao W., Sun X., Xie J., Zhao Z., Zhang Q., Tian Y., Xie Y., Adv. Mater. 2016, 28, 6940–6945; [DOI] [PubMed] [Google Scholar]
- 26b. Malina T., Poláková K., Skopalík J., Milotová V., Holá K., Havrdová M., Tománková K. B., Čmiel V., Šefc L., Zbořil R., Carbon 2019, 152, 434–443. [Google Scholar]
- 27. Rong M., Lin L., Song X., Zhao T., Zhong Y., Yan J., Wang Y., Chen X., Anal. Chem. 2015, 87, 1288–1296. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Zhou Z., Wang J., Yu J., Shen Y., Li Y., Liu A., Liu S., Zhang Y., J. Am. Chem. Soc. 2015, 137, 2179–2182; [DOI] [PubMed] [Google Scholar]
- 28b. Li D., Jing P., Sun L., An Y., Shan X., Lu X., Zhou D., Han D., Shen D., Zhai Y., Qu S., Zbořil R., Rogach A. L., Adv. Mater. 2018, 30, 1705913. [DOI] [PubMed] [Google Scholar]
- 29. Pan D., Zhang J., Li Z., Wu C., Yan X., Wu M., Chem. Commun. 2010, 46, 3681–3683. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Zhou J., Yang Y., Zhang C.-Y., Chem. Commun. 2013, 49, 8605–8607; [DOI] [PubMed] [Google Scholar]
- 30b. Long Y.-M., Zhou C.-H., Zhang Z.-L., Tian Z.-Q., Bao L., Lin Y., Pang D.-W., J. Mater. Chem. 2012, 22, 5917–5920. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary